

Abstract
Recent studies have made evident the fact that the 19S regulatory component of the proteasome has functions that extend beyond degradation, particularly in the regulation of transcription. Although 19S ATPases facilitate chromatin remodeling and acetylation events in yeast (Saccharomyces cerevisiae), it is unclear if they play similar roles in mammalian cells. We have recently shown that the 19S ATPase Sug1 positively regulates the transcription of the critical inflammatory gene for major histocompatibility complex class II (MHC-II) by stabilizing enhanceosome assembly at the proximal promoter. We now show that Sug1 is crucial for regulating histone H3 acetylation at the MHC-II proximal promoter. Sug1 binds to acetylated histone H3 and, in the absence of Sug1, histone H3 acetylation is dramatically decreased at the proximal promoter, with a preferential loss of acetylation at H3 lysine 18. Sug1 also binds to the MHC-II histone acetyltransferase CREB-binding protein (CBP) and is critical for the recruitment of CBP to the MHC-II proximal promoter. Our current study strongly implicates the 19S ATPase Sug1 in modifying histones to initiate MHC-II transcription and provides novel insights into the role of the proteasome in the regulation of mammalian transcription.
Major histocompatibility complex class II (MHC-II) molecules are cell surface glycoproteins which bind and present processed antigenic peptides to CD4+ T lymphocytes to initiate immune system protection against invading pathogens and tumors (57). Tight regulation of MHC-II expression is crucial to maintain a functional immune system and to limit the opportunity for the development of autoimmune diseases (32, 57). MHC-II is expressed constitutively on antigen-presenting cells and can be inducibly expressed on most nucleated cells by gamma interferon (IFN-γ) (7, 20). Constitutive and IFN-γ-inducible MHC-II expression is regulated at the level of transcription by a series of elements in the MHC-II promoter. Nuclear factor Y, regulatory factor X, and cyclic AMP response element binding protein (CREB) bind, respectively, to the Y and X elements of the MHC-II proximal promoter, forming a multiprotein enhanceosome complex, which is necessary but not sufficient for transcription initiation. Once the enhanceosome is assembled on the MHC-II promoter, the class II transactivator (CIITA) can be recruited. CIITA binding stabilizes the enhanceosome complex and recruits basal transcriptional components, including the CDK7 subunit of TFIIH and the CDK9 subunit of P-TEFb, which phosphorylate polymerase II and initiate the switch to an elongation complex (12, 41, 48, 50, 68). CIITA is also known to interact with a variety of transcriptional cofactors, including multiple histone acetyltransferases (HATs) and histone deacetylases (HDACs) (8, 29, 76, 81). Although much is known about the requirement of these basal and inducible transcription factors for MHC-II expression, less is known regarding the importance of epigenetic modifications required to open the chromatin structure and allow these transcription factors to bind.
Integral to eukaryotic chromatin structure are nucleosomes, which consist of an octameric histone protein core. There are four histone proteins in the octamer, H2A, H2B, H3, and H4, around which DNA winds to create an environment of reduced DNA accessibility. The N-terminal ends of histone proteins extend out of the nucleosome and are regions for posttranslational modifications (14). Potential covalent modifications of histone N-terminal tail regions include acetylation, phosphorylation, and methylation, which can occur at the approximately 30-amino-acid residues in these tails (14, 40, 47, 66). It is now widely accepted that it is the complex combination of these modifications that opens the chromatin structure and creates docking sites for the recruitment of effector proteins, thereby aiding in determining the expressivity of the gene (28, 30, 40, 49).
The most studied histone modification is the acetylation of lysine residues in the N-terminal tails of H3 and H4, which counteracts the compact nature of chromatin by relaxing the interaction between histone proteins and DNA (24, 31, 37). Epigenetic studies of the IFN-γ-inducible MHC-II promoter in HeLa cells have shown that prior to IFN-γ stimulation, histone H3 and histone H4 are acetylated at low to moderate levels, which coordinates with the assembly of regulatory factor X, nuclear factor Y, and CREB components of the enhanceosome complex (8). Following IFN-γ stimulation in HeLa cells, CIITA is expressed and bound to the proximal promoter, which recruits basal transcriptional machinery and enhances the recruitment of HATs, thus increasing acetylation and allowing the transcription of the MHC-II gene HLA-DRA (8, 76). The CREB-binding protein (CBP)/p300 is a transcriptional coactivator with potent HAT activity which is capable of acetylating all four core histones (6, 56). CBP is thought to have multiple functions on the MHC-II promoter, both as a HAT that acetylates histones H3 and H4 and as an integrator that links CIITA and CREB (15). In HeLa cells, CBP binding to the MHC-II proximal promoter is increased within 2 hours of IFN-γ stimulation, preceding CIITA promoter binding by several hours (80). In IFN-γ-inducible endothelial cells, CBP is associated with the MHC-II promoter at low levels prior to IFN-γ stimulation, and association increases rapidly upon IFN-γ stimulation (1). When CBP promoter binding is blocked, constitutive levels of MHC-II promoter acetylation are inhibited in unstimulated endothelial cells, indicating a closed chromatin conformation due to decreased CBP binding and promoter histone hypoacetylation that is independent of CIITA expression (1).
Although active expression of MHC-II genes is associated with robust histone H3 acetylation, it is less understood how this and other modifications coordinate gene expression (35, 51, 63, 68, 80). It is well established that many of the dramatic changes in acetylation occurring on histones H3 and H4 at the activated MHC-II promoter are CIITA dependent. That CIITA is required for MHC-II gene expression indicates that CIITA promoter binding likely directs HAT activity toward histone H3 and opens up a large portion of the promoter for transcriptional activation (8). However, as to the initial acetylation events that precede CIITA binding, only limited information is available on how histone-modifying enzymes are initially recruited to the MHC-II promoter in order to open the chromatin structure for CIITA binding, full enhanceosome formation, and transcription initiation and elongation.
The 26S proteasome is a large, multisubunit complex that functions to degrade polyubiquitinated proteins and has been determined to play an emerging role in transcriptional regulation (7, 17, 46, 55, 70). The 26S proteasome consists of two macromolecular complexes: the 20S catalytic core and the 19S regulatory complex. The 20S catalytic core degrades proteins into peptides in an energy-independent manner (7, 73). The 19S regulatory complex regulates the assembly of the 26S proteasome, recruits polyubiquitinated proteins to the proteasome, and shuttles the targeted proteins to the 20S core for degradation (22, 34, 69). The mammalian 19S consists of a lid comprised of eight non-ATPase proteins and a base comprised of six ATPase (S4, S6a, S6b, S7, S8 [Sug1], and S10b) and three non-ATPase (S1, S2, and S5a) proteins (34). Observations that yeast (Saccharomyces cerevisiae) 19S ATPases associate with the Gal4 activator, are recruited to the GAL promoter (36), and are critical for efficient transcriptional elongation by RNA polymerase II (26) firmly established a role for the 19S proteasome in yeast transcription. Specific alleles of Rpt6, the yeast ortholog of Sug1, rescue a class of Gal4 activation domain mutants (62), recruit transcription factors to TBP (72), and are associated with actively transcribing genes (36). Rpt6 also has recently been shown to link histone ubiquitination and histone methylation, which precede acetylation as important steps in initiating transcriptional elongation (25, 59, 65, 71). Reports from studies with yeast that the 19S proteasome enhances the recruitment of the HAT complex SAGA (Spt-Ada-Gcn5-acetyltransferase) to promoters and that Rpt6 associates with SAGA and is required for SAGA targeting further imply that this 19S ATPase is also important for targeting HATs to promoters (23, 26, 36, 46). In fact, the physical and functional interactions between 19S ATPases and chromatin structure seem to be quite widespread in yeast, as evidenced by several recent reports (5, 44, 67, 70).
The ubiquitin-proteasome pathway has also been linked to the regulation of transcription in mammalian cells (10, 21, 27, 38, 77). These emerging roles for the 19S ATPases in regulating transcription highlight the importance of understanding the biochemical function of Sug1 at mammalian promoters. Sug1 is recruited to p21waf1 promoters in response to UV-induced DNA damage (78) as well as to the human immunodeficiency virus type 1 promoter to enhance Tat-dependent transcription (45). Sug1 also associates with p53, with E1A, and with several nuclear receptors (46, 74); however, evidence for roles for 19S in regulating mammalian chromatin structure has been lacking. We have previously shown that Sug1 is rapidly recruited to the MHC-II promoter upon IFN-γ stimulation and that the decreased expression of Sug1 results in reduced CIITA recruitment to the MHC-II proximal promoter and decreased MHC-II gene expression (10). However, the function of Sug1 in CIITA recruitment and the potential for interactions between Sug1 and histone modifications in mammalian cells remain to be investigated. We therefore analyzed the role played by Sug1 in regulating histone H3 acetylation at HLA-DRA. Data presented here offer new insights into MHC-II transcription and establish a role for Sug1 in the deposition of histone acetylation modifications at the MHC-II proximal promoter. We show that Sug1 associates in vivo with acetylated histone H3 and that decreased expression of Sug1 and additional 19S ATPases decreases MHC-II promoter-specific H3 acetylation. We also show that recruitment of the HAT CBP to the MHC-II promoter is blocked in the absence of Sug1. Taken together, these studies offer novel insights into the regulation of MHC-II transcription and expand our knowledge of the role the proteasome plays in mediating the epigenetic regulation of mammalian genes.
MATERIALS AND METHODS
Cell lines.
HeLa (human epithelial) cells from ATCC (Manassas, VA) were maintained in Dulbecco's modified Eagle's medium (Mediatech Inc., Herndon, VA) supplemented with 10% fetal calf serum, 5 mM l-glutamine, and 5 mM penicillin-streptomycin at 37°C with 5% carbon dioxide.
Antibodies.
Histone H3, acetylated histone H3, acetylated histone H3 K9, and rabbit immunoglobulin G (IgG) isotype control antibodies were from Upstate (Lake Placid, NY). Acetylated H3 K18, p53, and Myc antibodies were from Abcam (Cambridge, MA). S5a, S6a, S7, and alpha 4 antibodies were from Biomol International, L.P. (Plymouth Meeting, PA). Sug1 antibody was from Novus Biologicals (Littleton, CO), CIITA antibody was from Rockland (Rockland, Gilbertsville, PA), and CBP antibody was from Santa Cruz (Santa Cruz, CA).
Plasmids.
The Myc Sug1 and Myc Sug1 ATPase mutant constructs were a generous gift from A. A. Wani (78).
Coimmunoprecipitations.
