

Abstract
High-risk human papillomaviruses (HPVs) are small nonenveloped DNA viruses with a strict tropism for squamous epithelium. The viruses are causative agents of cervical cancer and some head and neck cancers, but their differentiation-dependent life cycles have made them difficult to study in simple cell culture. Thus, many aspects of early HPV infection remain mysterious. We recently showed the high-risk HPV type 31 (HPV31) enters its natural host cell type via caveola-dependent endocytosis, a distinct mechanism from that of the closely related HPV16 (Smith et al., J. Virol. 81:9922-9931, 2007). Here, we determined the downstream trafficking events after caveolar entry of HPV31 into human keratinocytes. After initial plasma membrane binding, HPV31 associates with caveolin-1 and transiently localizes to the caveosome before trafficking to the early endosome and proceeding through the endosomal pathway. Caveosome-to-endosome transport was found to be Rab5 GTPase dependent. Although HPV31 capsids were observed in the lysosome, Rab7 GTPase was dispensable for HPV31 infection, suggesting that viral genomes escape from the endosomal pathway prior to Rab7-mediated capsid transport. Consistent with this, the acidic pH encountered by HPV31 within the early endosomal pathway induces a conformational change in the capsid resulting in increased DNase susceptibility of the viral genome, which likely aids in uncoating and/or endosomal escape. The entry and trafficking route of HPV31 into human keratinocytes represents a unique viral pathway by which the virions use caveolar entry to eventually access a low-pH site that appears to facilitate endosomal escape of genomes.
Human papillomaviruses (HPVs) are small, naked viruses that carry a circular doubled-stranded DNA genome and infect human squamous epithelial cells. The causal relationship between HPVs and cervical cancer is well established (3), and HPVs have been implicated in other epithelial cancers as well, including head and neck cancers (14, 36). The development of an effective vaccine against four HPV types promises to be a very valuable tool for preventing new HPV infections (1). However, the cost of this vaccine series may be prohibitive in the populations of women in developing countries who would benefit most (2). In addition, the long-term protection offered by the vaccine, an essential property given the role of HPVs in cancer, has not been established. Therefore, continued study of high-risk HPVs is essential to elucidate the many aspects of basic HPV biology that remain undefined.
Many viruses enter cells through endocytosis, hijacking the cellular machinery for entry, and invasion of the host cell (32). The majority of viruses have been demonstrated to use clathrin-mediated endocytosis for entry, whereas a few have been shown to enter through caveolae. Classically, ligands using clathrin-dependent endocytosis proceed through the endosomal pathway, while cargo entering via caveolae move to an intracellular caveolin-1-positive and pH-neutral structure known as the caveosome before trafficking to the endoplasmic reticulum (ER) (23). These pathways, previously believed to be distinct, are now found to display cross talk, whereby cargo can move between them. For example, the polyomavirus JC virus (JCV) has been shown to enter glial cells through clathrin-mediated endocytosis. However, after entry the virions localize to caveolin-1-positive structures, presumably the caveosome, and inhibition of caveolin-1 activity blocks a postentry trafficking event in JCV infection (26). Bovine papillomavirus type 1 (BPV1) was recently shown to enter and infect human HEK293 cells in the same manner (17). Conversely, after entry via caveolae-mediated endocytosis, simian immunodeficiency virus 40 (SV40) is observed to traffic between caveosomes and early endosomes. However, SV40 infection, as defined by nuclear delivery of active genomes, requires transport back to the caveosomes and on to the ER (22). In both cases, movement between the caveolar and endosomal pathways was found to be dependent on the small GTPase Rab5.
Pathogens that proceed through the endosomal pathway during trafficking in the host cell typically take advantage of the decreased pH of the endosomal compartments (15, 37). Acidic pH acts as a trigger for many viruses to undergo conformational changes, leading to any number of events that facilitate endosomal escape of virion proteins and/or viral genomes. Such events may include modification of the viral-receptor interaction, exposure of protease digestion motifs, viral envelope-endosomal membrane fusion, or partial to complete uncoating of the viral genome (12, 35). Although a C-terminal region of the HPV minor capsid protein L2 has been identified that displays pH-dependent membrane destabilizing activity (16), the exact mechanism by which this structure may assist in endosomal escape remains unclear.
Previous work showed that the high-risk HPV16 utilizes a clathrin-dependent entry mechanism (10). In contrast, we recently demonstrated that high-risk HPV31 infection of human keratinocytes, the natural host cell type for HPVs, occurs through caveola-mediated endocytosis, and the initial entry route is distinct from that of HPV16 (34). In this report, we investigated the trafficking of HPV31 virions following caveolar entry into human keratinocytes to identify the key cellular compartments involved in virus infection. Colocalization studies with various markers of cellular organelles reveal that, after entry, HPV31 traffics through the caveosome to the endosomal pathway, and this trafficking is dependent on the small GTPase Rab5. Consistent with passage through the endosomal pathway, HPV31 infection is dependent on the acidification of endosomes, and low-pH treatment of virions induces a conformational change in the HPV31 capsid that appears to promote genome uncoating.
MATERIALS AND METHODS
Cell culture, virion production, and infections.
