

Abstract
The protein encoded by open reading frame 50 (ORF50) of Kaposi's sarcoma-associated herpesvirus (KSHV) functions as a transcriptional activator and in lytic viral DNA replication to mediate the switch from latent viral infection to the lytic phase. Here we identify regulatory regions of ORF50 protein that independently control DNA binding and abundance of the protein. One region contains a DNA-binding inhibitory sequence (DBIS) located between amino acids (aa) 490 and 535 of ORF50. A cluster of basic amino acids in this sequence is important in inhibiting DNA binding. The DBIS can function at the N or C terminus or internally in the ORF50 protein. Since the DBIS is functional in ORF50 protein purified from Escherichia coli, it is likely to work through an intramolecular mechanism. The second regulatory region, a protein abundance regulatory signal (PARS), consists of two components. Component I of the PARS overlaps the DBIS but can be differentiated from the DBIS by specific substitution of basic amino acid residues. Component II of PARS is located between aa 590 and 650. Mutation or deletion of either component results in abundant expression of ORF50 protein. When the two-component PARS was fused to a heterologous protein, Glutathione S-transferase, the fusion protein was unstable. Mutations in the DBIS or PARS impair the capacity of ORF50 to activate direct and indirect target viral promoters. Since these overlapping regulatory motifs are located in the C-terminal transactivation domain, they are likely to be important in controlling many actions of ORF50 protein.
Many elaborate regulatory mechanisms operative in cells are mediated through DNA-binding transcription factors. To ensure the accuracy of gene expression, these transcription factors must be present at the right time, at the right place, and in the correct amount. Their DNA-binding activity and transcriptional activation functions must be tightly controlled to achieve the optimal expression of each downstream gene (10, 32).
Regulation of DNA binding and protein stability of transcription factors plays a crucial role in their functional activities. Several cellular transcription factors, such as Ets-1, p53, androgen receptor, TATA-binding protein, and IRF3, contain DNA-binding inhibitory sequences (DBISs) (12-14, 18, 19, 22, 42, 43). Complex protein-protein interactions or protein modifications may restore DNA-binding activity (25). In addition, many transcription factors are unstable in cells as a result of degradation by proteasome-mediated processes (33). The degradation signals found in these factors, including ATF6, E2F1, c-Fos, c-Jun, c-Myc, p53, HIF-1α, c-Rel, and STAT5, often overlap with their transcriptional activation domains (7, 23). These observations imply that multiple cis-regulatory regions in transcription factors reciprocally control their function.
The protein encoded by open reading frame 50 (ORF50) of the Kaposi's sarcoma-associated herpesvirus (KSHV) genome is a potent transcription activator with a DNA-binding domain and an activation domain (2, 3, 34). The 691-amino-acid (aa) KSHV ORF50 protein does not share obvious homology with cellular proteins. However, it is related to immediate-early transcriptional activator proteins of other gammaherpesviruses, such as ORF50a encoded by herpesvirus saimiri and Rta encoded by the BRLF1 gene of Epstein-Barr virus (EBV) (21, 31). The KSHV ORF50 protein has been shown to play a central role in the switch of the viral life cycle from latency to lytic replication. Ectopic expression of KSHV ORF50 is sufficient to disrupt viral latency and to activate a complete lytic replication cycle (11, 31). In addition to its role as a transcription activator, ORF50 protein also participates in directing the assembly of replication complexes by recruiting specific proteins required for lytic DNA synthesis (1, 36).
ORF50 activates downstream KSHV targets by at least two mechanisms: direct DNA binding and indirect access to DNA (4, 17). In transient-transfection assays, many viral promoters, including its own gene, polyadenylated nuclear (PAN) RNA, K12 (kaposin), ORF57, K6 (vMIP-1), K8 (K-bZIP), K9 (vIRF), ORF21 (thymidine kinase), K5, ORF6 (single-stranded DNA-binding protein), K14 (vOX-2), ORF74 (vGPCR), and K2 (vIL6), are activated by ORF50 protein (3, 5, 8, 9, 15, 20, 29, 35, 41). Some of these promoters are activated by ORF50 protein through a direct DNA-binding mechanism (3, 29, 30). DNA-binding-deficient ORF50 mutants which are defective in activating direct targets are nonetheless capable of activating some other promoters and driving the lytic cycle through interacting with ORF50 promoter itself (4). Thus, ORF50 protein must possess an elaborate mechanism for regulating its DNA binding function in cells.
Wild-type ORF50 protein expressed in mammalian cells shows a limited capacity to bind target DNA (2). However, we showed that mutations outside the DNA-binding domain of ORF50 protein (aa 1 to 390) markedly enhanced DNA-binding activity (2). A critical regulatory region was located within aa 520 to 535 of ORF50 protein, which contains a motif of basic amino acids KKRK. Basic-to-acidic substitutions of the KKRK motif in aa 527 to 530 of the ORF50 protein, such as ORF50(KK/EE) and ORF50(RK/DE), dramatically enhanced DNA-binding activity. The same mutations in the ORF50 gene also resulted in abundant expression of mutant proteins (2). In particular, a variant with accelerated electrophoretic mobility, which we designated ORF50B, was overexpressed after mutation of the KKRK motif.
Our previous studies left unanswered several questions that we attempt to address here. Can the amino acids of the KKRK motif which play a role in regulating DNA binding and those which affect protein abundance be clearly discriminated with point mutants of ORF50 protein? What are the boundaries of the functional region encompassing the KKRK motif that inhibits DNA binding and modulates the level of ORF50 protein? If we can identify these regulatory regions, are they position dependent or independent? What is the specificity of their action? Moreover, what are the possible mechanisms of inhibition of DNA binding and regulation of protein abundance controlled by these cis motifs?
Here, we identify two mobile overlapping regulatory regions encompassing the KKRK motif that independently control DNA binding and protein abundance. We designate these as a DBIS and a protein abundance regulatory signal (PARS). The DBIS is likely to operate by an intramolecular mechanism. The PARS consists of two components that are likely to destabilize ORF50 by promoting protein degradation.
MATERIALS AND METHODS
Cell culture and transfection.
HKB5/B5, a cell line formed by fusion of HH514-16 cells with 293T cells (3), was grown in RPMI 1640 medium with 8% fetal bovine serum. HKB5/B5 (2 × 106) cells in six-well plates were transfected with a constant amount of plasmid DNA (4 μg) by using the DMRIE-C reagent (Invitrogen) (3). In some experiments a 10 to 40 mM concentration of the proteasome inhibitor MG132, dissolved in dimethyl sulfoxide (DMSO), was applied to cells 4 h posttransfection, and the treated cells were incubated for 20 h.
Plasmid construction.
pCMV-FLAG-ORF50 and pCMV-FLAG-ORF50(KK/EE) have been described previously (2). Amino acid substitutions in ORF50(KK/AA), ORF50(KK/RR), ORF50(KRK/EDE), and ORF50(RSK/DSE) constructs were made by using a QuikChange site-directed mutagenesis kit (Stratagene). To make ORF50 C-terminal deletion mutants, various regions of the ORF50 gene were amplified from pCMV-FLAG-ORF50 or pCMV-FLAG-ORF50(KK/EE) by PCR and cloned into pFLAG-CMV-2 (Sigma) with EcoRI and XbaI sites. For overexpression of the recombinant ORF50 proteins in bacteria, a DNA fragment encoding ORF50 (aa 1 to 590) with or without the KK/EE mutation or a DNA fragment encoding ORF50 (aa 333 to 691) was PCR amplified and inserted into pET-22b (Novagen) at the NdeI and XhoI sites. The DNA fragments encoding putative ORF50 regulatory motifs were inserted into the 3′ or 5′ end of the ORF50 gene, which had been modified to create restriction enzyme sites at either end. A PstI site downstream of the ORF50 gene and a NotI site upstream of the ORF50 gene were used for cloning these DNA fragments. pE-R350 was constructed by inserting a DNA fragment encoding EBV Rta (aa 1 to 350) into pFLAG-CMV-2 with EcoRI and BglII sites. Similarly, the glutathione S-transferase (GST) or GST fusion construct was made by inserting the coding region into pFLAG-CMV-2.