HeLa cells were plated at a cell density of 8 ×105 in 10-cm tissue culture plates. Following adhesion, cells were transfected with 5 μg of the indicated plasmids by use of Fugene 6 (Roche, Indianapolis, IN) according to the manufacturer's protocol. Twenty-four hours following transfection, cells were harvested and lysed in radioimmunoprecipitation assay buffer (1 M Tris [pH 8.0], 5% deoxycholate, 10% Nonidet P-40, 5 M NaCl, 10% sodium dodecyl sulfate [SDS], 5 mM EDTA, 1 M dithiothreitol [DTT]) supplemented with complete EDTA-free protease inhibitors (Roche, Indianapolis, IN) on ice and then precleared with 30 μl of IgG beads (Sigma, Saint Louis, MO). Cell lysates were subjected to immunoprecipitation (IP) overnight with 5 μg anti-acetylated histone H3 (Upstate), anti-CBP (Santa Cruz Biotechnology), rabbit IgG isotype control (Upstate), or Myc-conjugated beads (Sigma). Immune complexes were isolated by incubating lysates with 50 μl of protein G beads (Pierce, Rockland, IL) for 2 h. IP proteins were denatured with Laemmli buffer (Bio-Rad, Hercules, CA) and subjected to SDS-polyacrylamide gel electrophoresis. Associations were detected by immunoblotting (IB) with Myc horseradish peroxidase-conjugated antibody (Abcam). Horseradish peroxidase was detected with Supersignal West Pico Chemiluminescent substrate (Pierce). Bradford assays were used to normalize for protein and equal loading was verified by IB.
siRNA constructs and transient transfections.
Short interfering RNA (siRNA) duplexes were used for transient knockdown of 19S ATPases Sug1, S6a, and S7. siRNA sequences were designed with a G+C content of 35 to 55% containing dTdT overhangs and were compared to the NCBI BLAST nucleotide database. The target sequences of Sug1 and S7 siRNA used were 5′-AAGGTACATCCTGAAGGTAAA-3′ and 5′-AACTGCGAGAAGTAGTTGAAA-3′, respectively (Qiagen, Valencia, CA). S6a siRNA was predesigned siRNA directed against human PSMC3 (Qiagen). siRNA for Lamin protein was used as positive control siRNA (Qiagen), and scrambled sequence siRNA was used as negative control siRNA (Qiagen). HeLa cells were transfected with scrambled sequence control siRNA (Qiagen; Santa Cruz Biotechnology) or ATPase-specific siRNA (Qiagen) and were treated with IFN-γ as indicated. Cells were lysed in NP-40 lysis buffer (1 M Tris [pH 8.0], 1 M KCl, 10% NP-40, 0.5 M EDTA, 5 M NaCl, 1 M DTT, distilled water [dH2O]) supplemented with complete EDTA-free protease inhibitors (Roche), and knockdown efficiency and specificity were assessed by Western blotting for ATPase expression as described above.
Histone extractions.
HeLa cells were treated with Sug1 siRNA (Qiagen) or scrambled sequence control siRNA. Forty-eight hours after siRNA transfection, 10% of the total cell volume was lysed with 1% Nonidet P-40 buffer (1 M Tris [pH 8.0], 1 M KCl, 10% NP-40, 0.5 M EDTA, 5 M NaCl, 1 M DTT, dH2O) with protease inhibitor and analyzed by Western blotting for ATPase knockdown as described above. The rest of the cell volume was lysed in Triton extraction solution (0.4 M sodium butyrate, 10% Triton X-100, 2% NaN3) supplemented with complete EDTA-free protease inhibitors (Roche) at 4°C. Histone proteins were isolated by incubation with 0.2 N HCl at 4°C for 4 h. Lysate samples were normalized for protein concentration, supplemented with 5% (vol/vol) β-mercaptoethanol, denatured with Laemmli buffer (Bio-Rad), and separated by SDS-polyacrylamide gel electrophoresis. Gels were transferred to nitrocellulose and subjected to IB using polyclonal acetylated histone H3 or acetylated histone H3 K18 primary antibodies (Upstate) and horseradish peroxidase-conjugated rabbit secondary antibody (Santa Cruz). Horseradish peroxidase was detected with Supersignal West Pico chemiluminescent substrate (Pierce). Bradford assays were used to normalize for protein.
ChIP.
Chromatin IP (ChIP) assays were performed as previously described (38). Briefly, HeLa cells were stimulated with 500 U/ml IFN-γ (Peprotech, Rocky Hill, NJ), 5 mM HDAC inhibitor sodium butyrate (Upstate), 5.2 μΜ proteasome inhibitor MG132 (EMD Biosciences, San Diego, CA), or 10 μΜ proteasome inhibitor lactacystin (BostonBiochem, Cambridge, MA) as indicated. Cells were cross-linked with 1% formaldehyde for 10 min at room temperature. Cross-linking was stopped by the addition of 0.125 M glycine for 5 min at room temperature. Cells were lysed in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris [pH 8.0], dH2O) with protease inhibitor for 30 min on ice and were sonicated at a constant pulse to generate an average of 500 to 750 bp of sheared DNA. Sonicated lysates were precleared with salmon sperm-coated agarose beads (Upstate), and half of the lysate was subjected to IP with 10 μg of polyclonal antibody against CIITA (Rockland), acetylated H3 (Upstate), acetylated H3 K18 (Abcam), acetylated H3 K9 (Upstate), S6a (Biomol), S7 (Biomol), S5a (Biomol), alpha 4 (Biomol), or CBP (Santa Cruz) overnight at 4°C. The remaining half of the lysate was used as a control and was subjected to IP with isotype control antibody (Upstate). Following an additional 2-h IP with 50 μl of salmon sperm-coated agarose beads, samples were washed for 5 min at 4°C with the following buffers: low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris [pH 8.0], 150 mM NaCl, dH2O), high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris [pH 8.0], 500 mM NaCl, dH2O), LiCl buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris [pH 8.0], dH2O), and 1× Tris-EDTA buffer and were eluted with SDS elution buffer (1% SDS, 0.1 M NaHCO3, dH2O). Following elution, cross-links were reversed overnight with 5 M NaCl at 65°C and IP DNA was isolated using a phenol-chloroform-isoamyl alcohol mix (Invitrogen) as per the manufacturer's instructions. Isolated DNA was analyzed by real-time PCR using primers spanning the W-X-Y box of the MHC class II HLA-DRA promoter, MHC class II HLA-DRA exon III (16), MHC class II HLA-DRA exon V (16), the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) promoter, and CIITA pIV (53). Sequences for primers and probe are located in Table S1 in the supplemental material. Values graphed were calculated based on the standard curves generated.
ChIP in ATPase knockdowns.
HeLa cells were transfected with ATPase-specific siRNA (Qiagen) or control siRNA (Qiagen). Cells were treated with 5 mM sodium butyrate (Upstate) and 500 U/ml IFN-γ as indicated, and 10% of the total cell volume was lysed with 1% Nonidet P-40 buffer (1 M Tris [pH 8.0], 1 M KCl, 10% NP-40, 0.5 M EDTA, 5 M NaCl, 1 M DTT, dH2O) with protease inhibitor and analyzed by Western blotting for ATPase knockdown as described above. The remaining fraction of the cells was subjected to a ChIP assay.
ChIP with ATPase overexpression.
HeLa cells were transfected with 5 μg of the indicated plasmids by use of Fugene 6 (Roche) according to the manufacturer's protocol. Cells were treated with 500 U/ml IFN-γ as indicated, and 10% of the total cell volume was lysed with 1% Nonidet P-40 buffer (1 M Tris [pH 8.0], 1 M KCl, 10% NP-40, 0.5 M EDTA, 5 M NaCl, 1 M DTT, dH2O) with protease inhibitor and analyzed by Western blotting for Sug1 overexpression as described above. The remaining fraction of the cells was subjected to a ChIP assay.
In vivo proteolytic activity.
HeLa cells were transfected with Sug1-specific siRNA (Qiagen) or control siRNA (Qiagen). Cells were treated with 5.2 μΜ MG132 proteasome inhibitor (EMD Biosciences) and 100 μΜ cycloheximide (Sigma) as indicated. The total cell volume was lysed with 1% Nonidet P-40 buffer with protease inhibitor and analyzed by Western blotting for ATPase knockdown efficiency and p53 degradation as described above.
RESULTS
Sug1 associates with acetylated histone H3 in vivo.
The 19S proteasome recruits polyubiquitinated proteins to the 20S catalytic core for degradation. Research in yeast has also suggested that the 19S proteasome functions independent of protein degradation to regulate transcription by aiding in the opening of the chromatin structure (27, 36, 45). In yeast, the 19S proteasome associates with the activated Gal1-10 promoter (36, 46), and in mammalian cells, components of the 19S proteasome are recruited to p21waf1 promoters in response to UV-induced DNA damage and to MHC-II HLA-DRA promoters in response to IFN-γ stimulation (10, 78). Furthermore, the yeast 19S ATPase Rpt6 has been implicated in regulating histone modifications, including histone acetylation and methylation (25, 46). We therefore sought to determine whether the mammalian ortholog of Rpt6, Sug1, plays a role in mediating posttranslational histone modifications in mammalian cells. To determine whether Sug1 associates with acetylated histone H3 in vivo, coimmunoprecipitations were performed with HeLa (human epithelial) cells transfected with Myc-tagged Sug1 (Fig. 1). Polyclonal antibody was used to subject endogenous acetylated histone H3 to IP. Sug1 and acetylated histone H3 precipitated together (Fig. 1, top, lane 3). Control samples were subjected to IP with Myc beads (Fig. 1, top, lane 1) or an isotype control (Fig. 1, top, lane 2). Equal loading was confirmed by IB analysis of lysates (Fig. 1, bottom).
FIG. 1.
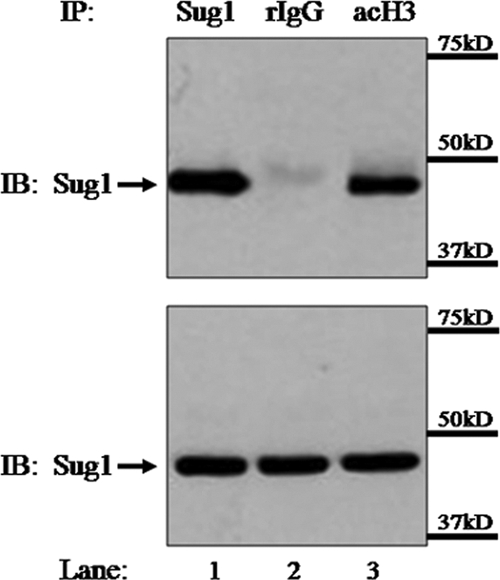
Sug1 associates with acetylated histone H3 in vivo. HeLa cells transfected with Myc-tagged Sug1 were lysed and subjected to IP with polyclonal antibody against acetylated histone H3 (acH3; lane 3, top). Control samples were subjected to IP with Sug1 antibody (lane 1, top) and isotype control rabbit IgG (rIgG; lane 2, top). IP and lysate control samples (bottom) were subjected to IB for Myc. Results reported are data representative of three experiments.
