

Abstract
In a forward genetic screen for interaction with mitochondrial iron carrier proteins in Saccharomyces cerevisiae, a hypomorphic mutation of the essential DRE2 gene was found to confer lethality when combined with Δmrs3 and Δmrs4. The dre2 mutant or Dre2-depleted cells were deficient in cytosolic Fe/S cluster protein activities while maintaining mitochondrial Fe/S clusters. The Dre2 amino acid sequence was evolutionarily conserved, and cysteine motifs (CX2CXC and twin CX2C) in human and yeast proteins were perfectly aligned. The human Dre2 homolog (implicated in blocking apoptosis and called CIAPIN1 or anamorsin) was able to complement the nonviability of a Δdre2 deletion strain. The Dre2 protein with triple hemagglutinin tag was located in the cytoplasm and in the mitochondrial intermembrane space. Yeast Dre2 overexpressed and purified from bacteria was brown and exhibited signature absorption and electron paramagnetic resonance spectra, indicating the presence of both [2Fe-2S] and [4Fe-4S] clusters. Thus, Dre2 is an essential conserved Fe/S cluster protein implicated in extramitochondrial Fe/S cluster assembly, similar to other components of the so-called CIA (cytoplasmic Fe/S cluster assembly) pathway although partially localized to the mitochondrial intermembrane space.
Fe/S clusters in eukaryotic cells are modular cofactors (4) that function in essential processes and reside in various cellular compartments (22). Mitochondrial Fe/S cluster proteins mediate electron transfer in the mitochondrial electron transport chain (e.g., complexes I, II, and III) and catalysis in the citric acid cycle (e.g., aconitase). In some settings, Fe/S clusters are involved in sensing of environmental signals (e.g., IRP1 or cytoplasmic aconitase, which acts as an iron sensor) (34). Recently discovered Fe/S cluster proteins have been implicated in DNA repair (FancJ and XPD) (36), DNA replication (primase priL) (17), and signal transduction for promoting neuronal growth in mammals (sprouty) (44). An essential role for the conserved [4Fe-4S] cluster protein Rli1 was discovered in ribosome maturation in the nucleus and translation initiation in the cytoplasm. Thus, mutations that interfere with Fe/S cluster synthesis have been associated with defects in ribosome processing and protein translation (16, 45).
In cells, biogenesis of Fe/S clusters requires a complex machinery including cysteine desulfurase, scaffold proteins for assembling intermediates, chaperones, and reductases (7, 13, 22). Mitochondria have all of these essential components and can synthesize clusters on their own when supplied with key ingredients (cysteine sulfur and iron) (2). Some of these biosynthetic proteins (e.g., Nfs1 and Isu) also reside outside mitochondria in some settings (8, 43). Nfs1, the cysteine desulfurase, has been shown to have essential functions both inside and outside mitochondria (29). In addition, a class of mutants has been discovered with deficiency of extramitochondrial Fe/S clusters and preservation of mitochondrial Fe/S clusters (15, 35). This class includes mutants of Atm1 and Erv1 in the mitochondria and Cfd1, Nbp35, Nar1, and Cia1 (the so-called CIA components) in the cytoplasm and nucleus (22). Atm1 is a mitochondrial ABC transporter oriented with its ATP binding site and putative substrate binding site in the mitochondrial matrix (19). Thus, the phenotype of Atm1 mutants, characterized by defective cytoplasmic and preserved mitochondrial Fe/S clusters, suggests that its unknown substrate, perhaps a key ingredient or a signaling molecule, might be required for cluster formation outside (22).
The mitochondrial carrier proteins, Mrs3 and Mrs4, have been implicated in iron delivery across the mitochondrial inner membrane for heme and Fe/S cluster synthesis occurring inside (26, 46, 47). Yet many aspects of these transporters remain mysterious, such as the identity of transport substrates going into mitochondria (or coming out). Some level of redundancy between the two transporters is suggested by the marked enhancement of the mutant phenotypes in the double mutant versus single mutant strains (Δmrs3 Δmrs4 versus Δmrs3 or Δmrs4). However, even the double mutant is viable and grows robustly unless subjected to severe iron deprivation. The Δmrs3 Δmrs4 mutant has other complex regulatory phenotypes, including lowered cytoplasmic iron and induced high-affinity plasma membrane iron uptake activities (21). With this as background, we initiated a synthetic lethal genetic screen, seeking mutations which result in lethality when combined with Δmrs3 Δmrs4. A mutant, J137, was obtained with deficient extramitochondrial Fe/S clusters. The corresponding gene was identified as DRE2, an essential gene encoding a conserved Fe/S cluster protein.
MATERIALS AND METHODS
Strains and plasmids.
Strains and plasmids are described in Tables 1 and 2, respectively.
TABLE 1.
Yeast strains used in this study
| Strain no. | Strain name | Relevant genotype |
|---|---|---|
| 84-1a | CDV38a cyh2 | MATaura3 his3 leu2 trp1 ade2 ade3 cyh2 |
| 84-9a | CDV39alpha cyh2 | MATα ura3 leu2 trp1 ade2 ade3 lys2 cyh2 |
| 90-7a | CDV38a Δmrs3 | MATaura3 his3 leu2 trp1 ade2 ade3 cyh2 Δmrs3::TRP1 |
| 90-8a | CDV39alpha Δmrs3 | MATα ura3 leu2 ade2 ade3 lys2 cyh2 Δmrs3::TRP1 |
| 91-71a | CDV38a Δmrs3/4 [pTSV31A] | MATaura3 leu2 ade2 ade3 cyh2 Δmrs3::TRP1 Δmrs4::HIS3[pTSV31A-URA3-ADE3] |
| 91-79a | CDV38a Δmrs3/4 | MATaura3 leu2 ade2 ade3 cyh2 Δmrs3::TRP1 Δmrs4::HIS3 |
| 96-47a | CDV38a Δmrs3/4 [pTSV31A-MRS4] | MATaura3 leu2 ade2 ade3 cyh2 Δmrs3::TRP1 Δmrs4::HIS3 [pTSV31A-URA3-ADE3-MRS4] |
| 93-71a | CDValpha Δmrs3/4 | MATα ura3 leu2 ade2 ade3 cyh2 Δmrs3::TRP1 Δmrs4::HIS3 |
| 96-56a | CDValpha Δmrs3/4 [pTSV31A-MRS4] | MATα leu2 ade2 ade3 cyh2 Δmrs3::TRP1 Δmrs4::HIS3 [pTSV31A-URA3-ADE3-MRS4] |
| 97-19a | J137 | MATaleu2 ade2 ade3 cyh2 Δmrs3::TRP1 Δmrs4::HIS3 [pTSV31A-URA3-ADE3-MRS4] dre2-137 |
| 97-21a | J137 [pRS318-MRS4] | MATaura3 ade2 ade3 cyh2 Δmrs3::TRP1 Δmrs4::HIS3 [pRS318-CYH2-LEU-MRS4] dre2-137 |
| 53-75b | YPH499cyh2 | MATaura3-52 lys2-801(amber) ade2-101 (ochre) trp1-Δ63 his3-Δ200 leu2-Δ1 cyh2 |
| 98-67b | Gal-Dre2 | MATa HisMX6-PGAL1-Dre2::DRE2 ura3-52 lys2-801(amber) ade2-101(ochre) trp1-Δ63 leu2-Δ1 |
| 83-25b | Gal-Atm1 | MATa HisMX6-PGAL1-Atm1::ATM1 ura3-52 lys2-801(amber) ade2-101(ochre) trp1-Δ63 leu2-Δ1 cyh2 |
| 98-39b | Gal-Cfd1 | MATa HisMX6-PGAL1-Cfd1::CFD1 ura3-52 lys2-801(amber) ade2-101(ochre) trp1-Δ63 leu2-Δ1 |
| 57-65b | Δisu1 | MATalys2-801(amber) ade2-101(ochre) trp1-Δ63 his3-Δ200 leu2-Δ1 Δisu1::URA3 |
| 87-13b | Δmrs3/4-25D | MATα trp1-Δ1 ade2-101(ochre) his3-Δ200 cyh2 Δmrs4::KAN Δmrs3::URA3 |
| 100-75b | DRE2-HA3 | MATaura3-52 lys2-801(amber) ade2-101(ochre) trp1-Δ63 leu2-Δ1 cyh2 Dre2-3HA-TADH1-His3MX6::DRE2 |
| 97-52a | 1A Δmrs3/4 | MATα ura3 leu2 ade2 ade3 cyh2 Δmrs3::TRP1 Δmrs4:HIS3 |
| 97-53a | 1B WT | MATα ura3 leu2 trp1 ade2 ade2 cyh2 |
| 97-54a | 1C dre2-137 Δmrs3/4 | MATaura3 leu2 ade2 ade3 cyh2 dre2-137 Δmrs3::TRP1 Δmrs4:HIS3 |
| 97-55a | 1D dre2-137 | MATaura3 leu2 trp1 ade2 ade3 cyh2 dre2-137 |
| 84-81 | BY4741Δleu1 | MATahis3D1 leu2D0 met15D0 ura3D0 leu1::KAN |
| 100-62b | YPH501cyh2/cyh2 DRE2/Δdre2::KAN | MATa/α ura3-52/ura3-52 lys2-801/lys2-801 ade2-101/ade2-101 trp1-Δ63/trp1-Δ63 his3-Δ200/his3-Δ200 leu2-Δ1 leu2-Δ1 cyh2/cyh2 dre2/dre2::kanMX |
| 100-69b | YPH501cyh2/cyh2 DRE2/Δdre2::KAN [pRS318-DRE2] | MATa/α ura3-52/ura3-52 lys2-801/lys2-801 ade2-101/ade2-101 trp1-Δ63/trp1-Δ63 his3-Δ200/his3-Δ200 leu2-Δ1 leu2-Δ1 cyh2/cyh2 dre2/dre2::kanMX [pRS318-DRE2prom-DRE2] |
| 101-19b | DRE2 shuffle (1A) | MATaura3-52 lys2-801(amber) ade2-101(ochre) trp1-Δ63 his3-Δ200 leu2-Δ1 cyh2 dre2::kanMX [pRS318-DRE2] |
CDV genetic background.