The reporter plasmids PANp/E4-Luc, vMIP-1p/E4-Luc, and ORF57p/E4-Luc were constructed by inserting double-stranded annealed oligonucleotides encompassing the ORF50 response elements of PAN, vMIP-1, and ORF57 promoters (4) into the XhoI and NheI sites of pE4-Luc, a luciferase reporter containing an adenovirus E4 minimal promoter (3).
Northern blot analysis.
Total cellular RNAs from 107 transfected cells were prepared with an RNeasy kit (Qiagen), fractionated on 1% formaldehyde-agarose gels, and transferred to nylon membranes (Hybond-N+; Amersham Pharmacia Biotech). Detection of ORF50 mRNA has been described previously (2). Hybridization was carried out in 6× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 5× Denhardt solution, 0.5% sodium dodecyl sulfate (SDS), and 100 μg of salmon DNA per ml at 60°C overnight. Membranes were washed in 2× SSC-0.5% SDS once for 10 min and in 0.1× SSC-0.5% SDS three times for 25 min at 60°C.
Electrophoretic mobility shift assays (EMSAs).
Transfected HKB5/B5 cells were suspended in lysis buffer (0.42 M NaCl, 20 mM HEPES [pH 7.5], 25% glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 2 μg of aprotinin per ml). Cell lysates were centrifuged at 90,000 rpm for 15 min in a benchtop ultracentrifuge, and supernatants were collected and frozen. Annealed double-stranded PANp oligonucleotide was end labeled with 32P using T4 polynucleotide kinase. Binding reactions contained 15 μg of protein extract in a solution containing 10 mM HEPES (pH 7.5), 50 mM NaCl, 2 mM MgCl2, 2.5 μM ZnSO4, 0.5 mM EDTA, 1 mM dithiothreitol, 15% glycerol, and 0.5 to 1.0 μg of poly(dI-dC) in a total volume of 20 μl. For competition assays, nonradioactive competitor DNA was added to the initial reaction mix. Antibody to the FLAG tag (Sigma) or ORF50 peptide (aa 230 to 250) was used for supershifts.
Western blot analysis.
Cell protein extracts were mixed with 3× SDS gel loading buffer and boiled for 5 min before being loaded on 8% polyacrylamide gels. After immunoblotting with antibody to FLAG, immunoreactive polypeptides were detected with 125I-labeled protein A.
Luciferase assays.
HKB5/B5 cells (7 × 105) in 24-well plates were transfected with 0.5 μg of pCMV or plasmids expressing wild-type or mutant ORF50 proteins and 0.5 μg of reporter DNA. Cells were harvested 48 h after transfection. Each transfection experiment was repeated at least three times, and individual samples in each experiment were in duplicate. Preparation of cell lysates and measurement of luciferase activity were performed according to the manufacturer's protocol for the luciferase reporter assay system (Promega). The fold activation was calculated as the luciferase activity in the presence of ORF50 or ORF50 mutants divided by luciferase activity in the presence of control vector pCMV.
Purification of recombinant ORF50 proteins from bacteria.
For preparation of ORF50 proteins, E. coli BL21 cells harboring the plasmid encoding ORF50(1-590), ORFR50(1-590)(KK/EE), or ORF50(333-691) with six histidine residues at the C terminus were grown to exponential phase at 37°C. After 2 h induction with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside), bacterial cells were resuspended in a lysis buffer (50 mM NaH2PO4, 300 mM NaCl, and 10 mM imidazole [pH 8.0]). Recombinant ORF50 proteins were purified by using nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography (Qiagen). Briefly, the cell lysates were incubated with Ni-NTA agarose at 4°C for 2 h. After extensive washing with buffer containing 50 mM NaH2PO4, 300 mM NaCl, and 50 mM imidazole (pH 8.0), the recombinant ORF50 proteins were eluted with buffer containing 50 mM NaH2PO4, 300 mM NaCl, and 250 mM imidazole (pH 8.0).
RESULTS
Mutations of the KKRK motif in the regulatory region of ORF50 protein discriminate between its effect on protein expression and DNA binding in mammalian cells.
In a previous study, we identified a multifunctional region, present in aa 520 to 535 of ORF50 protein, that regulates protein expression and DNA binding (2). Mutational analyses revealed that a basic tetrapeptide (KKRK) motif in this regulatory region was critical for mediating the phenotypes of protein expression and DNA binding (2). As shown in Fig. 1, substitution of glutamic acids (E) for K527 and K528 in this motif, as in the mutant ORF50(KK/EE), markedly enhanced the abundance of ORF50B protein. ORF50B appears to be a form of ORF50 that is decreased in posttranslational modification, whereas ORF50A represents a hypermodified form (2). Since ORF50 mRNA levels were similar in human cells transfected with the wild type and KK/EE mutant (Fig. 1B), the enhanced expression of ORF50B was controlled at a posttranscriptional level. Increased ORF50 DNA-binding activity was also observed by EMSA with the KK/EE mutant (Fig. 1D). Thus, the KKRK basic motif in ORF50 exerts a pivotal role in regulating both the expression level and the DNA-binding function of ORF50 protein.
FIG. 1.

Amino acid substitutions in the KKRK motif discriminate its function in regulating protein abundance and DNA binding. (A) Diagram of wild-type ORF50 and ORF50 mutants. Putative domains are indicated as follows: LZ, leucine zipper; black bar, regulatory region; AD, activation domain. (B) Northern blot analysis of ORF50 mRNA in HKB5/B5 cells. At 48 h after transfection total RNA from transfected HKB5/B5 cells was analyzed by Northern blotting with a specific probe to detect ORF50 mRNA. Hybridization with H1 RNA of RNaseP served as a loading control. (C) Expression of ORF50 and ORF50 mutant proteins. Extracts of transfected HKB5/B5 cells were analyzed by immunoblotting with anti-FLAG antibody. (D) DNA-binding activity of ORF50 and ORF50 mutants. The same cell extracts shown in panel C were used in an EMSA. The probe in the EMSA was the ORF50 response element of the PAN promoter. Antibody to FLAG was used for supershift tests.
To attempt to discriminate the functions of basic amino acid residues of the KKRK motif that were crucial in regulating ORF50 expression and DNA binding, we mutated the first two lysines of the KKRK motif into alanines (A) or arginines (R). Substitutions of either alanines or arginines for lysines enhanced the expression of ORF50 mutant proteins in a manner similar to the KK/EE mutation (Fig. 1A and C). However, the KK/RR mutant ORF50 protein was abundant but bound DNA only weakly (Fig. 1D). Amino acid changes of the basic motif did not alter the level of ORF50 mRNA (Fig. 1B). These results lead to two important conclusions. First, a conservative basic-to-basic mutation in the KKRK motif still retained an inhibitory function for ORF50 DNA binding. Second, since all mutants were abundantly expressed, it appeared clear that lysines per se in the KKRK motif were crucial for regulating protein expression. The specific point mutations demonstrated that inhibition of DNA binding and ORF50 protein level were under independent control by the KKRK motif.