Sug1 knockdown decreases MHC-II promoter-specific histone H3 acetylation.
We have previously shown that the 19S ATPase Sug1 is recruited to the MHC-II proximal promoter (10). In the absence of Sug1, IFN-γ stimulation fails to recruit CIITA to the MHC-II promoter and results in a dramatic loss of MHC-II gene expression (10). The molecular mechanism responsible for the loss of CIITA promoter binding in Sug1-deficient cells remains unknown. To investigate potential roles for Sug1 in regulating chromatin modifications responsible for stabilizing and/or recruiting CIITA to the MHC-II proximal promoter, we utilized a siRNA duplex to specifically knock down endogenous Sug1 expression in HeLa cells and then performed ChIP experiments to detect endogenous levels of H3 acetylation at the MHC-II proximal promoter (Fig. 2). siRNA-mediated knockdown of Sug1 resulted in an approximately 90% decrease in endogenous Sug1 expression (Fig. 2A, top) but did not impact the expression of additional proteasomal ATPases S6a and S7 (Fig. 2A, bottom). Initial ChIP mapping experiments demonstrated that constitutive levels of histone acetylation at the MHC-II promoter are enhanced upon IFN-γ stimulation (Fig. 2B, left). When siRNA was used to knock down Sug1 expression, dramatic effects on MHC-II promoter acetylation were observed. Upon IFN-γ stimulation, H3 acetylation was enhanced in control siRNA-treated cells (Fig. 2B, right) but was inhibited in Sug1 siRNA-treated cells (Fig. 2B, right). HDAC enzymes were inhibited by pretreating cells with HDAC inhibitor, which resulted in histone H3 hyperacetylation (compare Fig. 2C, left, to B, left). Even in the presence of HDAC inhibition and IFN-γ induced H3 acetylation, H3 acetylation was dramatically decreased in the absence of Sug1 (Fig. 2C, right) compared to what was seen for control siRNA-treated cells (Fig. 2C, right). These experiments indicate that Sug1 plays a critical role in regulating MHC-II transcription by regulating key epigenetic events at the proximal promoter.
FIG. 2.

Sug1 knockdown decreases MHC-II promoter histone H3 acetylation. (A) Sug1 siRNA specifically and efficiently decreases Sug1 protein expression in the presence or absence of IFN-γ stimulation. HeLa cells transfected with control or Sug1-specific siRNA were harvested and subjected to Western blot analysis of the endogenous expression of 19S ATPases Sug1, S6a, and S7. Western blotting shows >90% knockdown of Sug1 (top) and stable expression of S6a and S7 (middle and bottom). (B) Sug1 knockdown decreases histone H3 acetylation at the MHC-II proximal promoter. ChIP assays were carried out with HeLa cells stimulated with IFN-γ for 0 to 18 h (left) or with HeLa cells transfected with scrambled siRNA control or Sug1-specific siRNA duplexes and 24 h later stimulated with IFN-γ for 0 to18 h (right). Lysates were subjected to IP with control antibody or with antibody to endogenous acetylated histone H3. Associated DNA was isolated and analyzed via real-time PCR using primers spanning the W-X-Y box of the MHC-II HLA-DRA promoter. Real-time PCR values were normalized to the total amount of HLA-DRA promoter DNA added to the reaction (input). Input values represent 5% of the total cell lysate. IP values are presented as increases in the MHC-II promoter DNA relative to unstimulated acetylated histone H3 IP sample values. Control IP values were (1.2 ± 0.3)-fold. Control and acetylated histone H3 IP values represent the mean ± the standard error of the mean (SEM) of three independent experiments. (C) Sug1 knockdown decreases histone H3 acetylation at the MHC-II proximal promoter in the presence of HDAC inhibition. ChIP assays were carried out with untransfected (left) or siRNA-transfected (right) HeLa cells which were treated with HDAC inhibitors (20 h) and stimulated with IFN-γ for 0 to 18 h. Lysates were subjected to IP with control antibody or with antibody to endogenous acetylated histone H3, and associated DNA was isolated and analyzed via real-time PCR as described for panel B. IP values are presented as increases in the MHC-II promoter DNA relative to unstimulated acetylated histone H3 IP sample values. Control IP values were (2.7 ± 0.8)-fold. Control and acetylated histone H3 IP values represent the mean ± SEM of three independent experiments. ***, P < 0.001 versus control siRNA.
To determine if the loss of MHC-II promoter acetylation at histone H3 is indicative of a global decrease in levels of acetylated histone H3, siRNA was used to knock down Sug1 expression in HeLa cells. Histones were acid extracted and lysates were subjected to IB for acetylated histone H3. Although Sug1 was sufficiently knocked down (Fig. 3A, bottom), levels of acetylated histone H3 were unaffected by the loss of Sug1 (Fig. 3A, top). Serially diluted lysates verified that H3 acetylation is indeed maintained in the Sug1-deficient cells (see Fig. S1 in the supplemental material). To account for the potential loss of histone H3 at the MHC-II promoter upon IFN-γ stimulation and/or Sug1 knockdown, ChIP assays were repeated utilizing an antibody specific for endogenous histone H3. Neither IFN-γ treatment alone (Fig. 3B, left) nor IFN-γ treatment in combination with Sug1 siRNA transfection (Fig. 3B, right) affected levels of histone H3 at the MHC-II proximal promoter. To ascertain if additional IFN-γ-inducible promoters are also epigenetically regulated by Sug1, ChIP experiments were performed to detect endogenous levels of H3 acetylation at the IFN-γ-inducible CIITA promoter, pIV, in the absence of Sug1. ChIP mapping experiments demonstrated that levels of histone H3 acetylation at CIITA pIV are enhanced upon IFN-γ stimulation (Fig. 3C, left). Consistent with ChIP studies indicating Sug1 association with CIITA pIV (data not shown), Sug1 knockdown resulted in reduced CIITA pIV acetylation (Fig. 3C, right). We have previously shown that Sug1 knockdown decreases MHC-II transcript levels but not CIITA transcript levels (10), suggesting that despite binding multiple inducible promoters, Sug1 binding may differentially regulate specific genes. To further determine if Sug1 specifically regulates H3 acetylation at inducible promoters, we performed ChIP experiments to detect endogenous levels of H3 acetylation at the GAPDH promoter in the absence of Sug1. ChIP mapping experiments demonstrated that the levels of histone acetylation at the GAPDH promoter were comparable to previously published results (8, 53), indicating that IFN-γ stimulation results in only a marginal change in histone acetylation (Fig. 3D, left). Furthermore, siRNA-mediated Sug1 knockdown had no effect on GAPDH promoter acetylation (Fig. 3D, right). These studies strongly implicate the Sug1 19S ATPase in the specific epigenetic regulation of transcription at inducible genes.
FIG. 3.

Sug1 knockdown decreases histone H3 acetylation in a promoter-specific manner. (A) Global histone H3 acetylation is unaffected by Sug1 knockdown. HeLa cells were left untreated (NT) or were transfected with either scrambled control siRNA duplexes or Sug1-specific siRNA duplexes. Lysates were subjected to IB for acetylated histone H3 (top) or for endogenous Sug1 (bottom). Results reported are data representative of three independent experiments. (B) Sug1 knockdown does not impact levels of histone H3 at the MHC-II proximal promoter. ChIP assays were carried out with HeLa cells stimulated with IFN-γ for 0 to 18 h (left) or HeLa cells transfected with scrambled siRNA control or Sug1-specific siRNA duplexes and 24 h later stimulated with IFN-γ for 0 to 18 h (right). Lysates were subjected to IP with control antibody or with antibody to endogenous histone H3. Associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. IP values are presented as increases in the MHC-II promoter DNA relative to unstimulated histone H3 IP sample values. Control IP values were (1.15 ± 0.15)-fold. Control and histone H3 IP values represent the mean ± SEM of two or three independent experiments. (C) Sug1 knockdown decreases levels of acetylated histone H3 at the IFN-γ-inducible CIITA pIV. ChIP assays were carried out with HeLa cells stimulated with IFN-γ for 0 to 18 h (left) or HeLa cells transfected with scrambled control or Sug1-specific siRNA duplexes and 24 h later stimulated with IFN-γ for 0 to 18 h (right). Lysates were subjected to IP with control antibody or with antibody to endogenous acetylated histone H3. Associated DNA was isolated and analyzed via real-time PCR using primers spanning CIITA pIV. Real-time PCR values were normalized to the total amount of CIITA pIV DNA added to the reaction (input). Input values represent 5% of the total cell lysate. IP values are presented as increases in CIITA pIV DNA relative to unstimulated acetylated histone H3 IP sample values. Control IP values were (1.0 ± 0.1)-fold. Control and histone H3 IP values represent the mean ± SEM of three independent experiments. (D) Sug1 knockdown does not impact levels of acetylated histone H3 at the GAPDH promoter. ChIP assays were carried out with HeLa cells stimulated with IFN-γ for 0 to 18 h (left) or HeLa cells transfected with scrambled control or Sug1-specific siRNA duplexes and 24 h later stimulated with IFN-γ for 0 to 18 h (right). Lysates were subjected to IP with control antibody or antibody to endogenous acetylated histone H3. Associated DNA was isolated and analyzed via real-time PCR using primers spanning the GAPDH promoter. Real-time PCR values were normalized to the total amount of GAPDH promoter DNA added to the reaction (input). Input values represent 5% of the total cell lysate. IP values are presented as increases in GAPDH promoter DNA relative to unstimulated acetylated histone H3 IP sample values. Control IP values were (0.75 ± 0.25)-fold. Control and histone H3 IP values represent the mean ± SEM of three independent experiments. ***, P < 0.001 versus control siRNA.
Inhibition of proteasomal activity does not affect MHC-II promoter acetylation.