HaCaT cells are a spontaneously immortalized epithelial line derived from normal adult skin (4). 293T cells are derived from the human embryonic kidney cell line HEK293 and express SV40 large T antigen. Cells were maintained in Dulbecco modified Eagle medium-Ham's F-12 nutrient mixture containing 10% fetal calf serum, 4× amino acids, 2 mM glutamine, 100 U of penicillin/ml, and 1 μg of streptomycin/ml. HPV31 virions were produced and purified by density gradient centrifugation as previously described (25, 34); stocks were quantified by blot hybridization to determine viral genome equivalents (vge) per unit volume as reported (19, 20). Transmission electron microscopy (TEM; Hitachi 7500) was performed to visualize virus stocks at 80 kV after binding to a carbon-coated electron microscopic grid and negative staining with 2% uranyl acetate. Infections were performed as previously described (19, 20). Briefly, virion stocks were diluted in media and exposed to HaCaT cells for 1 h at 4°C with agitation to allow viral attachment without entry. Inocula were aspirated, and cells were washed and refed with fresh media. Cells were incubated for various amounts of time at 37°C before analysis.
RNA isolation and RT-qPCR analysis.
Total RNA was extracted at 48 h postinfection by using Tri-Reagent (Sigma). Nucleic acid concentrations were determined by spectrophotometry. Reverse transcription (RT) of 2 μg of total RNA and triplicate quantitative PCR (qPCR) were performed by using GeneAmp RNA PCR reagents and AmpliTaq Gold DNA polymerase (Applied Biosystems) with primers, probes, and conditions as previously described (19, 21). Error bars represent standard error of the mean (SEM).
Virion labeling with fluorescent dyes.
Approximately 1 μg of Alexa Fluor 594 (AF594), carboxylic acid, and succinimidyl ester (Molecular Probes) was mixed with 108 virions, followed by incubation for 1 h at room temperature. Labeled stocks were washed three times with 1× HSB (25 mM HEPES [pH 7.5], 500 mM NaCl, 0.02% Brij58, 1 mM MgCl2, 100 μM EDTA, 0.5% ethanol) and recovered by using a Millipore Amicon Ultra-4 centrifugation filter to remove unincorporated dye.
Colocalization studies.
The following antibodies were used for immunofluorescence at the indicated dilutions: anti-caveolin-1 (1:500; BD Biosciences clone 2297), anti-protein disulfide isomerase (PDI; 1:500; Stressgen clone 1D3), anti-golgin 97 (1:500; Molecular Probes clone CDF4), anti-early endosome antigen 1 (EEA1; 1:1,000; Abcam ab15846), anti-lysosomal-associated membrane protein 1 (LAMP1; 1:500; Abcam clone H4A3), AF488-goat anti-mouse immunoglobulin G (IgG; 1:500; Molecular Probes), and AF680-goat anti-mouse IgG (1:500; Molecular Probes). Cells were seeded at 5,000 to 10,000 cells per glass coverslip in 60-mm dishes 1 day prior to virus exposure. AF594-HPV31 virions were diluted in media to 5,000 to 10,000 vge/cell and exposed to cells for 1 h at 4°C. Cells were washed, refed with fresh media, and incubated at 37°C for various times. For caveolin-1 staining, cells were fixed with 3.7% paraformaldehyde and permeabilized with cold methanol. For PDI and golgin-97 staining, cells were fixed with 3.7% paraformaldehyde. For EEA1 and LAMP1 staining, cells were fixed and permeabilized with cold acetone. After fixation, slides were blocked in 4% bovine serum albumin and phosphate-buffered saline (PBS) and then incubated with the appropriate primary antibody diluted 1:100 in 4% bovine serum albumin-PBS for 1 h at room temperature. Cells were washed extensively with PBS and then incubated with AF488- or AF680-anti-mouse IgG diluted to 5 μg/ml in PBS. Cells were washed and mounted with VectaShield containing DAPI (4′,6′-diamidino-2-phenylindole; Vector Labs). Photomicroscopy was performed by using a Zeiss LSM 510 META confocal microscope with a ×63 objective and the appropriate filters.
Rab GTPase transfections.
Wild-type (wt) and dominant-negative (dn) Rab5, Rab7, and Rab11 proteins fused with green fluorescent protein (GFP-Rab5wt, GFP-Rab5 S34N, GFP-Rab7wt, GFP-Rab7 T22N, GFP-Rab11wt, and GFP-Rab11 S25N) were previously reported (7, 8, 13, 27, 28). HaCaT cells were transfected with each construct by using the Amaxa Nucleofector V protocol as previously described (34). At 48 h posttransfection, cells were exposed to virions in triplicate (as described above); colocalization studies were performed at various times postattachment. After identification of transfected cells by GFP expression, AF680-stained caveolin-1, EEA1, and LAMP1 were pseudocolored green, and ∼100 AF594-labeled virion signals were assessed for colocalization with each respective immunostained organelle.
DNase susceptibility assay.
Approximately 108 virions in triplicate were treated with 10 mM dithiothreitol (DTT) overnight at room temperature or at the indicated pH for 2 h at 37°C. Each sample was then washed over a Millipore Amicon Ultra 4K centrifugation filter and exchanged into DNase I buffer (40 mM Tris-Cl [pH 7.9], 10 mM NaCl, 6 mM MgCl2, 1 mM CaCl2). Samples were equally divided and either mock treated or treated with 5 U of DNase I for 1 h at 37°C. Samples were then blotted onto a GeneScreen membrane and probed for HPV31 genome as previously described (19). Genome copy number was quantified by densitometry, and the percentage of DNase-resistant genome was calculated by normalization of DNase treated to mock-treated samples.
RESULTS
HPV31 traffics through caveosomes to the endosomal pathway.