YPH499/500/501 genetic background.
TABLE 2.
Plasmids used in this studya
| Plasmid no. | Plasmid name | Features |
|---|---|---|
| 13-29 | pTSV31A | 2μm URA3 ADE3 |
| 17-1 | pTSV31A-MRS4 | 2μm URA3 ADE3 MRS4 genomic fragment (700 bp 5′ UTR-MRS4 ORF-200 bp 3′ UTR) |
| 17-37 | pRS318-MRS4 | CEN/ARS LEU2 CYH2 MRS4 genomic fragment (700 bp 5′ UTR-MRS4 ORF-200 bp 3′ UTR) |
| 17-2 | pRS415-MRS4 | CEN/ARS LEU2 MRS4 genomic fragment (700 bp 5′ UTR-MRS4 ORF-200 bp 3′ UTR) |
| 17-5 | pRS415-MRS3 | CEN/ARS LEU2 MRS3 genomic fragment (700 bp 5′ UTR-MRS3 ORF-200 bp 3′ UTR) |
| YCplac111 yeast genomic library | CEN/ARS LEU2, yeast genomic fragments | |
| B28-32 | YCplac111-DRE2 A | CEN/ARS LEU2, yeast genomic fragments chromosome XI 572486-575667 |
| B28-33 | YCplac111-DRE2 B | CEN/ARS LEU2, yeast genomic fragments chromosome XI 572940-577784 |
| B28-34 | YCplac111-DRE2(Acc1) | CEN/ARS LEU2, B 28-32 digested with AccI followed by religation |
| 17-59 | YCplac111-DRE2(minimal) | CEN/ARS LEU2, B 28-32 digested with XhoI followed by religation |
| 17-64 | pET21b-DRE2-His6 | T7 promoter-driven yeast DRE2 ORF, PCR amplified from plasmid B 28-32 |
| 17-65 | pET21b-DRE2-His6 | T7 promoter-driven yeast DRE2 ORF, yeast colony PCR amplified from CDV38a (84-1) |
| 17-66 | pET21b-DRE2Mut-His6 | T7 promoter-driven yeast DRE2 ORF, yeast colony PCR amplified from J137 (97-19) |
| 18-30 | pRS415-DRE2 | CEN/ARS LEU2 DRE2 genomic fragment (700 bp 5′ UTR-Dre2 ORF-200 bp 3′ UTR) |
| 18-39 | pRS318-DRE2 | CEN/ARS LEU2 DRE2 genomic fragment (700 bp 5′ UTR-Dre2 ORF-200 bp 3′ UTR) |
| 18-3 | YCplac22-GPDprom-IRP1 | CEN/ARS TRP1 GPD promoter-driven human IRP1 ORF |
| 15-78 | pRS426-GPDprom-LEU1-His6 | 2μm URA3 GPD promoter-driven Leu1 |
| B29-64 | pRS316-RPS2eGFP | CEN/ARS URA3, gift from Eduard Hurt |
| B29-63 | pRS316-RPL25eGFP | CEN/ARS URA3, gift from Eduard Hurt |
| 13-64 | YCplac22-GPDprom | CEN/ARS TRP1, GPD promoter |
| 18-35 | YCplac22-GPDprom-ScDre2 | CEN/ARS TRP1, GPD promoter-driven yeast Dre2 |
| B29-70 | YCplac22-GPDprom-huDre2 | CEN/ARS TRP1, GPD-promoter driven human Dre2 |
| 18-67 | YCplac22-GPDprom-dre2-137 | CEN/ARS TRP1, GPD promoter-driven yeast Dre2 with dre2-137 (lys [codon 26] mutated to stop) |
| 19-13 | YCplac22-GPDprom-Dre2 (1-25aa) | CEN/ARS TRP1, GPD promoter-driven yeast Dre2 N-terminal fragment (residues 1-25) |
| 19-15 | YCplac22-GPDprom-Dre2(Δ26) | CEN/ARS TRP1, GPD promoter-driven yeast -Δ26 Dre2 (missing first 26 residues on N terminus) |
| 19-17 | YCplac22-GPDprom-Dre2(Δ119) | CEN/ARS TRP1, GPD promoter-driven yeast -Δ119 Dre2 (missing first 119 residues on N terminus) |
Abbreviations: UTR, untranslated region; ORF, open reading frame.
Sectoring screen and genetic methods.
The sectoring strain 96-47 was grown in defined medium minus uracil to maintain selection for the plasmid and then subjected to UV mutagenesis (2 × 108 cells in a watch glass in 2 ml of water) to 50% lethality. The cells were plated on yeast extract-peptone-dextrose (YPD) agar with low adenine (5 mg/liter). After 1 week, a mutant was identified by intense red color and lack of white sectors. Treatment with fluoroorotic acid (counterselection against URA3) or 10 μg/ml cycloheximide (counterselection against CYH2) was performed according to standard methods (9). Crosses were performed by zygote manipulation, and tetrad dissection was performed as described previously (9). The GAL1 promoter swaps used a PCR-generated cassette with a HIS3 marker (23).
Medium composition.
YPAD and complete defined medium were made as described previously (37). Iron starvation ferrozine plates have been described previously (41). Inducing conditions for the GAL1 promoter utilized 2% raffinose plus 0.5% galactose. Repressing conditions utilized 2% raffinose or 2% glucose.
Enzyme assays.
Spectrophotometric assays for aconitase (14), succinate dehydrogenase (27), malate dehydrogenase (39), and isopropylmalate isomerase (Leu1) (18) have been described previously. An in-gel activity assay for aconitase was performed as described previously (2, 43).
Microscopy for ribosomal protein localization.
Gal-Dre2 (98-67) cells transformed with reporter plasmid pRS316-Rps2-eGFP or pRS316-Rpl25-eGFP (45), gifts from Eduard Hurt, were propagated in defined medium minus uracil with raffinose-galactose as carbon source. Cells were shifted to noninducing conditions for 16 h, harvested, fixed in ice-cold 70% ethanol for 10 min, briefly stained with DAPI (4′,6′-diamidino-2-phenylindole), and examined using a fluorescence microscope (E800; Nikon) with a 60× Plan Apo objective. The images were acquired using Image-Pro Plus software (Media Cybernetics, Inc.).
Cellular and mitochondrial fractionation.
A triple hemagglutinin (HA3) tag was placed in the genome in frame with the carboxyl terminus of the Dre2 open reading frame, generating strain 100-75. The cells with the epitope tag were grown in rich raffinose-based medium, Dre2-HA3 cells were treated with zymolyase to remove the cell wall, and a fraction was loaded on the gel. Following homogenization with a Dounce homogenizer in 0.6 M sorbitol, 20 mM HEPES-KOH, pH 7.5, and protease inhibitors, postmitochondrial supernatant (PMS) and crude mitochondria (CM) were separated by differential centrifugation. CM were then subjected to Nycodenz density gradient centrifugation to isolate gradient-purified mitochondria (GM) (33). The cell fractions (total [T], PMS, CM, and GM) were treated with proteinase K (25 μg/ml) for 30 min on ice, neutralized, and subjected to trichloroacetic acid precipitation prior to gel separation and blotting for HA. In a separate experiment, CM were treated with trypsin (0.2 mg/ml) or proteinase K (25 μg/ml) prior to neutralization and separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). CM with Dre2-HA3 were suspended in hypotonic buffer (0.12 M sorbitol) and incubated on ice for 10 min followed by pipetting up and down to rupture the outer membrane. After centrifugation at 9,000 × g for 10 min, the supernatant (intermembrane space [IMS]) and the mitoplast pellet were recovered. Purification of CM (28), protease protection (3), and subfractionation of mitochondrial compartments (24) have been described elsewhere. Rabbit polyclonal antibodies to Yfh1, Ccp1, and Tom70 were prepared against the corresponding open reading frames expressed in bacteria. Monoclonal antibodies to Pgk1, penta-His, and HA epitope were purchased from Molecular Probes, Qiagen, and Covance, respectively. The antibody directed against IRP1 was a polyclonal rabbit antipeptide antibody, a gift from Tracey Rouault.
Protein expression and purification.
Escherichia coli BL21(DE3) (codon plus) with pET21b-Dre2-His6 (strain 17-65) was grown in 6 liters LB with ampicillin (100 μg/ml), chloramphenicol (34 μg/ml), and supplementary iron (125 μM ferric-nitrilotriacetate) at 25°C until the optical density at 600 nm (OD600) reached 0.8 to 1.0. Protein expression was induced by addition of 0.2 mM isopropyl-β-d-thiogalactoside for 3 h. Cells were recovered, washed with water, and resuspended in 180 ml degassed lysis buffer (50 mM Tris-HCl, pH 8, and 10% glycerol). Lysozyme (final concentration, 133 μg/ml) was added and incubated on ice for 35 min. Cells were disrupted by sonication in a glass beaker on ice (30 s, six times). Sonicated samples were centrifuged at 9,000 × g for 30 min at 4°C, and the supernatant was subjected to nickel-nitrilotriacetic acid (Ni-NTA) affinity purification. The supernatant was tumbled end over end with 2 ml of Ni-NTA agarose beads in lysis buffer in air at 4°C. The protein was eluted with 400 mM imidazole, which was subsequently removed, and the eluted protein was concentrated using Amicon filters.
Iron and sulfide quantitation.
The iron chelator bathophenanthrolinedisulfonic acid disodium salt (BPS) was used to determine the iron content of purified Dre2 protein, mitochondria, and cell lysates. Samples were treated with 0.5% SDS, 1 mM sodium dithionite, and 1 mM BPS. Absorbance was measured at 515 nm and compared with a standard curve generated from NH4Fe(SO4)2 ferric ammonium sulfate at 0, 5, 10, 20, 40, 60, 80, and 100 μM. Sulfide content of purified Dre2 protein was determined by measuring the characteristic absorbance of a blue complex after reaction with N,N-dimethyl-p-phenylenediamine dihydrochloride (DPD). Samples were diluted in 600 μl water and 50 μl 6% NaOH. One hundred twenty-five microliters 0.1% DPD reagent (1 mg DPD dissolved in 1 ml 5 N HCl and 50 μl 11.5 mM FeCl3) was added to the reaction mixture, which was then incubated for 20 min at 30°C. After vortexing and centrifugation for 5 min at maximum speed, the OD670 of the supernatant was measured (40).