Sug1, a component of the 19S proteasome, works with the other 19S ATPases (S4, S6a, S6b, S7, and S10b) to recruit polyubiquitinated proteins for degradation (18, 34). To determine the impact of Sug1 knockdown on proteasome function, we assayed in vivo proteolytic activities in HeLa cells transfected with Sug1-specific or control siRNA over a time course of cycloheximide treatment to prevent de novo translation. Because it is well established that p53 is polyubiquitinated and degraded via the 26S proteasome (3, 4, 52, 78), cells were lysed and analyzed for expression of this relatively short-lived protein. Consistent with published reports (13, 75), control siRNA-transfected cells showed a loss of p53 expression by 90 min after cycloheximide treatment (Fig. 4A, top). As a control, one set of control siRNA-transfected cells was pretreated with proteasome inhibitor MG132, which resulted in p53 accumulation upon cycloheximide treatment (Fig. 4A, bottom). Not surprisingly, Sug1 knockdown impacted proteasome activity. Although p53 accumulation was not as drastic as that in MG132-treated samples, cells transfected with Sug1 siRNA also showed elevated p53 expression upon cycloheximide treatment (Fig. 4A, middle). To determine if non-ATPase components of the 19S proteasome as well as components of the 20S core are also associated with the MHC-II proximal promoter, ChIP assays were performed to detect the 19S non-ATPase S5a and the 20S alpha 4 subunit. The 19S non-ATPase S5a associated with the MHC-II proximal promoter, and this low association was enhanced and maintained upon cytokine stimulation (Fig. 4B). The alpha 4 subunit of the 20S catalytic core also showed low-level binding upon cytokine stimulation that appeared to dissipate by 18 h poststimulation (Fig. 4C). The binding of these additional subunits to the MHC-II proximal promoter might indicate a role for the intact proteasome in regulating epigenetics at the MHC-II promoter. Therefore, we sought to ascertain if the loss of histone acetylation observed for Sug1-deficient cells was due in part to a lack of proteasome function. Cells were treated with either of the proteasome inhibitors MG132 and lactacystin, and ChIP experiments were performed to detect endogenous levels of H3 acetylation at the MHC-II proximal promoter. MG132-treated HeLa cells (Fig. 4D) or lactacystin-treated HeLa cells (Fig. 4E) stimulated with IFN-γ showed levels of H3 acetylation that were comparable to those observed for untreated cells (Fig. 2B, left). These experiments emphasize that although Sug1 function is required for proteolysis mediated by the 26S proteasome, the role played by Sug1 in regulating MHC-II proximal promoter acetylation is independent of proteolysis.
FIG. 4.

Sug1-dependent regulation of MHC-II promoter histone acetylation is proteolysis independent. (A) Sug1 is critical for degradation mediated by the 26S proteasome. HeLa cells were transfected with Sug1-specific or control siRNA and were treated with proteasome inhibitor MG132 as indicated. Following cycloheximide treatment, cells were lysed and lysates analyzed by Western blotting for p53 degradation. Results reported are data representative of three independent experiments. (B and C) 19S non-ATPase S5a and 20S core protein alpha 4 are recruited to the MHC-II proximal promoter upon IFN-γ stimulation. ChIP assays were carried out with HeLa cells stimulated with IFN-γ for 0 to 18 h. Lysates were subjected to IP with control antibody or antibody to endogenous S5a (B) or alpha 4 (C), and associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. IP values are presented as increases in the MHC-II promoter DNA relative to unstimulated S5a or alpha 4 IP sample values. Control IP values were (0.8 ± 0.1)-fold. Control and IP values represent the mean ± SEM of two independent experiments. (D and E) MHC-II proximal promoter histone H3 acetylation is unaffected by proteasome inhibition. ChIP assays were carried out with HeLa cells stimulated with IFN-γ for 0 to 18 h and treated with proteasome inhibitors MG132 (D) or lactacystin (E) for 4 h prior to harvesting. Lysates were subjected to IP with control antibody or antibody to endogenous acetylated histone H3, and associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. IP values are presented as increases in the MHC-II promoter DNA relative to unstimulated acetylated histone H3 IP sample values. Control IP values were (1.2 ± 0.25)-fold. Control and acetylated histone H3 IP values represent the mean ± SEM of two or three independent experiments.
Sug1 knockdown decreases lysine 18 acetylation at the MHC-II proximal promoter.
Acetylation occurs on lysine residues in histone tails. Epigenetic mapping of the MHC-II promoter has shown that lysines (K) K9, K14, K18, and K27 of histone H3 are acetylated (35, 63). Acetylation of H3 K9, K18, and K27 is CIITA dependent, whereas H3 K14 acetylation is also found constitutively in B cells lacking functional CIITA (RJ2.2.5). H3 K18 acetylation is robustly elevated at the activated MHC-II proximal promoter (35). To determine whether Sug1 preferentially associates with acetylated H3 K18 in vivo, coimmunoprecipitations were performed with HeLa cells transfected with Myc-tagged Sug1 (Fig. 5A). Polyclonal antibody was used to subject endogenous histone H3, acetylated H3, or acetylated H3 K18 to IP. As expected, Sug1 and histone H3 as well as acetylated histone H3 precipitated together (Fig. 5A, top, lanes 2 and 4). Enhanced binding was observed for cells subjected to IP with acetylated H3 K18 (Fig. 5A, top, lane 5). Control samples were subjected to IP with Myc beads (Fig. 5A, top, lane 1) or an isotype control (Fig. 5A, top, lane 3). Equal loading was confirmed by IB analysis of lysates (Fig. 5A, bottom). To evaluate H3 K18 acetylation levels at the MHC-II proximal promoter, HeLa cells were stimulated with IFN-γ, subjected to IP with antibody to endogenous acetylated H3 K18, and analyzed by real-time PCR with primers spanning the MHC-II HLA-DRA proximal promoter. ChIP assays showed elevated levels of H3 K18 acetylation at the MHC-II promoter upon IFN-γ stimulation that were substantially enhanced upon HDAC inhibition (Fig. 5B). While cells transfected with control siRNA showed an ∼40-fold increase in histone H3 acetylation upon IFN-γ stimulation and HDAC inhibition (Fig. 5C, left), similarly treated cells that were transfected with Sug1-specific siRNA showed dramatically decreased histone H3 K18 acetylation at the MHC-II promoter (Fig. 5C, left). To further determine if Sug1 specifically regulates H3 K18 acetylation at the MHC-II proximal promoter, we performed ChIP experiments to detect endogenous levels of H3 K18 acetylation at the GAPDH promoter. In the absence of Sug1, GAPDH promoter H3 K18 acetylation levels (Fig. 5C, right) were comparable to those in cells treated with control siRNA (Fig. 5C, right). Because H3 K18 acetylation appears to be preferentially impacted by Sug1 knockdown, we sought to determine the extent to which this interaction extends into the coding sequence of MHC-II genes. Therefore, we repeated real-time PCR using primers and probes specific for regions within MHC-II exon III and MHC-II exon V (Fig. 5D) in the ChIP studies for acetylated H3 K18 in Sug1-specific or control siRNA-transfected cells. Upon cytokine stimulation and in the presence of HDAC inhibition, H3 K18 acetylation was diminished within both exon III (Fig. 5E) and exon V (Fig. 5F) in Sug1 knockdown cells compared to what was seen for control samples (Fig. 5E and F).
FIG. 5.

Sug1 knockdown decreases histone H3 lysine 18 acetylation at the MHC-II proximal promoter. (A) Sug1 associates with acetylated H3 K18 in vivo. HeLa cells transfected with Myc-tagged Sug1 were lysed and subjected to IP with polyclonal antibody against histone H3 (lane 2, top), acetylated histone H3 (lane 4, top), or acetylated H3 K18 (lane 5, top). Control samples were subjected to IP with Myc beads (lane 1, top) and isotype control IgG (lane 3, top). IP and lysate control samples (bottom) were subjected to IB for Myc. Results reported are data representative of three experiments. (B) Histone H3 K18 acetylation at the MHC-II proximal promoter is enhanced upon IFN-γ stimulation and HDAC inhibition. ChIP assays were carried out with HeLa cells treated with IFN-γ for 0 to 18 h or HDAC inhibitor (20 h) and IFN-γ (18 h). Lysates were subjected to IP with control antibody or antibody to endogenous acetylated histone H3 K18, and associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. IP values are presented as increases in MHC-II promoter DNA relative to unstimulated acetylated H3 K18 IP sample values. Control IP values were (0.8 ± 0.2)-fold. Control and acetylated histone H3 K18 IP values represent the mean ± SEM of three independent experiments. (C) Sug1 knockdown specifically decreases H3 K18 acetylation at the MHC-II proximal promoter. HeLa cells were transfected with scrambled siRNA control or with Sug1-specific siRNA duplexes and 24 h later were treated with HDAC inhibitor (20 h) and stimulated with IFN-γ for 0 to 18 h. Lysates were subjected to IP with control antibody or antibody to endogenous acetylated H3 K18, and associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. Data are presented as increases in the MHC-II promoter DNA (left) or GAPDH promoter DNA (right) relative to unstimulated acetylated H3 K18 IP sample values. Control IP values were (1.0 ± 0.4)-fold. Control and acetylated histone H3 K18 IP values represent the mean ± SEM of two to four independent experiments. (D) Real-time PCR primer locations in the MHC-II gene. (E and F) Sug1 knockdown decreases H3 K18 acetylation within the MHC-II coding region. ChIP assays were performed with HeLa cells transfected with scrambled control or Sug1-specific siRNA duplexes and stimulated as described for Fig. 2B. Isolated DNA was analyzed via real-time PCR using primers and probes specific for MHC-II exon III (E) or exon V (F). Data are presented as increases in MHC-II exon DNA relative to unstimulated acetylated H3 K18 IP sample values. Control IP values were (0.9 ± 0.3)-fold. Control and acetylated histone H3 K18 IP values represent the mean ± SEM of three independent experiments. ***, P < 0.001 versus control siRNA.
To determine if the loss of MHC-II H3 K18 acetylation is indicative of a global decrease in levels of histone H3 K18 acetylation, siRNA was used to knock down Sug1 expression in HeLa cells. Histones were acid extracted and lysates were subjected to IB for acetylated H3 K18. Although Sug1 was sufficiently knocked down (Fig. 6A, bottom), levels of acetylated H3 K18 were unaffected by the loss of Sug1 (Fig. 6A, top). Serial dilutions of lysates verified that H3 K18 acetylation is indeed maintained in the Sug1-deficient cells (see Fig. S2 in the supplemental material). To verify that this phenomenon is lysine residue specific, ChIP studies were performed with Sug1 knockdown cells by use of endogenous antibody against acetylated histone H3 lysine 9. HeLa cells stimulated with IFN-γ showed elevated levels of H3 K9 acetylation at the MHC-II promoter that were enhanced upon treatment with HDAC inhibitors (Fig. 6B). H3 K9 acetylation was not decreased by Sug1 siRNA transfection (Fig. 6C) compared to what was seen for control-transfected cells (Fig. 6C).
