

Abstract
Emerging studies suggest an important role for the innate immune response in replication-defective adenovirus (Ad)-mediated acute liver toxicity. Specifically, classical innate immune cells (including NK cells, neutrophils, and Kupffer cells) have all been implicated in the development of Ad-mediated acute liver toxicity. The nonclassical innate immune T cell, the γδT cell, has been implicated in the pathophysiology of several viral infections that predominantly affect the mucosa and brain, but the specific role in the pathology of AdLacZ-mediated acute liver inflammation and injury as well as accompanying vector clearance is largely unknown. In the present study, we demonstrated that a CXCL9-CXCR3-dependent mechanism governed the accumulation of γδT cells in the livers of mice infected with Ad expressing the Escherichia coli LacZ gene (AdLacZ). We also showed a critical role for γδT cells in initiating acute liver toxicity after AdLacZ administration, driven in part by the ability of γδT cells to promote the recruitment of the conventional T cell, the CD8+ T cell, into the liver. Furthermore, reduced hepatic injury in AdLacZ-infected γδT-cell-deficient mice was associated with lower hepatic levels of gamma interferon (IFN-γ) and CXCL9, an IFN-γ-inducible chemokine. Finally, our study highlighted a key role for IFN-γ and CXCL9 cross talk acting in a feedback loop to drive the proinflammatory effects of γδT cells during AdLacZ-mediated acute liver toxicity. Specifically, intracellular IFN-γ produced by activated hepatic γδT cells interacts with hepatocytes to mediate hepatic CXCL9 production, with the consequent accumulation of CXCR3-bearing γδT cells in the liver to cause acute liver damage without vector clearance.
Gene therapy is an innovative and potentially very important clinical strategy to treat many inherited and acquired human diseases. In 2007, over 1,100 gene therapy clinical trials were performed worldwide for the treatment of various ailments (including cardiovascular diseases, cystic fibrosis, and cancer), with 66% of these trials being conducted in the United States (www.wiley.co.uk/genmed/clinical). Of the 28 vectors used in gene therapy in 2007, replication-deficient adenovirus (Ad) was the most popular vector, accounting for 25% of all clinical trials (www.wiley.co.uk/genmed/clinical). However, broad clinical application of Ad vectors in gene therapy is limited by the induction of a robust immune response against the Ad vector (25, 39, 40, 75), with associated loss of the Ad transgene and the induction of liver inflammation and injury in rodents (25, 40, 75), nonhuman primates (45, 61), and humans (56). Various approaches to suppress the ability of the host inflammatory and cellular immune responses to eliminate the Ad vector initially focused on manipulating the adaptive immune system (80-83).
The current paradigm suggests that the Ad vector is cleared from the blood within minutes of systemic administration and sequestered primarily in the liver (30, 33, 65, 74), where the viral gene is extensively expressed (30, 33, 45, 78). Specifically, published reports demonstrate that there is widespread expression of the viral DNA in the liver within 24 h of systemic Ad administration, with ∼90% of the viral DNA being cleared from the liver by week 3 (46, 78, 82, 83, 87). Initial studies primarily focused on defining the role of the adaptive immune response in liver Ad clearance (80-83). However, a decade ago, Ronald Crystal's group (76, 78) demonstrated that hepatic elimination of the Ad vector may also be mediated by the innate immune response. Moreover, it is widely believed that the innate immune response to Ad is associated with hepatic NF-κΒ activation (37, 48) and subsequent secretion of cytokines/chemokines by transduced liver cells (including hepatocytes, Kupffer cells, and endothelial cells) (37, 38, 40, 48), which in turn promotes the hepatic recruitment and/or activation of the innate immune cells, neutrophils (17, 36, 48) and NK cells (43, 52, 59). The accompanying inflammation results in liver injury/damage and rapid and early elimination of the administered vector. In view of the evidence implicating NK cells (43, 52, 59), Kupffer cells (38), and neutrophils (17, 36, 48) in initiating the early liver injury associated with systemically administered Ad, the innate immune response against the Ad vector (rather than that mediated by the adaptive immune response) and accompanying liver toxicity are widely believed to be the biggest hurdle to the broad clinical application of Ad vectors in gene therapy. Therefore, a detailed characterization of immunological studies governing liver inflammation and injury mediated by systemically administered Ad is urgently needed to provide a knowledge base for the development of therapies that ameliorate early liver toxicity and thus improve the efficacy of the Ad vector in gene therapy. Most attention has been devoted to demonstrating that innate immune cells such as Kupffer cells, neutrophils, and NK cells promote early liver injury associated with the Ad vector because these leukocytes are considered to be “classical innate immune cells.” Therefore, it is not surprising that little is known about the role that the nonclassical innate immune T cell, the γδT cell (see below for an overview), may play in mediating early liver toxicity associated with systemically administered Ad.
γδT cells represent a unique T-cell lineage that possesses a distinct T-cell receptor (TCR), which is composed of two glycoprotein chains called γ and δ (15, 16, 23, 50). Although, γδT cells are less widespread in tissues than conventional αβT cells, they can be found in a number of different anatomical sites, with the highest frequency in the mucosa (15, 16, 23, 34, 50). An important feature of γδT cells is their ability to regulate the innate and adaptive immune systems since these cells can be polarized to produce Th1- and/or Th2-type cytokines (14-16, 24, 47, 49, 50). In addition to the immunoregulatory effects of γδT cells via cytokine production, these cells can also exert cytotoxic-mediated killing of a variety of target cells via Fas/FasL (27, 53). In the last decade, numerous studies have utilized γδT-cell-deficient mice or γδT-cell-depleting antibodies to provide evidence that demonstrates a crucial role for γδT cells in the pathophysiology of infectious diseases. For instance, increased numbers of γδT cells in the peripheral blood and/or localization to sites of infection has been documented in several human viral infections mediated by human immunodeficiency virus (31), hepatitis C virus (71), Epstein-Barr virus (58), and cytomegalovirus (18). In addition, a protective role for γδT cells has been documented in murine models of viral infection affecting the mucosa (as shown for herpes simplex virus type 1 [62], vaccinia virus [63], and influenza virus [9]) infections) and brain (as seen for murine West Nile viral infection [72]). Although γδT cells are relatively few in number in normal liver (in comparison to other organs), increased expression of this T-cell subset is observed in the livers of patients infected with the hepatitis C virus (71). In addition, the number of γδT cells in the liver is significantly increased following systemic administration of Ad (32). In view of the significant evidence implicating γδT cells in the pathophysiology of numerous viral infections, the present study specifically assessed the role of γδT cells in the pathophysiology of acute liver inflammation and injury in response to systemically administered Ad vector.
MATERIALS AND METHODS
Mice.
Male C57BL/6J mice, TCR-δ-deficient mice (C57BL/6J background), and gamma interferon (IFN-γ)-deficient mice (C57BL/6J background), 6 to 8 weeks old, were all purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were maintained under specific-pathogen-free conditions and were kept in a conventional animal facility at the Louisiana State University Health Sciences Center at Shreveport. All procedures in this study were conducted in accordance with institutional guidelines for animal care and use.
Virus infection and in vivo treatment protocol.
Endotoxin-free, replication-deficient E1- and E3-deleted type 5 Ad vectors expressing the Escherichia coli LacZ gene (denoted AdLacZ) or green fluorescent protein (GFP) gene (denoted AdGFP) as reporter genes driven by the cytomegalovirus promoter were purchased from Vector Development Laboratory (Baylor College of Medicine, Houston, TX). For in vivo experiments, mice were injected intraperitoneally (30, 59, 60) with AdLacZ or AdGFP (1011 virus particles [7, 17, 43, 48, 64-66, 78]) or an equivalent volume of phosphate-buffered saline (PBS; vehicle). At days 1 and 6 after AdLacZ or AdGFP administration, mice were anesthetized with a mixture of xylazine and ketamine hydrochloride; blood serum was collected, whereas livers were perfused with ice-cold sterile PBS to remove blood elements.
For the antibody blocking experiment, anti-murine CXCR3 serum, anti-CXCL9 serum, or control serum (0.5 ml/mouse, intraperitoneally) was administered to naive mice 16 h before AdLacZ administration and an additional dose was given every 48 h until termination of the experiment (13, 69). All mice were sacrificed 6 days after AdLacZ administration. Anti-murine CXCR3 and anti-CXCL9 sera were obtained from Robert Strieter (University of Virginia, Charlottesville). Control serum was purchased from Sigma Chemical Company (St. Louis, MO).
β-Galactosidase activity assay.
The E. coli LacZ gene is a very common reporter gene that codes for an active subunit of β-galactosidase. This enzyme is commonly used to assess transfection/transduction efficiency in cells and tissues because it is very stable and resistant to proteolytic degradation and can be conveniently assayed in situ. Specifically, when the β-galactosidase gene is expressed in transfected cells/tissues, it converts the colorless substrate o-nitrophenyl-β-d-galactopyranoside into a colored reaction product that can be easily quantified spectrophotometrically. Thus, we used β-galactosidase activity as a biochemical marker to evaluate AdLacZ transgene expression in the livers of wild-type (WT) mice and γδT-cell-deficient (knockout [KO]) mice after AdLacZ administration. Briefly, perfused liver was immediately homogenized in lysis buffer, and the lysate was assayed for β-galactosidase activity using a β-galactosidase assay kit (OZ Biosciences, France) according to the manufacturer's instructions. Recombinant β-galactosidase was used to construct a standard curve and also served as a positive control, whereas liver lysate from vehicle-treated mice was used as a negative control. The final reaction mixture was measured using a spectrophotometer (Bio-Rad Laboratories Inc., CA), and the results were calculated as units β-galactosidase per milligram total liver protein. Protein concentration was determined using bicinchoninic acid protein assay reagent (Pierce Biotechnology, Rockford, IL).
Evaluation of liver injury.