UV-visible spectra.
Spectra (wavelength from 260 to 700 nm) were collected using an Olis Cary-14 spectrophotometer for a concentrated sample of bacterially expressed and recently purified Dre2. After the initial recording, the sample was reduced with freshly prepared sodium dithionite (10 mM dissolved in 50 mM Tris-HCl, pH 8, bubbled with argon gas).
Electron paramagnetic spectroscopy.
Electron paramagnetic resonance (EPR) samples were immediately prepared after Dre2 purification. Anaerobically prepared neutralized sodium dithionite solution (0.5 M) was used at a final concentration of 10 mM. The protein solutions were transferred to EPR tubes, and after 5 to 10 min of incubation at room temperature the samples were quickly frozen in a dry ice-ethanol mixture and then stored in liquid nitrogen until EPR analyses. EPR spectra were recorded by a Bruker ESP 300E spectrometer at X-band (9.4 GHz) using an Oxford Instrument ESR900 helium flow cryostat, a Hewlett-Packard 5350B microwave frequency counter, and an ITC4 temperature controller to control sample temperature. EPR spectra of the Fe/S clusters were simulated by SimFonia (Bruker, Germany), and spin quantitations were carried out under nonsaturating conditions using 0.5 mM Cu(II) EDTA or 0.5 mM Cu(II) perchlorate as standard (1).
RESULTS
A synthetic lethal screen was undertaken with the Δmrs3 Δmrs4 mutant as a starting point (Fig. 1). Briefly, Δmrs3 Δmrs4 mutations were introduced into a strain with ade2 ade3 mutations and MRS4 was put back on a 2μm plasmid also carrying ADE3 and URA3 (Fig. 1A). The parent strain 96-47 exhibited red to white sectoring on rich YPD medium with low adenine. Following UV mutagenesis, a red colony, J137 (97-19), was identified that could not lose the covering plasmid due to the presence of a synthetic lethal mutation (Fig. 1B). The synthetic lethality was confirmed by nonviability following fluoroorotic acid treatment (Fig. 1C) and rescued following plasmid swap with completely different LEU2-based plasmids carrying MRS4 or MRS3 (Fig. 1D). The synthetic lethal mutation was recessive (Fig. 1E) and could be recovered as a single locus in a backcross with the wild-type parental strain. A genomic library was used to complement the nonsectoring phenotype of J137, and the complementing activity was located on a fragment carrying MET1, YKR070W, and DRE2 open reading frames (Fig. 2, row 1). The AccI digestion and religation of the complementing plasmid abrogated complementation, indicating that DRE2 was responsible (Fig. 2, row 4). The genomic copy of DRE2 in J137 (dre1-137) was rescued and found to contain a point mutation changing codon 26 to a stop.
FIG. 1.

Sectoring screen for synthetic lethals with Δmrs3 Δmrs4. (A) Strain. The parent strain 96-47 contains Δmrs3 Δmrs4 Δade2 Δade3 mutations and 2μm plasmid 17-1 with MRS4, URA3, and ADE3. This strain produces red and white sectors on YPD with low adenine. (B) Mutant selection. Following UV mutagenesis, a colony/clone called J137 that failed to sector and remained red was identified. (C) Test for plasmid dependence. The parent 96-47 and mutant J137 97-19 were exposed to fluoroorotic acid (FOA) to counterselect against the plasmid, resulting in viability of the former and nonviability of the latter. (D) Plasmid swap. Parent and mutant were transformed with plasmid 17-37 pRS318-MRS4-LEU2-CHY2, and both were rendered FOA resistant (not shown). The treatment with cycloheximide (CHX) to remove the newly introduced plasmid led to viability of the parent and nonviability of the mutant. (E) Genetics. The J137 (97-19) mutant was analyzed by crossing with a Δmrs3 Δmrs4 Δade2 Δade3 strain of the opposite mating type, 93-71. The diploid homozygous for Δmrs3 Δmrs4 but heterozygous for the dre2-137 mutation was streaked on FOA plates. The diploid was able to grow, indicating that the mutation in J137 was recessive.
FIG. 2.

Further genetic analysis of synthetic lethal mutant J137. The wild-type copy of the mutated gene in J137 was cloned by complementation with a genomic library in plasmid YCplac111. The transformants formed small nonsectoring red colonies for the most part, with the exception of two single large white colonies. (Rows 1 to 4) The complementing plasmids (rows 1 and 2) were rescued, and complementing activity was further localized by deletions inactivating YKR070W (row 3) or DRE2 (row 4). Only 17-59 retained complementing activity, showing that DRE2 was responsible. (Row 5) The mutation in the genome of J137, called dre2-137, was identified following amplification of the DRE2 open reading frame and flanking regions. A single A-to-T change was present, altering codon 26 from a lysine to a stop codon. The dre2-137 strain, called 97-55, was recovered following backcross of J137 with parental strain 84-9, sporulation, and tetrad dissection. This strain was slow growing but viable and exhibited sensitivity to iron deprivation. (Row 6) A shuffle strain was constructed by heterozygous gene deletion in the diploid, transformation with a Dre2-expressing plasmid with CYH2 marker (18-39), and sporulation. The shuffle strain (101-19) with a Δdre2::kanMX deletion covered by the Dre2 plasmid was completely nonviable following counterselection against the plasmid with cycloheximide. (Row 7) By contrast, the shuffle strain with Dre2 under the control of a strong GPD promoter reinserted on YCplac22 (plasmid 18-65) was viable on cycloheximide. (Row 8) The identical plasmid with the human Dre2 homolog expressed from the GPD promoter (plasmid B29-70) conferred robust growth on the shuffle strain on cycloheximide. (Row 9) The YCplac22-GPD-Dre2 plasmid was mutated, introducing the dre2-137 point mutation (codon 26 changed from lysine to stop), and this plasmid (18-67) was introduced into the shuffle strain, conferring viable growth on cycloheximide. (Row 10) The YCplac22-GPD-Dre2(1-25aa) plasmid (19-13) was transformed into the shuffle strain, resulting in transformants that were not able to grow on a cycloheximide plate. (Rows 11 and 12) However, the plasmids containing N-terminal-truncated Dre2 forms, YCplac22-GPD-Dre2(Δ26) (row 11) and YCplac22-GPD-Dre2(Δ119) (row 12), were able to permit growth on cycloheximide. FOA, fluoroorotic acid.
DRE2 was annotated in the Saccharomyces Genome Database as an essential gene of unknown function. Synthetic lethal interaction with pol3-13, a mutated allele of DNA polymerase delta, was described previously (6), and a recent publication demonstrated an effect of a temperature-sensitive allele on chromosome transmission fidelity, although a mechanism of this effect has not been described (5). In order to confirm the essentiality of DRE2, a shuffle strain was constructed, and counterselection against the covering plasmid was shown to produce nonviability (Fig. 3B). The nonviability could not be rescued by anaerobic growth or iron addition to the medium (not shown). The mutant allele in J137 was reconstructed by site-directed mutagenesis of a plasmid-borne copy of DRE2, and this was able to rescue the Δdre2 lethality. Interestingly, the amino-terminal 26 amino acids possessed no complementing activity whereas the carboxy-terminal portions from amino acid 27 to the end or from an internal methionine 120 to the end were able to complement lethality in a shuffle strain (Fig. 3B). The data suggest that viability of the J137 strain containing the dre2-137 allele with a stop codon at position 27 was sustained by a low level of downstream initiation from an internal methionine at position 120 in the open reading frame.
FIG. 3.

Sequence alignment and shuffle strain complementation. (A) Alignment of human Dre2 (Q6F181, also called CIAPIN1 or anamorsin) and yeast Dre2 amino acid sequences using Clustal W 2.0. Underneath the alignment, asterisks represent identical residues, and dots or colons represent lesser degrees of conservative changes. Cysteines are colored red, with eight of nine cysteines perfectly aligned. Above the alignment a black circle over residue 26 indicates the location of the mutation in dre2-137 changing K to stop. Arrows indicate the translational starts of truncated constructs (arrow 1 including positions 27 to 348 labeled Δ26; arrow 2 including positions 120 to 348 labeled Δ119) used in panel B. Overlines indicate possible functional domains: amino-terminal domain (a), acidic and serine-rich patch (b), basic domain (c), CX2CXC domain (d), twin CX2C motifs (e), and region of amino acid identity of unknown significance (f). (B) Complementation of shuffle strain with various Dre2 constructs and human homolog. The shuffle (strain 101-19) with the Δdre::kanMX deletion covered by DRE2 (plasmid 18-39) carrying wild-type DRE2 and CYH2 counterselectable marker was transformed with various Dre2 constructs carried on plasmid YCplac22 and driven by the GPD promoter. The YCplac22 constructs shown in the top panel are as follows: Dre2(Δ26) (1 and 2), Dre2(Δ119) (3 and 4), Dre2(1-25aa) (5 and 6), YCplac22 alone (7), Dre2 open reading frame (8), dre2-137 (9), and human Dre2 open reading frame (10). The transformants with both the counterselectable Dre2-CYH2 and the test Dre2-TRP1 were grown and photographed (middle panel). Then the covering plasmid was removed by plating on cycloheximide (CHX), leaving only the test Dre2-TRP1 plasmid (bottom panel). The results show that DRE2 is required for viability, and the deletion can be rescued by expression of the yeast wild-type protein, the human homolog, the point mutant dre2-137, or the Δ26 and Δ119 truncations but not the fragment with N-terminal amino acids 1 to 25.