FIG. 6.

Sug1 knockdown decreases histone H3 lysine 18 acetylation in a promoter-specific manner. (A) Global histone H3 K18 acetylation is unaffected by Sug1 knockdown. HeLa cells were left untreated (NT) or were transfected with either scrambled control or Sug1-specific siRNA duplexes. Histones were subjected to acid extraction. Lysates were subjected to IB for acetylated H3 K18 (top) or for endogenous Sug1 (bottom). Results reported are data representative of two independent experiments. (B) Histone H3 K9 acetylation at the MHC-II proximal promoter is enhanced upon IFN-γ stimulation and HDAC inhibition. ChIP assays were carried out with HeLa cells stimulated with IFN-γ for 0 to 18 h in combination with HDAC inhibitor (20 h) as indicated. Lysates were subjected to IP with control antibody or with antibody to endogenous acetylated histone H3 K9, and associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. IP values are presented as increases in the MHC-II promoter DNA relative to unstimulated acetylated H3 K9 IP sample values. Control IP values were (1.15 ± 0.1)-fold. Control and acetylated histone H3 IP values represent the mean ± SEM of three independent experiments. (C) H3 K9 acetylation at the MHC-II proximal promoter is unaffected by the loss of Sug1 expression. HeLa cells were transfected with scrambled control or Sug1-specific siRNA duplexes and 24 h later were treated with HDAC inhibitor and stimulated with IFN-γ for 0 to 4 h. Lysates were subjected to IP with control antibody or antibody to endogenous acetylated H3 K9, and associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. Data are presented as increases in the MHC-II promoter DNA relative to unstimulated acetylated H3 K9 IP sample values. Control IP values were (1.6 ± 0.27)-fold. Control and acetylated histone H3 K9 IP values represent the mean ± SEM of four independent experiments.
Sug1 knockdown decreases recruitment of the HAT CBP to the MHC-II promoter.
An open chromatin conformation is required for the initiation of transcription. This restructuring event involves the recruitment of HAT enzymes which add acetyl groups to lysine residues of the N-terminal tail region of histones and loosen protein-DNA interactions. One of the most studied HATs in yeast is GCN5, which is the catalytic subunit of the SAGA complex. The SAGA complex associates with various transcriptional activators in vivo and is recruited to promoters where GCN5 is able to acetylate nearby histones (11, 61). Proteasomal ATPases interact with the SAGA complex in yeast, and decreasing expression of the 19S ATPases reduces global histone acetylation and SAGA recruitment to actively transcribing promoters (23, 46). We therefore sought to determine if the Sug1 ATPase associates with HATs in mammalian cells. Sug1-deficient cells show dramatically decreased levels of histone H3 lysine 18 acetylation (Fig. 5C, left, and E and F), a modification that can be mediated by the HAT CBP (2, 64). To determine whether Sug1 associates with CBP in vivo, coimmunoprecipitations were performed with HeLa cells transfected with Myc-tagged Sug1, Myc-tagged ATPase mutant Sug1, or a Myc control plasmid (Fig. 7A). Polyclonal antibody was used to subject endogenous CBP to IP, and associations were detected by IB of the samples with Myc antibody. Sug1 and CBP precipitated together (Fig. 7A, top, lane 3), whereas the empty Myc plasmid showed no association with CBP (Fig. 7A, top, lane 2). This interaction appeared independent of ATPase activity, as overexpressed ATPase mutant Sug1 also associated with CBP (Fig. 7A, top, lane 4). A positive control sample was subjected to IP with Myc beads (Fig. 7A, top, lane 1). Equal loading was confirmed by IB analysis of lysates (Fig. 7A, bottom).
FIG. 7.

Sug1 knockdown decreases HAT recruitment to the MHC-II promoter. (A) Sug1 associates with CBP in HeLa cells. HeLa cells were transfected with Myc-tagged Sug1, Myc-tagged ATPase mutant Sug1, or a Myc control plasmid as indicated. Cells were lysed and subjected to IP with polyclonal antibody against CBP (lanes 2 to 4, top). A control sample was subjected to IP with Myc beads (lane 1, top). IP and lysate control (bottom) samples were subjected to IB for Myc. Results reported are data representative of three experiments. (B and C) CBP association with the MHC-II proximal promoter precedes CIITA and is enhanced upon IFN-γ stimulation. ChIP assays were carried out with HeLa cells stimulated with IFN-γ for 0 to 18 h. Lysates were subjected to IP with control antibody or antibody to endogenous CBP (B) or CIITA (C). Associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. IP values are presented as increases in MHC-II promoter DNA relative to unstimulated CBP or CIITA IP sample values. Control IP values were (1 ± 0.4)-fold. Control and acetylated histone H3 IP values represent the mean ± SEM of two to four independent experiments. (D) Sug1 knockdown decreases CBP association with the MHC-II proximal promoter. HeLa cells were transfected with control or Sug1-specific siRNA duplexes and 24 h later were stimulated with IFN-γ for 0 to 18 h. Lysates were subjected to IP with control antibody or with antibody to endogenous CBP, and associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. Data are presented as increases in the MHC-II promoter DNA relative to unstimulated CBP IP sample values. Control IP values were (1.2 ± 0.3)-fold. Control and CBP IP values represent the mean ± SEM of three independent experiments. ***, P < 0.001 versus control siRNA.
To determine the role played by Sug1 in recruiting CBP to the MHC-II proximal promoter, we performed ChIP experiments to detect endogenous levels of CBP at the HLA-DRA proximal promoter. Initial ChIP studies confirmed endogenous levels of CBP recruitment that rapidly increase upon IFN-γ stimulation in HeLa cells (29, 43, 79, 80). CBP interacts with CREB, a component of the MHC-II enhanceosome complex, and thus can be found associated with the promoter region in unstimulated HeLa cells and in stimulated cells prior to CIITA recruitment (Fig. 7B and C). CBP binding is enhanced upon prolonged cytokine stimulation, when CIITA is also bound (Fig. 7B and C). To determine if the decreased acetylation of histone H3 at the MHC-II HLA-DRA promoter in the absence of Sug1 is due to the decreased recruitment of CBP, we performed ChIP experiments in the presence of siRNA-mediated knockdown of Sug1. Cells transfected with control siRNA showed a threefold increase in CBP recruitment following IFN-γ stimulation (Fig. 7D), while cells transfected with Sug1-specific siRNA lacked this enhanced IFN-γ-dependent recruitment of CBP to the MHC-II promoter (Fig. 7D).
ATPase activity is not required for Sug1-mediated MHC-II chromatin remodeling.
Because the Sug1 ATPase mutant associated with CBP (Fig. 7A, top, lane 4), we sought to determine the effects this Sug1 ATPase mutant had on H3 K18 acetylation. ChIP assays for endogenous acetylated H3 K18 at the MHC-II HLA-DRA proximal promoter were performed with HeLa cells transfected with either Myc-tagged Sug1 or Myc-tagged ATPase mutant Sug1 and stimulated with IFN-γ. Although the absence of wild-type levels of Sug1 results in an almost complete loss of the H3 K18 acetylation necessary for chromatin remodeling at the MHC-II proximal promoter (Fig. 5C, left), the overexpression of a single 19S ATPase (Sug1) showed only a marginal increase in the levels of H3 K18 acetylation observed at the MHC-II proximal promoter (Fig. 8). Consistent with CBP binding to ATPase mutant Sug1 (Fig. 7A, top, lane 4), the lack of Sug1 ATPase activity did not impact H3 K18 acetylation (Fig. 8).
FIG. 8.
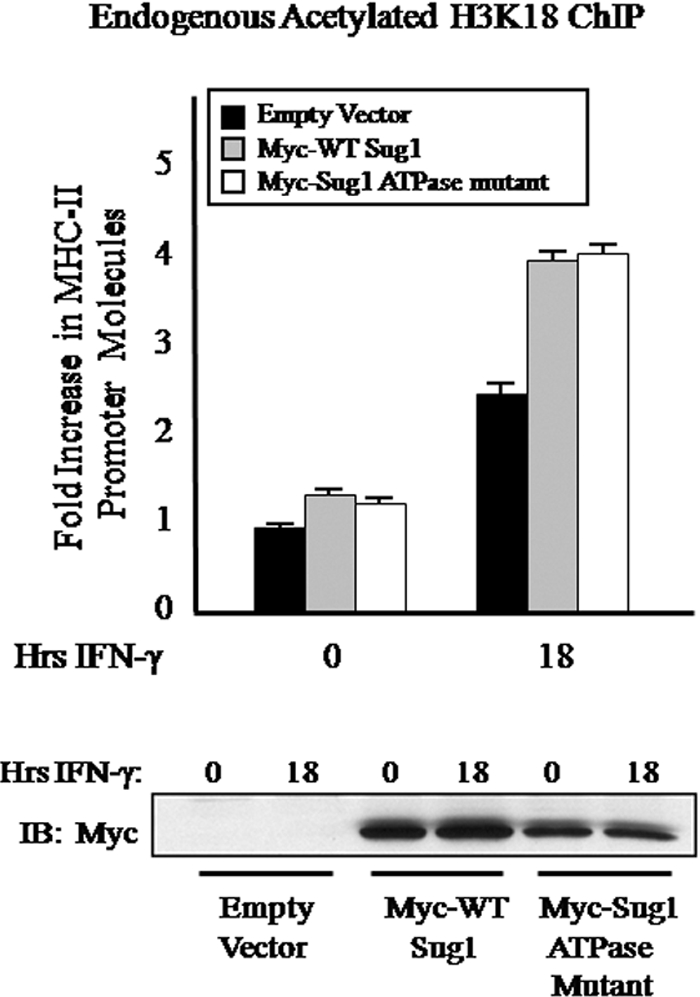
ATPase activity is not required for Sug1-mediated MHC-II chromatin remodeling. HeLa cells were transfected with Myc-tagged wild-type (WT) Sug1, Myc-tagged ATPase mutant Sug1, or a Myc control plasmid as indicated and treated with IFN-γ for 0 to 18 h. Ten percent of the total cell volume was lysed and analyzed by Western blotting for Sug1 overexpression (bottom) as described for Fig. 1. The remaining fraction of cells was subjected to a ChIP assay. Lysates were subjected to IP with control antibody or antibody to endogenous acetylated H3 K18, and associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. Data are presented as increases in the MHC-II promoter DNA relative to unstimulated empty vector transfected acetylated H3 K18 IP sample values. Control IP values were (1.0 ± 0.1)-fold. Control and acetylated histone H3 K18 IP values represent the mean ± SEM of four independent experiments.