The C-terminal domain of ORF50 protein encompasses a two-component PARS.
In our previous experiments the DNA-binding inhibitory region was identified through a series of C-terminal deletions of ORF50 protein (2). However, we could not systematically compare the protein expression level of each C-terminal truncated ORF50 mutant protein because we lacked antibodies to the N-terminal region of ORF50. To solve this problem, we produced ORF50 C-terminal deletions in constructs with an N-terminal FLAG tag (Fig. 2A). As shown in Fig. 2B, the expression level of FLAG-tagged wild-type ORF50 protein (F-691) was lower than F-ORF50(KK/EE), as had been previously demonstrated comparing untagged wild-type and ORF50(KK/EE) mutant proteins (2). When the expression level of C-terminal deleted ORF50 proteins was analyzed, we found that deletion of the C-terminal 41 aa did not markedly change protein level compared to wild-type ORF50 protein (Fig. 2B). Both F-ORF50(F-691) and F-ORF50(F-650) were expressed at a low level. However, after deletion of 101 aa or more to aa 590, 564, or 490, the ORF50 deletion proteins became abundant (Fig. 2B). These results suggested that in addition to the KKRK motif, another regulatory component, located between aa 650 and 590, controlled protein abundance. To confirm this hypothesis, we made an internal deletion in ORF50 protein (Fig. 2C). The ORF50 mutant protein lacking aa 590 to 650, F-Δ590-650, was abundantly expressed (Fig. 2C). Thus, there are two components of the PARS in the C terminus of ORF50 protein. One component (I) is located in the KKRK-containing region; the other component (II) is located within aa 590 to 650.
FIG. 2.

Abundant ORF50 expression controlled by the KKRK motif is linked to a second component located in aa 590 to 650 of ORF50 protein. (A) Diagram of ORF50 and ORF50 deletion mutants. (B and C) Expression of ORF50 deletion mutant proteins without or with the KK/EE mutation. (D) DNA binding of ORF50 mutants was analyzed in EMSA using the same cell extracts as in panel C.
To determine whether the second component of PARS influenced DNA binding by ORF50 protein, we carried out the EMSA experiments using C-terminal deletion mutants with or without the KK/EE mutation (Fig. 2D). Whenever the KK/EE mutation was present in ORF50 proteins, the proteins displayed enhanced DNA binding. The enhanced DNA-binding activity conferred by the KK/EE mutation could be shown to be independent of protein expression. The protein expression level of constructs F-590 and F-Δ590-650, both of which lack component II of the PARS, was similar to that of their counterparts with the KK/EE mutation (Fig. 2C). Yet only constructs containing the KK/EE mutation bound DNA. These results confirm the conclusion that the KKRK basic motif located in the C terminus regulates DNA binding independently of abundant protein expression.
Inhibition of binding by ORF50 protein to DNA is mediated through an intrinsic mechanism.
The results from Fig. 1 and Fig. 2 showed that the expression level of ORF50 protein and its capacity to bind DNA were controlled independently, although the KKRK motif was essential for regulating both activities. The KKRK motif in ORF50 protein could regulate DNA binding by an intrinsic intramolecular mechanism or by interaction with other cellular proteins. To distinguish these alternatives, ORF50 proteins lacking and containing the KK/EE mutations were expressed and purified from E. coli (Fig. 3A). The ORF50(1-590) constructs were selected for these experiments because in earlier experiments in mammalian cells they were expressed to equal levels since they lacked the second essential component of PARS (Fig. 2B and C). ORF50(1-590) and ORF50(1-590)(KK/EE) were also expressed to equal levels in bacteria (Fig. 3A). Nonetheless, the ORF50(KK/EE) mutant displayed significantly stronger binding to the ORF50 response element in the PAN promoter than did the wild-type protein (Fig. 3B). The specificity of binding by ORF50(1-590)(KK/EE) was confirmed by oligonucleotide competition and supershifting with a specific antibody to ORF50. A 100-fold excess of cold duplex oligonucleotide containing the ORF50 response element efficiently competed for binding, while an oligonucleotide with a mutation in the ORF50 binding site (3) did not compete (Fig. 3C). The C-terminal region of ORF50 (aa 333 to 691) that itself contains the DNA-binding inhibitory region did not bind specifically to the PAN promoter (Fig. 3B). This protein was purified from E. coli in parallel with the ORF50(1-590) constructs and expressed to comparable levels (data not shown).
FIG. 3.

ORF50(1-590) purified from E. coli displays limited DNA-binding activity compared to its counterpart with the KK/EE mutation. (A) Coomassie blue staining and immunoblotting (WB) of purified ORF50(1-590) without or with the KK/EE mutation. (B) Binding of purified ORF50(1-590), ORF50(1-590)(KK/EE), or ORF50(333-691) to the PAN promoter. Increasing amounts (50, 150, 450, and 1,500 ng) of purified protein were used in an EMSA. (C) Binding specificity of purified ORF50(1-590) with the KK/EE mutation to DNA. Wild-type or mutant PAN elements served as a cold competitor (20×, 50×, and 100×) in EMSA. Antibody to ORF50(230-250) was used for supershift analysis.
ORF50(490-535) is a functional DBIS.
Although the experiments illustrated in Fig. 1 and 3 indicated that the KKRK-containing region of ORF50 protein was critical for inhibiting binding of ORF50 protein to DNA, we wished to define the boundary of a minimal DBIS that functioned in mammalian cells. Various portions of ORF50 protein were fused to the DNA-binding domain found in aa 1 to 390 (3) (Fig. 4). A functional DBIS was defined as a portion of ORF50 protein that would block binding of ORF50(1-390) to the high-affinity ORF50 binding site in the PAN promoter. Moreover, the KK/EE mutation in the transferred fragment should abolish the DNA-binding inhibitory function (Fig. 4A). Based on these criteria, the region spanning aa 490 to 535 constituted a minimal DBIS (Fig. 4A and C). This region overlapped component I of the PARS. Although ORF50(520-564) included the KKRK motif, it only slightly blocked DNA binding when fused to ORF50(1-390) (Fig. 4B;lane 5). The capacity of any chimera containing ORF50 protein aa 1 to 390 plus candidate inhibitory sequences to bind DNA was independent of protein expression. All of the constructs lacked component II of PARS, and all proteins were expressed to high levels.
FIG. 4.

aa 490 to 535 define a functional DBIS of ORF50 protein. (A) Summary of DNA-binding activity of ORF50 deletion mutants without or with the KK/EE substitution. NA, not applicable. (B and C, top panels) EMSAs were performed using cell extracts of HKB5/B5 cells transfected with the indicated plasmids. (Bottom panels) Expression of ORF50 deletion mutant proteins in cell extracts was detected by antibody to FLAG.
Additional basic motifs in the 46-aa DBIS of ORF50 contribute to the DNA-binding inhibitory function.
In addition to the KKRK motif (aa 527 to 530), other clusters of basic amino acids are also found in the DBIS; these include KRK (aa 516 to 518) and RSK (aa 520 to 522) (Fig. 5A). When these basic amino acids were mutated to aspartate (D) or glutamate (E) in the context of FLAG-ORF50(1-564) (Fig. 5B) or full-length ORF50 (Fig. 5C), they all restored DNA-binding activity. In the context of FLAG-ORF50(1-564), which lacks component II of PARS, all of the constructs were expressed equally (Fig. 5B, bottom panel). In contrast, in the context of full-length ORF50, only the KK/EE mutant (K527E and K528E) resulted in abundant expression of ORF50B (Fig. 5C, bottom panel). These additional basic amino acids in the DBIS regulate DNA binding but not protein abundance. These results further confirm the independent function of the DBIS and PARS.