We showed that HPV31 enters human keratinocytes through a caveola-mediated and dynamin 2-dependent pathway in which the bulk of particles enter the cells with a very slow half time of 14 h (34). However, some particles enter more rapidly, as HPV31 infection detected by early gene expression occurs as early as 4 h after inducing internalization by warming the cells to 37°C (19). More recently, we found that the prolonged infection process of the majority of particles is primarily due to active and continual filopodium-mediated uptake of virions originally bound to the extracellular matrix (ECM) occurring over many hours (J. Smith, D. Lidke, and M. Ozbun, unpublished data). The net result is that entry and the process of infection is quite asynchronous; a fact that complicates studies of virion localization within the cell. To investigate the trafficking route of HPV31 particles during and after entry into human keratinocytes, nonsaturating numbers of AF594-labeled virions were allowed to bind the plasma membrane and ECM of HaCaT cells and enter for various amounts of time before counterstaining for several cellular organelles. Due to the asynchronicity of the internalization and trafficking, we viewed each “colocalization” experiment described below from 0 to 24 h postinitiation of entry at 37°C. We define colocalization as any observed merge of the red fluorescent virions with the green fluorescent detection of the organelles. Although many of the figures show only a few merged spots, they are representative of results from numerous cells under the given experimental conditions. No colocalization was ever observed between HPV31 particles and caveolin-1 immediately after attachment (Fig. 1A); however, as early as 30 min after attachment, HPV31 and caveolin-1 were observed in adjacent locales with some colocalization (Fig. 1B). This suggests that incubation at 37°C is required for HPV31-caveola association and is similar to what we observe for the organization of HPV31 into detergent-resistant microdomains/lipid rafts (34). At later time points internalized HPV31 was only found associated with caveolin-1-positive structures near the cell periphery (Fig. 1C), whereas virions located in perinuclear regions were no longer localized with caveolin-1 (Fig. 1D). Thus, HPV31 appears to transiently associate with caveolin-1 at the plasma membrane and at a peripheral caveolin-1-positive structure consistent with the caveosome before trafficking to a caveolin-1 free intracellular compartment.
FIG. 1.

HPV31 transiently localizes to caveolin-1 structures at the plasma membrane and intracellularly. HaCaT cells were exposed to AF594-labeled HPV31 virions at 10,000 vge/cell for 1 h at 4°C. Cells were washed and incubated at 37°C for 0 h (A), 30 min (B), or 16 h (C and D). Cells were fixed and stained for caveolin-1 (AF488; green), and nuclei were visualized with DAPI. The microscopy is representative, and the focal plane is mid-cell body/nucleus. Right panels are enlarged views of the boxed areas in the left panels.
Cargo that traffics through the caveosome is typically transported to the Golgi apparatus and/or ER (23). To determine whether HPV31 traffics to either of these structures, cells infected with AF594-HPV31 were stained at 16 and 24 h postattachment for PDI, an ER marker (Fig. 2A), or golgin-97, a marker of the Golgi apparatus (Fig. 2B). Surprisingly, at no time during infection was colocalization observed between HPV31 and either of these markers, indicating that HPV31 does not traffic to the Golgi or ER. However, high levels of HPV31 capsids were found consistently colocalized with markers of the endosomal pathway at later times in infection (Fig. 2C and D). This included markers of the early endosome, EEA1 (Fig. 2C), or the lysosome, LAMP1 (Fig. 2D) shown at 16 and 24 h postattachment. In addition, colocalization between HPV31 capsids and LAMP1 increased over time, indicating that particles accumulate in this structure. These data demonstrate that HPV31 virions enter cells via caveolae and transiently localize to the caveosome before trafficking to the endosomal pathway.
FIG. 2.

HPV31 localizes to early endosomal pathway at late times postentry. HaCaT cells were exposed to AF594-labeled HPV31 virions at 10,000 vge/cell for 1 h at 4°C. Unbound virions were removed, and cells were incubated at 37°C for 16 h or 24 h. Cells were fixed and immunostained (AF488; green) for PDI (A), Golgin-97 (B), EEA1(C), or LAMP1 (D) as described in Materials and Methods. Nuclei were visualized with DAPI. The microscopy is representative, and the focal plane is mid-cell body/nucleus. Right panels are enlarged views of the boxed areas in the left panels.
The role of pH in HPV31 trafficking and uncoating.
Many ligands and viruses that traffic through the endosomal pathway require the acidic pH of these compartments for proper transport or to otherwise facilitate function or infection (15, 37). We investigated the importance of acidic pH during HPV31 infection of human keratinocytes by treating cells with three inhibitors of endosomal acidification, including the lysosomotropic agents ammonium chloride and chloroquine, as well as bafilomycin A, which inhibits the vacuolar H+ ATPase. At 48 h postinfection, we assessed infection levels by RT-qPCR quantification of the spliced viral transcript E1∧E4 as previously reported (19, 21). All three inhibitors of endosomal acidification blocked HPV31 infection (Fig. 3A), indicating that the acidic pH of endosomal vesicles is required during this process.
FIG. 3.