Additional 19S ATPases also play a role in remodeling MHC-II chromatin.
Overexpressing Sug1 did not result in substantial enhancement of histone H3 acetylation at the MHC-II proximal promoter (Fig. 8). Therefore, we sought to determine the contribution of additional 19S ATPases to histone H3 acetylation. First, ChIP assays were used to determine that both S6a (Fig. 9A) and S7 (Fig. 9B) are recruited to the MHC-II promoter upon cytokine stimulation, preceding CIITA recruitment (Fig. 7C). Next, specific siRNAs were generated to knock down 19S ATPases S6a (Fig. 9C, bottom) and S7 (Fig. 9D, bottom). Similar to what was seen in experiments with Sug1 knockdown cells (Fig. 2B, right), acetylated H3 ChIP assays performed with S6a siRNA (Fig. 9C, top)- or S7 siRNA (Fig. 9D, top)-transfected cells exhibited impaired H3 acetylation at the MHC-II promoter compared to what was seen for control siRNA-transfected cells (Fig. 9C and D, top).
FIG. 9.

Additional 19S ATPases also mediate MHC-II promoter histone acetylation. (A and B) 19S ATPases S6a and S7 are recruited to the MHC-II proximal promoter upon IFN-γ stimulation. ChIP assays were carried out with HeLa cells stimulated with IFN-γ for 0 to 18 h. Lysates were subjected to IP with control antibody or antibody to endogenous S6a (A) or S7 (B), and associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. IP values are presented as increases in the MHC-II promoter DNA relative to unstimulated S6a or S7 IP sample values. Control IP values were (1.2 ± 0.2)-fold. Control and ATPase IP values represent the mean ± SEM of two to four independent experiments. (C and D) S6a and S7 knockdowns diminish H3 acetylation at the MHC-II proximal promoter. ChIP assays were carried out with HeLa cells transfected with S6a, S7, or control siRNA duplexes and 24 h later stimulated with IFN-γ for 0 to18 h. Ten percent of the total cell volume was lysed and analyzed by Western blotting for S6a or S7 knockdown (bottom). The remaining fractions of cells were subjected to a ChIP assay. Lysates were subjected to IP with control antibody or antibody to endogenous acetylated H3, and associated DNA was isolated and analyzed via real-time PCR as described for Fig. 2B. IP values are presented as increases in the MHC-II promoter DNA relative to unstimulated acetylated histone H3 IP sample values. Control IP values were (0.85 ± 0.4)-fold. Control and acetylated histone H3 IP values represent the mean ± SEM of two independent experiments. ***, P < 0.001 versus control siRNA.
DISCUSSION
Our results have provided novel evidence that the 19S proteasome plays an important role in mediating the epigenetic regulation of MHC-II transcription. Several pieces of data above argue that the 19S ATPase Sug1 recruits the HAT CBP, and potentially additional histone-modifying enzymes, to the MHC-II proximal promoter to enhance activation-induced promoter acetylation and to establish a necessary platform for CIITA binding, additional HAT recruitment, and robust histone H3 acetylation. We have previously demonstrated that following IFN-γ stimulation, Sug1 rapidly binds the MHC-II proximal promoter (10). We now show that Sug1 associates in vivo with histone H3 and acetylated histone H3 and that when Sug1 is knocked down, levels of histone H3 acetylation are markedly reduced at the activated MHC-II proximal promoter. Despite our observations of several proteasomal subunits binding to the MHC-II promoter, the effects of Sug1 are independent of proteasome proteolytic function, as proteasomal inhibition does not impact MHC-II promoter H3 acetylation. Furthermore, the effects observed on histone H3 acetylation by Sug1 knockdown are specific, as whole-cell lysates show no global change in levels of acetylated histone H3 upon Sug1 knockdown. The effects of Sug1 are also lysine specific, as histone H3 lysine 18 acetylation is diminished by Sug1 knockdown at the activated MHC-II promoter, whereas H3 lysine 9 acetylation remains unaffected. The impact of Sug1 on MHC-II transcription is substantial and extends beyond the proximal promoter, as exons III and V also show markedly decreased histone H3 lysine 18 acetylation in the absence of Sug1. These observations correlate with decreased binding of the HAT CBP to the MHC-II promoter, data consistent with those from experiments that indicate an in vivo association between Sug1 and CBP.
The seminal discovery that 19S ATPases are recruited to activated yeast promoters in the absence of proteolytic components of the proteasome first suggested a nonproteolytic role for the 19S proteasome in transcriptional regulation (36). We have recently demonstrated a role in mammalian transcription for the 19S proteasome as a positive regulator of MHC-II transcription initiation. Studies by Bhat et al. indicate that the 19S ATPase Sug1 is recruited to the MHC-II proximal promoter prior to CIITA, that knocking down Sug1 decreases MHC-II but not CIITA expression, and that in the absence of Sug1, markedly reduced levels of CIITA are recruited to the MHC-II HLA-DRA proximal promoter (10). Research in yeast has recently indicated novel roles for the ortholog of Sug1, Rpt 6, in epigenetically regulating transcription. The ATPase activity of Rpt 6 is required to target the Saccharomyces cerevisiae SAGA HAT complex to a DNA-bound activator and to globally regulate the acetylation of histone H3 (23, 46). Our results illustrate similar regulatory activity in mammalian cells and indicate that in the absence of wild-type levels of Sug1, necessary chromatin remodeling does not occur at the MHC-II proximal promoter, even in the presence of sustained HDAC inhibition. As seen in our study, in yeast cells ATP hydrolysis is less important for SAGA binding to promoters but is required for optimal dissociation of SAGA from the 19S proteasome (46). It will be useful next to determine the role played by energy utilization in the actions of the 19S proteasome and Sug1 on chromatin at the MHC-II proximal promoter.
Levels of histone H3 K18 acetylation are substantially reduced and the MHC-II promoter is rendered hypoacetylated in the absence of Sug1. H3 K18 acetylation occurs at the MHC-II proximal promoter within 4 h of IFN-γ treatment and is sustained for 48 h (35). The rapid occurrence of H3 K18 acetylation correlates with our demonstrated CBP promoter recruitment, and CBP can generate this modification in vitro (64). Previous studies have shown that the association of CIITA with the MHC-II proximal promoter correlates with dramatic increases in histone H3 and H4 acetylation (8, 51). As CIITA binding to the MHC-II promoter is reduced in the absence of Sug1, it is not surprising that this specific CIITA-driven modification would also be targeted. What is unexpected is the dramatic loss of MHC-II proximal promoter histone H3 acetylation in the absence of Sug1. The hypoacetylated state of the MHC-II promoter following IFN-γ stimulation and HDAC inhibition indicates that even the moderate levels of acetylation normally seen prior to CIITA expression are blocked in Sug1 knockdown cells. That CBP is the likely target of Sug1 is supported by data demonstrating CBP binding to the MHC-II promoter prior to CIITA and by a failure to increase CBP binding upon IFN-γ stimulation in the Sug1 knockdown cells. However, our data do not rule out the interesting possibility that Sug1 interacts with additional HATs to regulate histone modifications at the MHC-II promoter prior to, or in addition to, interactions with CBP. Future studies will enable a determination of the full extent of the mechanisms by which Sug1 and additional 19S ATPases regulate HAT recruitment to the MHC-II promoter.
The finding that levels of H3 acetylation at the MHC-II proximal promoter are dramatically reduced in the absence of Sug1 suggests a model whereby Sug1 controls histone modifications associated with IFN-γ-induced HLA-DRA gene activation. An interesting recent report suggests that histone modifications introduced at the MHC-II proximal promoter are likely important in activities such as promoter clearance, transcriptional elongation, and transcriptional memory rather than in transcription initiation (63). If so, Sug1 and the 19S ATPases may ultimately provide spatial and temporal regulation of mammalian transcription by controlling transcription elongation through the recruitment of histone-modifying activators. It remains to be determined if additional histone-modifying enzymes interact with 19S ATPases to coordinate MHC-II transcription. A complex series of specific histone modifications occurs to open the MHC-II chromatin structure and allow CIITA binding, but what links these modifying enzymes to each other and to other cofactors is not known. Active expression of MHC-II is associated with histone H3 acetylation, histone H4 acetylation, histone H3 methylation, and histone H2B ubiquitination (35, 51, 63, 68, 80). Multiple possible scenarios exist for Sug1 and the 19S proteasome functioning in the mediation of these histone modifications at the MHC-II promoter. Similar to the role played by the 19S proteasome in targeting SAGA to yeast promoters, Sug1 may mediate promoter recruitment of CBP and additional HATs, like the elongator complex, that move along chromatin by binding elongating polymerase II (19, 39, 42). Indeed, ATPases of the 19S proteasome have been demonstrated to bind to coding sequences of yeast genes and to be required for transcriptional elongation (26, 36, 44, 60). Our demonstration of decreased histone H3 lysine 18 acetylation in exons III and V of HLA-DRA supports a similar role for Sug1 in elongation in mammalian cells. The yeast ortholog of Sug1, Rpt 6, is recruited to promoters by the ubiquitination of histone H2B and is necessary for the methylation of histone H3 (25). Evidence that mammalian H3 acetylation is coupled to prior H2B ubiquitination and H3 K4 methylation provides the interesting possibility that Sug1 and the 19S proteasome will recruit histone-modifying enzymes linked to these modifications as well (9, 58). Our observations that 19S ATPases S6a and S7 also bind MHC-II promoters and modulate MHC-II histone H3 acetylation provide evidence that multiple 19S ATPases play important roles in regulating mammalian transcription. Our demonstration that the 19S non-ATPase S5a and 20S subunit alpha 4 also bind the MHC-II proximal promoter suggest that, despite the lack of a requirement for proteasome activity in the Sug1-dependent transcriptional regulation of HLA-DRA genes, additional components of the 26S proteasome are present at this promoter. These results support findings in the yeast literature that an intact, albeit proteolytically inactive, proteasome binds yeast promoters (20, 25, 46, 54). However, the observation that the binding of non-ATPase proteasome components is relatively low compared to that seen for 19S ATPases does not rule out the possibility that APIS (19S ATPase proteins independent of 20S) structures also bind the MHC-II promoter as unique complexes, a finding which has also been supported by the yeast literature (27, 33, 67, 70).