FIG. 5.

Basic amino acids upstream of the KKRK motif contribute to the DNA-binding inhibitory function. (A) Amino acid sequence of the minimal DNA-binding inhibitory sequence in the ORF50 protein. The basic motifs in the regulatory region are boxed. (B and C, top panels) DNA binding of ORF50 mutants. EMSAs were carried out by ORF50 mutants in the context of ORF50(1-564) (B) or full-length ORF50 (C). The expression of ORF50 mutants detected by antibody to FLAG is shown in the bottom panels.
The 46-aa DBIS of ORF50 protein is dominant and functions at different positions in ORF50 protein.
To study the dominance and positional effects of the DBIS, ORF50(1-564) was used as a parental construct. This protein binds DNA very weakly or undetectably; the KK/EE mutation markedly enhances DNA binding without affecting protein expression (Fig. 2B, 4, 5B, 6B, and 6C). The addition of a second wild-type DBIS at the C terminus eliminated DNA binding of ORF50(1-564)(KK/EE). However, if the added DBIS contained a KK/EE mutation, the resulting chimeric protein still bound strongly to the ORF50 response element in the PAN promoter (Fig. 6B). A DBIS placed at the N terminus of the protein also markedly reduced the capacity of ORF50(1-564)(KK/EE) to bind DNA (Fig. 6C). The inhibitory effect was abolished when the KK/EE mutation was introduced into the DBIS. These experiments demonstrated that a single copy of the wild-type DBIS could function dominantly in cis. Moreover, the inhibitory effect was demonstrated when the DBIS was placed at the N or C terminus of the protein.
FIG. 6.

The 46-aa DBIS is dominant and position independent. (A) Diagram of ORF50 fusion constructs used in EMSA. (B and C, top panels) EMSAs were carried out with extracts prepared from HKB5/B5 cells transfected with the indicated plasmids. (B and C, bottom panels) Expression of ORF50 mutant proteins in cell extracts detected by antibody to FLAG.
To determine whether the DBIS could block DNA-binding activity of a related heterologous DNA-binding protein, it was fused to the DNA-binding domain of EBV Rta (aa 1 to 350) (6). Parental EBV Rta(1-350), EBV Rta(1-350)+DBIS, and EBV Rta(1-350)+DBIS(KK/EE) all bound with the same affinity to a duplex DNA oligonucleotide probe of the Rta response element from the EBV BMLF1 promoter (Fig. 7). This result suggested that the DBIS of ORF50 was specific to the ORF50 protein itself.
FIG. 7.

The DBIS of ORF50 protein does not alter the binding of EBV Rta to DNA. (A) Diagram of EBV Rta and fusion constructs. The DNA-binding domain (DBD) of EBV Rta was from aa 1 to 350 (E-R350). The coding sequences between aa 440 and 564 containing the DNA-binding inhibitory region of the ORF50 protein without or with the KK/EE mutation were added downstream of the Rta DBD. (B) EMSA using the Rta response element from the BMLF1 promoter as a probe. (C) Protein expression of the fusion constructs detected by antibody to FLAG.
Component I of the PARS is dominant over the KK/EE mutation and functions in a position-independent manner to reduce ORF50 expression.
The 46-aa DBIS in ORF50 protein overlaps component I of the PARS. Since the DBIS was dominant for inhibiting the DNA-binding function and functioned at different positions in ORF50 protein, similar experiments were carried out examining effects on protein expression of full-length ORF50 mutants (Fig. 8). An extra copy of the DBIS was appended downstream of full-length ORF50(KK/EE), which contains a mutated component I and a functional component II of PARS (Fig. 8). When the 46-aa DBIS was fused to ORF50(KK/EE), the expression level of the fusion protein was as low as the wild-type ORF50 protein (Fig. 8B, lane 4). However, the same construct with KK/EE mutations in the added DBIS expressed high level of the protein (Fig. 8B, lane 5). A 9-aa sequence containing the KKRK motif partially reduced expression of ORF50(KK/EE) (Fig. 8B, lane 6). However, the 4-aa sequence (KKRK) at the C terminus of ORF50(KK/EE) was not sufficient to mediate the reduction of ORF50(KK/EE) expression. Although the 46-aa DBIS containing component I of PARS was dominant in inhibiting the DNA-binding activity of the fusion constructs in an EMSA experiment, 9- and 4-aa peptides containing KKRK did not posses this inhibitory activity (Fig. 8C).
FIG. 8.

An extra copy of the 46-aa regulatory region of ORF50 protein mediates inhibition of expression of full-length ORF50 protein containing the KK/EE mutation. (A) Diagram of wild-type ORF50, ORF50(KK/EE), and constructs in which the minimal regulatory sequence or portions thereof were fused to FLAG-tagged ORF50(KK/EE). (B) Protein expression of ORF50 derivatives detected by anti-FLAG antibody. (C) An EMSA was performed with the extracts shown in panel B.
To determine whether component I (aa 490 to 535) of PARS was functional at the N terminus of full-length ORF50, it was transferred upstream of ORF50(KK/EE). A second copy of component I (aa 490 to 535) appended at the N terminus of ORF50(KK/EE) destabilized the fusion protein (Fig. 9B, lane 4), but the addition of component I with a KK/EE mutation did not promote ORF50(KK/EE) degradation (Fig. 9B, lane 5). A second wild-type copy of the regulatory region also markedly reduced DNA binding of F564(KK/EE), whereas a second copy of the regulatory region with the KK/EE mutation did not alter F564(KK/EE) binding to DNA.
FIG. 9.

The 46-aa regulatory region promotes autodegradation when positioned at the N terminus of ORF50(KK/EE). (A) Diagram of FLAG-tagged ORF50 constructs. A second copy of the regulatory sequence without or with the KK/EE mutant was added to full-length ORF50(KK/EE). The minimal DBIS (or component I of PARS) is shaded. Protein expression and the DNA-binding function of the ORF50 constructs were analyzed by immunoblotting (B) and by EMSA (C).
The results of Fig. 6, 7, 8 through 9 can be summarized as follows: the region of ORF50 from aa 490 to 535 contains the DBIS and PARS component I. An single extra wild-type copy of this region is dominant over the KK/EE mutation or deletions that enhance ORF50 DNA binding (Fig. 6) or protein expression (Fig. 8 and 9). Moreover, both regulatory activities of the region were functional at either terminus of the protein (Fig. 6, 8, and 9). The DBIS is functional on a minimal DNA-binding domain of KSHV ORF50 but not on a comparable domain of EBV Rta (Fig. 7).
The entire PARS of ORF50 protein is transferable to a heterologous protein.
Low-level expression of ORF50 protein, presumably as the result of destabilization, relies on two components located between aa 490 and 535 (component I) and aa 590 and 650 (component II). To determine whether the protein destabilization function of PARS could be transferred to a heterologous protein, we first created FLAG-tagged constructs in which the N-terminal 20 amino acids of ORF50, containing the nuclear localization signal (NLS), were fused to ORF50(490-691). These constructs, encompassing both components of the PARS, were made without or with the KK/EE mutation (Fig. 10A). GST protein in frame with the NLS of ORF50 protein served as a target (Fig. 10). This experiment demonstrated that the PARS of ORF50 could function in cis on a heterologous protein. Fusion of GST to the PARS resulted in low-level expression of the chimeric protein (GST+PARS). When the PARS contained the KK/EE mutation, higher levels of the GST fusion protein were detected. Proteins containing the ORF50 NLS and PARS alone, without GST, were comparably stable, with or without the KK/EE mutation. These results suggested that GST contains a target for the PARS, which is not present in the PARS itself.