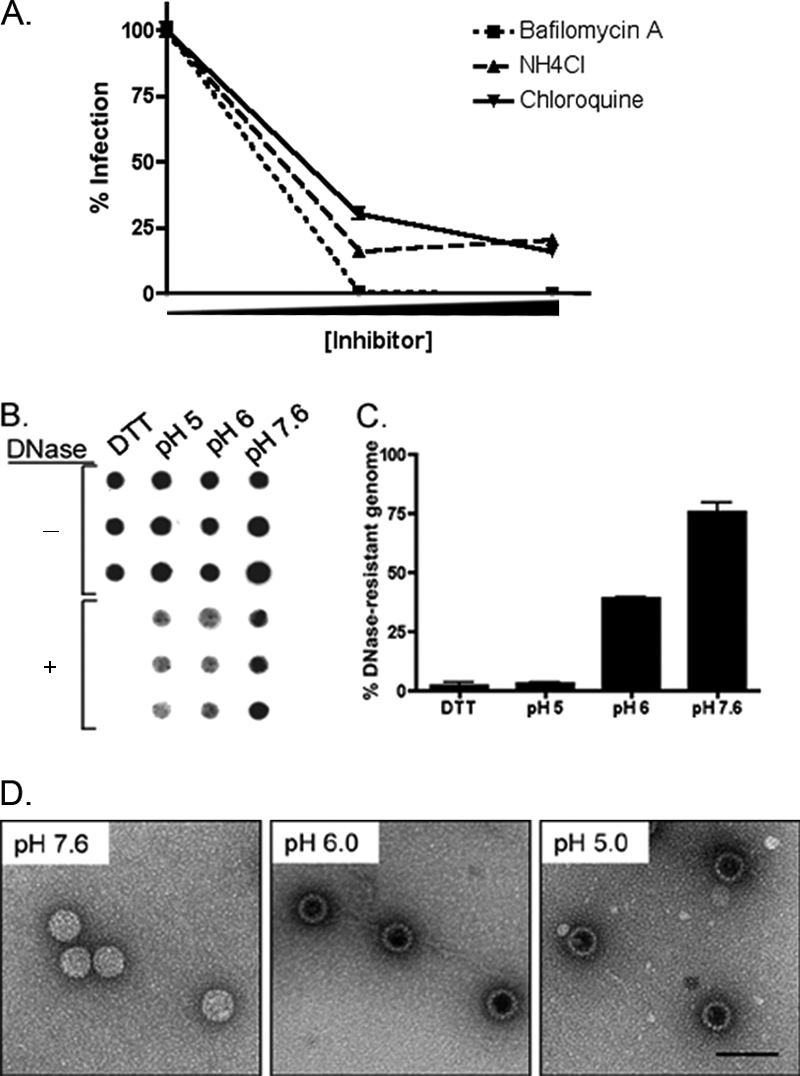
HPV31 infection requires endosomal acidification and low pH induces conformational changes in the HPV31 virion. (A) HaCaT cells were pretreated for 30 min with bafilomycin A (75 or 100 nM; dotted line), NH4Cl (15 or 20 mM; dashed line), or chloroquine (5 or 10 μM; solid line) and then infected with HPV31 at 100 vge/cell in the presence of inhibitors for 48 h. Cells were monitored for viability. Total RNA was analyzed for HPV31 E1∧E4 by RT-qPCR. Values are expressed as the percentage of infection normalized to mock-treated cells. Error bars represent the SEM. (B) HPV31 virions (∼108 vge) were treated in triplicate in vitro with 10 mM DTT at room temperature overnight or at the indicated pH for 2 h at 37°C. Samples were equally divided and mock or DNase I treated for 1 h at 37°C, blotted onto GeneScreen membrane, and probed for intact HPV31 genomes. (C) Quantification of panel B, where values are expressed as the percent DNase-resistant genome normalized to the corresponding mock-treated sample. Error bars represent the SEM. (D) A total of ∼108 HPV31 virions were treated in vitro at indicated pH for 2 h at 37°C. Samples were bound to carbon-coated electron microscopy grids and stained with uranyl acetate before visualization by TEM.
The acidic milieu of the endosomal compartment is a common trigger for several viruses, often causing conformational changes in the capsid that promote genome uncoating and/or endosomal escape (6, 12, 40). To investigate how acidic pH affects HPV31 capsid structure, we exposed virions to lower pH in vitro and measured the effects visually and by access of the viral genome to DNase I. The tight conformation of HPV capsids formed during virion morphogenesis cause viral genomes encapsidated in mature virions to be DNase resistant (5, 25). Treatment with DTT, which reduces intercapsomeric bonds in papillomavirus particles, causes near complete susceptibility of the viral genome to DNase (18) (Fig. 3B). Whereas neutral pH had no significant effect on viral genome DNase sensitivity, lower pH had substantial effects. Exposure of virions to pH 6 (equivalent to early-late endosomes) or pH 5 (equivalent to lysosomes) rendered the virion-associated genomes increasingly more susceptible to DNase (Fig. 3B). Quantification after each treatment (Fig. 3C) revealed that more than 98% of the viral genomes were susceptible to DNase digestion after pH 5 treatment, similar to the susceptibility of DTT-treated virions. Incubation at pH 6 resulted in a 60% loss of viral genome after DNase treatment, while >75% of viral genome remained DNase resistant after treatment at neutral pH (Fig. 3C). To observe changes in virion morphology upon exposure to a lower pH, virions were stained with uranyl acetate and visualized by TEM (Fig. 3D). Virions treated at neutral pH displayed normal morphology. However, after treatment at pH 5 or pH 6, particles exhibited increased uptake of uranyl acetate dye, as evidenced by the darker staining in the center of virions. This change in staining suggests that acidic pH-treated particles have a looser conformation, which is consistent with the increased DNase susceptibility observed for these samples (Fig. 3C). These data demonstrate that incubation at pH levels equivalent to those encountered by HPV31 virions during trafficking through the early endosomal pathway results in a conformational change in the virion, which likely contributes to uncoating and endosomal escape of the viral genome.
HPV31 trafficking between the caveosome and early endosomes is Rab5 dependent.