Liver injury was determined by biochemical and histological means. Biochemical liver damage in control and AdLacZ-infected mice was determined by measuring serum levels of the liver enzyme alanine aminotransferase (ALT) using a commercial kit (Thermo Electron, Waltham, MA) according to the manufacturer's instructions. For histological evaluation, perfused liver tissues from control and AdLacZ-infected mice were fixed overnight in 10% neutral buffered formalin, dehydrated in graded concentrations of ethanol, and then embedded in paraffin and sectioned. Liver sections (5 μm thick) were stained with hematoxylin and eosin (H&E) according to standard protocols and analyzed by light microscopy in a blinded fashion by a pathologist (P.A.A.). The degrees of inflammation of the liver and hepatocyte damage were graded as mild, moderate, or severe using a combination of the severity of the inflammation and the degree of hepatocyte degenerative changes, including ballooning degeneration, hepatocyte necrosis, and frequency of acidophilic bodies (12, 22).
Immunohistochemical localization of γδT cells.
The localization of γδT cells in the livers of AdLacZ-infected mice was performed on frozen liver sections using a specific γδT-cell antibody (clone GL3; BD Pharmingen) according to the manufacturer's instructions. Liver sections from AdLacZ-infected mice were also stained with isotype control antibody to demonstrate the specificity of the γδT-cell antibody.
Isolation of hepatic lymphocytes and flow cytometry.
Hepatic lymphocytes were isolated using our published protocols (2, 3). Briefly, perfused whole livers were obtained from uninfected (i.e., naive) or AdLacZ- or AdGFP-infected mice and minced in digestive medium containing 0.05% type 2 collagenase (Worthington Biochemical, NJ) and 0.002% DNase I (Roche Diagnostics, Indianapolis, IN). Following gentle agitation at 37°C for 30 min, the digest was passed through a nylon mesh (Small Parts Inc., Miami, FL) and then washed twice with cold sterile PBS. Cells were then subjected to density gradient centrifugation on Lympholyte-M (Cedarlane Laboratories, Canada) to isolate the lymphoid cell population. Isolated hepatic lymphocytes were resuspended in cold sterile PBS, and cell viability was determined using trypan blue exclusion dye. For the specific identification of γδT cells, isolated hepatic lymphocytes were preincubated with anti-mouse CD16/32 monoclonal antibody (MAb; clone 2.4G2; BD Pharmingen) to block FcγRs and then incubated simultaneously with phycoerythrin (PE)- or fluorescein isothiocyanate-labeled TCRγδ antibody (Ab; clone GL3; BD Pharmingen) and peridinin chlorophyll protein-labeled CD3ɛ MAb (clone 145-2C11; BD Pharmingen). Three-color staining was used to assess intracellular IFN-γ expression by hepatic γδT cells. Briefly, fluorochrome-labeled TCRγδ-CD3+ double-positive T cells were permeabilized with Cytofix/Cytoperm Plus (BD Pharmingen) and then stained intracellularly with fluorescein isothiocyanate-labeled murine IFN-γ MAb (clone XMG1.2) according to the manufacturer's instructions (1, 6). Three-color staining was also used to determine cell surface expression of CXCR3 or FasL on isolated hepatic γδT cells. Specifically, fluorochrome-labeled TCRγδ-CD3+ double-positive T cells were stained extracellularly with a specific murine allophycocyanin-labeled CXCR3 Ab (clone 220803; R & D Systems, Minneapolis, MN) or PE-labeled FasL Ab (clone Kay-10; BD Pharmingen).
For the specific identification of NK cells and CD8+ T cells, isolated hepatic lymphocytes were preincubated with anti-mouse CD16/32 MAb (clone 2.4G2; BD Pharmingen) to block FcγRs and then incubated with fluorochrome-labeled NK1.1 MAb (clone PK136; BD Pharmingen) or fluorochrome-labeled CD8a MAb (clone 53-6.7; BD Pharmingen).
To determine intracellular IFN-γ expression by hepatic NKT cells, isolated hepatic lymphocytes were preincubated with anti-mouse CD16/32 MAb (clone 2.4G2; BD Pharmingen) to block FcγRs and then incubated simultaneously with allophycocyanin-labeled TCRβ MAb (clone H57-597; BD Pharmingen) and PE-labeled NKT tetramer (CD1d-PBS57; NIH Tetramer Core Facility, Atlanta, GA). Intracellular IFN-γ expression by hepatic NKT cells was determined as described above for γδT cells. In all experiments, corresponding isotype control antibodies were used to set analysis gates. In addition, viable lymphocyte populations were gated using forward and side scatter characteristics and analyzed using a FACSCalibur and FACScan Diva software (BD Biosciences).
Purification of γδT cells.
A single-cell suspension was prepared from the spleens of naive male C57BL/6 mice by conventional methods under sterile conditions. Briefly, splenocytes were isolated by grinding the spleen between two sterile frosted slides, passing it through a 100-μm cell strainer (BD Pharmingen), and lysing red blood cells with sterile ammonium chloride buffer. The lysed splenocytes were quickly washed with RPMI 1640 medium (supplemented with 10% fetal calf serum, nonessential amino acids, l-glutamine, 2-mercaptoethanol, sodium pyruvate, and penicillin-streptomycin). Cell viability was determined using trypan blue exclusion dye. Next, γδT cells were purified from viable splenic cells using a mouse TCRγδ T-cell isolation kit (Miltenyi Biotec, CA) in accordance with the instructions provided by the manufacturer (44). γδT-cell purification was confirmed by flow cytometric analysis, and cell viability was greater than 95%.
Ad binding and transduction of γδT cells in vitro.
The following experiments were performed to determine whether the Ad vector can directly bind and infect γδT cells. A fluorescence microscopy-based technique was used to determine the potential binding of Ad to γδT cells. Specifically, purified γδT cells (1 × 105 cells per well) were incubated with AdGFP particles at a concentration of 104 virus particles/cell (17, 77, 79, 85, 87) for 60 min at 37°C. After the incubation period, cells were washed and transferred onto glass slides (by Cytospin), followed by fixation with 3.7% paraformaldehyde for 15 min at room temperature. The cells were then treated with propidium iodide (at a final concentration of 2.5 μg/ml in PBS; Sigma-Aldrich, St. Louis, MO) and 0.1% Triton X-100 for 10 min at room temperature to stain the nuclei (29, 70). To minimize quenching of fluorescence, slides were mounted in antifade gel mount medium (Biomeda, CA) and covered with a coverslip. An Olympus IX70 epifluorescence microscope was used for imaging of the samples.
To evaluate the transduction of γδT cells by Ad, purified γδT cells (1 × 105 cells per well) were incubated with AdLacZ particles (104 virus particles/cell, as above) for 24 h at 37°C (77). After this, the cells were lysed in lysis buffer, supernatant was collected, and the LacZ transgene product, β-galactosidase, was measured using a β-galactosidase activity assay kit as described previously (7, 46, 64-66).
Hepatic IFN-γ and CXCL9 protein levels.
Murine CXCL9 and IFN-γ protein levels in the livers of AdLacZ- or vehicle-treated mice were determined using the Bio-Plex array system (Bio-Rad Laboratories) (10), which is based on the Luminex technology, in accordance with the manufacturer's instructions. Briefly, perfused livers were homogenized in cold sterile PBS buffer containing protease cocktail inhibitor (Sigma-Aldrich). Liver homogenates were processed and stored at −80°C until used for the determination of CXCL9 and IFN-γ levels. Total protein levels in liver were determined using the bicinchoninic acid protein assay reagent (Pierce Biotechnology). Data are expressed as picograms of chemokine per milligram of total liver protein.
Statistical analysis.
All data are shown as means ± standard errors of the means (SEM). For comparisons of means between two experimental groups, a Student unpaired t test was used. Comparison among three or more experimental groups was performed using one-way analysis of variance, followed by either Dunnett's multiple-comparison test or the Newman-Kuels post hoc test. A P value of <0.05 was considered significant.
RESULTS
Kinetics of γδT-cell accumulation in AdLacZ-infected liver.
In agreement with previous reports (42, 43, 46, 52, 66), liver damage in response to systemically administered AdLacZ was characterized by a progressive increase in serum ALT levels beginning at day 1, with a significant increase documented at day 6 relative to that observed in the uninfected control (i.e., day 0), as shown in Fig. 1A. Next, we used flow cytometric analysis to determine the kinetics of γδT-cell (TCRγδ-CD3+ double-positive T-cell) accumulation in the liver following the systemic administration of AdLacZ. We observed that γδT cells constitute a minor population of T cells in the livers of uninfected mice (i.e., day 0) since γδT cells were not readily evident (Fig. 1B and C). With AdLacZ administration, the frequency (>3.5-fold) and absolute number (>50-fold) of γδT cells in the liver were significantly increased on day 6 (but not day 1) compared to values for uninfected mice (Fig. 1B and C). Immunohistochemical examination of AdLacZ-infected liver showed that γδT cells were predominantly localized among the hepatocytes (Fig. 1D). Our observation of increased accumulation of γδT cells in the liver in response to systemically administered AdLacZ is consistent with a previously reported study (32).
FIG. 1.

Kinetics of acute liver damage and hepatic γδT-cell accumulation following AdLacZ administration. (A) C57BL/6 mice were given vehicle or infected with AdLacZ. Serum was collected at the indicated times and used to measure ALT levels (as described in Materials and Methods). Data are shown as means ± SEM (n = 5 to 8 mice per group; *, P ≤ 0.05 versus uninfected group, i.e., day 0). (B and C) To determine γδT-cell accumulation in the livers of C57BL/6 mice systemically challenged with AdLacZ or vehicle, hepatic lymphoid cells were isolated at the indicated times, stained with specific fluorochrome-labeled TCRγδ and CD3 MAbs, and then analyzed by flow cytometry to reveal the percentages (B) and absolute numbers (C) of γδT cells (i.e., TCRγδ-CD3 double-positive T cells) per liver. Data are presented as means ± SEM (n = 5 or 6 mice per group; *, P ≤ 0.05 versus uninfected group, i.e., day 0). (D) To reveal the localization of γδT cells in the liver, frozen liver sections were obtained from AdLacZ-infected C57BL/6 mice 6 days after infectivity and immunohistochemical localization of γδT cells was performed. Liver sections were stained with isotype control Ab or anti-γδT-cell Ab. γδT cells (white arrows) and hepatocytes (red arrows) are shown. Original magnification, ×400.