Ours is the first report demonstrating a role for the 19S proteasome and the Sug1 ATPase in aiding in the transition to an open chromatin structure in a mammalian system and suggests an evolutionarily conserved role for 19S in histone modifications. The finding that Sug1 modulates CBP promoter recruitment and the acetylation of histones at the MHC-II proximal promoter advances our knowledge of the function of 19S in histone modifications. Evidence that promoter acetylation is dramatically reduced in the absence of Sug1 implicates Sug1 as being deeply involved in a mammalian histone modification pathway. Chromatin must be opened before transcription factors can bind and initiate transcription, and it is a complex set of histone modifications that allows for this opening. A full understanding of the contributions of Sug1 and the 19S proteasome to the epigenetic regulation of MHC-II transcription will require further studies into the molecular interactions occurring at this and other promoters.
Supplementary Material
Acknowledgments
This work is supported by grants from the Georgia Cancer Coalition and Georgia State University (to S. F. Greer) and the Georgia State University Molecular Basis of Disease Program (to O. I. Koues).
We thank A. Wani (Department of Radiology, The Ohio State University, Columbus, OH) for generously providing the Myc-Sug1 constructs (78).
Footnotes
Published ahead of print on 28 July 2008.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Adamski, J., Z. Ma, S. Nozell, and E. N. Benveniste. 2004. 17β-Estradiol inhibits class II major histocompatibility complex (MHC) expression: influence on histone modifications and cbp recruitment to the class II MHC promoter. Mol. Endocrinol. 181963-1974. [DOI] [PubMed] [Google Scholar]
- 2.Agalioti, T., G. Chen, and D. Thanos. 2002. Deciphering the transcriptional histone acetylation code for a human gene. Cell 111381-392. [DOI] [PubMed] [Google Scholar]
- 3.Alarcon-Vargas, D., and Z. Ronai. 2002. p53-Mdm2—the affair that never ends. Carcinogenesis 23541-547. [DOI] [PubMed] [Google Scholar]
- 4.Athanassiou, M., Y. Hu, L. Jing, B. Houle, H. Zarbl, and A. M. Mikheev. 1999. Stabilization and reactivation of the p53 tumor suppressor protein in nontumorigenic revertants of HeLa cervical cancer cells. Cell Growth Differ. 10729-737. [PubMed] [Google Scholar]
- 5.Auld, K. L., C. R. Brown, J. M. Casolari, S. Komili, and P. A. Silver. 2006. Genomic association of the proteasome demonstrates overlapping gene regulatory activity with transcription factor substrates. Mol. Cell 21861-871. [DOI] [PubMed] [Google Scholar]
- 6.Bannister, A. J., and T. Kouzarides. 1996. The CBP co-activator is a histone acetyltransferase. Nature 384641-643. [DOI] [PubMed] [Google Scholar]
- 7.Baumeister, W., J. Walz, F. Zuhl, and E. Seemuller. 1998. The proteasome: paradigm of a self-compartmentalizing protease. Cell 92367-380. [DOI] [PubMed] [Google Scholar]
- 8.Beresford, G. W., and J. M. Boss. 2001. CIITA coordinates multiple histone acetylation modifications at the HLA-DRA promoter. Nat. Immunol. 2652-657. [DOI] [PubMed] [Google Scholar]
- 9.Bernstein, B. E., M. Kamal, K. Lindblad-Toh, S. Bekiranov, D. K. Bailey, D. J. Huebert, S. McMahon, E. K. Karlsson, E. J. Kulbokas III, T. R. Gingeras, S. L. Schreiber, and E. S. Lander. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120169-181. [DOI] [PubMed] [Google Scholar]
- 10.Bhat, K. P., J. D. Turner, S. E. Myers, A. D. Cape, J. P. Ting, and S. F. Greer. 2008. The 19S proteasome ATPase Sug1 plays a critical role in regulating MHC class II transcription. Mol. Immunol. 452214-2224. [DOI] [PubMed] [Google Scholar]
- 11.Bhaumik, S. R., T. Raha, D. P. Aiello, and M. R. Green. 2004. In vivo target of a transcriptional activator revealed by fluorescence resonance energy transfer. Genes Dev. 18333-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boss, J. M., and P. E. Jensen. 2003. Transcriptional regulation of the MHC class II antigen presentation pathway. Curr. Opin. Immunol. 15105-111. [DOI] [PubMed] [Google Scholar]
- 13.Buschmann, T., O. Potapova, A. Bar-Shira, V. N. Ivanov, S. Y. Fuchs, S. Henderson, V. A. Fried, T. Minamoto, D. Alarcon-Vargas, M. R. Pincus, W. A. Gaarde, N. J. Holbrook, Y. Shiloh, and Z. Ronai. 2001. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol. Cell. Biol. 212743-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carey, M., and S. T. Smale. 2000. Transcriptional regulation in eukaryotes. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 15.Chan, H. M., and N. B. La Thangue. 2001. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 1142363-2373. [DOI] [PubMed] [Google Scholar]
- 16.Chou, S. D., and T. B. Tomasi. 2008. Spatial distribution of histone methylation during MHC class II expression. Mol. Immunol. 45971-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciechanover, A. 1998. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 177151-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ciechanover, A. 1994. The ubiquitin-proteasome proteolytic pathway. Cell 7913-21. [DOI] [PubMed] [Google Scholar]
- 19.Close, P., N. Hawkes, I. Cornez, C. Creppe, C. A. Lambert, B. Rogister, U. Siebenlist, M. P. Merville, S. A. Slaugenhaupt, V. Bours, J. Q. Svejstrup, and A. Chariot. 2006. Transcription impairment and cell migration defects in elongator-depleted cells: implication for familial dysautonomia. Mol. Cell 22521-531. [DOI] [PubMed] [Google Scholar]
- 20.Collins, G. A., and W. P. Tansey. 2006. The proteasome: a utility tool for transcription? Curr. Opin. Genet. Dev. 16197-202. [DOI] [PubMed] [Google Scholar]
- 21.Conaway, R. C., C. S. Brower, and J. W. Conaway. 2002. Emerging roles of ubiquitin in transcription regulation. Science 2961254-1258. [DOI] [PubMed] [Google Scholar]
- 22.Coux, O., K. Tanaka, and A. L. Goldberg. 1996. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 65801-847. [DOI] [PubMed] [Google Scholar]
- 23.Daniel, J. A., and P. A. Grant. 2007. Multi-tasking on chromatin with the SAGA coactivator complexes. Mutat. Res. 618135-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eberharter, A., and P. B. Becker. 2002. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 3224-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ezhkova, E., and W. P. Tansey. 2004. Proteasomal ATPases link ubiquitylation of histone H2B to methylation of histone H3. Mol. Cell 13435-442. [DOI] [PubMed] [Google Scholar]
- 26.Ferdous, A., F. Gonzalez, L. Sun, T. Kodadek, and S. A. Johnston. 2001. The 19S regulatory particle of the proteasome is required for efficient transcription elongation by RNA polymerase II. Mol. Cell 7981-991. [DOI] [PubMed] [Google Scholar]
- 27.Ferdous, A., T. Kodadek, and S. A. Johnston. 2002. A nonproteolytic function of the 19S regulatory subunit of the 26S proteasome is required for efficient activated transcription by human RNA polymerase II. Biochemistry 4112798-12805. [DOI] [PubMed] [Google Scholar]
- 28.Fish, J. E., C. C. Matouk, A. Rachlis, S. Lin, S. C. Tai, C. D'Abreo, and P. A. Marsden. 2005. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J. Biol. Chem. 28024824-24838. [DOI] [PubMed] [Google Scholar]
- 29.Fontes, J. D., S. Kanazawa, D. Jean, and B. M. Peterlin. 1999. Interactions between the class II transactivator and CREB binding protein increase transcription of major histocompatibility complex class II genes. Mol. Cell. Biol. 19941-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Francis, J., S. K. Chakrabarti, J. C. Garmey, and R. G. Mirmira. 2005. Pdx-1 links histone H3-Lys-4 methylation to RNA polymerase II elongation during activation of insulin transcription. J. Biol. Chem. 28036244-36253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freiman, R. N., and R. Tjian. 2003. Regulating the regulators: lysine modifications make their mark. Cell 11211-17. [DOI] [PubMed] [Google Scholar]
- 32.Gerloni, M., and M. Zanetti. 2005. CD4 T cells in tumor immunity. Springer Semin. Immunopathol. 2737-48. [DOI] [PubMed] [Google Scholar]
- 33.Gillette, T. G., F. Gonzalez, A. Delahodde, S. A. Johnston, and T. Kodadek. 2004. Physical and functional association of RNA polymerase II and the proteasome. Proc. Natl. Acad. Sci. USA 1015904-5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glickman, M. H., D. M. Rubin, H. Fu, C. N. Larsen, O. Coux, I. Wefes, G. Pfeifer, Z. Cjeka, R. Vierstra, W. Baumeister, V. Fried, and D. Finley. 1999. Functional analysis of the proteasome regulatory particle. Mol. Biol. Rep. 2621-28. [DOI] [PubMed] [Google Scholar]
- 35.Gomez, J. A., P. Majumder, U. M. Nagarajan, and J. M. Boss. 2005. X box-like sequences in the MHC class II region maintain regulatory function. J. Immunol. 1751030-1040. [DOI] [PubMed] [Google Scholar]
- 36.Gonzalez, F., A. Delahodde, T. Kodadek, and S. A. Johnston. 2002. Recruitment of a 19S proteasome subcomplex to an activated promoter. Science 296548-550. [DOI] [PubMed] [Google Scholar]
- 37.Gorisch, S. M., M. Wachsmuth, K. F. Toth, P. Lichter, and K. Rippe. 2005. Histone acetylation increases chromatin accessibility. J. Cell Sci. 1185825-5834. [DOI] [PubMed] [Google Scholar]
- 38.Greer, S. F., E. Zika, B. Conti, X. S. Zhu, and J. P. Ting. 2003. Enhancement of CIITA transcriptional function by ubiquitin. Nat. Immunol. 41074-1082. [DOI] [PubMed] [Google Scholar]
- 39.Hawkes, N. A., G. Otero, G. S. Winkler, N. Marshall, M. E. Dahmus, D. Krappmann, C. Scheidereit, C. L. Thomas, G. Schiavo, H. Erdjument-Bromage, P. Tempst, and J. Q. Svejstrup. 2002. Purification and characterization of the human elongator complex. J. Biol. Chem. 2773047-3052. [DOI] [PubMed] [Google Scholar]