FIG. 10.

The C-terminal 200 aa of ORF50 protein inhibits expression of a heterologous protein. (A) Diagram of FLAG-tagged ORF50 deletion mutants, FLAG-tagged GST, and chimeric GST-ORF50. The constructs contained ORF50 aa 490 to 691, which encompasses both components of the PARS. (B) Expression of ORF50 deletions and GST fusion proteins detected by anti-FLAG antibody.
A proteasome inhibitor reproduces effects of mutation in component I of PARS.
One possible role of the PARS might be to target ORF50 protein for degradation by the proteasome pathway. In wild-type ORF50 the “A” form is more abundant than the “B” form, whereas in the KK/EE mutant the converse is true (Fig. 1C and Fig. 2C, compare lanes 2 and 3). The KK/EE mutation might protect ORF50 from degradation via the proteasome. To investigate this possibility, wild-type ORF50 and ORF50 (KK/EE) were expressed in the absence or presence of the proteasome inhibitor MG132 (Fig. 11). MG132 markedly enhanced the ratio of ORF50B to ORF50A. This enhancement was 3- to 4-fold for wild-type ORF50 protein and 1.5- to 2.7-fold for the KK/EE mutant. These results are consistent with the hypothesis that relatively low levels of wild-type ORF50B protein could be due to degradation through a proteasome pathway. The experiments also suggest that the KK/EE mutation may not completely protect against degradation by the proteasome since the inhibitor also increased the ORF50B/ORF50A ratio of the mutant protein.
FIG. 11.
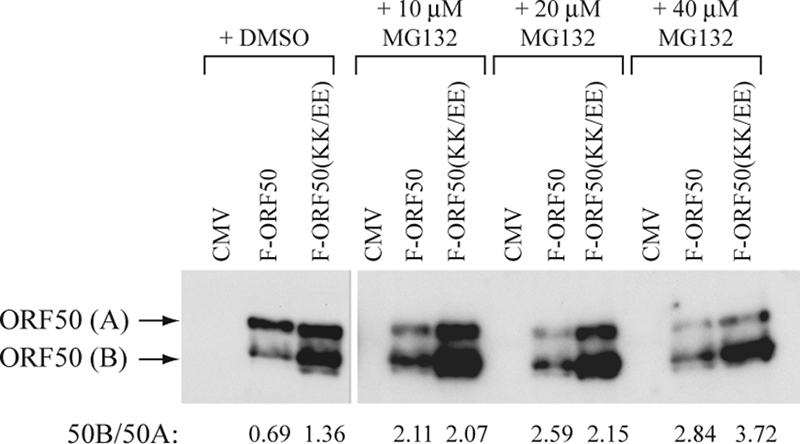
A proteasome inhibitor stabilizes ORF50B expression. HKB5/B5 cells were transfected with plasmids expressing FLAG-tagged wild-type ORF50 or KK/EE mutant. Four hours after transfection, cells were suspended in medium containing DMSO or different amounts of MG132 dissolved in DMSO. After 20 h cell the lysates were resolved by SDS-polyacrylamide gel electrophoresis and analyzed by immunoblotting with anti-FLAG antibody. The ratio of expression of ORF50B to ORF50A in each sample was determined by densitometry.
Effect of mutations in the DIBS and PARS on the capacity of ORF50 to activate different classes of promoters of KSHV lytic cycle genes.
There are two classes of KSHV lytic cycle gene promoters. One class, represented by the promoters of PAN and kaposin, is activated by ORF50 binding directly to DNA (3). Another class, represented by promoters of vMIP-1 and ORF57, is regulated by ORF50 protein interacting with RBPJK (4). Figure 12 shows that mutations that increase the abundance of ORF50 protein and mutations that enhance DNA binding by ORF50 protein both reduce the capacity of the protein to activate direct and indirect target promoters. For four mutants that enhance DNA binding, this reduction in activity was 68, 51, and 64% for the PAN, vMIP-1, and ORF57 promoters, respectively. The mutant KK/RR, which was enhanced in protein abundance but not altered in DNA binding, was reduced in activity by 35 to 45% on the three promoters. These data indicate that both the DBIS and the PARS are important for optimal biologic function of the ORF50 protein.
FIG. 12.

Activation of viral lytic cycle promoters by wild-type and mutant ORF50 proteins. The target promoters containing ORF50 response elements were fused to the minimal adenovirus E4 promoter and luciferase. The viral promoters were PAN (A), a direct target, and vMIP-1 (B) and ORF57 (C), which are activated via interactions with RBP-Jκ. Whether the mutation increased (+) or did not affect (−) ORF50 abundance or DNA binding relative to the wild type is indicated below each mutant. The values of fold activation represent at least three transfections with duplicate samples in each transfection.
DISCUSSION
ORF50 protein of KSHV is a multifunctional transcription factor and replication protein that plays a central role in the switch of the viral life cycle from latency to lytic replication (11, 31). To achieve optimal KSHV lytic-cycle gene expression and viral DNA synthesis, the many functions of the ORF50 protein must be subject to exquisite control. In the present study, we show that the DNA-binding activity and protein abundance of ORF50 protein are controlled by several cis-regulatory motifs (Fig. 1 and 2). One regulatory region, a DBIS that represses the DNA-binding function of ORF50, was mapped to aa 490 to 535 (Fig. 4). The same region overlaps the essential component I of the PARS (Fig. 1, 2, 8, and 9). Destabilization or other mechanisms of maintaining low levels of ORF50 protein requires a second cis-component located within aa 590 to 650 (Fig. 2). Although the region between aa 490 and 535 (DBIS; component I of PARS) regulates both DNA binding and abundance of ORF50 protein, these phenomena are separable based on specific point mutants and chimeras (Fig. 1, 2, 4, and 5 and Table 1). Furthermore, both the DBIS and the component I of PARS in ORF50 are functional in a position-independent manner (Fig. 6, 8, and 9). The entire functional PARS can be transferred to a heterologous protein. Mutations that inactivate the DBIS or PARS reduce the activity of ORF50 protein.
TABLE 1.