AdLacZ interaction with γδT cells.
It is not known if AdLacZ could directly interact with γδT cells in vivo. Therefore, mice were administered AdGFP and hepatic γδT cells were isolated on days 1 and 6 postinfection. By using AdGFP, we were able to assess the ability of the vector to directly interact with γδT cells by screening for the expression of GFP-positive γδT cells using flow cytometry. As depicted in Fig. 2A, GFP expression on isolated hepatic γδT cells was observed at days 1 and 6 after AdGFP administration, showing that Ad could directly interact with hepatic γδT cells.
FIG. 2.
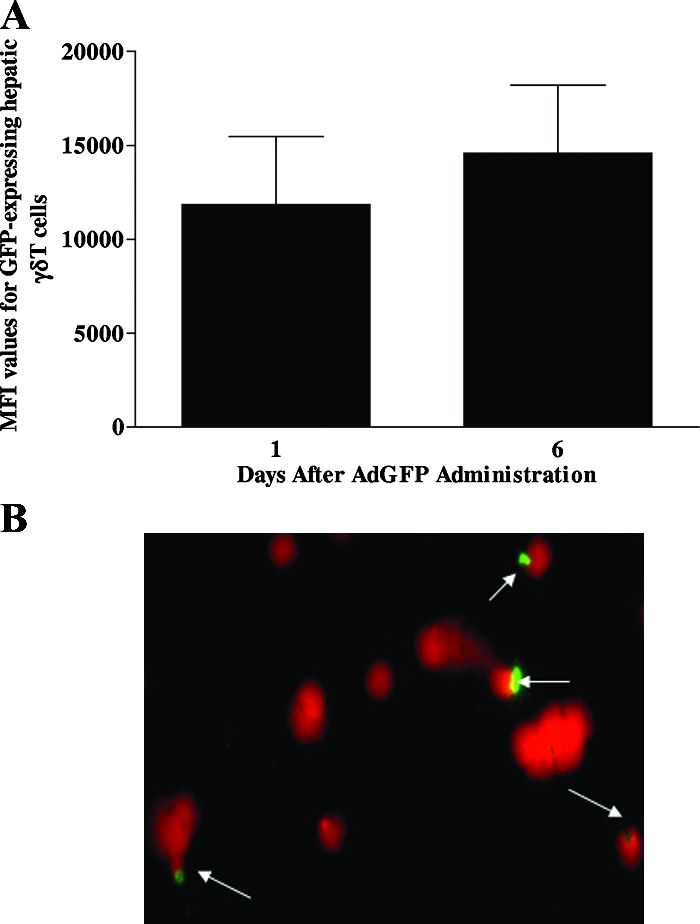
Ad interacts with γδT cells. (A) Male C57BL/6 mice were injected with AdGFP, and isolated hepatic fluorochrome-labeled γδT cells were screened for the presence of GFP by flow cytometry. The median fluorescence intensity (MFI) values for GFP-positive hepatic γδT cells are shown. Data are reported as means ± SEM (n = 4 mice per group). (B) To determine if Ad could interact with γδT cells, splenocytes were isolated from naive C57BL/6 mice and γδT cells were purified using a mouse γδT-cell isolation kit. Purified splenic γδT cells were then incubated in vitro with GFP-labeled Ad (104 virus particles) for 60 min. A fluorescence microscopic technique was used to evaluate Ad interaction with splenic γδT cells. Ad-GFP is shown in green, whereas γδT-cell nuclei counterstained with propidium iodide are shown in red.
To confirm direct interaction of the Ad vector with γδT cells, purified splenic γδT cells from naive mice were cultured in vitro with AdGFP and the cells were analyzed by fluorescence microscopy. Immunofluorescence imaging of the γδT cells revealed that green fluorescence associated with Ad particle was observed in the nuclei, specifically around the nuclear envelopes, of γδT cells (Fig. 2B), a confirmation that the Ad vector is capable of interacting with γδT cells. Next, we assessed the ability of AdLacZ to transduce γδT cells. Purified splenic γδT cells from naive mice were cultured in vitro with AdLacZ, and β-galactosidase activity was determined (see Materials and Methods). β-Galactosidase activity in AdLacZ-treated γδT cells in vitro was similar to that seen in vehicle-treated cells (data not shown). These results show that γδT cells can directly interact with the Ad vector but are not transduced by the vector.
Effects of γδT-cell deficiency on AdLacZ-mediated acute liver inflammation and injury.
To specifically define the functional role of γδT cells in the development of acute liver inflammation and injury associated with systemically administered AdLacZ, we used γδT-cell-deficient mice (i.e., γδT-cell KO mice). For our functional study, a time point of day 6 was chosen due to considerable liver damage, as shown by increased serum ALT levels (42, 43, 46, 52). WT and γδT-cell KO mice that received vehicle had comparable baseline levels of serum ALT (Fig. 3A). As shown in Fig. 3A, a significant increase in serum ALT level was observed in WT mice following AdLacZ administration (relative to that for vehicle-treated WT mice). In contrast, γδT-cell KO mice were highly resistant to liver injury, as shown by significantly lower (80% reduction) serum ALT levels in these mice after AdLacZ administration than in WT mice infected with AdLacZ (Fig. 3A). It is noteworthy that serum ALT levels in AdLacZ-infected γδT-cell KO mice did not significantly differ from the levels observed in vehicle-treated γδT-cell KO mice (Fig. 3A). In parallel with the biochemical finding, liver sections from WT mice exhibited extensive hepatocyte damage following AdLacZ administration (Fig. 3B) whereas liver sections from γδT-cell KO mice displayed reduced inflammatory cell infiltrates (such as CD8+ T cells; see Fig. 6) and little or no hepatocyte damage in response to systemically administered AdLacZ (Fig. 3B). Furthermore, the hepatic level of IFN-γ was also significantly reduced by γδT-cell deficiency at day 6 after AdLacZ administration (hepatic IFN-γ: 0.372 ± 0.02 mg/pg total protein in WT mice versus 0.24 ± 0.03 mg/pg total protein in γδT-cell KO mice; P < 0.05). In summary, our data demonstrate an important proinflammatory role for γδT cells in promoting the development of acute hepatic inflammation and injury in mice systemically challenged with AdLacZ.
FIG. 3.

Effects of γδT-cell deficiency on acute liver inflammation and injury in AdLacZ-infected mice. WT and γδT-cell KO mice were infected with AdLacZ or vehicle for 6 days. (A) Serum samples were obtained for the determination of ALT levels. All results are presented as means ± SEM (n = 6 mice per group; *, P ≤ 0.05 versus all vehicle-treated controls; #, P ≤ 0.05 versus AdLacZ-infected WT mice). (B) Photomicrograph of H&E-stained representative liver sections depicting diffuse and severe acute hepatic injury with swelling of the hepatocytes, obliteration of the sinusoid spaces, hepatocellular necrosis, and numerous acidophilic bodies in AdLacZ-infected WT mice compared to the minimal distortion of lobular architecture and absence of inflammatory cell infiltrates in liver sections from γδT-cell KO mice after 6 days of AdLacZ infection. Original magnification, ×200. In contrast, H&E-stained liver sections from vehicle-treated WT and γδT-cell KO mice were normal.
FIG. 6.

Effects of γδT-cell deficiency on NK cell and CD8+ T-cell accumulation in the livers of AdLacZ-infected mice. WT and γδT-cell KO mice were infected with AdLacZ, and hepatic lymphoid cells were isolated 6 days later. Isolated hepatic lymphoid cells were then stained with fluorochrome-labeled NK1.1 MAb and fluorochrome-labeled CD8a MAb and analyzed by flow cytometry to identify accumulation of hepatic NK and CD8+ T cells, respectively. The absolute numbers of NK cells and CD8+ T cells are shown. All results are presented as means ± SEM (n = 5 or 6 mice per group; *, P ≤ 0.05 versus AdLacZ-infected WT mice).
Mechanism(s) of γδT-cell induced acute liver inflammation and injury in AdLacZ-infected mice.
Next, we evaluated the specific mechanisms underlying the proinflammatory effects of γδT cells during acute liver inflammation and injury in mice systemically challenged with AdLacZ.
It is well established that activated γδT cells may secrete the Th1 cytokine IFN-γ during viral infections to modulate the ongoing inflammatory response (49, 54, 63, 72, 84). Moreover, IFN-γ is a cytokine that has been implicated in liver damage associated with experimental autoimmune (35) and toxin (41) liver diseases. Since IFN-γ level is increased in the livers and circulation of AdLacZ-infected mice (43, 59), we determined by intracellular staining and flow cytometric analysis whether hepatic γδT cells represent an important cellular source of IFN-γ after AdLacZ administration. As depicted in a representative fluorescence-activated cell sorter (FACS) histogram in Fig. 4A, increased intracellular IFN-γ expression by isolated hepatic γδT cells was observed at days 1 and 6 after AdLacZ administration compared to that seen in uninfected mice (i.e., day 0). The preceding results demonstrate that activated hepatic γδT cells are a source of IFN-γ in mice systemically challenged with AdLacZ (Fig. 4A), so we hypothesized that γδT cells may be an essential participant in liver inflammation and injury after AdLacZ administration via IFN-γ release. It is noteworthy that hepatic NK cells (43) and NKT cells (Fig. 4B) are also important producers of IFN-γ during AdLacZ-mediated liver toxicity.
FIG. 4.