Rab GTPases regulate many steps of membrane traffic, including vesicle formation, vesicle movement along actin and tubulin networks, and membrane fusion. Rab5 is one of the more intensely studied members of the diverse family of Rab GTPases (9). Functional studies demonstrate Rab5 to be a crucial regulator of early endocytosis, where it is involved in clathrin-coated vesicle formation, fusion between early endosomes, endosomal cargo recruitment, and endosomal movement (38, 41). Rab7 regulates cargo transport from the early to late endosome, and Rab11 controls exocytic and recycling endosome transport between the trans-Golgi network and the plasma membrane (30). To investigate the importance of each of these Rab GTPases in HPV31 trafficking in human keratinocytes, cells were transfected with well-characterized fluorescent-tagged wild-type and dominant-negative forms of Rab5, Rab7, and Rab11. The cells were then infected with AF594-labeled HPV31 virions for 24 h and immunostained for markers for the caveosome (caveolin-1 [cav-1]), early endosome (EEA1), and lysosome (LAMP1; Fig. 4A). Rab-transfected cells were identified by fluorescent protein expression, and colocalization between HPV31 (AF594; red), and each of the organelle markers (AF680; pseudocolored green) was quantified in a blinded analysis (Fig. 4B). Expression of the dominant-negative Rab5 S34N had the most profound effect on HPV31 localization. A 43% increase of HPV31 colocalization in caveosomes resulted from the expression of the dominant-negative Rab5 GTPase, which kinetically delays transport, compared to the more transient HPV31 localization with caveolin-1 in wild-type Rab5 transfected cells. There was little obvious corresponding decrease in the colocalization with endosomal or lysosomal markers where labeled HPV31 capsids accumulate in the lysosome. This shows that limited transport between the caveosome and endosomal pathway continues in the presence of the kinetically delayed Rab5 mutant as expected. This demonstrates that HPV31 particle transport between the caveosome and the endosomal pathway is regulated by Rab5 GTPase, a route similar to that shown for SV40 and cholera toxin B (22).
FIG. 4.

Trafficking of HPV31 between the caveosome and endosomal pathway is Rab5 GTPase dependent and essential for the infection of human keratinocytes. (A) HaCaT cells were transfected with fluorescence-tagged Rab5 wild-type (Rab5wt), Rab5 S34N dominant-negative mutant (Rab11dn), Rab7wt, Rab7dn T22N, Rab11wt, or Rab11dn S25N constructs. At 48 h, AF594-HPV31 was exposed to cells at 10,000 vge/cell for 1 h at 4°C, washed, refed, and incubated at 37°C for 24 h. Cells were fixed and immunostained for caveolin-1 (cav-1; top row), EEA1 (middle row), or LAMP1 (bottom row) (AF680; pseudocolored green), and nuclei were visualized with DAPI. Images were captured on a Zeiss META confocal microscope with a ×63 objective and are representative. The focal plane was not limited to mid-cell body but was variable to assess colocalization. (B) Blinded quantification of HPV31 virions (red; AF594) colocalizing with caveolin-1-, EEA1-, or LAMP1-positive structures (pseudocolored green). Values are expressed as the percentage of HPV31 virions per cell colocalized with indicated marker compared to total virions. (C) Triplicate-transfected HaCaT cell cultures were infected with HPV31 at 100 vge/cell. Total RNA was harvested at 48 h postinfection and quantified in triplicate by RT-qPCR for spliced viral E1∧E4 transcripts. Values are expressed as the percentage of infection compared to wild-type control transfected cells. Error bars represent the SEM of triplicate reactions. *, P > 0.01.
Rab7 regulates transport between early and late endosomes, and cells expressing a kinetically delayed Rab7 T22N GTPase mutant demonstrated a similar, but less dramatic increase in HPV31 localization in caveosomes (Fig. 4A and B). This result further supports a model whereby HPV31 circulation between caveosomes and early endosomes occurs regulated by Rab5: as virion traffic backs up into the early endosome due to Rab7 inhibition, Rab5-mediated circulation sustains particle cycling resulting in a net accumulation in the caveosome. As predicted, dominant-negative Rab11 GTPase, which is well known to control exocytic transport between the Golgi and plasma membrane (7, 8, 39), had no effect on HPV31 localization, confirming that Golgi trafficking is not involved in virus entry.
If Rab proteins are necessary in the HPV31 infection process prior to genome uncoating and nuclear transport, the delayed transport induced by mutant Rabs would be expected to cause a reduction in our infectious entry assay which measures HPV31 early transcription after virion entry. Therefore, to firmly establish a role for Rab GTPases in HPV31 early infection, we quantified the abilities of wild-type and mutant Rab5, Rab7, and Rab11 proteins to block HPV31 early transcription as a measure of entry, uncoating, and nuclear viral genome delivery (Fig. 4C). At 48 h posttransfection with the Rab constructs, cells were exposed to HPV31 and incubated at 37°C for an additional 48 h to allow viral entry, uncoating, and initiation of early transcription. Infection was assessed by RT-qPCR to measure levels of spliced viral E1∧E4 RNAs (Fig. 4C). As expected, blocking exocytic Golgi to plasma membrane trafficking by dominant-negative Rab11 did not alter HPV31 infection. Surprisingly, mutant Rab5 GTPase was the only protein to cause significant inhibition of HPV31 infection. Despite the change in localization of HPV31 capsids observed in mutant Rab7 transfected cells (Fig. 4A and B), no infection inhibition was detected in these cells. This finding, along with the fact that viral genomes are susceptible to DNase at an early endosomal pH, is consistent with a model in which the uncoated HPV31 genome exits the endosomal pathway before Rab7-mediated trafficking of capsids occurs. Therefore, the infection data indicate that the signal from AF594-labeled particles in the lysosome likely represents empty capsids (Fig. 2D and 4A).