Homologs of Saccharomyces cerevisiae Dre2 are found in fungi, plants, and animals (called CIAPIN1 or anamorsin) but apparently not in bacteria. The similarity between yeast and human proteins is shown in the Clustal alignment (Fig. 3A) and is characterized by alignment of eight of nine cysteines and conserved elements distributed throughout the proteins. Therefore, an effort was made to test the complementing activity of the human protein expressed in yeast. The Q6F181 open reading frame encoding the human Dre2 homolog (huDre2) was cloned behind a strong yeast promoter on a TRP1 marked plasmid. The human protein-expressing, yeast protein-expressing, or empty plasmids were transformed into the Dre2 shuffle strain, and the transformants were treated with cycloheximide to remove the shuffle plasmid. The huDre2 and yeast Dre2 complemented the lethality under these conditions, whereas the strain with empty plasmid showed no growth. The huDre2 protein was highly expressed by immunoblotting, although biochemical complementation was incomplete as indicated by uncorrected Leu1 activity in these cells (see below).
A promoter swap of the chromosomal DRE2 promoter with the GAL1 promoter was performed in a different yeast genetic background (23), and after backcross, recombinants with Δmrs3 Δmrs4 were isolated (Fig. 4). The induced genomic copy of DRE2 combined with Δmrs3 Δmrs4 grew slightly worse than the individual mutants did (Fig. 4B), consistent with synthetic dosage lethality, but the repressed DRE2 combined with Δmrs3 Δmrs4 was much worse, thus recapitulating the synthetic lethal interaction phenotype of the dre2 mutant allele from the genetic screen (Fig. 4B and C). The Δmrs3 Δmrs4 strain was crossed with other mutants or GAL1-regulated strains, and recombinants were isolated (Fig. 5). Recombinants with Δmrs3 Δmrs4 and repressed Gal-Atm1 or Gal-Cfd1 (indicated by an asterisk in Fig. 5) formed smaller colonies than did the mutants separately, showing synthetic lethal or synthetic sick interactions. No such interactions were observed when Δmrs3 Δmrs4 was combined with mitochondrial matrix components involved in Fe/S cluster assembly (Gal-Ssq1 or Δisu1) (indicated by a number sign in Fig. 5). A cross of Δmrs3 Δmrs4 with nfs1-14, a hypomorphic mutant of the essential cysteine desulfurase (20), shown to function both inside and outside of mitochondria (29), showed no synthetic interaction. A cross of Δmrs3 Δmrs4 with Δyfh1, the frataxin homolog presumed to be involved in iron delivery for Fe/S cluster synthesis, showed marked synthetic interaction (47). In summary, these genetic results were complex but provided some suggestion that Dre2 could be grouped with components of the extramitochondrial Fe/S cluster assembly pathway.
FIG. 4.
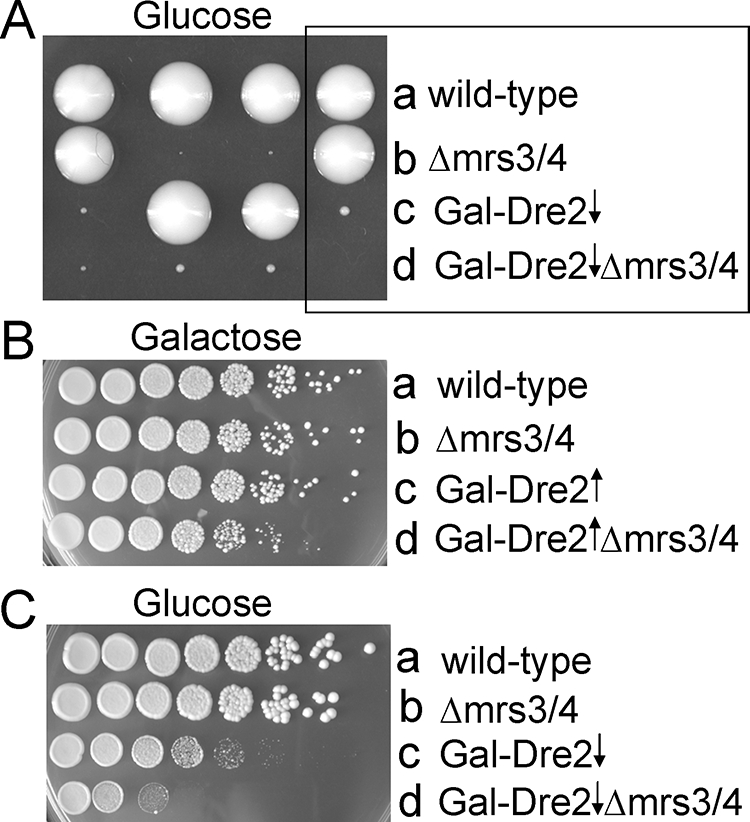
Recapitulation of synthetic interaction using Gal-Dre2. Gal-Dre2 (98-67) was crossed with the Δmrs3 Δmrs4 strain (87-13), sporulated, and dissected on glucose or galactose plates. (A) Shown are tetrad spore clones from dissection on a glucose plate. The boxed tetrad is a tetratype with indicated genotypes. The spore clone in row d presumed to represent Gal-Dre2 Δmrs3 Δmrs4 was microscopically visible and was arrested at fewer than 20 cells. (B and C) Tetrad clones of various genotypes were rescued from a galactose plate and diluted in water, and serial dilutions were spotted on a galactose plate (B) or glucose plate (C). Note the slight growth defect of induced Gal-Dre2 Δmrs3 Δmrs4 on the galactose plate, indicating a synthetic dosage effect. On the glucose plate, the triple mutant (repressed Gal-Dre2 Δmrs3 Δmrs4) was more severely compromised than the individual mutants, the Gal-Dre2 or the Δmrs3 Δmrs4 strain.
FIG. 5.

Genetic interaction phenotypes. Strains with gene deletions or GAL1 promoter swaps were crossed with a congenic strain with Δmrs3 Δmrs4 deletions. Following sporulation and dissection, tetrad clones were evaluated for colony size and genotype. Triple mutant colonies were smaller than single or double mutants for Gal-Cfd1 or Gal-Atm1 (indicated by asterisks), but not for Gal-Ssq1 or Δisu1 (indicated by number signs). WT, wild type.
Dre2 was isolated in a screen with mutants of Mrs3 and Mrs4, presumed to be mitochondrial iron transporters, and therefore, a link to iron metabolism was sought. A backcross of J137 allowed isolation of recombinants of various genotypes, surprisingly including the triple mutant (Δmrs3 Δmrs4 dre2-137) although the latter was extremely slow growing. The strains of different genotypes were evaluated for iron-dependent growth on plates with a range of available iron concentrations (Fig. 6). As noted previously (21), the Δmrs3 Δmrs4 strain showed compromised growth at the lowest iron concentration (Fig. 6A, Δmrs3/4) and restoration of normal growth at higher iron concentrations (Fig. 6B to D, Δmrs3/4). The dre2-137 single mutant also showed iron-dependent growth, although this was only partially corrected by iron addition. The Δmrs3 Δmrs4 dre2-137 triple mutant was more severely impaired than either the Δmrs3 Δmrs4 or the dre2-137 mutant individually on low-iron plates (Fig. 6 A to C) but not on high-iron plates (Fig. 6D). These data demonstrate a synthetic lethal or synthetic slow growth interaction between Δmrs3 Δmrs4 and dre2-137 and also show that the effect is mitigated by addition of iron to the growth medium.
FIG. 6.

Iron-dependent growth of mutants. The backcross of J137 (97-19) with the MRS3 MRS4 parental strain (84-9) was sporulated, and tetrads were dissected and analyzed, generating viable clones of various genotypes: wild type (97-53), Δmrs3/4 (97-52), dre2-137 (97-55), and dre2-137 Δmrs3/4 (97-54). Cells were washed with water, and 1:5 serial dilutions of 5 × 105 cells were plated on ferrozine-buffered plates. The chelator plates contained 50 mM morpholineethanesulfonic acid buffer, pH 6.1, and 1 mM iron chelator ferrozine (A) or the same with 20 μM ferrous ammonium sulfate (B), 100 μM ferrous ammonium sulfate (C), or 350 μM ferrous ammonium sulfate (D). Compared with wild type, Δmrs3/4 and dre2-137 strains showed iron-dependent growth, and the triple mutant was the most severely compromised on low-iron plates.
Dre2 function in cytosolic Fe/S cluster assembly was examined using the human cytosolic aconitase (IRP1) as a reporter. Previously, IRP1 was expressed in the cytoplasm of yeast and found to be active, indicating that yeast is capable of expressing the human protein and inserting its [4Fe-4S] cluster (35). Here a plasmid containing IRP1 under the strong GPD (glyceraldehyde-3-phosphate dehydrogenase) promoter was transformed into the dre2-137 mutant and corresponding wild type. The transformants were grown in defined glucose medium with different iron concentrations, and lysates were evaluated for aconitase activity by using an in-gel aconitase assay (Fig. 7) (2, 43). Two bands were visualized corresponding to the endogenous mitochondrial aconitase (Aco1) and the cytoplasmic heterologous aconitase expressed from the plasmid (IRP1). The mitochondrial aconitase was uniformly expressed in all samples, but IRP1 showed dependence on iron addition to the medium in both wild-type and mutant strains. However, in the dre2-137 mutant, the levels were markedly decreased and even at the highest iron level did not achieve the activity of the wild type. A similar experiment was performed with the Gal-Dre2 strain following shift from inducing conditions for Dre2 expression (defined medium with raffinose and galactose) to noninducing conditions (raffinose with no galactose). In this experiment (Fig. 8), Aco1 activity was present in excess over IRP1 activity, perhaps due to carbon source effect. The cells were grown in raffinose-galactose or raffinose and thus were not subjected to catabolite repression, allowing for higher mitochondrial aconitase activity. IRP1 expressed from the GPD promoter was less highly expressed also due to the carbon source effect. However, IRP1 activity, present during the time course of Dre2 depletion at the zero time point and 2 or 4 h following shift to noninducing conditions, was decreased at 6 h and virtually absent thereafter (Fig. 8A). By contrast, mitochondrial Aco1 activity increased at 6 h and was markedly increased at the 8- and 10-h time points before declining slightly (Fig. 8A). IRP1 protein level in Gal-Dre2 repressed lysates was minimally decreased, and Pgk1 was not altered (Fig. 8B), indicating that Dre2-depleted cells retained expression of IRP1 apoprotein. In sum, the results show that IRP1 cytoplasmic aconitase activity was dependent on Dre2 in various carbon sources in both the hypomorphic mutant and wild-type depletion experiments.