Summary of abundance and DNA-binding activity of ORF50 proteins with deletions and point mutations
| Construct | ORF50 aa | Point mutant(s) | Abundant ORF50 proteina | DNA bindingb | Figure(s) |
|---|---|---|---|---|---|
| F-ORF50/F-691 | 1-691 | No | No | No | 1, 2 |
| F-ORF50/F-691 (KK/EE) | 1-691 | K527E, K528E | Yes | Yes | 1, 2 |
| F-ORF50(KK/AA) | 1-691 | K527A, K528A | Yes | Yes | 1 |
| F-ORF50(KK/RR) | 1-691 | K527R, K528R | Yes | No | 1 |
| F-650 | 1-650 | No | No | No | 2 |
| F-650(KK/EE) | 1-650 | K527E, K528E | Yes | Yes | 2 |
| F-590 | 1-590 | No | Yes* | No | 2 |
| F-590(KK/EE) | 1-590 | K527E, K528E | Yes* | Yes | 2 |
| F-564 | 1-564 | No | Yes* | Trace | 2, 4, 5 |
| F-564(KK/EE) | 1-564 | K527E, K528E | Yes* | Yes | 2, 4, 5 |
| F-490 | 1-490 | No | Yes* | (Yes) | 2 |
| F-D590-650 | 1-589; 651-691 | No | Yes* | No | 2 |
| F-D590-650(KK/EE) | 1-589; 651-691 | K527E, K528E | Yes* | Yes | 2 |
| F-390 | 1-390 | No | Yes* | Yes | 4 |
| F-390+(440-564) | 1-390; 440-564 | No | (Yes*)c | (No) | 4 |
| F-390+(440-564)(KK/EE) | 1-390; 440-564 | K527E, K528E | (Yes*) | (Yes) | 4 |
| F-390+(490-564) | 1-390; 440-564 | No | Yes* | Weak | 4 |
| F-390+(490-564)(KK/EE) | 1-390; 490-564 | K527E, K528E | Yes* | Yes | 4 |
| F-390+(520-564) | 1-390; 520-564 | No | Yes* | Yes | 4 |
| F-390+(520-564)(KK/EE) | 1-390; 520-564 | K527E, K528E | Yes* | Yes | 4 |
| F-390+(535-564) | 1-390; 535-564 | No | Yes* | Yes | 4 |
| F-390+(490-535) | 1-390; 490-535 | No | Yes* | Weak | 4 |
| F-390+(490-535)(KK/EE) | 1-390; 490-535 | K527E, K528E | Yes* | Yes | 4 |
| F-564(KRK/EDE) | 1-564 | K516E, R517D, K518E | Yes* | Yes | 5 |
| F-564(RSK/DSE) | 1-564 | R520D, K522E | Yes* | Yes | 5 |
| F-ORF50(KRK/EDE) | 1-691 | K516E, R517D, K518E | No | Yes | 5 |
| F-ORF50(RSK/DSE) | 1-691 | R520D, K522E | No | Yes | 5 |
| F-564(KK/EE)+IS | 1-564; 490-535 | K527E, K528E | Yes* | No | 6 |
| F-564(KK/EE)+IS(KK/EE) | 1-564; 490-535 | K527E, K528E (2×)d | Yes* | Yes | 6 |
| F-IS + 564(KK/EE) | 490-535; 1-564 | K527E, K528E | Yes* | Weak | 6 |
| F-IS(KK/EE)+564(KK/EE) | 490-535; 1-564 | K527E, K528E (2×) | Yes* | Yes | 6 |
| F-ORF50(KK/EE)+IS | 1-691; 490-435 | K527E, K528E | No | No | 8 |
| F-ORF50(KK/EE)+IS-E | 1-691; 490-435 | K527E, K528E (2×) | Yes | Yes | 8 |
| F-ORF50(KK/EE)+KK9 | 1-691; 525-533 | K527E, K528E | No | Yes | 8 |
| F-ORF50(KK/EE)+EE9 | 1-691; 525-533 | K527E, K528E (2×) | Yes | Yes | 8 |
| F-ORF50(KK/EE)+KKRK | 1-691; 527-530 | K527E, K528E | Yes | Yes | 8 |
| F-ORF50(KK/EE)+EERK | 1-691; 527-530 | K527E, K528E (2×) | Yes | Yes | 8 |
| F-N-IS-ORF50(KK/EE) | 490-535; 1-691 | K527E, K528E | No | Weak | 9 |
| F-N-IS(KK/EE)-ORF50(KK/EE) | 490-535; 1-691 | K527E, K528E (2×) | Yes | Yes | 9 |
| F-PARS | 1-20; 490-691 | No | Yes* | NAe | 10 |
| F-PARS(KK/EE) | 1-20; 490-691 | K527E, K528E | Yes* | NA | 10 |
*, We were unable to determine whether the abundant form of ORF50 expressed by truncation mutants was ORF50A or ORF50B.
“(Yes)” and “(No)” data for DNA binding are not shown in this study.
“(Yes*)” data for protein abundance are not shown in this study.
2×, Two occurrences.
NA, not applicable.
Regulation of ORF50's DNA-binding function.
The ORF50 transcription factor protein contains a basic DNA-binding domain located between aa 1 and 390. Although the DNA-binding domain of ORF50 protein is important for its transactivation function, direct binding to target DNA is not the exclusive or dominant strategy utilized by ORF50 to activate its responsive genes (4). Only a few genes, including PAN and K12, are classified as direct targets of ORF50. Most ORF50 responsive genes seem to be regulated through an indirect DNA-binding mechanism (4). Thus, the non-DNA-binding form of ORF50 protein may be predominant in lytically infected cells. Our discovery that wild-type ORF50 protein expressed in mammalian cells does not display detectable DNA-binding capacity in vitro is consistent with this hypothesis (2). We now show that inhibition of ORF50 DNA-binding activity is controlled by a cis-active motif, DBIS (Fig. 1 and 4).
Several possibilities may explain why the DBIS in cis inhibits the DNA-binding function of ORF50 protein. First, the DBIS may interact with cellular repressors that block the DNA-binding function of ORF50. Second, the DBIS may promote ORF50 posttranslational modifications, which mediate inhibition of the DNA-binding function. Third, the DBIS may autoregulate the DNA-binding function through intrinsic intramolecular repression. When we analyzed the ORF50 proteins purified from E. coli, we found that wild-type ORF50(1-590) still displayed limited DNA-binding capability compared to its KK/EE counterpart (Fig. 3). These results suggest that inhibition of DNA binding by wild-type ORF50 was independent of eukaryotic cellular proteins, including those that interact with ORF50 protein or those that confer posttranslational modifications of ORF50 protein. Thus, intrinsic intramolecular repression is likely to be the major mechanism for inhibiting DNA binding by ORF50.
Thus far, we do not understand the details of the mechanism by which the DBIS intrinsically inhibits the function of the ORF50's DNA-binding domain. Three possible mechanisms include the following: (i) the DBIS may interfere with ORF50 homodimerization, which is a prerequisite for DNA binding; (ii) the DBIS may directly interact with the DNA-binding domain and inhibit its DNA-binding function; and (iii) the basic DBIS may possess a nonspecific DNA-binding activity that competes for specific binding by ORF50's DNA-binding domain. There is a stretch of basic residues found within aa 516 to 530 of the DBIS. The sequence in this region is KRKQRSKERSSKKRK; 10 of 15 amino acids are basic (indicated in italics). In addition to the KKRK motif, this sequence contains two upstream clusters of the basic amino acids KRK and RSK. Acidic amino acid substitutions in all three motifs restored DNA-binding activity (Fig. 5), while basic-to-basic substitution in the KKRK motif still retained the DNA-binding inhibitory function of ORF50 (Fig. 1D). These results imply that basic residues are critical for inhibition of ORF50 binding to DNA. The importance of basic amino acids in the function of the DBIS is consistent with the hypothesis that the DBIS possesses a general nonspecific DNA-binding activity that overrides the specific binding of ORF50 protein to DNA.
A DBIS is found in a lysine- and arginine-rich motif at the extreme C terminus of the p53 protein. This basic region functions as a sequence-nonspecific nucleic acid-binding region. In the “sliding model” of regulation of DNA binding by p53 the C-terminal basic region allows p53 to search DNA for its specific response elements that are ultimately bound in a sequence-specific manner. The basic C-terminal domain also reduces the stability of p53 protein bound specifically to DNA (16, 28).