Role of IFN-γ in the development of acute liver inflammation and injury in mice systemically challenged with AdLacZ. (A) To determine if γδT cells are a source of hepatic IFN-γ after AdLacZ infectivity, C57BL/6 mice were systemically challenged with AdLacZ or vehicle and hepatic lymphoid cells were isolated at the indicated times. Isolated hepatic lymphoid cells were first stained with specific fluorochrome-labeled TCRγδ and CD3 MAbs to identify γδT cells, and the cells were then permeabilized prior to staining with fluorochrome-labeled IFN-γ MAb to determine γδT-cell intracellular IFN-γ after flow cytometric analysis. A representative FACS histogram demonstrating intracellular IFN-γ expression by isolated hepatic γδT cells after AdLacZ administration is shown. (B) Next, we determined if hepatic NKT cells are also an early source of intracellular IFN-γ after AdLacZ administration. C57BL/6 mice were systemically challenged with AdLacZ or vehicle, and hepatic lymphoid cells were isolated 24 h later. Isolated hepatic lymphoid cells were first stained with specific fluorochrome-labeled TCRβ MAb and the NKT cell tetramer CD1d-PBS57 to identify NKT cells (see Materials and Methods), and the cells were then permeabilized prior to staining with fluorochrome-labeled IFN-γ MAb. A representative FACS histogram demonstrating intracellular IFN-γ expression by isolated hepatic NKT cells after AdLacZ or vehicle administration at day 1 posttreatment is shown. (C) To determine the specific contribution of IFN-γ to acute liver injury mediated by systemically administered AdLacZ, WT and IFN-γ KO mice were infected with AdLacZ for 6 days. Serum samples were obtained for the determination of ALT levels. All results are presented as means ± SEM (n = 5 or 6 mice per group; *, P ≤ 0.05 versus all vehicle-treated controls; #, P ≤ 0.05 versus AdLacZ-infected WT mice). (D) Photomicrograph of H&E-stained representative liver sections depicting diffuse and severe acute hepatic injury (inflammation and hepatocellular necrosis with numerous acidophilic bodies) in AdLacZ-infected WT mice compared with essentially normal liver histology in AdLacZ-infected IFN-γ KO mice. Original magnification, ×200.
To this effect, we evaluated the role of IFN-γ in promoting acute liver injury during AdLacZ infection. Vehicle-treated WT and IFN-γ KO mice had comparable baseline levels of serum ALT (Fig. 4C). Infection of WT mice with AdLacZ caused a significant increase in serum ALT level in comparison to that in vehicle-treated WT mice, an effect that was significantly attenuated (∼60% decrease) by IFN-γ deficiency (Fig. 4C). Of note, the serum ALT level in AdLacZ-infected IFN-γ KO mice was not statistically different from the level documented in vehicle-treated IFN-γ KO mice (Fig. 4C). In agreement with the biochemical findings, liver sections from AdLacZ-infected IFN-γ KO mice (Fig. 4D) showed essentially normal histology, with no noticeable hepatocellular damage in comparison to prominent liver damage in liver sections obtained from WT mice after AdLacZ administration (Fig. 4D). A similar finding was documented in studies that utilized anti-murine IFN-γ antibody (data not shown). Overall, these results identify the potential of γδT cells to initiate acute liver inflammation and injury after AdLacZ administration via IFN-γ release.
The FasL/Fas death pathway has been implicated in the effector function of γδT cells in vivo (27, 53). In support, we have evidence that γδT-cell KO mice resist acute liver failure compared to WT mice at 3.5 h after the intraperitoneal administration of agonistic anti-Fas antibody (clone Jo2; BD Pharmingen; at a dose of 0.5 μg/g of body weight). Figure 5A and B show a significant attenuation (58% reduction) of serum ALT level and a marked improvement in hepatic histology in γδT-cell KO mice treated with Jo2, relative to the finding for Jo2-treated WT mice. Thus, hepatic γδT cells are capable of contributing to Fas-mediated acute liver apoptosis. Next, we assessed if the FasL/Fas-mediated death pathway may be an additional potential mechanism through which γδT cells promote acute liver damage in mice systemically challenged with AdLacZ. A previous study has demonstrated that mice deficient in FasL or Fas ameliorate liver inflammation and injury associated with systemically administered AdLacZ (42). Specifically, we assessed by flow cytometric analysis whether hepatic γδT cells express FasL during AdLacZ liver infection. The results show that FasL cell surface expression on γδT cells was increased by AdLacZ administration relative to that seen in uninfected mice (Fig. 5C). In addition, the absolute number of extracellular FasL-expressing γδT cells in the liver was significantly increased at day 6 after AdLacZ infection of mice: 9.97 × 103 ± 1.21 × 103 cells versus 0.56 × 103 ± 0.04 × 103 cells in uninfected mice.
FIG. 5.

Role of the Fas/FasL-mediated death pathway in the proinflammatory effect of γδT cells. (A and B) Agonistic anti-Fas antibody was used to specifically determine if γδT cells are capable of promoting acute liver damage via the Fas-mediated death pathway. WT and γδT-cell KO mice were administered the agonistic anti-Fas antibody Jo2 (0.5 μg/g body weight; intraperitoneally), and acute liver injury (i.e., ALT levels and histology) was assessed 3.5 h later. Data are presented as means ± SEM (n = 4 to 7 mice per group; *, P ≤ 0.05 versus WT mice). Photomicrograph of H&E-stained representative liver sections depicting diffuse and severe acute liver failure (inflammation and widespread hepatocellular necrosis with numerous apoptosis) in Jo2-treated WT mice compared with patchy necrosis and reduced inflammation in a liver sections from γδT-cell KO mice administered Jo2. Original magnification, ×200. (C) C57BL/6 mice were administered vehicle or AdLacZ, and hepatic lymphoid cells were isolated from these mice 6 days after infectivity. Isolated hepatic lymphoid cells were initially stained with fluorochrome-labeled TCRγδ and CD3 MAbs to identify γδT cells and then stained with fluorochrome-labeled FasL MAb to determine FasL cell surface expression on hepatic γδT cells. A representative FACS histogram demonstrating extracellular FasL expression on γδT cells after AdLacZ administration is depicted.
In addition to the aforementioned mechanisms, we evaluated if the proinflammatory effects of γδT cells during acute liver inflammation and injury following AdLacZ administration may also be attributed to the recruitment of functional effector NK cells as well as CD8+ T cells into the liver. Figure 6 revealed that γδT-cell deficiency was associated with a significant decrease (41% reduction) in the number of CD8+ T-cell infiltrates in the livers of AdLacZ-infected γδT-cell KO mice relative to the number in WT mice after AdLacZ administration. In contrast, γδT-cell deficiency did not alter the number of NK cells accumulating in the liver after AdLacZ administration. Collectively, these results demonstrate that the recruitment of γδT cells is paramount for the development of acute liver inflammation and injury and that γδT cells are an essential mediator for the recruitment of CD8+ T cells (but not NK cells) into the livers of AdLacZ-infected mice.
Molecular determinant of γδT-cell accumulation in AdLacZ-infected liver.
In Fig. 4A, we demonstrated that γδT cells are an important source of hepatic IFN-γ during AdLacZ infection. A previous study provided evidence implicating the IFN-γ-inducible chemokine CXCL9 in the recruitment of CD8+ T cells into the liver following AdLacZ administration (7). Based on that report (7), we initially confirmed that AdLacZ liver infection is associated with significant increases in the hepatic level of CXCL9 at days 1 and 6 after AdLacZ administration (Fig. 7A). In contrast, CXCL10, another CXCR3 ligand, was significantly increased in the liver only at day 6 (not at day 1) after AdLacZ administration (M. N. Ajuebor, unpublished observation). Furthermore, we found that hepatic CXCL9 production in response to AdLacZ administration was significantly impaired by γδT-cell depletion as well as by IFN-γ deficiency (Fig. 7A). Next, we postulated that CXCL9 via its specific receptor, CXCR3, may play an important role in promoting the accumulation of γδT cells in the liver after AdLacZ administration since the frequency and absolute number of hepatic γδT cells expressing CXCR3 receptors were significantly increased in AdLacZ-infected mice (Fig. 7B and C). In line with this supposition, we observed that anti-CXCR3 serum and anti-CXCL9 serum treatments were both associated with significant decreases in hepatic γδT-cell accumulation after AdLacZ administration relative to that in AdLacZ-infected mice given control serum (Fig. 7D). Furthermore, liver CD8+ T-cell (but not NK cell) accumulation was significantly inhibited by both treatments (Fig. 7D). Additional studies demonstrated that blockade of the CXCL9-CXCR3 axis was also associated with improved hepatic injury as shown by lower ALT levels (>75% reduction) compared to that for AdLacZ-infected mice given control serum (Fig. 7E). In agreement with the biochemical finding, liver sections from mice given control serum exhibited extensive hepatocyte damage at day 6 after AdLacZ infection (Fig. 7F). In comparison, liver sections from mice that received anti-CXCR3 serum or anti-CXCL9 serum displayed little or no hepatocyte damage and had no significant hepatocellular necrosis or inflammatory-cell infiltrates following AdLacZ administration (Fig. 7F). Overall, these studies suggest that IFN-γ-inducible CXCL9 drives the accumulation of CXCR3-expressing γδT cells in the liver, an effect that promotes acute liver inflammation and injury via multiple mechanisms including increased IFN-γ production and the recruitment of effector CD8+ T cells.
FIG. 7.