FIG. 7.

Heterologous IRP1 activities under various iron growth conditions in wild type and dre2-137 mutant strains. The CDV38a wild type (strain 84-1) and dre2-137 mutant (strain 97-55) were transformed with a plasmid (YCplac22-GPDprom-IRP1, plasmid 18-3) for expression of human cytosolic aconitase IRP1. Cells were grown to stationary phase in defined selective glucose medium to maintain the IRP1 plasmid and diluted to an OD600 of 0.1 in growth medium of the same composition supplemented with various amounts of iron added as ferrous ammonium sulfate. After four doublings, cells were harvested and digested with zymolyase, and 100 μg of cell lysate was separated by native gel electrophoresis. Aconitase activities on lysates separated by native gel electrophoresis were revealed by an in-gel assay as described previously (2, 43). Positions of the endogenous mitochondrial aconitase activity (Aco1) and plasmid-expressed cytoplasmic aconitase activity (IRP1) are indicated. Lanes 1, 2, 3, 4, and 5 are wild-type lysates grown in 0, 100 nM, 1 μM, 10 μM, and 20 μM added iron, respectively. Lanes 6, 7, 8, 9, and 10 are dre2-137 strain lysates grown in 0, 100 nM, 1 μM, 10 μM, and 20 μM added iron, respectively. Lane 11 contains 100 μg of purified mitochondria.
FIG. 8.
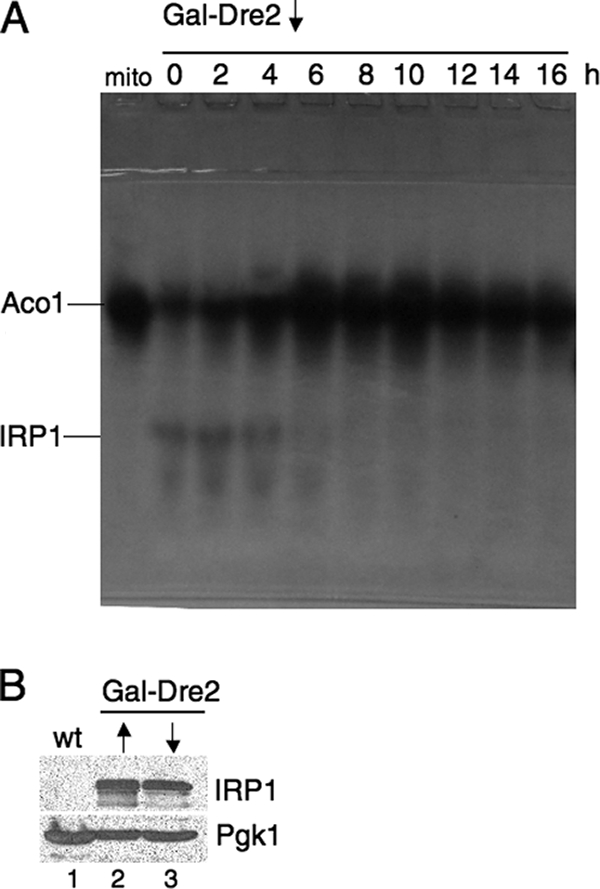
Dre2-depleted cells lack IRP1 activity. The Gal-Dre2 strain (98-67) was transformed with YCplac22-GPDprom-IRP1 (plasmid 18-3). Cells were grown in the presence of galactose (defined medium with 2% raffinose and 0.5% galactose), centrifuged, and resuspended in the absence of galactose for the indicated times, thereby depleting Dre2. (A) Depletion time course: cells were collected immediately following shift (zero time) and 2, 4, 6, 8, 10, 12, 14, and 16 h later. Lysates generated by zymolyase digestion were resolved by native gel electrophoresis, and an in-gel assay for aconitase was performed. Mitochondrial aconitase activity (Aco1) and plasmid-expressed cytoplasmic aconitase activity (IRP1) are indicated. The leftmost lane (“mito”) contains 100 μg of purified mitochondria from untransformed parental strain YPH499. (B) Immunoblots for IRP1 protein. Whole-cell lysates from untransformed parental strain YPH499 (lane 1), induced Gal-Dre2 expressing IRP1 (lane 2), and noninduced Gal-Dre2 expressing IRP1 following 16 h of growth (lane 3) were analyzed by SDS-PAGE and immunoblotted with anti-IRP1 or cytoplasmic marker Pgk1 as a control (100 μg of protein for each lane). wt, wild type.
Isopropylmalate isomerase (Leu1) activity has also been used as an indicator of cytoplasmic Fe/S cluster status (15). This cytoplasmic enzyme contains a [4Fe-4S] cluster and catalyzes the second step in leucine biosynthesis in the yeast cytoplasm (18). A plasmid for overexpression of Leu1 with a C-terminal His6 tag was introduced into the Gal-Dre2 strain, which permitted tracking of Leu1 protein levels. The transformed Gal-Dre2 strain was grown in inducing or noninducing defined medium for 16 h. Cytoplasmic lysates were prepared and fractionated by sequential ammonium sulfate precipitations prior to measurement of Leu1 activity. For the induced cells, the Leu1-His6 protein and isomerase activity were found in the 50 to 65% fraction. Activity was recorded as the time-dependent decrease in OD235, resulting from consumption of citraconate (18). Dre2-induced lysate showed a continuous decrease in absorbance with similar rates for three sequential tracings (Fig. 9A, left panel), whereas Dre2-repressed lysate showed no change in absorbance, indicating undetectable Leu1 activity (Fig. 9A, right panel). The activity was also measured over 30 min for different amounts of lysate. Dre2-induced samples showed dependence on the amount of input lysate, whereas Dre2-repressed samples showed no activity in any case (Fig. 9B). A Western blot assay with anti-His6 antibody showed equivalent amounts of Leu1-His6-tagged protein in the repressing (R) or inducing (I) conditions (Fig. 9B, inset). Thus, Dre2-depleted cell lysates were devoid of Leu1 activity, and yet Leu1 protein, probably apoprotein, was still present.
FIG. 9.

Dre2-depleted cells lack isopropylmalate isomerase (Leu1) activity. The Gal-Dre2 strain with plasmid pRS426-Leu1-His6 was grown in selective defined medium with either galactose-raffinose or raffinose to deplete Dre2. (A) Isopropylmalate isomerase (Leu1) activity was recorded as the decrease in OD235 resulting from isomerization of the artificial substrate citraconate in Dre2-induced (left panel) or -depleted (right panel) partially purified lysates. Tracings were recorded at room temperature for 3 min and twice repeated for each 10-μl lysate, equivalent to 100 μg of protein. (B) Leu1 activities (decrease in OD235 per 30 min) for Dre2-repressed (R) or -induced (I) lysates were measured for 50, 100, or 200 μg of cell lysate. The assays were performed three times with similar results and negligible activity for the repressed samples in each case. Inset: immunoblotting was performed with anti-His antibody. Activity and protein were measured in the 50 to 65% ammonium sulfate fraction with 100 μg of protein from repressed and induced samples.
Mutants with defective Fe/S cluster formation, such as CIA mutants (e.g., Cfd1, Nbp35, Nar1, and Cia1 mutants), were recently found to exhibit retention of ribosomal protein green fluorescent protein (GFP) reporters in the nucleus, perhaps as a consequence of Rli1 dysfunction (16, 45). Rli1 is an Fe/S cluster protein, which shuttles between nucleus and cytoplasm and mediates processing of the ribosomes in the nucleus (16, 45). The plasmids Rpl25-eGFP and Rps2-eGFP, expressing, respectively, ribosomal large and small subunit proteins fused to a GFP reporter, were kindly given to us by Eduard Hurt for evaluating the Dre2 phenotype, as it resembles other CIA components in many respects (45). The Gal-Dre2 cells, transformed with the reporter plasmids, were grown in defined medium with or without galactose for 16 h and then examined microscopically for DAPI and GFP signals. The Dre2-expressing cells (Fig. 10A, upper panels) were stained with DAPI but exhibited negligible diffuse GFP signals. By contrast, in Dre2-depleted cells (Fig. 10A, lower panels), a bright Rpl25-eGFP signal was noted in the nucleus of some of the cells. Other cells showed more of a nucleolar signal, forming a bright dot or crescent within the larger DAPI-stained nucleus. Many but not all cells (24 of 105 cells from multiple microscopic fields) showed the mutant phenotype; no cells showed this mutant phenotype when Dre2 was expressed (0 of 155 cells) (Fig. 10A and B). In parallel experiments with a fusion of the small ribosomal protein subunit Rps2-eGFP, again only the Dre2-depleted cells showed intense nuclear staining (data not shown). Thus, ribosomal large and small subunit proteins were retained in the nucleus of Dre2-depleted cells. In addition, similarly to CIA mutants, Dre2-depleted cells accumulated somewhat more cellular iron than did expressing cells (2.45 ± 0.73 versus 1.23 ± 0.24 pmol/μg protein) but failed to accumulate iron in mitochondria (2.65 ± 0.1 versus 3.13 ± 0.1 pmol/μg protein).
FIG. 10.

Nuclear retention of ribosomal proteins in Dre2-depleted cells. The plasmid pRS316-Rpl25-eGFP was transformed into the Gal-Dre2 strain. Dre2 protein was expressed or depleted by growth in defined medium with raffinose-galactose or raffinose alone, respectively, for 16 h. (A) DAPI and Rpl25-eGFP fluorescent images are shown for the Gal-Dre2-induced (top) and Gal-Dre2-depleted (bottom) cells. (B) Individual cell from Gal-Dre2 culture following Dre2 depletion, showing differential interference contrast (DIC) (a), DAPI (b), and Rpl25-eGFP (c) images. Nuclear accumulation of GFP fluorescent signal appeared only under conditions of Dre2 depletion.