The results from Fig. 4 and 6 showed that the 46-aa DBIS was functional at multiple positions in the ORF50 protein. An extra wild-type copy was dominant over a single copy with the KK/EE mutation. Such a strong inhibitory region may repress the DNA-binding function of other transactivators. The Rta protein of EBV is a homolog of KSHV ORF50 protein, which controls the lytic switch of EBV from latency (26, 27). Recently, a DNA-binding inhibitory region has been identified in the C terminus of EBV Rta (6). Although the KSHV DBIS was active in ORF50 protein (Fig. 4 and 6), it did not affect the DNA binding of EBV Rta protein (Fig. 7). This result suggests that the ORF50 DBIS may specifically regulate ORF50 DNA binding, although a broader range of DNA-binding target proteins needs to be studied.
Regulation of abundance of ORF50 protein.
In almost all unstable transcription factors found in eukaryotes and eubacteria, transcriptional activation domains and protein degradation signals overlap (23, 32). Our results showed that the PARS of ORF50 is located in the C-terminal activation domain (Fig. 2, 8, and 9). There are two components of PARS in ORF50 protein (Fig. 2). Mutations in either component led to abundant expression of ORF50 protein. Recently, Yu et al. reported that ORF50 protein could function as an ubiquitin ligase (E3) and autoregulate its own polyubiquitination and stability. The destabilization signal characterized by Yu et al. was located in a Cys-plus-His-rich region (aa 118 to 207) in the N-terminal domain (40). Since this region does not overlap either of the elements of PARS that we have defined, there may be two or more destabilization systems in ORF50 protein. As shown in Fig. 10, deletion of the N-terminal region (aa 20 to 490) in ORF50 protein, F-PARS or F-PARS(KK/EE), resulted in stable protein expression at a level similar to the GST protein. However, when the GST protein was fused to the C-terminal PARS (encompassing component I plus component II), the resulting level of protein expression was decreased compared to its KK/EE counterpart (Fig. 10). These results suggest that the C-terminal destabilization signal of ORF50 protein is critical and sufficient to promote the degradation of an unrelated protein.
Ubiquitin-mediated proteolysis has been proposed to play a central role for protein degradation (24). Regulation of ORF50 protein stability may be controlled by a ubiquitin-mediated mechanism. Thus, a proposed hypothesis is that one or more lysines in component I of the PARS may function as an acceptor site for ubiquitination, and a ubiquitin ligase (E3) or ubiquitin-conjugating enzymes (E2) may dock on component II. Although ORF50 protein could be demonstrated to be ubiquitinated in vitro (40), we have not yet been able to demonstrate ubiquitination of ORF50 protein in vivo. Based on the current model of protein ubiquitination (37), spacing between the enzyme-docking signal and acceptor sites for ubiquitin should be critical for the polyubiquitination of target proteins. However, our results showed that component I of PARS functioned to promote protein destabilization not only at its natural location but also at either the N or the C terminus of ORF50 protein (Fig. 8 and 9). It is unclear whether the fully folded conformation of ORF50 allows the accessibility of the docking enzymes to the KKRK motif or whether degradation of ORF50 is mediated through another mechanism that does not involve ubiquitin.
The results presented in Fig. 11 suggest that the mutations K527E/K528E in component I of PARS impair proteasome-mediated degradation of ORF50B. Expression of wild-type ORF50B protein was markedly enhanced relative to ORF50A in the presence of the proteasome inhibitor MG132. The experiments, however, do not exclude mechanisms other than regulation of protein stability to account for enhanced ORF50 protein expression when the PARS is mutated. Amino acid sequences themselves, interactions of the nascent polypeptide with other proteins, or interaction of the mRNA with micro RNAs may influence the translation rate (38, 39).
How the DBIS and PARS might impact the biological functions of ORF50.
As shown previously (4), ORF50 mutants with enhanced DNA-binding activity and increased protein expression were somewhat diminished in their overall transcriptional function whether assayed by reporters or by the capacity to activate expression of viral early lytic genes such as PAN. We have considered several possibilities to explain the defect of the ORF50 mutants in activating the PAN promoter: (i) the KK/EE mutant might have aberrant location in the cell, (ii) the KK/EE mutant might impair the transcriptional activation domain, (iii) the expression of ORF50B might affect protein-protein interaction or possess dominant effects, and (iv) increased DNA binding affinity of the mutants might alter the on-off rate.
The basic KKRK motif could be a NLS, and mutation of the KKRK motif may decrease importation of ORF50 protein into the nucleus. However, analysis of fractionated cell lysates showed that ORF50 protein with the KK/EE mutation did not influence its location in nucleus (data not shown). Furthermore, the ORF50(KK/EE) mutant displayed enhanced DNA binding to the PAN and K12 promoters in ChIP assays (2). Thus, loss of the transcription function of ORF50 mutants, at least for activating the PAN gene, is not due to sequestration of ORF50 protein in the cytoplasm or to failure to interact with target promoters.
Since the stretch of basic amino acids (516 to 530) overlaps the C-terminal activation domain, mutation of the KKRK, KRK, or RSK motifs may decrease an essential protein-protein interaction with a coactivator or a component of the basal transcriptional machinery. Thus, weaker transcription function of the ORF50 mutants may be due to impairment of the function of the activation domain. Mutations in these three motifs which increase protein abundance or enhance DNA binding or both significantly impair the activity of the protein (Fig. 12). These results show that both the PARS and the DBIS must be intact for optimal transcriptional activity.
The events of gene activation and activator degradation may be coupled. A growing body of evidence shows that nonubiquitylated activators are stable but inactive (7, 23). The connection between transcription and proteolysis raises the possibility that abundant protein expression of the ORF50 mutants may result in accumulation of nonfunctional ORF50 protein. The extremely high-affinity binding of ORF50 protein to DNA may reduce the on-off rate and usage of the bound protein.
In conclusion, we provide here evidence that emphasizes that DNA binding, protein abundance, and transcriptional activation function of the ORF50 protein are tightly controlled and linked. An optimal level of protein expression and DNA-binding activity appears to be required for maximal biologic activity. The cis-regulatory motifs we identified in ORF50 protein may represent general phenomena. Other transcription factors may posses similar but as-yet-unidentified motifs to control DNA binding, protein stability, and gene activation. Thus, understanding the mechanisms that regulate the ORF50 protein is not only relevant to the oncogenic human gammaherpesviruses, it has important implications for understanding the control of cellular transcription factors.
Acknowledgments
This study was supported by grants CA16038 and CA12055 from the National Institutes of Health to G.M. and in part by medical research grant CMRPD650012 from the Chang-Gung Memorial Hospital (Taiwan) and by grant NSC95-2320-B-182-054-MY3 from the National Science Council (Taiwan) to P.-J.C.
We thank Jill Countryman, Ayman El-Guindy, and Jinajiang Ye for helpful comments on the manuscript.
Footnotes
Published ahead of print on 23 July 2008.