Role of the CXCR3-CXCL9 pathway in the effector function of γδT cells during AdLacZ-induced liver inflammation and injury. (A) Kinetics of hepatic CXCL9 production during AdLacZ-mediated liver toxicity was evaluated using a Bioplex array system (see Materials and Methods). The effects of γδT-cell deficiency and IFN-γ deficiency on hepatic CXCL9 production after AdLacZ administration were also evaluated. Results are presented as means ± SEM (n = 5 or 6 mice per group; *, P ≤ 0.05 versus uninfected group, i.e., day 0; #, P ≤ 0.05 versus AdLacZ-infected WT mice; **, P ≤ 0.05 versus AdLacZ-infected WT mice). (B and C) Flow cytometric analysis was used to determine the kinetics of CXCR3 expression on hepatic γδT cells during AdLacZ infection. C57BL/6 mice were uninfected (day 0) or infected with AdLacZ, and hepatic lymphoid cells were isolated at the indicated times. Isolated cells were then stained with specific fluorochrome-labeled TCRγδ, CD3, and CXCR3 MAbs to reveal the percentage (B) and absolute number (C) of hepatic γδT cells expressing CXCR3 receptors. Data are presented as means ± SEM (n = 5 or 6 mice per group; *, P ≤ 0.05 versus uninfected group, i.e., day 0). (D) To determine the functional role of CXCR3 and CXCL9 in the recruitment of γδT cells, NK cells, and CD8+ T cells to the liver following AdLacZ administration, anti-CXCR3 serum and anti-CXCL9 serum were used. C57BL/6 mice were given control serum, anti-CXCR3 serum, or anti-CXCL9 serum intraperitoneally 16 h before AdLacZ infection, with additional doses given every 48 h until termination of the experiment. All mice were sacrificed at day 6 after AdLacZ administration for the determination of hepatic γδT-cell, NK cell, and CD8+ T-cell accumulation by flow cytometry. Data are presented as means ± SEM (n = 5 or 6 mice per group; *, P ≤ 0.05 versus control serum; #, P ≤ 0.05 versus control serum). (E and F) The effects of anti-CXCR3 serum and anti-CXCL9 serum treatment on liver injury 6 days after AdLacZ infection were also determined biochemically by measuring ALT levels (E) and by histology (F). ALT values are depicted as means ± SEM (n = 6 to 8 mice per group; *, P ≤ 0.05 versus control serum). Panel F shows a representative photomicrograph of liver section from AdLacZ-infected mice treated with control serum, which was characterized by diffuse and severe acute hepatic injury, whereas liver sections from anti-CXCR3 serum-treated and anti-CXCL9-treated mice were characterized by nearly normal liver histology with no hepatocellular injury following the administration of AdLacZ. Original magnification, ×200.
Effect of γδT-cell deficiency on AdLacZ expression.
β-Galactosidase activity was used to determine the role of γδT cells in AdLacZ clearance. Hepatic β-galactosidase activity in γδT-cell KO mice did not significantly differ from that seen in WT mice at day 6 after AdLacZ liver infection. For AdLacZ-infected WT mice and γδT-cell KO mice, β-galactosidase activities (means ± SEM; n = 4 to 6 mice per group) were 21.4 ± 8.0 and 15.4 ± 6.9 units of β-galactosidase activity/mg of total liver protein, respectively.
DISCUSSION
The γδT cell is an innate immune T cell that has been shown to exert a protective response in viral diseases that predominantly affect the mucosa and brain (15, 16, 23, 34, 50). However, γδT cells may also promote a pathogenic effect during autoimmune diseases by initiating tissue injury in experimental arthritis (57) and multiple sclerosis (55, 68). Although increased γδT-cell infiltrates are observed during AdLacZ liver infection (32), the mechanisms underlying increased γδT-cell accumulation in the liver in response to systemically administered AdLacZ are unknown. Besides, the functional role of γδT cells in the development of acute liver inflammation and injury associated with systemically administered AdLacZ is not defined. Our study demonstrates a novel, unique, and central effector role for γδT cells in promoting acute inflammation and injury during AdLacZ-induced liver infection without altering AdLacZ transgene expression.
Using flow cytometry-based techniques, we observed that the progressive increase in acute liver damage in response to AdLacZ administration was also associated with increased γδT-cell accumulation in the liver. However, significant accumulation of γδT cells in the livers of AdLacZ-infected mice was a delayed event (occurring at day 6 but not day 1). In contrast, we established that hepatic γδT-cell activation following AdLacZ administration was an early event since increased intracellular IFN-γ expression by hepatic γδT cells was observed initially at day 1, a time point before a significant accumulation of γδT cells in the liver. Despite our evidence that hepatic γδT cells are activated prior to a significant accumulation of γδT cells in the livers of AdLacZ-infected mice, we found (by immunofluorescence imaging) that γδT cells can directly interact with the Ad vector but are not transduced by the vector based on our in vitro β-galactosidase assay that utilized purified splenic γδT cells. This may be due to the potential deficiency or low levels of the Ad receptor CAR on γδT cells, as documented for dendritic cells (77) and neutrophils (17). Nonetheless, our study provided compelling evidence that hepatic γδT cells played a critical role in the development of acute liver damage since the dramatic increase in acute liver injury in WT mice in response to AdLacZ administration was almost completely abolished by γδT-cell deficiency. The mechanism(s) responsible for early activation but delayed accumulation of hepatic γδT cells during AdLacZ-induced acute liver toxicity was further investigated (discussed below).
Next, we determined the potential mechanisms by which γδT cells could initiate and sustain hepatic inflammation and damage following AdLacZ administration. The first mechanism assessed was the role of IFN-γ. We have discussed previously that IFN-γ, a major cytokine produced by γδT cells in several viral disease model systems (49, 54, 63, 72, 84), is capable of promoting acute liver damage during experimental autoimmune and toxin liver injuries (35, 41), possibly by directly exerting a cytotoxic effect on hepatocytes which express the IFN-γ receptor on their cell surfaces (11). As a result, we hypothesized that γδT cells can be induced to express intracellular IFN-γ, which subsequently promotes the development of acute liver inflammation and injury following AdLacZ administration. Indeed, we have provided several lines of evidence suggesting an important role for endogenous IFN-γ in contributing to the proinflammatory effects of γδT cells during AdLacZ-induced acute liver injury. Specifically, we have demonstrated that (i) a significant increase in intracellular IFN-γ expression by hepatic γδT cells is seen following AdLacZ administration and (ii) a reduction in hepatic level of IFN-γ in AdLacZ-infected γδT-cell KO mice (compared to WT mice) correlated with reduced hepatic injury. Finally, we reported that IFN-γ-deficient mice were resistant to acute liver injury following AdLacZ administration in comparison to infected WT mice. Collectively, these data strongly support an important role for IFN-γ in the proinflammatory effects of γδT cells in the development of acute liver injury associated with systemically administered AdLacZ. While γδT cells may produce interleukin-4 (IL-4) (21, 73) and interleukin-17 (44) in vivo, these cytokines were not increased in the livers of AdLacZ-infected mice (M. N. Ajuebor, unpublished observation), thus excluding a potential role for the aforementioned cytokines in the pathogenic effect of γδT cells after AdLacZ liver infection. However, it is unlikely that IFN-γ is the sole mediator contributing to the proinflammatory effect of γδT cells since the acute liver damage in IFN-γ KO mice following AdLacZ administration was not completely attenuated. For this reason, we also assessed the role of the Fas/FasL-mediated death pathway in the pathogenic effects of hepatic γδT cells in AdLacZ-infected mice.
The Fas-mediated death pathway is activated by binding FasL or agonistic anti-Fas antibody to Fas, triggering trimerization and intracellular signaling. The seminal concept that the liver is highly sensitive to Fas-mediated apoptosis was initially demonstrated by Ogasawara and colleagues in 1993 (51). Specifically, systemic administration of agonistic anti-Fas antibody (Jo2) was associated with acute liver failure and mouse death within a few hours due to diffuse hemorrhage and massive apoptosis of hepatocytes (51). This finding has been confirmed by numerous studies (19, 20, 67, 86). As mentioned previously, there are reports implicating the Fas/FasL-mediated death pathway in the effector function of γδT cells during tissue injury affecting the brain and heart (27, 53). Moreover, we have observed that γδT cells do kill hepatocytes by a Fas-mediated apoptotic mechanism since γδT-cell KO mice were less susceptible to acute liver failure following agonistic anti-Fas antibody (i.e., Jo2) treatment than Jo2-treated WT mice. The Fas/FasL-mediated death pathway has been shown to promote acute liver injury after AdLacZ administration (42), and we observed increased FasL cell surface expression on hepatic γδT cells following AdLacZ administration. Therefore, we proposed that FasL/Fas-mediated killing of hepatocytes is the second potential mechanism by which γδT cells may promote the development of acute liver injury after AdLacZ administration.
In the final mechanism, we evaluated the role of the immune cells, NK and CD8+ T cells, in the effector function of γδT cells during acute liver toxicity associated with systemically administered AdLacZ. Due to the fact that NK and CD8+ T cells contribute to the liver toxicity associated with systemically administered AdLacZ (43, 46, 52), we examined the effect of γδT-cell deficiency on NK and CD8+ T-cell recruitment into the liver after AdLacZ administration. Flow cytometry-based evaluation of inflammatory cell infiltrates in the liver after AdLacZ administration revealed that CD8+ T-cell (but not NK cell) accumulation in the liver was severely impaired by γδT-cell deficiency. In summary, our data demonstrate that γδT cells play an important and a previously unrecognized role in governing AdLacZ-induced CD8+ T-cell recruitment to the liver and the subsequent development of acute liver toxicity.
We then turned our attention to the role of γδT cells in AdLacZ gene expression. A β-galactosidase activity assay (a marker for AdLacZ gene expression) revealed that γδT cells do not promote AdLacZ clearance since hepatic β-galactosidase activity in γδT-cell KO mice did not significantly differ from that detected in WT mice after AdLacZ administration. Our observation indicates that, unlike classical innate immune cells (i.e., NK and Kupffer cells), hepatic γδT cells do not contribute to AdLacZ clearance. Thus, γδT cells do not affect the transduction efficiency of the AdLacZ vector during the acute hepatic inflammatory response.
An important finding that was addressed in our study was the mechanism underlying increased γδT-cell accumulation in the liver at day 6 (but not at day 1) in AdLacZ-infected mice. Chemokines are well-defined chemoattractants for leukocytes (4, 5). Furthermore, a previous study reports that the CXCR3 ligands CXCL9 and CXCL10 are induced by IFN-γ acting on hepatocytes during AdLacZ-induced acute liver toxicity (7) and that these chemokines promote the recruitment of CD8+ T cells into the livers of AdLacZ-infected mice (7). Accordingly, we initially confirmed that the CXCL9 level in the liver is increased following AdLacZ administration. In addition, we also confirmed that hepatic CXCL9 production was IFN-γ dependent by demonstrating that CXCL9 production in the liver is severely impaired by IFN-γ deficiency after AdLacZ administration. Since hepatic γδT cells are enriched in intracellular IFN-γ after AdLacZ administration and γδT-cell deficiency is associated with lower hepatic levels of IFN-γ, we hypothesized that hepatic γδT cells (via intracellular IFN-γ) may play an essential role in the induction of hepatic CXCL9 after AdLacZ administration. Indeed, we observed that hepatic CXCL9 production was also significantly suppressed by γδT-cell deficiency at days 1 and 6 after AdLacZ infection. However, the nearly complete inhibition of hepatic CXCL9 levels at day 6 (i.e., late phase) in AdLacZ-treated γδT-cell KO mice but partial and significant decrease in CXCL9 hepatic level in γδT-cell KO mice at day 1 (i.e., early phase) after AdLacZ administration may suggest that IFN-γ from other innate immune cells (such as NK and NKT cells) may also contribute to early hepatic CXCL9 production. Taken together, our data identify a key role for intracellular IFN-γ specifically expressed by activated hepatic γδT cells in positively regulating early-phase (day 1) and late-phase (day 6) CXCL9 production in the liver during AdLacZ-induced acute liver toxicity.