In order to localize Dre2 protein in cells, an HA3 epitope tag was inserted at the C terminus of Dre2 in the genome (23). This modified strain was indistinguishable from the parental control in terms of growth and Fe/S cluster assembly activities. Subcellular fractionation was performed, and the tagged Dre2 was found in cytoplasm and mitochondria (Fig. 11). Cells carrying the Dre2-HA3 were treated with zymolyase to remove the cell wall, followed by Dounce homogenization to rupture the cells. The total lysate (T) showed a strong HA signal migrating at 47 kDa, and this was undiminished following low-speed centrifugation to pellet unbroken cells and nuclei (data not shown). The lysate was then fractionated into PMS and CM. The CM underwent an additional purification step on a Nycodenz gradient (GM). The Dre2-HA3 protein was found in the PMS and also in mitochondrial fractions, both crude and gradient purified (Fig. 11A). The pattern of HA3 in the cellular fractions was equivocal in terms of demonstrating mitochondrial localization. Although a signal for HA3 was found in purified mitochondria, so was a signal for phosphoglycerate kinase (PGK), a cytoplasmic marker. However, the presence of Dre2 in mitochondria was demonstrated by relative resistance of HA3 to protease treatments. Cellular fractions were treated with proteinase K, the protease was neutralized, and remaining proteins were visualized by immunoblotting. Mitochondrial Dre2-HA3 in the CM and GM was protected, whereas the extramitochondrial protein was not seen and was presumably digested (Fig. 11B, left panel). Protection from protease digestion is consistent with localization of a portion of Dre2 within a membrane-bound compartment within mitochondria. The experiment was repeated with CM treated with trypsin or proteinase K. Similar results were obtained, showing protease protection of a portion of Dre2-HA3. By contrast, Tom70, an exposed mitochondrial outer membrane protein, was digested by either protease whereas the matrix protein Yfh1 was not digested (Fig. 11B, right panel). The mitochondrial Dre2-HA3 was further localized within mitochondria. CM were subjected to hypotonic shock, and following centrifugation, the IMS contents were collected and separated from the mitoplast pellet. The mitochondrial Dre2-HA3 signal was found almost entirely in the supernatant fraction, indicating that the protein resides in the IMS. The protein behaved in a manner similar to Ccp1, a known IMS protein. Loading of more IMS protein was associated with increased Ccp1 signal and increased Dre2-HA3 signal. By contrast, the IMS reactivity for the matrix protein, Yfh1, was not increased, consistent with the presence of a small amount of matrix contamination in this fraction (Fig. 11C). Thus, Dre2-HA3 is found in the cytoplasm and in the mitochondrial IMS.
FIG. 11.

Cellular localization of Dre2-HA3. (A) Equivalent protein amounts (100 μg) from total cell lysate (T), PMS, CM, and GM were separated by SDS-PAGE and blotted, and the blots were developed with antibodies to HA tag, Yfh1 (mitochondria), or Pgk1 (cytoplasm). (B) The cell fractions were treated with proteinase K for 30 min on ice and analyzed by HA blotting (left panel). CM were treated with proteinase K or trypsin and analyzed by HA blotting or with control antibodies against Tom70 or Yfh1 (right panel). (C) CM were treated with hypotonic shock, and IMS (100, 200, or 500 μg, designated 1X, 2X, or 5X, respectively) or the mitoplasts (100 μg, designated 1X) were recovered and analyzed by blotting with HA, Ccp1, or Yfh1 antibodies. The amido black-stained membrane shows the protein pattern. MP, mitoplast pellet.
Following initial studies suggesting that Dre2 protein expressed in bacteria was brown, the possibility that a cofactor might be involved was considered, and conditions were optimized to favor iron availability and protein solubility. Dre2 with a His6 tag was expressed in E. coli at low temperature (25°C) for 3 h, and iron was added to the growth medium before and during induction. The Dre2 protein was purified on Ni-NTA agarose under conditions designed to minimize oxidant damage, using degassed buffers and supplementation of all solutions with 10% glycerol. The brown color of the purified protein (Fig. 12, inset) suggested the presence of a cofactor(s), and chemical analysis of the protein following acid treatment indicated the presence of labile iron and sulfide. Purified protein at a concentration of 13 mg/ml or 300 μM released 265 μM iron and 396 μM sulfide, and thus, the iron-to-sulfur stoichiometry was roughly 1:1 with likely incomplete Fe/S cluster loading under these bacterial expression conditions.
FIG. 12.
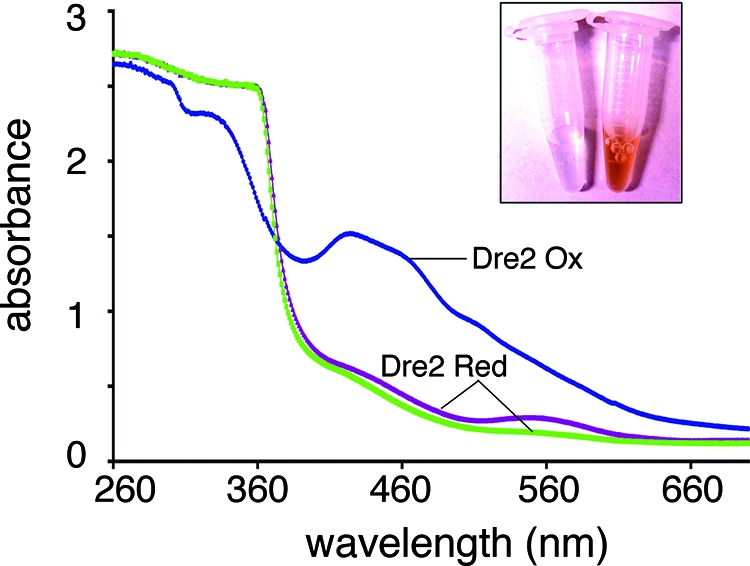
UV-visible spectra of purified Dre2. The Dre2-His6 protein was rapidly purified under mild conditions, minimizing oxidant damage and air exposure, with degassed buffers containing 10% glycerol. Spectra were recorded immediately after purification (blue tracing) and following addition of freshly made 10 mM dithionite (brown tracing) or 10 min later (green tracing). The peaks at 314, 420, 458, and 500 nm seen with the air-oxidized specimen disappeared immediately with dithionite reduction, whereas a 560-nm peak disappeared more slowly. Inset: purified Dre2-His6 protein (13-mg/ml concentration) has a dark brown color (right side) compared with buffer (left side).
UV-visible absorbance spectra were recorded immediately after purification, showing broad absorption peaks/shoulders around 314, 420, 458, and 500 nm (Fig. 12, Dre2 Ox). The spectra of Dre2 as purified did not change for several hours at room temperature, suggesting that the Dre2 protein is relatively stable under aerobic conditions. Following dithionite reduction, the absorbance in the entire visible region was quenched (Fig. 12, Dre2 Red). The data suggest that Dre2 contains Fe/S clusters that are redox active. EPR studies confirmed the presence of Fe/S clusters in the Dre2 protein isolated from bacteria (Fig. 13). At higher temperatures (45 K) with microwave power at 5 mW, the dithionite-reduced protein exhibited the EPR spectrum of a [2Fe-2S] cluster with g values (gz,y,x = 2.00, 1.96, and 1.92, respectively) (Fig. 13A). At low temperature (6 K) and 5 mW, the reduced Dre2 protein showed signals characteristic of a [4Fe-4S] cluster. However, there were still small signals from the [2Fe-2S] cluster in the spectra (Fig. 13B). Therefore, the g values (gz,y,x = 2.00, 1.95, and 1.90, respectively) for the [4Fe-4S] cluster signals were defined from the simulation data, which were obtained from Dre2 mutants in conserved cysteine residues (unpublished results). When microwave power was increased to 20 mW, the EPR signals from the [4Fe-4S] cluster were greatly enhanced. In addition, newly appearing signals around g = 2.10 and 1.80 became noticeable, indicating some interactions between the [4Fe-4S] cluster and probably the [2Fe-2S] cluster or other unidentified species. Further detailed investigation is now under way. Very small EPR signals arising from a [3Fe-4S]+ cluster were detected in the oxidized sample.
FIG. 13.

EPR spectra. EPR spectra for purified Dre2 protein at 45 K and 5 mW (A) and at 6 K and 5 mW (B). The sample (13 mg/ml) in 50 mM Tris-HCl at pH 7.5 containing 50 mM NaCl and 10% glycerol was reduced with 10 mM dithionite. EPR conditions: microwave frequency, 9.45 GHz; modulation amplitude, 8.034 G; modulation frequency, 100 kHz; time constant, 82 ms. Principal g values are indicated.
DISCUSSION
Dre2 was identified in a genetic screen for synthetic lethality with mitochondrial carriers, Mrs3 and Mrs4, implicated in iron delivery into mitochondria. The dre2-137 mutant carried a hypofunctioning allele of an essential gene. The phenotypes of the mutant and Dre2-depleted cells were similar, characterized by deficiency in cytoplasmic Fe/S cluster protein activities, with preservation of mitochondrial Fe/S clusters. The iron dependence of the synthetic lethality (worse in low iron) may be explained by partial iron-dependent restoration of deficient cytoplasmic Fe/S cluster assembly in the Dre2 mutant. Cytoplasmic iron deficiency, associated with the Δmrs3 Δmrs4 mutant as a consequence of activated vacuolar iron uptake (21), may have combined with crippled cytoplasmic Fe/S cluster assembly in the dre2 mutant assembly to produce lethality.
The phenotypes of the dre2 mutant or Dre2-depleted cells resemble those of so-called CIA components (22, 35). Fe/S cluster-containing proteins outside mitochondria were deficient, whereas Fe/S clusters inside mitochondria (aconitase and succinate dehydrogenase) were maintained or in some cases increased. Isopropylmalate isomerase (Leu1) and IRP1 (the human cytosolic aconitase expressed in yeast cytoplasm) were markedly compromised in both dre2 mutant and Dre2-depleted cells. Also, GFP-tagged proteins of the large and small ribosomal subunits, Rpl25 and Rps2, were retained in the nucleus and nucleolus in the Dre2-depleted cells, indicating defective ribosome assembly functions, perhaps due to Rli1 deficiency. Rli1 is an Fe/S cluster protein of cytoplasm and nucleus, which is required for ribosomal maturation and translation initiation (16, 45). Interestingly a publication describing a comprehensive yeast temperature-sensitive mutant resource implicated a temperature-sensitive Dre2 allele in chromosome transmission fidelity (5). Recently identified Fe/S cluster proteins involved in DNA repair (Rad3, Pri2, and Ntg2) might be mediators of the effect, but this has not been shown experimentally.