DISCUSSION
Trafficking of cargo after caveolar entry for many years was believed to be a parallel but separate pathway from transport following entry by clathrin-dependent endocytosis into the endosomal pathway. Typically, ligands and viruses that enter via caveolae move through the caveosome to the Golgi apparatus or ER, avoiding the endocytic route (24). However, more complex interactions and cross talk between these two pathways have recently become evident. For example, the polyomavirus SV40 has been shown to traffic between the caveosome and the early endosome in a Rab5-dependent manner. However, SV40 remains tightly associated with its receptor and is not released into the endosomal pathway. Instead, the virus is transported to the ER, where resident proteins direct genome uncoating (24, 29). In this regard, caveola-to-endosome transport is not required for SV40 infection. In contrast, polyomavirus JCV enters host cells via clathrin-mediated endocytosis, but then virions travel through the caveosome en route to the ER in a Rab5-, caveolin-1-, cholesterol-, and pH-dependent manner (26). Similarly, BPV1 entry into human 293 cells is clathrin dependent but requires caveolin-1 intracellularly (17). Finally, cholera toxin B circulates between the caveosome and early endosome and uses the acidic pH of the early endosome as a trigger for disassociation from its receptor to allow continued trafficking within the endosomal pathway (22).
To our knowledge, the trafficking route of HPV31 in natural host keratinocytes is the first example of a virus that requires the caveosome-to-endosome pathway for infectivity (Fig. 5). The protracted and unusually asynchronous uptake of HPV31 from the ECM to the plasma membrane and/or directly from the plasma membrane into host cells makes it challenging to assay for infection-related intracellular trafficking, since colocalization studies require investigation at many and extensive times postattachment. Functionally, the drawn-out entry means that virions are never simultaneously present at the same locale or confined to a single organelle at any single point in time. However, our infectious entry assays using unlabeled virions, which depend upon nuclear delivery of and transcription from viral genomes postinfection, corroborate each of the informative localization results with labeled virions. Oncogenic HPV31, which we previously demonstrated to enter human keratinocytes through caveola-dependent endocytosis, first traffics to the caveosome as expected (Fig. 5A). The shuttling of HPV31 between the caveosome and early endosomal compartment is Rab5 dependent (Fig. 5B), and the infectious route continues along the endosomal pathway (Fig. 5C and D). Although Rab5-dependent vesicle transfer between the caveosome and endosomal compartments has been described (24, 26), this represents a novel productive trafficking route for a virus.
FIG. 5.

Model of HPV31 infectious entry in human keratinocytes. (A) HPV31 enters via caveola- and dynamin 2-mediated endocytosis and traffics to caveosomes. (B) Thereafter, virions are transported from the caveosome to the early endosome dependent on Rab5 GTPase. (C) As HPV31 proceeds through the endosomal pathway, the decreasing pH of the endosomal compartments causes a conformational change in the viral capsid, which results in DNase I sensitivity of genomes. We propose that this conformational change leads to endosomal escape of the viral genome or genome/L2 complex (D) before Rab7 GTPase-mediated capsids are transported to the lysosome (E). Empty/disassembled capsids can be visualized in the lysosome.
An interesting role for the Rab7 GTPase in HPV31 particle trafficking was also identified. The kinetically delayed Rab7 mutant altered localization of capsids by increasing residence in the caveosome, supporting a model in which particles delayed in leaving the early endosome by Rab7 inhibition are transported back to the caveosome. However, infection measured by synthesis of early transcripts, an event that requires endosomal escape of the viral genome and transport to the nucleus, was found to be independent of Rab7. Thus, the infectious pathway of HPV31 diverges from the endosomal pathway before Rab7-mediated transport. Viral uncoating and endosomal escape in the intermediate/late endosome is consistent with this pathway, suggesting that lysosomal localization of fluorescent signal at late times postinfection reflects empty viral capsids (Fig. 5D and E). Consistent with this idea, significant changes in capsid structure and DNase susceptibility of viral genomes were observed after virion exposure to an early endosomal pH of 6.0. This suggests that the environment of endosomal pathway can trigger capsid conformational changes, promoting uncoating and endosome escape.
To date, the requirement for endosomal acidification during HPV infections has been tenuous, since most studies have used nonkeratinocyte cells. Furthermore, many reports claim endosomal involvement, but few demonstrate HPV virion-endosome colocalization or confirm findings with infectivity assays. BPV1 clearly requires endosomal acidification for infection and localizes to endosomal structures in nonhost mouse C127 cells following clathrin-dependent entry (10). HPV33 pseudovirions need endosomal acidification in COS-7 monkey kidney cells (31). In the present study, we show localization of HPV31 particles with early and late endosomal compartments, genetically demonstrate the requirement for endosomal trafficking effectors, and show acidification is necessary for infectivity and capsid alterations that likely lead to uncoating.
We recently demonstrated that the closely related high-risk types HPV16 and HPV31 enter human keratinocytes through distinct pathways (34). Our present findings that downstream trafficking of HPV31 requires the acidic compartments of the endosomal pathway is, however, similar to what we observed for HPV16 in human keratinocytes (S. K. Campos and M. A. Ozbun, unpublished results) and what was proposed to occur following HPV16-BPV1 colocalization during entry into C127 mouse cells (10). Thus, it appears that the trafficking routes of the two oncogenic types, despite utilizing distinct entry mechanisms at the plasma membrane, converge in the endosomal pathway, sharing a requirement for low pH during the infectious process.