The role of CXCR3, the cognate receptor for CXCL9, in the recruitment of immune cells, including T-cell subsets and NK cells, during inflammation is well documented (8, 13, 26, 28). In this study, we observed that CXCR3 cell surface expression on γδT cells accumulating in the livers of AdLacZ-infected mice is significantly increased. However, our observation that hepatic γδT cells are activated at day 1 after AdLacZ administration may suggest that CXCR3-expressing γδT cells in the liver are activated (at day 1) prior to expansion (at day 6) following the systemic administration of AdLacZ. Next, we hypothesized that CXCL9, via interaction with CXCR3, may promote the accumulation of γδT cells in the liver following AdLacZ administration. Indeed, we provided direct evidence that hepatic CXCL9 drives increased accumulation of CXCR3-expressing γδT cells in the livers of mice systemically challenged with AdLacZ since pretreatment of mice with a specific anti-CXCL9 serum or anti-CXCR3 serum markedly attenuated the accumulation of γδT cells in the liver, an effect that also reduced hepatic injury since biochemical and histological liver damage was almost completely abolished. Although we cannot completely exclude a role for other CXCR3 ligands (i.e., CXCL10 and CXCL11) in hepatic γδT-cell accumulation, our observation that anti-CXCL9 and anti-CXCR3 serum treatments caused comparable reductions in both γδT-cell accumulation and liver injury may suggest that the effect of CXCL9 during AdLacZ-mediated acute liver toxicity is not redundant. Of note, anti-CXCL9 and anti-CXCR3 serum treatments also suppressed CD8+ T-cell accumulation in the liver, a response that could be attributed to reduced accumulation of γδT cells in the liver since γδT-cell deficiency is associated with reduced hepatic CD8+ T-cell accumulation. In summary, our results directly indicate that the γδT-cell inflammatory response mounted against AdLacZ is to a large extent dependent on the biological function of the CXCL9-CXCR3 axis.
This study characterized a key molecular mechanism that governs the accumulation of γδT cells in the livers of AdLacZ-infected mice. We also showed a critical role for γδT cells in initiating acute liver toxicity after AdLacZ administration, driven in part by the ability of γδT cells to promote the recruitment of CD8+ T cells, an event that occurs without vector clearance. Our study also highlighted a key role for cytokine-chemokine cross talk in driving the pathogenic function of γδT cells during AdLacZ-mediated acute liver toxicity. Thus, we proposed that the combined events schematized in Fig. 8 play a crucial role in the effector function of γδT cells during AdLacZ liver infection. Specifically, we propose that (i) AdLacZ directly interacts with resident hepatic γδT cells localized among the hepatocytes; (ii) intracellular IFN-γ produced by activated γδT cells (and possibly NKT and NK cells) then act on hepatocytes to mediate early hepatic CXCL9 production observed at day 1; (iii) early hepatic CXCL9 subsequently drives the accumulation of CXCR3-bearing γδT cells into the liver at day 6; (iv) CXCR3-bearing γδT cells are enriched in intracellular IFN-γ, so this cytokine may directly exert acute liver damage and may also act on hepatocytes to induce late (i.e., day 6) hepatic CXCL9 production; (v) CXCL9 consequently initiates acute liver damage and also sustains the continuous accumulation of IFN-γ-enriched CXCR3-expressing γδT cells in the liver; and (vi) CXCR3-bearing γδT cells may also sustain or contribute to acute liver damage by recruiting effector CD8+ T cells. In addition, a direct interaction between activated hepatic γδT cells expressing high levels of FasL and hepatocytes enriched in Fas (67) could result in further acute liver damage in AdLacZ-infected mice.
FIG. 8.

Schematic overview of the potential mechanisms that promote the proinflammatory effect of γδT cells during AdLacZ-mediated acute liver toxicity (see Discussion).
Acknowledgments
This study was funded by an LSUHSC Intramural Preclinical Gene Therapy Research award (grant 130001040A) to M.N.A.
We are grateful to Deborah Chervenak (Flow Cytometry Core), Angela Dooley (Histopathology Core, Dept. of Physiology), Catherine Hickman (Breeding Colony), and Suzanne Dempsy (Dept. of Pathology), all at LSUHSC-Shreveport, and Maria Burdick (University of Virginia) for technical assistance. We are also highly indebted to Qiang-Jin Zhang (Department of Cellular Biology & Anatomy at LSUHSC-Shreveport) for giving us free access to the AutoMACS.
Footnotes
Published ahead of print on 30 July 2008.
REFERENCES
- 1.Ajuebor, M. N., A. I. Aspinall, F. Zhou, T. Le, Y. Yang, S. J. Urbanski, S. Sidobre, M. Kronenberg, C. M. Hogaboam, and M. G. Swain. 2005. Lack of chemokine receptor CCR5 promotes murine fulminant liver failure by preventing the apoptosis of activated CD1d-restricted NKT cells. J. Immunol. 1748027-8037. [DOI] [PubMed] [Google Scholar]
- 2.Ajuebor, M. N., C. M. Hogaboam, T. Le, A. E. Proudfoot, and M. G. Swain. 2004. CCL3/MIP-1α is pro-inflammatory in murine T cell-mediated hepatitis by recruiting CCR1-expressing CD4+ T cells to the liver. Eur. J. Immunol. 342907. [DOI] [PubMed] [Google Scholar]
- 3.Ajuebor, M. N., C. M. Hogaboam, T. Le, and M. G. Swain. 2003. C-C chemokine ligand 2/monocyte chemoattractant protein-1 directly inhibits NKT cell IL-4 production and is hepatoprotective in T cell-mediated hepatitis in the mouse. J. Immunol. 1705252-5259. [DOI] [PubMed] [Google Scholar]
- 4.Ajuebor, M. N., and M. G. Swain. 2002. Role of chemokines and chemokine receptors in the gastrointestinal tract. Immunology 105137-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ajuebor, M. N., M. G. Swain, and M. Perretti. 2002. Chemokines as novel therapeutic targets in inflammatory diseases. Biochem. Pharmacol. 631191-1196. [DOI] [PubMed] [Google Scholar]
- 6.Ajuebor, M. N., Z. Wondimu, C. M. Hogaboam, T. Le, A. E. Proudfoot, and M. G. Swain. 2007. CCR5 deficiency drives enhanced natural killer cell trafficking to and activation within the liver in murine T cell-mediated hepatitis. Am. J. Pathol. 1701975-1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arai, K., Z. X. Liu, T. Lane, and G. Dennert. 2002. IP-10 and Mig facilitate accumulation of T cells in the virus-infected liver. Cell. Immunol. 21948-56. [DOI] [PubMed] [Google Scholar]
- 8.Belperio, J. A., M. P. Keane, M. D. Burdick, J. P. Lynch III, Y. Y. Xue, K. Li, D. J. Ross, and R. M. Strieter. 2002. Critical role for CXCR3 chemokine biology in the pathogenesis of bronchiolitis obliterans syndrome. J. Immunol. 1691037-1049. [DOI] [PubMed] [Google Scholar]
- 9.Benton, K. A., J. A. Misplon, C. Y. Lo, R. R. Brutkiewicz, S. A. Prasad, and S. L. Epstein. 2001. Heterosubtypic immunity to influenza A virus in mice lacking IgA, all Ig, NKT cells, or gamma delta T cells. J. Immunol. 1667437-7445. [DOI] [PubMed] [Google Scholar]
- 10.Bergheim, I., J. P. Luyendyk, C. Steele, G. K. Russell, L. Guo, R. A. Roth, and G. E. Arteel. 2006. Metformin prevents endotoxin-induced liver injury after partial hepatectomy. J. Pharmacol. Exp. Ther. 3161053-1061. [DOI] [PubMed] [Google Scholar]
- 11.Boehm, U., T. Klamp, M. Groot, and J. C. Howard. 1997. Cellular responses to interferon-gamma. Annu. Rev. Immunol. 15749-795. [DOI] [PubMed] [Google Scholar]
- 12.Bukowski, J. F., B. A. Woda, S. Habu, K. Okumura, and R. M. Welsh. 1983. Natural killer cell depletion enhances virus synthesis and virus-induced hepatitis in vivo. J. Immunol. 1311531-1538. [PubMed] [Google Scholar]
- 13.Burdick, M. D., L. A. Murray, M. P. Keane, Y. Y. Xue, D. A. Zisman, J. A. Belperio, and R. M. Strieter. 2005. CXCL11 attenuates bleomycin-induced pulmonary fibrosis via inhibition of vascular remodeling. Am. J. Respir. Crit. Care Med. 171261-268. [DOI] [PubMed] [Google Scholar]
- 14.Cai, J. L., and P. W. Tucker. 2001. Gamma-delta T cells: immunoregulatory functions and immunoprotection. Chem. Immunol. 7999-138. [DOI] [PubMed] [Google Scholar]
- 15.Carding, S. R., and P. J. Egan. 2002. Gammadelta T cells: functional plasticity and heterogeneity. Nat. Rev. Immunol. 2336-345. [DOI] [PubMed] [Google Scholar]
- 16.Carding, S. R., and P. J. Egan. 2000. The importance of gamma delta T cells in the resolution of pathogen-induced inflammatory immune responses. Immunol. Rev. 17398-108. [DOI] [PubMed] [Google Scholar]