Synthesis of cytoplasmic Fe/S clusters must involve delivery of iron and sulfur, principal constituents of the clusters themselves. Additional roles for scaffolds, chaperones, and reductants are likely as for synthesis of mitochondrial Fe/S clusters (22). Iron and sulfur for cytoplasmic Fe/S clusters might originate inside mitochondria and be transported out, or these constituents might originate outside mitochondria, or both. The sole cysteine desulfurase that supplies sulfide for Fe/S cluster synthesis has dual localization, both inside and outside mitochondria. Other components, even those usually or predominantly found in the mitochondrial matrix, have been shown to have alternative extramitochondrial localizations in some cells (8, 29, 43). The CIA proteins (Cfd1, Nbp35, Nar1, and Cia1) are required for Fe/S clusters made outside mitochondria. The proteins themselves are primarily found in the cytoplasm and nucleus, and mitochondrial localizations have not been reported for these proteins. They are Fe/S cluster proteins themselves (except Cia1), coordinating [4Fe-4S] clusters (22). Two of the proteins, Cfd1 and Nbp35, are homologous and strongly interact with each other, forming a scaffold for transiently associated clusters that can be transferred to substrate proteins such as Leu1 (30). The roles of the other assembly proteins have not been as well characterized, but they probably act after Cfd1 and Nbp35 in the assembly process (30).
The role of Dre2 in this process is not at all defined, although the localization of a portion of the protein in the mitochondrial IMS is intriguing. If a key component for Fe/S cluster assembly is trafficked from mitochondria to cytoplasm, as suggested elsewhere (15), this might indicate that Dre2 in the mitochondrial IMS functions prior to the other CIA components in the cytoplasm. Dre2 in mitochondria might generate a component or signal that is permissive for the subsequent assembly steps in the cytoplasm. Another unresolved question relates to the function of the clusters in the Fe/S cluster assembly components. Different Fe/S clusters apparently have different functions. For example, the Cfd1-Nbp35 clusters were shown to be labile and easily transferred to recipient apoprotein, suggesting that they are scaffold components. By contrast, Nar1 clusters were required for in vivo but not for in vitro Fe/S cluster assembly (30). The presence of the [2Fe-2S] cluster in Dre2 is unique, as the Fe/S clusters in the other assembly components are all of the [4Fe-4S] type. Furthermore, the clusters of Dre2 are apparently more stable than those of the other components, as they did not fall apart even after hours of air exposure during purification. One can imagine a special role for Dre2 in iron binding and iron reduction, perhaps involving the acidic patch domain and the Fe/S clusters, although this is very speculative. A source of electrons for such a reductase activity would still need to be identified.
Mitochondria consist of different subcompartments, outer membrane, IMS, inner membrane, and matrix. Dre2 is nucleus encoded and yet has a dual localization, being found in both cytoplasm and the mitochondrial IMS. The question arises of how this dual localization is accomplished. Mitochondrial proteins must be directed to the IMS by targeting signals that allow passage through the import translocase. Subsequently, IMS proteins must be retained within this compartment by folding or interaction with partner proteins (31). In some cases the folding step is mediated by a dedicated oxidase machinery consisting of Mia40 and Erv1, forming disulfides and trapping the substrate in the IMS. Most of the known substrates of Mia40-Erv1 are small proteins with characteristic cysteine motifs (twin CX3C, twin CX9C, or very recently discovered twin CX2C) (12). Interestingly, Dre2 has two CX2C motifs, similar to some other IMS proteins, although Dre2 is larger than other Mia40-Erv1 substrates. The dual cellular protein distribution might be explained by a kinetic process in which folding of the Dre2 precursor outside the mitochondria competes with oxidative folding (and trapping) inside the IMS (25). A similar protein distribution has been described for Sod1, the copper zinc superoxide dismutase (42). In the case of Sod1, the precursor is trapped in the IMS by interaction with the Ccs chaperone mediating formation of a critical disulfide in this space and chaperone-assisted cofactor insertion (42). Dre2 might undergo similar types of processing, oxidative folding, and cofactor insertion in the IMS. The IMS volume is very small, perhaps less than 0.5% of the cellular volume (42), and thus, although Dre2 (like Sod1) appears to be present in low amounts compared with the total cell protein population, the local concentration within the IMS is probably quite high.
Another unique feature of Dre2 is that it is a soluble Fe/S cluster protein occurring in the IMS. Although it is not directly proven that the clusters are present in the IMS-localized form, this would be quite novel and raise a number of new questions regarding Fe/S cluster biogenesis in various cell compartments. The proteins present in the mitochondrial IMS must enter this compartment via the outer membrane TOM translocase. The latter admits only unfolded proteins, and thus, proteins with cofactors that reside in the IMS are thought to acquire their cofactors while folding inside this compartment (31). In analogous fashion, Dre2 may be translated on cytoplasmic ribosomes, imported in an unfolded state into the IMS, and then loaded with its Fe/S clusters within the IMS. The existence of an IMS machinery or small amounts of the components of the CIA machinery in the IMS must then be considered. The only other example, in terms of IMS Fe/S cluster proteins, is the Rieske protein of complex III. This protein contains a Cx2C motif and an intramolecular disulfide, suggesting that it might interact with the oxidative trap pathway (12). As distinct from Dre2, however, the Rieske protein is tethered to the inner membrane and is associated with other protein components of respiratory complex III. Assembly of the Rieske protein with complex III requires a special chaperone called Bcs1 (32). Biogenesis of its Fe/S cluster has not been examined in terms of the genetic and biochemical requirements, and components for the Rieske protein Fe/S cluster, as for Dre2 biogenesis, might come from inside mitochondria and/or from the cytoplasm.
Dre2 sequence and sequence motifs are conserved between yeast and human, and the human protein was able to correct the lethality of a yeast Δdre2 deletion. Complementation was not complete in terms of biochemical correction of cytoplasmic Fe/S cluster assembly functions, however. The data may indicate that the Fe/S cluster assembly function of yeast Dre2 is partially but inefficiently provided by the human protein or alternatively that yeast Dre2 and human Dre2 have both overlapping and distinct functions. Studies of the mammalian homologs have focused on their antiapoptotic roles, and these homologous proteins have not yet been associated with Fe/S cluster assembly or shown to carry Fe/S clusters. The human homolog, named CIAPIN1 (cytokine-induced apoptosis inhibitor 1), was found to be upregulated in the context of cancer drug resistance. In addition, its overexpression in the HL60 cell line conferred resistance to cancer drugs (adriamycin and vincristine) (10). In cell fractionation studies, the human protein was noted to be in the cytoplasm, nucleus, and nucleolus, but mitochondria were not specifically examined (11). The mouse homolog of Dre2 (anamorsin) was found to prevent apoptosis of the pre-B-cell line Ba/F3, occurring upon cytokine withdrawal (38). Also the mouse knockout of anamorsin was nonviable as a consequence of a defect in definitive hematopoiesis. Myeloid and erythroid colony-forming activities in response to cytokines were severely disrupted, perhaps due to abnormal apoptosis (38). The mechanism of action of human or mouse Dre2 homologs in blocking apoptosis and promoting development of hematopoietic cells has not been clarified. A possibility is that the mammalian Dre2 homologs interact and negatively regulate a key apoptotic component in the IMS (e.g., cytochrome c). Alternatively, if the mammalian homolog, like yeast Dre2, is required for Fe/S cluster synthesis, then its antiapoptotic activity might be mediated by an unknown Fe/S cluster protein.
Acknowledgments
We thank Eduard Hurt of the Biochemie-Zentrum der Universitat Heidelberg (BZH), Heidelberg, Germany, for the plasmids pRS316-Rpl25-eGFP and pRS316-Rps2-eGFP. We thank Tracey Rouault of the Cell Biology and Metabolism Branch of NICHD, NIH, Bethesda, MD, for the antibody to IRP1. We thank Gunter B. Kohlhaw of Purdue University, West Lafayette, IN, for helpful discussions regarding the isopropylmalate isomerase assay and for sharing enzyme substrate with us. We thank John Stevens, Tufts University, and Qifan (Jenny) Zhang, Stuyvesant High School, New York, NY, for assistance with the genetic screening.
D. Pain is supported by AHA 0655946T and NIH R01 AG030504; F. Daldal is supported by NIH GM 38239 and DOE ER20052; E. Bi is supported by NIH R01 GM59216; T. Ohnishi is supported by NIH R01 GM30736; A. Dancis is supported by NIH R01 DK53953.
Footnotes
Published ahead of print on 14 July 2008.