REFERENCES
- 1.AuCoin, D. P., K. S. Colletti, S. A. Cei, I. Papouskova, M. Tarrant, and G. S. Pari. 2004. Amplification of the Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 318542-555. [DOI] [PubMed] [Google Scholar]
- 2.Chang, P. J., and G. Miller. 2004. Autoregulation of DNA binding and protein stability of Kaposi's sarcoma-associated herpesvirus ORF50 protein. J. Virol. 7810657-10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang, P. J., D. Shedd, L. Gradoville, M. S. Cho, L. W. Chen, J. Chang, and G. Miller. 2002. Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol. 763168-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang, P. J., D. Shedd, and G. Miller. 2005. Two subclasses of Kaposi's sarcoma-associated herpesvirus lytic cycle promoters distinguished by open reading frame 50 mutant proteins that are deficient in binding to DNA. J. Virol. 798750-8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, J., K. Ueda, S. Sakakibara, T. Okuno, and K. Yamanishi. 2000. Transcriptional regulation of the Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor gene. J. Virol. 748623-8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, L. W., P. J. Chang, H. J. Delecluse, and G. Miller. 2005. Marked variation in response of consensus binding elements for the Rta protein of Epstein-Barr virus. J. Virol. 799635-9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins, G. A., and W. P. Tansey. 2006. The proteasome: a utility tool for transcription? Curr. Opin. Genet. Dev. 16197-202. [DOI] [PubMed] [Google Scholar]
- 8.Deng, H., M. J. Song, J. T. Chu, and R. Sun. 2002. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 768252-8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng, H., A. Young, and R. Sun. 2000. Auto-activation of the rta gene of human herpesvirus-8/Kaposi's sarcoma-associated herpesvirus. J. Gen. Virol. 81(Pt. 12)3043-3048. [DOI] [PubMed] [Google Scholar]
- 10.Ejkova, E., and W. P. Tansey. 2002. Old dogs and new tricks: meeting on mechanisms of eukaryotic transcription. EMBO Rep. 3219-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gradoville, L., J. Gerlach, E. Grogan, D. Shedd, S. Nikiforow, C. Metroka, and G. Miller. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 746207-6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagman, J., and R. Grosschedl. 1992. An inhibitory carboxyl-terminal domain in Ets-1 and Ets-2 mediates differential binding of ETS family factors to promoter sequences of the mb-1 gene. Proc. Natl. Acad. Sci. USA 898889-8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hupp, T. R., A. Sparks, and D. P. Lane. 1995. Small peptides activate the latent sequence-specific DNA binding function of p53. Cell 83237-245. [DOI] [PubMed] [Google Scholar]
- 14.Jayaraman, J., and C. Prives. 1995. Activation of p53 sequence-specific DNA binding by short single strands of DNA requires the p53 C terminus. Cell 811021-1029. [DOI] [PubMed] [Google Scholar]
- 15.Jeong, J., J. Papin, and D. Dittmer. 2001. Differential regulation of the overlapping Kaposi's sarcoma-associated herpesvirus vGCR (orf74) and LANA (orf73) promoters. J. Virol. 751798-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko, L. J., and C. Prives. 1996. p53: puzzle and paradigm. Genes Dev. 101054-1072. [DOI] [PubMed] [Google Scholar]
- 17.Liang, Y., J. Chang, S. J. Lynch, D. M. Lukac, and D. Ganem. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jκ (CSL), the target of the Notch signaling pathway. Genes Dev. 161977-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin, R., Y. Mamane, and J. Hiscott. 1999. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol. Cell. Biol. 192465-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu, G. Z., H. Wang, and Z. Wang. 2003. Identification of a highly conserved domain in the androgen receptor that suppresses the DNA-binding domain-DNA interactions. J. Biol. Chem. 27814956-14960. [DOI] [PubMed] [Google Scholar]
- 20.Lukac, D. M., L. Garibyan, J. R. Kirshner, D. Palmeri, and D. Ganem. 2001. DNA binding by Kaposi's sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 756786-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF50 transactivator, a homolog of the EBV R protein. Virology 252304-312. [DOI] [PubMed] [Google Scholar]
- 22.Luo, J., M. Li, Y. Tang, M. Laszkowska, R. G. Roeder, and W. Gu. 2004. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1012259-2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muratani, M., and W. P. Tansey. 2003. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell. Biol. 4192-201. [DOI] [PubMed] [Google Scholar]
- 24.Pickart, C. M. 2001. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70503-533. [DOI] [PubMed] [Google Scholar]
- 25.Pufall, M. A., and B. J. Graves. 2002. Autoinhibitory domains: modular effectors of cellular regulation. Annu. Rev. Cell Dev. Biol. 18421-462. [DOI] [PubMed] [Google Scholar]
- 26.Ragoczy, T., and G. Miller. 2001. Autostimulation of the Epstein-Barr virus BRLF1 promoter is mediated through consensus Sp1 and Sp3 binding sites. J. Virol. 755240-5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ragoczy, T., and G. Miller. 1999. Role of the Epstein-Barr virus RTA protein in activation of distinct classes of viral lytic cycle genes. J. Virol. 739858-9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sauer, M., A. C. Bretz, R. Beinoraviciute-Kellner, M. Beitzinger, C. Burek, A. Rosenwald, G. S. Harms, and T. Stiewe. 2008. C-terminal diversity within the p53 family accounts for differences in DNA binding and transcriptional activity. Nucleic Acids Res. 361900-1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song, M. J., H. J. Brown, T. T. Wu, and R. Sun. 2001. Transcription activation of polyadenylated nuclear RNA by Rta in human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus. J. Virol. 753129-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song, M. J., H. Deng, and R. Sun. 2003. Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 779451-9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 9510866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tansey, W. P. 2001. Transcriptional activation: risky business. Genes Dev. 151045-1050. [DOI] [PubMed] [Google Scholar]
- 33.Thomas, D., and M. Tyers. 2000. Transcriptional regulation: kamikaze activators. Curr. Biol. 10R341-R343. [DOI] [PubMed] [Google Scholar]
- 34.Wang, S., S. Liu, M. Wu, Y. Geng, and C. Wood. 2001. Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF50 gene product contains a potent C-terminal activation domain which activates gene expression via a specific target sequence. Arch. Virol. 1461415-1426. [DOI] [PubMed] [Google Scholar]
- 35.Wang, S., S. Liu, M. H. Wu, Y. Geng, and C. Wood. 2001. Identification of a cellular protein that interacts and synergizes with the RTA (ORF50) protein of Kaposi's sarcoma-associated herpesvirus in transcriptional activation. J. Virol. 7511961-11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang, Y., H. Li, M. Y. Chan, F. X. Zhu, D. M. Lukac, and Y. Yuan. 2004. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 788615-8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu, G., G. Xu, B. A. Schulman, P. D. Jeffrey, J. W. Harper, and N. P. Pavletich. 2003. Structure of a b-TrCP1-Skp1-b-catenin complex: destruction motif binding and lysine specificity of the SCFb-TrCP1 ubiquitin ligase. Mol. Cell 111445-1456. [DOI] [PubMed] [Google Scholar]
- 38.Yin, Y., B. Manoury, and R. Fahraeus. 2003. Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science 3011371-1374. [DOI] [PubMed] [Google Scholar]
- 39.Yin, Y., C. W. Stephen, M. G. Luciani, and R. Fahraeus. 2002. p53 stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat. Cell Biol. 4462-467. [DOI] [PubMed] [Google Scholar]
- 40.Yu, Y., S. E. Wang, and G. S. Hayward. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 2259-70. [DOI] [PubMed] [Google Scholar]
- 41.Zhang, L., J. Chiu, and J. C. Lin. 1998. Activation of human herpesvirus 8 (HHV-8) thymidine kinase (TK) TATAA-less promoter by HHV-8 ORF50 gene product is SP1 dependent. DNA Cell Biol. 17735-742. [DOI] [PubMed] [Google Scholar]
- 42.Zhao, X., and W. Herr. 2003. Role of the inhibitory DNA-binding surface of human TATA-binding protein in recruitment of human TFIIB family members. Mol. Cell. Biol. 238152-8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao, X., L. Schramm, N. Hernandez, and W. Herr. 2003. A shared surface of TBP directs RNA polymerase II and III transcription via association with different TFIIB family members. Mol. Cell 11151-161. [DOI] [PubMed] [Google Scholar]