- 17.Cotter, M. J., A. K. Zaiss, and D. A. Muruve. 2005. Neutrophils interact with adenovirus vectors via Fc receptors and complement receptor 1. J. Virol. 7914622-14631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dechanet, J., P. Merville, A. Lim, C. Retiere, V. Pitard, X. Lafarge, S. Michelson, C. Meric, M. M. Hallet, P. Kourilsky, L. Potaux, M. Bonneville, and J. F. Moreau. 1999. Implication of gammadelta T cells in the human immune response to cytomegalovirus. J. Clin. Investig. 1031437-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faouzi, S., B. E. Burckhardt, J. C. Hanson, C. B. Campe, L. W. Schrum, R. A. Rippe, and J. J. Maher. 2001. Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-κB-independent, caspase-3-dependent pathway. J. Biol. Chem. 27649077-49082. [DOI] [PubMed] [Google Scholar]
- 20.Feng, G., and N. Kaplowitz. 2000. Colchicine protects mice from the lethal effect of an agonistic anti-Fas antibody. J. Clin. Investig. 105329-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrick, D. A., M. D. Schrenzel, T. Mulvania, B. Hsieh, W. G. Ferlin, and H. Lepper. 1995. Differential production of interferon-gamma and interleukin-4 in response to Th1- and Th2-stimulating pathogens by gamma delta T cells in vivo. Nature 373255-257. [DOI] [PubMed] [Google Scholar]
- 22.Goodman, Z. D. 2007. Grading and staging systems for inflammation and fibrosis in chronic liver diseases. J. Hepatol. 47598-607. [DOI] [PubMed] [Google Scholar]
- 23.Hayday, A., and R. Tigelaar. 2003. Immunoregulation in the tissues by gammadelta T cells. Nat. Rev. Immunol. 3233-242. [DOI] [PubMed] [Google Scholar]
- 24.Hayday, A. C., S. Roberts, and E. Ramsburg. 2000. gammadelta cells and the regulation of mucosal immune responses. Am. J. Respir. Crit. Care Med. 162S161-S163. [DOI] [PubMed] [Google Scholar]
- 25.Hitt, M. M., and F. L. Graham. 2000. Adenovirus vectors for human gene therapy. Adv. Virus Res. 55479-505. [DOI] [PubMed] [Google Scholar]
- 26.Hokeness, K. L., E. S. Deweerd, M. W. Munks, C. A. Lewis, R. P. Gladue, and T. P. Salazar-Mather. 2007. CXCR3-dependent recruitment of antigen-specific T lymphocytes to the liver during murine cytomegalovirus infection. J. Virol. 811241-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huber, S., C. Shi, and R. C. Budd. 2002. γδ T cells promote a Th1 response during coxsackievirus B3 infection in vivo: role of Fas and Fas ligand. J. Virol. 766487-6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang, D., J. Liang, J. Hodge, B. Lu, Z. Zhu, S. Yu, J. Fan, Y. Gao, Z. Yin, R. Homer, C. Gerard, and P. W. Noble. 2004. Regulation of pulmonary fibrosis by chemokine receptor CXCR3. J. Clin. Investig. 114291-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin, Y., S. J. Atkinson, J. A. Marrs, and P. J. Gallagher. 2001. Myosin ii light chain phosphorylation regulates membrane localization and apoptotic signaling of tumor necrosis factor receptor-1. J. Biol. Chem. 27630342-30349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson, M., S. Huyn, J. Burton, M. Sato, and L. Wu. 2006. Differential biodistribution of adenoviral vector in vivo as monitored by bioluminescence imaging and quantitative polymerase chain reaction. Hum. Gene Ther. 171262-1269. [DOI] [PubMed] [Google Scholar]
- 31.Kabelitz, D., and D. Wesch. 2001. Role of gamma delta T-lymphocytes in HIV infection. Eur. J. Med. Res. 6169-174. [PubMed] [Google Scholar]
- 32.Kafrouni, M. I., G. R. Brown, and D. L. Thiele. 2001. Virally infected hepatocytes are resistant to perforin-dependent CTL effector mechanisms. J. Immunol. 1671566-1574. [DOI] [PubMed] [Google Scholar]
- 33.Kass-Eisler, A., E. Falck-Pedersen, D. H. Elfenbein, M. Alvira, P. M. Buttrick, and L. A. Leinwand. 1994. The impact of developmental stage, route of administration and the immune system on adenovirus-mediated gene transfer. Gene Ther. 1395-402. [PubMed] [Google Scholar]
- 34.Klugewitz, K., D. H. Adams, M. Emoto, K. Eulenburg, and A. Hamann. 2004. The composition of intrahepatic lymphocytes: shaped by selective recruitment? Trends Immunol. 25590-594. [DOI] [PubMed] [Google Scholar]
- 35.Kusters, S., F. Gantner, G. Kunstle, and G. Tiegs. 1996. Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology 111462-471. [DOI] [PubMed] [Google Scholar]
- 36.Li, Y., D. A. Muruve, R. G. Collins, S. S. Lee, and P. Kubes. 2002. The role of selectins and integrins in adenovirus vector-induced neutrophil recruitment to the liver. Eur. J. Immunol. 323443-3452. [DOI] [PubMed] [Google Scholar]
- 37.Lieber, A., C. Y. He, L. Meuse, C. Himeda, C. Wilson, and M. A. Kay. 1998. Inhibition of NF-κB activation in combination with Bcl-2 expression allows for persistence of first-generation adenovirus vectors in the mouse liver. J. Virol. 729267-9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lieber, A., C. Y. He, L. Meuse, D. Schowalter, I. Kirillova, B. Winther, and M. A. Kay. 1997. The role of Kupffer cell activation and viral gene expression in early liver toxicity after infusion of recombinant adenovirus vectors. J. Virol. 718798-8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu, Q., and D. A. Muruve. 2003. Molecular basis of the inflammatory response to adenovirus vectors. Gene Ther. 10935-940. [DOI] [PubMed] [Google Scholar]
- 40.Liu, Q., A. K. Zaiss, P. Colarusso, K. Patel, G. Haljan, T. J. Wickham, and D. A. Muruve. 2003. The role of capsid-endothelial interactions in the innate immune response to adenovirus vectors. Hum. Gene Ther. 14627-643. [DOI] [PubMed] [Google Scholar]
- 41.Liu, Z. X., S. Govindarajan, and N. Kaplowitz. 2004. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology 1271760-1774. [DOI] [PubMed] [Google Scholar]
- 42.Liu, Z. X., S. Govindarajan, S. Okamoto, and G. Dennert. 2000. Fas- and tumor necrosis factor receptor 1-dependent but not perforin-dependent pathways cause injury in livers infected with an adenovirus construct in mice. Hepatology 31665-673. [DOI] [PubMed] [Google Scholar]
- 43.Liu, Z. X., S. Govindarajan, S. Okamoto, and G. Dennert. 2000. NK cells cause liver injury and facilitate the induction of T cell-mediated immunity to a viral liver infection. J. Immunol. 1646480-6486. [DOI] [PubMed] [Google Scholar]
- 44.Lockhart, E., A. M. Green, and J. L. Flynn. 2006. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J. Immunol. 1774662-4669. [DOI] [PubMed] [Google Scholar]
- 45.Lozier, J. N., G. Csako, T. H. Mondoro, D. M. Krizek, M. E. Metzger, R. Costello, J. G. Vostal, M. E. Rick, R. E. Donahue, and R. A. Morgan. 2002. Toxicity of a first-generation adenoviral vector in rhesus macaques. Hum. Gene Ther. 13113-124. [DOI] [PubMed] [Google Scholar]
- 46.Minagawa, M., H. Kawamura, Z. Liu, S. Govindarajan, and G. Dennert. 2005. Suppression of adenoviral gene expression in the liver: role of innate vs adaptive immunity and their cell lysis mechanisms. Liver Int. 25622-632. [DOI] [PubMed] [Google Scholar]
- 47.Moser, B., and M. Eberl. 2007. Gammadelta T cells: novel initiators of adaptive immunity. Immunol. Rev. 21589-102. [DOI] [PubMed] [Google Scholar]
- 48.Muruve, D. A., M. J. Barnes, I. E. Stillman, and T. A. Libermann. 1999. Adenoviral gene therapy leads to rapid induction of multiple chemokines and acute neutrophil-dependent hepatic injury in vivo. Hum. Gene Ther. 10965-976. [DOI] [PubMed] [Google Scholar]
- 49.Nanno, M., T. Shiohara, H. Yamamoto, K. Kawakami, and H. Ishikawa. 2007. Gammadelta T cells: firefighters or fire boosters in the front lines of inflammatory responses. Immunol. Rev. 215103-113. [DOI] [PubMed] [Google Scholar]
- 50.O'Brien, R. L., C. L. Roark, N. Jin, M. K. Aydintug, J. D. French, J. L. Chain, J. M. Wands, M. Johnston, and W. K. Born. 2007. Gammadelta T-cell receptors: functional correlations. Immunol. Rev. 21577-88. [DOI] [PubMed] [Google Scholar]
- 51.Ogasawara, J., R. Watanabe-Fukunaga, M. Adachi, A. Matsuzawa, T. Kasugai, Y. Kitamura, N. Itoh, T. Suda, and S. Nagata. 1993. Lethal effect of the anti-Fas antibody in mice. Nature 364806-809. [DOI] [PubMed] [Google Scholar]
- 52.Peng, Y., E. Falck-Pedersen, and K. B. Elkon. 2001. Variation in adenovirus transgene expression between BALB/c and C57BL/6 mice is associated with differences in interleukin-12 and gamma interferon production and NK cell activation. J. Virol. 754540-4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ponomarev, E. D., and B. N. Dittel. 2005. Gamma delta T cells regulate the extent and duration of inflammation in the central nervous system by a Fas ligand-dependent mechanism. J. Immunol. 1744678-4687. [DOI] [PubMed] [Google Scholar]
- 54.Ponomarev, E. D., M. Novikova, M. Yassai, M. Szczepanik, J. Gorski, and B. N. Dittel. 2004. Gamma delta T cell regulation of IFN-gamma production by central nervous system-infiltrating encephalitogenic T cells: correlation with recovery from experimental autoimmune encephalomyelitis. J. Immunol. 1731587-1595. [DOI] [PubMed] [Google Scholar]