The internalization receptor(s) for HPV16 and HPV31 remain unknown. Whereas HPV16 binding to and infection of human keratinocytes requires binding to heparan sulfonated proteoglycans, HPV31 infections do not (21). Furthermore, HPV16's entry half time is at least twice the rate as that of HPV31 (34). The different binding requirements and entry mechanisms of these two HPV types may therefore reflect distinct receptor usage. One might imagine a scenario wherein the use of one receptor species and/or entry mechanism might confer a particular viral type with an evolutionary advantage or disadvantage in specific host cells, whether cervical epithelium, oral epithelium, or some other cell reservoir. For example, the high levels of signaling induced within lipid rafts during caveolar entry (33), which we found for HPV31 (J. L. Smith and M. A. Ozbun, unpublished data) could contribute to differential innate antiviral responses for HPV31, explaining in part why HPV31 infections are far less prevalent and pathogenic compared to HPV16 infections (11). Regardless of initial entry mechanisms, the convergence of the two HPV types in the endosomal pathway strongly suggests that these viruses take advantage of the existing properties of this pathway (i.e., acidic pH), as has been demonstrated for many other viruses (15, 37). The unique nature of HPV31 infectious trafficking following caveolar entry into human keratinocytes described here represents yet another cellular pathway exploited by viruses to facilitate entry into and infection of host cells, as well as an additional means viruses may be used as tools to better understand cellular processes.
Acknowledgments
We thank N. Fusenig (DKFZ, Heidelberg, Germany) for HaCaT cells and T. Kanda (National Institute of Infectious Diseases, Tokyo, Japan) for the HPV31 L1/L2-expressing plasmid. We thank Rebecca Lee and Genevieve Phillips for assistance with confocal microscopy and Stephen Jett for assistance with electron microscopy.
All authors designed the study and analyzed results. J.L.S. and S.K.C. performed the research; J.L.S. and M.A.O. wrote the paper.
This study was supported by NIH grant CA085747 (M.A.O.), NSF grant MCB0446179 (A.W.-N.), NRSA postdoctoral fellowship F32 CA123842 (S.K.C.), and NIH training grant T32 AI07538 (J.L.S. and S.K.C.). Fluorescent images in this study were generated in the University of New Mexico Cancer Center Fluorescence Microscopy Facility, supported as detailed online (http://kugrserver.health.unm.edu:16080/microscopy/facility.html).
Footnotes
Published ahead of print on 30 July 2008.
REFERENCES
- 1.Barr, E., C. K. Gause, O. M. Bautista, R. A. Railkar, L. C. Lupinacci, R. P. Insinga, H. L. Sings, and R. M. Haupt. 2008. Impact of a prophylactic quadrivalent human papillomavirus (types 6, 11, 16, 18) L1 virus-like particle vaccine in a sexually active population of North American women. Am. J. Obstet. Gynecol. 198261.e1-261.e11. [DOI] [PubMed] [Google Scholar]
- 2.Bosch, F. X., X. Castellsagué, and S. de Sanjosé. 2008. HPV and cervical cancer: screening or vaccination? Br. J. Cancer 9815-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosch, F. X., M. M. Manos, N. Muñoz, M. Sherman, A. M. Jansen, J. Peto, M. H. Schiffman, V. Moreno, R. Kurman, K. V. Shah, and I. S. Group. 1995. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. J. Natl. Cancer Inst. 87796-802. [DOI] [PubMed] [Google Scholar]
- 4.Boukamp, P., R. T. Petrussevska, D. Breitkreutz, J. Hornung, A. Markham, and N. E. Fusenig. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106761-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buck, C. B., C. D. Thompson, Y.-Y. S. Pang, D. R. Lowy, and J. T. Schiller. 2005. Maturation of papillomavirus capsids. J. Virol. 792839-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandran, K., D. L. Farsetta, and M. L. Nibert. 2002. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein μ1 mediates membrane disruption. J. Virol. 769920-9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, W., Y. Feng, D. Chen, and A. Wandinger-Ness. 1998. Rab11 is required for trans-Golgi network-to-plasma membrane transport and a preferential target for GDP dissociation inhibitor. Mol. Biol. Cell 93241-3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, W., and A. Wandinger-Ness. 2001. Expression and functional analyses of Rab8 and Rab11a in exocytic transport from trans-Golgi network. Methods Enzymol. 329165-175. [DOI] [PubMed] [Google Scholar]
- 9.Colicelli, J. 2004. Human RAS superfamily proteins and related GTPases. Sci. STKE. 2004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day, P. M., D. R. Lowy, and J. T. Schiller. 2003. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 3071-11. [DOI] [PubMed] [Google Scholar]
- 11.de Sanjosé, S., M. Diaz, X. Castellsagué, G. Clifford, L. Bruni, N. Muñoz, and F. X. Bosch. 2007. Worldwide prevalence and genotype distribution of cervical human papillomavirus DNA in women with normal cytology: a meta-analysis. Lancet Infect. Dis. 7453-459. [DOI] [PubMed] [Google Scholar]
- 12.Doms, R. W., and A. Helenius. 1986. Quaternary structure of influenza virus hemagglutinin after acid treatment. J. Virol. 60833-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng, Y., B. Press, W. Chen, J. Zimmerman, and A. Wandinger-Ness. 2001. Expression and properties of Rab7 in endosome function. Methods Enzymol. 329175-187. [DOI] [PubMed] [Google Scholar]