REFERENCES
- 1.Aasa, R., T. Vanngard, and H. B. Dunford. 1975. EPR studies on compound I of horseradish peroxidase. Biochim. Biophys. Acta 391259-264. [DOI] [PubMed] [Google Scholar]
- 2.Amutha, B., D. M. Gordon, Y. Gu, E. R. Lyver, A. Dancis, and D. Pain. 2008. GTP is required for iron-sulfur cluster biogenesis in mitochondria. J. Biol. Chem. 2831362-1371. [DOI] [PubMed] [Google Scholar]
- 3.Amutha, B., and D. Pain. 2003. Nucleoside diphosphate kinase of Saccharomyces cerevisiae, Ynk1p: localization to the mitochondrial intermembrane space. Biochem. J. 370805-815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beinert, H., R. H. Holm, and E. Munck. 1997. Iron-sulfur clusters: nature's modular, multipurpose structures. Science 277653-659. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Aroya, S., C. Coombes, T. Kwok, K. A. O'Donnell, J. D. Boeke, and P. Hieter. 2008. Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol. Cell 30248-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chanet, R., and M. Heude. 2003. Characterization of mutations that are synthetic lethal with pol3-13, a mutated allele of DNA polymerase delta in Saccharomyces cerevisiae. Curr. Genet. 43337-350. [DOI] [PubMed] [Google Scholar]
- 7.Fontecave, M., and S. Ollagnier-de-Choudens. 2008. Iron-sulfur cluster biosynthesis in bacteria: mechanisms of cluster assembly and transfer. Arch. Biochem. Biophys. 474226-237. [DOI] [PubMed] [Google Scholar]
- 8.Goldberg, A. V., S. Molik, A. D. Tsaousis, K. Neumann, G. Kuhnke, F. Delbac, C. P. Vivares, R. P. Hirt, R. Lill, and T. M. Embley. 2008. Localization and functionality of microsporidian iron-sulphur cluster assembly proteins. Nature 452624-628. [DOI] [PubMed] [Google Scholar]
- 9.Guthrie, C., and G. R. Fink (ed.). 1991. Methods in enzymology, vol. 194. Guide to yeast genetics and molecular biology. Academic Press, Inc., San Diego, CA. [PubMed]
- 10.Hao, Z., X. Li, T. Qiao, R. Du, L. Hong, and D. Fan. 2006. CIAPIN1 confers multidrug resistance by upregulating the expression of MDR-1 and MRP-1 in gastric cancer cells. Cancer Biol. Ther. 5261-266. [DOI] [PubMed] [Google Scholar]
- 11.Hao, Z., X. Li, T. Qiao, R. Du, G. Zhang, and D. Fan. 2006. Subcellular localization of CIAPIN1. J. Histochem. Cytochem. 541437-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herrmann, J. M., and R. Kohl. 2007. Catch me if you can! Oxidative protein trapping in the intermembrane space of mitochondria. J. Cell Biol. 176559-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson, D. C., D. R. Dean, A. D. Smith, and M. K. Johnson. 2005. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 74247-281. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy, M. C., and H. Beinert. 1988. The state of cluster SH and S2- of aconitase during cluster interconversions and removal. A convenient preparation of apoenzyme. J. Biol. Chem. 2638194-8198. [PubMed] [Google Scholar]
- 15.Kispal, G., P. Csere, C. Prohl, and R. Lill. 1999. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 183981-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kispal, G., K. Sipos, H. Lange, Z. Fekete, T. Bedekovics, T. Janaky, J. Bassler, D. J. Aguilar Netz, J. Balk, C. Rotte, and R. Lill. 2005. Biogenesis of cytosolic ribosomes requires the essential iron-sulphur protein Rli1p and mitochondria. EMBO J. 24589-598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klinge, S., J. Hirst, J. D. Maman, T. Krude, and L. Pellegrini. 2007. An iron-sulfur domain of the eukaryotic primase is essential for RNA primer synthesis. Nat. Struct. Mol. Biol. 14875-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohlhaw, G. B. 1988. Isopropylmalate dehydratase from yeast. Methods Enzymol. 166423-429. [DOI] [PubMed] [Google Scholar]
- 19.Leighton, J. 1995. ATP-binding cassette transporter in Saccharomyces cerevisiae mitochondria. Methods Enzymol. 260389-396. [DOI] [PubMed] [Google Scholar]
- 20.Li, J., M. Kogan, S. A. Knight, D. Pain, and A. Dancis. 1999. Yeast mitochondrial protein, Nfs1p, coordinately regulates iron-sulfur cluster proteins, cellular iron uptake, and iron distribution. J. Biol. Chem. 27433025-33034. [DOI] [PubMed] [Google Scholar]
- 21.Li, L., and J. Kaplan. 2004. A mitochondrial-vacuolar signaling pathway in yeast that affects iron and copper metabolism. J. Biol. Chem. 27933653-33661. [DOI] [PubMed] [Google Scholar]
- 22.Lill, R., R. Dutkiewicz, H. P. Elsasser, A. Hausmann, D. J. Netz, A. J. Pierik, O. Stehling, E. Urzica, and U. Muhlenhoff. 2006. Mechanisms of iron-sulfur protein maturation in mitochondria, cytosol and nucleus of eukaryotes. Biochim. Biophys. Acta 1763652-667. [DOI] [PubMed] [Google Scholar]
- 23.Longtine, M. S., A. McKenzie III, D. J. Demarini, N. G. Shah, A. Wach, A. Brachat, P. Philippsen, and J. R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14953-961. [DOI] [PubMed] [Google Scholar]
- 24.Martin, H., C. Eckerskorn, F. Gartner, J. Rassow, F. Lottspeich, and N. Pfanner. 1998. The yeast mitochondrial intermembrane space: purification and analysis of two distinct fractions. Anal. Biochem. 265123-128. [DOI] [PubMed] [Google Scholar]
- 25.Morgan, B., and H. Lu. 2008. Oxidative folding competes with mitochondrial import of the small Tim proteins. Biochem. J. 411115-122. [DOI] [PubMed] [Google Scholar]
- 26.Muhlenhoff, U., J. A. Stadler, N. Richhardt, A. Seubert, T. Eickhorst, R. J. Schweyen, R. Lill, and G. Wiesenberger. 2003. A specific role of the yeast mitochondrial carriers MRS3/4p in mitochondrial iron acquisition under iron-limiting conditions. J. Biol. Chem. 27840612-40620. [DOI] [PubMed] [Google Scholar]
- 27.Munujos, P., J. Coll-Canti, F. Gonzalez-Sastre, and F. J. Gella. 1993. Assay of succinate dehydrogenase activity by a colorimetric-continuous method using iodonitrotetrazolium chloride as electron acceptor. Anal. Biochem. 212506-509. [DOI] [PubMed] [Google Scholar]
- 28.Murakami, H., D. Pain, and G. Blobel. 1988. 70-kD heat shock-related protein is one of at least two distinct cytosolic factors stimulating protein import into mitochondria. J. Cell Biol. 1072051-2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakai, Y., M. Nakai, H. Hayashi, and H. Kagamiyama. 2001. Nuclear localization of yeast Nfs1p is required for cell survival. J. Biol. Chem. 2768314-8320. [DOI] [PubMed] [Google Scholar]
- 30.Netz, D. J., A. J. Pierik, M. Stumpfig, U. Muhlenhoff, and R. Lill. 2007. The Cfd1-Nbp35 complex acts as a scaffold for iron-sulfur protein assembly in the yeast cytosol. Nat. Chem. Biol. 3278-286. [DOI] [PubMed] [Google Scholar]
- 31.Neupert, W., and J. M. Herrmann. 2007. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 76723-749. [DOI] [PubMed] [Google Scholar]
- 32.Nobrega, F. G., M. P. Nobrega, and A. Tzagoloff. 1992. BCS1, a novel gene required for the expression of functional Rieske iron-sulfur protein in Saccharomyces cerevisiae. EMBO J. 113821-3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Onder, O., H. Yoon, B. Naumann, M. Hippler, A. Dancis, and F. Daldal. 2006. Modifications of the lipoamide-containing mitochondrial subproteome in a yeast mutant defective in cysteine desulfurase. Mol. Cell. Proteomics 51426-1436. [DOI] [PubMed] [Google Scholar]
- 34.Rouault, T. A. 2006. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2406-414. [DOI] [PubMed] [Google Scholar]
- 35.Roy, A., N. Solodovnikova, T. Nicholson, W. Antholine, and W. E. Walden. 2003. A novel eukaryotic factor for cytosolic Fe-S cluster assembly. EMBO J. 224826-4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rudolf, J., V. Makrantoni, W. J. Ingledew, M. J. Stark, and M. F. White. 2006. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol. Cell 23801-808. [DOI] [PubMed] [Google Scholar]
- 37.Sherman, F. 2002. Getting started with yeast. Methods Enzymol. 3503-41. [DOI] [PubMed] [Google Scholar]
- 38.Shibayama, H., E. Takai, I. Matsumura, M. Kouno, E. Morii, Y. Kitamura, J. Takeda, and Y. Kanakura. 2004. Identification of a cytokine-induced antiapoptotic molecule anamorsin essential for definitive hematopoiesis. J. Exp. Med. 199581-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siegel, L., and S. Englard. 1962. Beef-heart malic dehydrogenases. III. Comparative studies of some properties of M-malic dehydrogenase and S-malic dehydrogenase. Biochim. Biophys. Acta 64101-110. [DOI] [PubMed] [Google Scholar]
- 40.Siegel, L. M. 1965. A direct microdetermination for sulfide. Anal. Biochem. 11126-132. [DOI] [PubMed] [Google Scholar]
- 41.Stearman, R., D. S. Yuan, Y. Yamaguchi-Iwai, R. D. Klausner, and A. Dancis. 1996. A permease-oxidase complex involved in high-affinity iron uptake in yeast. Science 2711552-1557. [DOI] [PubMed] [Google Scholar]
- 42.Sturtz, L. A., K. Diekert, L. T. Jensen, R. Lill, and V. C. Culotta. 2001. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 27638084-38089. [DOI] [PubMed] [Google Scholar]
- 43.Tong, W. H., and T. A. Rouault. 2006. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 3199-210. [DOI] [PubMed] [Google Scholar]
- 44.Wu, X., P. B. Alexander, Y. He, M. Kikkawa, P. D. Vogel, and S. L. McKnight. 2005. Mammalian sprouty proteins assemble into large monodisperse particles having the properties of intracellular nanobatteries. Proc. Natl. Acad. Sci. USA 10214058-14062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yarunin, A., V. G. Panse, E. Petfalski, C. Dez, D. Tollervey, and E. C. Hurt. 2005. Functional link between ribosome formation and biogenesis of iron-sulfur proteins. EMBO J. 24580-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang, Y., E. R. Lyver, S. A. Knight, E. Lesuisse, and A. Dancis. 2005. Frataxin and mitochondrial carrier proteins, Mrs3p and Mrs4p, cooperate in providing iron for heme synthesis. J. Biol. Chem. 28019794-19807. [DOI] [PubMed] [Google Scholar]
- 47.Zhang, Y., E. R. Lyver, S. A. Knight, D. Pain, E. Lesuisse, and A. Dancis. 2006. Mrs3p, Mrs4p, and frataxin provide iron for Fe-S cluster synthesis in mitochondria. J. Biol. Chem. 28122493-22502. [DOI] [PubMed] [Google Scholar]