- 55.Rajan, A. J., Y. L. Gao, C. S. Raine, and C. F. Brosnan. 1996. A pathogenic role for gamma delta T cells in relapsing-remitting experimental allergic encephalomyelitis in the SJL mouse. J. Immunol. 157941-949. [PubMed] [Google Scholar]
- 56.Raper, S. E., N. Chirmule, F. S. Lee, N. A. Wivel, A. Bagg, G. P. Gao, J. M. Wilson, and M. L. Batshaw. 2003. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 80148-158. [DOI] [PubMed] [Google Scholar]
- 57.Roark, C. L., J. D. French, M. A. Taylor, A. M. Bendele, W. K. Born, and L. O'Brien, R. 2007. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing γδ T cells. J. Immunol. 1795576-5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roncella, S., G. Cutrona, M. Truini, I. Airoldi, A. Pezzolo, A. Valetto, D. Di Martino, P. Dadati, A. De Rossi, M. Ulivi, I. Fontana, A. Nocera, U. Valente, M. Ferrarini, and V. Pistoia. 2000. Late Epstein-Barr virus infection of a hepatosplenic gamma delta T-cell lymphoma arising in a kidney transplant recipient. Haematologica 85256-262. [PubMed] [Google Scholar]
- 59.Ruzek, M. C., B. F. Kavanagh, A. Scaria, S. M. Richards, and R. D. Garman. 2002. Adenoviral vectors stimulate murine natural killer cell responses and demonstrate antitumor activities in the absence of transgene expression. Mol. Ther. 5115-124. [DOI] [PubMed] [Google Scholar]
- 60.Sakurai, F., K. Kawabata, N. Koizumi, N. Inoue, M. Okabe, T. Yamaguchi, T. Hayakawa, and H. Mizuguchi. 2006. Adenovirus serotype 35 vector-mediated transduction into human CD46-transgenic mice. Gene Ther. 131118-1126. [DOI] [PubMed] [Google Scholar]
- 61.Santra, S., M. S. Seaman, L. Xu, D. H. Barouch, C. I. Lord, M. A. Lifton, D. A. Gorgone, K. R. Beaudry, K. Svehla, B. Welcher, B. K. Chakrabarti, Y. Huang, Z. Y. Yang, J. R. Mascola, G. J. Nabel, and N. L. Letvin. 2005. Replication-defective adenovirus serotype 5 vectors elicit durable cellular and humoral immune responses in nonhuman primates. J. Virol. 796516-6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sciammas, R., P. Kodukula, Q. Tang, R. L. Hendricks, and J. A. Bluestone. 1997. T cell receptor-gamma/delta cells protect mice from herpes simplex virus type 1-induced lethal encephalitis. J. Exp. Med. 1851969-1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Selin, L. K., P. A. Santolucito, A. K. Pinto, E. Szomolanyi-Tsuda, and R. M. Welsh. 2001. Innate immunity to viruses: control of vaccinia virus infection by gamma delta T cells. J. Immunol. 1666784-6794. [DOI] [PubMed] [Google Scholar]
- 64.Shayakhmetov, D. M., A. Gaggar, S. Ni, Z. Y. Li, and A. Lieber. 2005. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J. Virol. 797478-7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shayakhmetov, D. M., Z. Y. Li, S. Ni, and A. Lieber. 2004. Analysis of adenovirus sequestration in the liver, transduction of hepatic cells, and innate toxicity after injection of fiber-modified vectors. J. Virol. 785368-5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shayakhmetov, D. M., Z. Y. Li, S. Ni, and A. Lieber. 2005. Interference with the IL-1-signaling pathway improves the toxicity profile of systemically applied adenovirus vectors. J. Immunol. 1747310-7319. [DOI] [PubMed] [Google Scholar]
- 67.Song, E., S. K. Lee, J. Wang, N. Ince, N. Ouyang, J. Min, J. Chen, P. Shankar, and J. Lieberman. 2003. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat. Med. 9347-351. [DOI] [PubMed] [Google Scholar]
- 68.Spahn, T. W., S. Issazadah, A. J. Salvin, and H. L. Weiner. 1999. Decreased severity of myelin oligodendrocyte glycoprotein peptide 33-35-induced experimental autoimmune encephalomyelitis in mice with a disrupted TCR delta chain gene. Eur. J. Immunol. 294060-4071. [DOI] [PubMed] [Google Scholar]
- 69.Stiles, L. N., M. P. Hosking, R. A. Edwards, R. M. Strieter, and T. E. Lane. 2006. Differential roles for CXCR3 in CD4+ and CD8+ T cell trafficking following viral infection of the CNS. Eur. J. Immunol. 36613-622. [DOI] [PubMed] [Google Scholar]
- 70.Suzuki, T., K. Fujikura, T. Higashiyama, and K. Takata. 1997. DNA staining for fluorescence and laser confocal microscopy. J. Histochem. Cytochem. 4549-53. [DOI] [PubMed] [Google Scholar]
- 71.Tseng, C. T., E. Miskovsky, M. Houghton, and G. R. Klimpel. 2001. Characterization of liver T-cell receptor gammadelta T cells obtained from individuals chronically infected with hepatitis C virus (HCV): evidence for these T cells playing a role in the liver pathology associated with HCV infections. Hepatology 331312-1320. [DOI] [PubMed] [Google Scholar]
- 72.Wang, T., E. Scully, Z. Yin, J. H. Kim, S. Wang, J. Yan, M. Mamula, J. F. Anderson, J. Craft, and E. Fikrig. 2003. IFN-gamma-producing gamma delta T cells help control murine West Nile virus infection. J. Immunol. 1712524-2531. [DOI] [PubMed] [Google Scholar]
- 73.Wen, L., D. F. Barber, W. Pao, F. S. Wong, M. J. Owen, and A. Hayday. 1998. Primary gamma delta cell clones can be defined phenotypically and functionally as Th1/Th2 cells and illustrate the association of CD4 with Th2 differentiation. J. Immunol. 1601965-1974. [PubMed] [Google Scholar]
- 74.Wickham, T. J., P. Mathias, D. A. Cheresh, and G. R. Nemerow. 1993. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 73309-319. [DOI] [PubMed] [Google Scholar]
- 75.Wilson, J. M. 1996. Adenoviruses as gene-delivery vehicles. N. Engl. J. Med. 3341185-1187. [DOI] [PubMed] [Google Scholar]
- 76.Wolff, G., S. Worgall, N. van Rooijen, W. R. Song, B. G. Harvey, and R. G. Crystal. 1997. Enhancement of in vivo adenovirus-mediated gene transfer and expression by prior depletion of tissue macrophages in the target organ. J. Virol. 71624-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Worgall, S., A. Busch, M. Rivara, D. Bonnyay, P. L. Leopold, R. Merritt, N. R. Hackett, P. W. Rovelink, J. T. Bruder, T. J. Wickham, I. Kovesdi, and R. G. Crystal. 2004. Modification to the capsid of the adenovirus vector that enhances dendritic cell infection and transgene-specific cellular immune responses. J. Virol. 782572-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Worgall, S., G. Wolff, E. Falck-Pedersen, and R. G. Crystal. 1997. Innate immune mechanisms dominate elimination of adenoviral vectors following in vivo administration. Hum. Gene Ther. 837-44. [DOI] [PubMed] [Google Scholar]
- 79.Yamaguchi, T., K. Kawabata, N. Koizumi, F. Sakurai, K. Nakashima, H. Sakurai, T. Sasaki, N. Okada, K. Yamanishi, and H. Mizuguchi. 2007. Role of MyD88 and TLR9 in the innate immune response elicited by serotype 5 adenoviral vectors. Hum. Gene Ther. 18753-762. [DOI] [PubMed] [Google Scholar]
- 80.Yang, Y., H. C. Ertl, and J. M. Wilson. 1994. MHC class I-restricted cytotoxic T lymphocytes to viral antigens destroy hepatocytes in mice infected with E1-deleted recombinant adenoviruses. Immunity 1433-442. [DOI] [PubMed] [Google Scholar]
- 81.Yang, Y., Q. Su, and J. M. Wilson. 1996. Role of viral antigens in destructive cellular immune responses to adenovirus vector-transduced cells in mouse lungs. J. Virol. 707209-7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang, Y., and J. M. Wilson. 1995. Clearance of adenovirus-infected hepatocytes by MHC class I-restricted CD4+ CTLs in vivo. J. Immunol. 1552564-2570. [PubMed] [Google Scholar]
- 83.Yang, Y., Z. Xiang, H. C. Ertl, and J. M. Wilson. 1995. Upregulation of class I major histocompatibility complex antigens by interferon gamma is necessary for T-cell-mediated elimination of recombinant adenovirus-infected hepatocytes in vivo. Proc. Natl. Acad. Sci. USA 927257-7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yin, Z., C. Chen, S. J. Szabo, L. H. Glimcher, A. Ray, and J. Craft. 2002. T-Bet expression and failure of GATA-3 cross-regulation lead to default production of IFN-gamma by gammadelta T cells. J. Immunol. 1681566-1571. [DOI] [PubMed] [Google Scholar]
- 85.Zaiss, A. K., Q. Liu, G. P. Bowen, N. C. Wong, J. S. Bartlett, and D. A. Muruve. 2002. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 764580-4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang, H., J. Cook, J. Nickel, R. Yu, K. Stecker, K. Myers, and N. M. Dean. 2000. Reduction of liver Fas expression by an antisense oligonucleotide protects mice from fulminant hepatitis. Nat. Biotechnol. 18862-867. [DOI] [PubMed] [Google Scholar]
- 87.Zhang, Y., N. Chirmule, G. P. Gao, R. Qian, M. Croyle, B. Joshi, J. Tazelaar, and J. M. Wilson. 2001. Acute cytokine response to systemic adenoviral vectors in mice is mediated by dendritic cells and macrophages. Mol. Ther. 3697-707. [DOI] [PubMed] [Google Scholar]