- 40.Jenuwein, T., and C. D. Allis. 2001. Translating the histone code. Science 2931074-1080. [DOI] [PubMed] [Google Scholar]
- 41.Kanazawa, S., T. Okamoto, and B. M. Peterlin. 2000. Tat competes with CIITA for the binding to P-TEFb and blocks the expression of MHC class II genes in HIV infection. Immunity 1261-70. [DOI] [PubMed] [Google Scholar]
- 42.Kim, J. H., W. S. Lane, and D. Reinberg. 2002. Human Elongator facilitates RNA polymerase II transcription through chromatin. Proc. Natl. Acad. Sci. USA 991241-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kretsovali, A., T. Agalioti, C. Spilianakis, E. Tzortzakaki, M. Merika, and J. Papamatheakis. 1998. Involvement of CREB binding protein in expression of major histocompatibility complex class II genes via interaction with the class II transactivator. Mol. Cell. Biol. 186777-6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laribee, R. N., Y. Shibata, D. P. Mersman, S. R. Collins, P. Kemmeren, A. Roguev, J. S. Weissman, S. D. Briggs, N. J. Krogan, and B. D. Strahl. 2007. CCR4/NOT complex associates with the proteasome and regulates histone methylation. Proc. Natl. Acad. Sci. USA 1045836-5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lassot, I., D. Latreille, E. Rousset, M. Sourisseau, L. K. Linares, C. Chable-Bessia, O. Coux, M. Benkirane, and R. E. Kiernan. 2007. The proteasome regulates HIV-1 transcription by both proteolytic and nonproteolytic mechanisms. Mol. Cell 25369-383. [DOI] [PubMed] [Google Scholar]
- 46.Lee, D., E. Ezhkova, B. Li, S. G. Pattenden, W. P. Tansey, and J. L. Workman. 2005. The proteasome regulatory particle alters the SAGA coactivator to enhance its interactions with transcriptional activators. Cell 123423-436. [DOI] [PubMed] [Google Scholar]
- 47.Liang, G., J. C. Lin, V. Wei, C. Yoo, J. C. Cheng, C. T. Nguyen, D. J. Weisenberger, G. Egger, D. Takai, F. A. Gonzales, and P. A. Jones. 2004. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc. Natl. Acad. Sci. USA 1017357-7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mach, B., V. Steimle, E. Martinez-Soria, and W. Reith. 1996. Regulation of MHC class II genes: lessons from a disease. Annu. Rev. Immunol. 14301-331. [DOI] [PubMed] [Google Scholar]
- 49.Martens, J. H., M. Verlaan, E. Kalkhoven, and A. Zantema. 2003. Cascade of distinct histone modifications during collagenase gene activation. Mol. Cell. Biol. 231808-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Masternak, K., E. Barras, M. Zufferey, B. Conrad, G. Corthals, R. Aebersold, J. C. Sanchez, D. F. Hochstrasser, B. Mach, and W. Reith. 1998. A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat. Genet. 20273-277. [DOI] [PubMed] [Google Scholar]
- 51.Masternak, K., N. Peyraud, M. Krawczyk, E. Barras, and W. Reith. 2003. Chromatin remodeling and extragenic transcription at the MHC class II locus control region. Nat. Immunol. 4132-137. [DOI] [PubMed] [Google Scholar]
- 52.McVean, M., H. Xiao, K. Isobe, and J. C. Pelling. 2000. Increase in wild-type p53 stability and transactivational activity by the chemopreventive agent apigenin in keratinocytes. Carcinogenesis 21633-639. [DOI] [PubMed] [Google Scholar]
- 53.Morris, A. C., G. W. Beresford, M. R. Mooney, and J. M. Boss. 2002. Kinetics of a gamma interferon response: expression and assembly of CIITA promoter IV and inhibition by methylation. Mol. Cell. Biol. 224781-4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morris, M. C., P. Kaiser, S. Rudyak, C. Baskerville, M. H. Watson, and S. I. Reed. 2003. Cks1-dependent proteasome recruitment and activation of CDC20 transcription in budding yeast. Nature 4231009-1013. [DOI] [PubMed] [Google Scholar]
- 55.Muratani, M., and W. P. Tansey. 2003. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 4192-201. [DOI] [PubMed] [Google Scholar]
- 56.Ogryzko, V. V., R. L. Schiltz, V. Russanova, B. H. Howard, and Y. Nakatani. 1996. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87953-959. [DOI] [PubMed] [Google Scholar]
- 57.Parham, P. 2005. The immune system. Garland Science, New York, NY.
- 58.Pavri, R., B. Zhu, G. Li, P. Trojer, S. Mandal, A. Shilatifard, and D. Reinberg. 2006. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell 125703-717. [DOI] [PubMed] [Google Scholar]
- 59.Pokholok, D. K., C. T. Harbison, S. Levine, M. Cole, N. M. Hannett, T. I. Lee, G. W. Bell, K. Walker, P. A. Rolfe, E. Herbolsheimer, J. Zeitlinger, F. Lewitter, D. K. Gifford, and R. A. Young. 2005. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122517-527. [DOI] [PubMed] [Google Scholar]
- 60.Rasti, M., R. J. Grand, A. F. Yousef, M. Shuen, J. S. Mymryk, P. H. Gallimore, and A. S. Turnell. 2006. Roles for APIS and the 20S proteasome in adenovirus E1A-dependent transcription. EMBO J. 252710-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roth, S. Y., J. M. Denu, and C. D. Allis. 2001. Histone acetyltransferases. Annu. Rev. Biochem. 7081-120. [DOI] [PubMed] [Google Scholar]
- 62.Rubin, D. M., O. Coux, I. Wefes, C. Hengartner, R. A. Young, A. L. Goldberg, and D. Finley. 1996. Identification of the gal4 suppressor Sug1 as a subunit of the yeast 26S proteasome. Nature 379655-657. [DOI] [PubMed] [Google Scholar]
- 63.Rybtsova, N., E. Leimgruber, Q. Seguin-Estevez, I. Dunand-Sauthier, M. Krawczyk, and W. Reith. 2007. Transcription-coupled deposition of histone modifications during MHC class II gene activation. Nucleic Acids Res. 353431-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schiltz, R. L., C. A. Mizzen, A. Vassilev, R. G. Cook, C. D. Allis, and Y. Nakatani. 1999. Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J. Biol. Chem. 2741189-1192. [DOI] [PubMed] [Google Scholar]
- 65.Schubeler, D., D. M. MacAlpine, D. Scalzo, C. Wirbelauer, C. Kooperberg, F. van Leeuwen, D. E. Gottschling, L. P. O'Neill, B. M. Turner, J. Delrow, S. P. Bell, and M. Groudine. 2004. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 181263-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shilatifard, A. 2006. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu. Rev. Biochem. 75243-269. [DOI] [PubMed] [Google Scholar]
- 67.Sikder, D., S. A. Johnston, and T. Kodadek. 2006. Widespread, but non-identical, association of proteasomal 19 and 20 S proteins with yeast chromatin. J. Biol. Chem. 28127346-27355. [DOI] [PubMed] [Google Scholar]
- 68.Spilianakis, C., A. Kretsovali, T. Agalioti, T. Makatounakis, D. Thanos, and J. Papamatheakis. 2003. CIITA regulates transcription onset via Ser5-phosphorylation of RNA Pol II. EMBO J. 225125-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Strickland, E., K. Hakala, P. J. Thomas, and G. N. DeMartino. 2000. Recognition of misfolding proteins by PA700, the regulatory subcomplex of the 26 S proteasome. J. Biol. Chem. 2755565-5572. [DOI] [PubMed] [Google Scholar]
- 70.Sulahian, R., D. Sikder, S. A. Johnston, and T. Kodadek. 2006. The proteasomal ATPase complex is required for stress-induced transcription in yeast. Nucleic Acids Res. 341351-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun, Z. W., and C. D. Allis. 2002. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418104-108. [DOI] [PubMed] [Google Scholar]
- 72.Swaffield, J. C., K. Melcher, and S. A. Johnston. 1995. A highly conserved ATPase protein as a mediator between acidic activation domains and the TATA-binding protein. Nature 37488-91. [DOI] [PubMed] [Google Scholar]
- 73.Voges, D., P. Zwickl, and W. Baumeister. 1999. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 681015-1068. [DOI] [PubMed] [Google Scholar]
- 74.vom Baur, E., C. Zechel, D. Heery, M. J. Heine, J. M. Garnier, V. Vivat, B. Le Douarin, H. Gronemeyer, P. Chambon, and R. Losson. 1996. Differential ligand-dependent interactions between the AF-2 activating domain of nuclear receptors and the putative transcriptional intermediary factors mSUG1 and TIF1. EMBO J. 15110-124. [PMC free article] [PubMed] [Google Scholar]
- 75.Wesierska-Gadek, J., D. Schloffer, V. Kotala, and M. Horky. 2002. Escape of p53 protein from E6-mediated degradation in HeLa cells after cisplatin therapy. Int. J. Cancer 101128-136. [DOI] [PubMed] [Google Scholar]
- 76.Wright, K. L., and J. P. Ting. 2006. Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol. 27405-412. [DOI] [PubMed] [Google Scholar]
- 77.Yu, P., and T. Kodadek. 2007. Dynamics of the hypoxia-inducible factor-1-vascular endothelial growth factor promoter complex. J. Biol. Chem. 28235035-35045. [DOI] [PubMed] [Google Scholar]
- 78.Zhu, Q., G. Wani, J. Yao, S. Patnaik, Q. E. Wang, M. A. El-Mahdy, M. Praetorius-Ibba, and A. A. Wani. 2007. The ubiquitin-proteasome system regulates p53-mediated transcription at p21(waf1) promoter. Oncogene 264199-4208. [DOI] [PubMed] [Google Scholar]
- 79.Zhu, X. S., and J. P. Ting. 2001. A 36-amino-acid region of CIITA is an effective inhibitor of CBP: novel mechanism of gamma interferon-mediated suppression of collagen α2(I) and other promoters. Mol. Cell. Biol. 217078-7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zika, E., L. Fauquier, L. Vandel, and J. P. Ting. 2005. Interplay among coactivator-associated arginine methyltransferase 1, CBP, and CIITA in IFN-gamma-inducible MHC-II gene expression. Proc. Natl. Acad. Sci. USA 10216321-16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zika, E., S. F. Greer, X. S. Zhu, and J. P. Ting. 2003. Histone deacetylase 1/mSin3A disrupts gamma interferon-induced CIITA function and major histocompatibility complex class II enhanceosome formation. Mol. Cell. Biol. 233091-3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.