- 14.Gillison, M. L., W. M. Koch, R. B. Capone, M. Spafford, W. H. Westra, L. Wu, M. L. Zahurak, R. W. Daniel, M. Viglione, D. E. Symer, K. V. Shah, and D. Sidransky. 2000. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 92709-720. [DOI] [PubMed] [Google Scholar]
- 15.Gruenberg, J., and F. G. van der Goot. 2006. Mechanisms of pathogen entry through the endosomal compartments. Nat. Rev. Mol. Cell. Biol. 7495-504. [DOI] [PubMed] [Google Scholar]
- 16.Kämper, N., P. M. Day, T. Nowak, H.-C. Selinka, L. Florin, J. Bolscher, L. Hilbig, J. T. Schiller, and M. Sapp. 2006. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J. Virol. 80759-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laniosz, V., K. A. Holthusen, and P. I. Meneses. 2008. Bovine papillomavirus type 1: from clathrin to caveolin. J. Virol. 826288-6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li, M., P. Beard, P. A. Estes, M. K. Lyon, and R. L. Garcea. 1998. Intercapsomeric disulfide bonds in papillomavirus assembly and disassembly. J. Virol. 722160-2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ozbun, M. A. 2002. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J. Virol. 7611291-11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozbun, M. A. 2002. Infectious human papillomavirus type 31b: purification and infection of an immortalized human keratinocyte cell line. J. Gen. Virol. 832753-2763. [DOI] [PubMed] [Google Scholar]
- 21.Patterson, N. A., J. L. Smith, and M. A. Ozbun. 2005. Human papillomavirus type 31b infection of human keratinocytes does not require heparan sulfate. J. Virol. 796838-6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pelkmans, L., T. Bürli, M. Zerial, and A. Helenius. 2004. Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell 118767-780. [DOI] [PubMed] [Google Scholar]
- 23.Pelkmans, L., and A. Helenius. 2002. Endocytosis via caveolae. Traffic 3311-320. [DOI] [PubMed] [Google Scholar]
- 24.Pelkmans, L., J. Kartenbeck, and A. Helenius. 2001. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 3473-483. [DOI] [PubMed] [Google Scholar]
- 25.Pyeon, D., P. F. Lambert, and P. Ahlquist. 2005. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc. Natl. Acad. Sci. USA 1029311-9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Querbes, W., B. A. O'Hara, G. Williams, and W. J. Atwood. 2006. Invasion of host cells by JC virus identifies a novel role for caveolae in endosomal sorting of noncaveolar ligands. J. Virol. 809402-9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts, R. L., M. A. Barbieri, K. M. Pryse, M. Chua, J. H. Morisaki, and P. D. Stahl. 1999. Endosome fusion in living cells overexpressing GFP-rab5. J. Cell Sci. 1123667-3675. [DOI] [PubMed] [Google Scholar]
- 28.Roberts, R. L., M. A. Barbieri, J. Ullrich, and P. D. Stahl. 2000. Dynamics of rab5 activation in endocytosis and phagocytosis. J. Leukoc. Biol. 68627-632. [PubMed] [Google Scholar]
- 29.Schelhaas, M., J. Malmström, L. Pelkmans, J. Haugstetter, L. Ellgaard, K. Grünewald, and A. Helenius. 2007. Simian virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 131516-529. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz, S. L., C. Cao, O. Pylypenko, A. Rak, and A. Wandinger-Ness. 2007. Rab GTPases at a glance. J. Cell Sci. 1203905-3910. [DOI] [PubMed] [Google Scholar]
- 31.Selinka, H.-C., T. Giroglou, and M. Sapp. 2002. Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions. Virology 299279-287. [DOI] [PubMed] [Google Scholar]
- 32.Sieczkarski, S. B., and G. R. Whittaker. 2005. Viral entry. Curr. Top. Microbiol. Immunol. 2851-23. [DOI] [PubMed] [Google Scholar]
- 33.Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell. Biol. 131-39. [DOI] [PubMed] [Google Scholar]
- 34.Smith, J. L., S. K. Campos, and M. A. Ozbun. 2007. Human papillomavirus type 31 uses a caveolin 1- and dynamin 2-mediated entry pathway for infection of human keratinocytes. J. Virol. 819922-9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stegmann, T., H. W. M. Morselt, J. Scholma, and J. Wilschut. 1987. Fusion of influenza virus in an intracellular acidic compartment measured by fluorescence dequenching. BBA Biomembr. 904165-170. [DOI] [PubMed] [Google Scholar]
- 36.Sturgis, E. M., and P. M. Cinciripini. 2007. Trends in head and neck cancer incidence in relation to smoking prevalence. Cancer 1101429-1435. [DOI] [PubMed] [Google Scholar]
- 37.Tsai, B. 2007. Penetration of nonenveloped viruses into the cytoplasm. Annu. Rev. Cell Dev. Biol. 2323-43. [DOI] [PubMed] [Google Scholar]
- 38.van der Bliek, A. M. 2005. A sixth sense for Rab5. Nat. Cell Biol. 7548-550. [DOI] [PubMed] [Google Scholar]
- 39.Wang, X., R. Kumar, J. Navarre, J. E. Casanova, and J. R. Goldenring. 2000. Regulation of vesicle trafficking in Madin-Darby canine kidney cells by Rab11a and Rab25. J. Biol. Chem. 27529138-29146. [DOI] [PubMed] [Google Scholar]
- 40.Wiethoff, C. M., H. Wodrich, L. Gerace, and G. R. Nemerow. 2005. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 791992-2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zerial, M., and H. McBride. 2001. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell. Biol. 2107-117. [DOI] [PubMed] [Google Scholar]