

Abstract
CCN2/CTGF is a multifunctional factor that plays a crucial role in the growth and differentiation of chondrocytes. The chicken ccn2 gene is regulated not only at the transcriptional level but also by the interaction between a posttranscriptional element in the 3′ untranslated region (3′-UTR) and a cofactor. In the present study, we identified a nucleophosmin (NPM) (also called B23) as this cofactor. Binding of NPM to the element was confirmed, and subsequent analysis revealed a significant correlation between the decrease in cytosolic NPM and the increased stability of the ccn2 mRNA during chondrocyte differentiation in vivo. Furthermore, recombinant chicken NPM enhanced the degradation of chimeric RNAs containing the posttranscriptional cis elements in a chicken embryonic fibroblast extract in vitro. It is noteworthy that the RNA destabilization effect by NPM was far more prominent in the cytosolic extract of chondrocytes than in that of fibroblasts, representing a chondrocyte-specific action of NPM. Stimulation by growth factors to promote differentiation changed the subcellular distribution of NPM in chondrocytes, which followed the expected patterns from the resultant change in the ccn2 mRNA stability. Therefore, the present study reveals a novel aspect of NPM as a key player in the posttranscriptional regulation of ccn2 mRNA during the differentiation of chondrocytes.
CCN2 (connective tissue growth factor [CTGF]/hypertrophic chondrocyte-specific gene product 24 [Hcs24]) is a cysteine-rich secretory protein of 36 to 38 kDa that has four distinct modules, i.e., insulin-like growth factor-binding protein-likes, von Willebrand factor type C repeat, thrombospondin type 1 repeat, and C-terminal modules (4, 6, 54, 68, 69). CCN2 is a member of the CCN family (reviewed in references 6, 38, 53, 54, 68, and 69), which comprises ccn1 (cef-10/cyr61 [37, 66]), ccn2 (ctgf/hcs24/fisp12 [5, 46, 57]), ccn3 (nov [25]), ccn4 (elm-1/wisp-1 [20, 52]), ccn5 (ctgf-l/wisp-2/cop1 [52, 75]), and ccn6 (wisp-3 [52]). CCN2 was initially isolated from angioendothelial cells as a growth factor related to platelet-derived growth factor (PDGF) and was revealed to have PDGF-like mitogenic and chemotactic activities toward fibroblasts (17, 23, 28, 33, 54). However, recent studies have revealed that CCN2 is a multifunctional factor that regulates the growth and/or differentiation, chemotaxis, adhesion of various cells, and extracellular matrix formation by various cells, including vascular endothelial cells (2, 29, 54, 64). Furthermore, we showed earlier that CCN2 plays an important role in the growth and differentiation of chondrocytes and osteoblasts during endochondral ossification (46-48).
As to CCN2, it has been suggested that its gene expression is regulated at multiple steps, such as transcriptional, posttranscriptional, and translational stages (1), for playing its multiple roles mentioned above. For instance, transforming growth factor β (TGF-β) induces the expression of CCN2 (28, 46), and a few TGF-β response elements have been found in the ccn2 promoter region (11-13, 19, 39, 55). In addition to such transcriptional regulation, we also reported earlier that expression of the gene is regulated by its 3′ untranslated region (3′-UTR) at posttranscriptional stages (30-32, 34-36, 44). Furthermore, we recently reported (45) that a cis element in the 3′-UTR of chicken ccn2 mRNA and its putative trans-factor counterpart collaboratively play an important role in the posttranscriptional regulation by determining the stability of ccn2 mRNA. In fact, the affinity of binding between them is altered during chondrocytic differentiation, thus suggesting the involvement of this RNA-protein interaction in the precise posttranscriptional regulation of ccn2 mRNA during endochondral ossification. However, the identity of the putative trans factor and its mechanism of action were not clarified at that time.
In the present study, we finally purified and identified this trans-factor protein as nucleophosmin (NPM) (also called B23), which actually bound to the minimal repressive cis element in the 3′-UTR of ccn2 mRNA. With the recombinant protein we produced, we could successfully reconstruct the posttranscriptional regulatory events in vitro that were observed during endochondral ossification in vivo. We also obtained data showing that alteration of nucleocytoplasmic shuttling of the trans factor is critical for the regulation of chicken ccn2 expression during the differentiation of chondrocytes.
MATERIALS AND METHODS
Cell isolation and culture.
Chicken embryonic fibroblasts (CEFs) were isolated from a whole 10-day-old chicken embryo and maintained in Dulbecco's modified Eagle minimum essential medium supplemented with 10% fetal bovine serum (FBS) in an atmosphere of humidified air containing 5% CO2 at 37°C. Chicken upper sternum chondrocytes (US cells) and lower sternum chondrocytes (LS cells) were isolated from chicken embryo sternum cartilage on day 18 as described previously (44).
For growth factor stimulation, the medium was exchanged for FBS-free medium, and 24 h later, the cells were treated with 10 ng/ml of human TGF-β (Biomedical Technologies Inc., Stoughton, MA), 200 ng/ml of recombinant human bone morphogenetic protein 2 (BMP 2) (R&D Systems, Minneapolis, MN), 10 ng/ml of human PDGF (Sigma Aldrich, St. Louis, MO), or 30 ng/ml of recombinant human CCN2 (47) for a further 24 h in the absence of FBS.
Preparation of nuclear, cytoplasmic, and total cellular extracts.
Nuclear and cytoplasmic extracts were prepared by using a commercial kit, the CelLytic NuCLEAR extraction kit (Sigma Aldrich), according to the manufacturer's protocol. The protein concentrations of both fractions were determined by use of a bicinchoninic acid protein assay kit (Pierce, Rockford, IL) (67). Total cell extracts were prepared with Cell Lytic-M lysis reagent (Sigma). In a few experiments, the DNA concentrations in nuclear and total cell extracts were quantified by measuring the fluorescence emitted from 5 μl of the extract in the presence of 100 ng/ml of bisbenzimide trihydrochloride (Hoechst 33258) at a wavelength of 458 nm (excitation wavelength, 356 nm).
Isolation and purification of the trans-factor protein.
The trans-factor protein that bound to the 3′-UTR of chicken ccn2 mRNA was isolated by utilizing an RNA affinity column according to a previous report (62) with slight modifications. All of the procedures were carried out at 4°C. In the first step, a heparin-Sepharose column (HiTrap heparin HP [5 ml]; Amersham Bioscience, Piscataway, NJ) was prewashed with 50 ml of buffer B (50 mM Tris-HCl [pH 7.5] containing 2 mM EDTA, 5% glycerol, 7 mM 2-mercaptoethanol [2-ME], and 100 mM NaCl). Two grams of the nuclear extract of CEFs was then applied to the column. Then, the column was washed with 100 ml of buffer B containing 100 mM NaCl, and the bound proteins were eluted from the column by a step gradient of NaCl concentrations (100 to 1,000 mM) in buffer B.
These fractions were assayed for binding to the minimal repressive cis region of the 3′-UTR of chicken ccn2 mRNA (3′-100/50) (36) by using a UV cross-linking method. The fractions that showed the maximal binding to the 3′-100/50 fragment were eluted at NaCl concentrations of 200 to 600 mM and then pooled. The pooled fractions were subsequently loaded onto an RNA affinity chromatography column that had been prepared by cross-linking N-hydroxysuccinimide (NHS)-activated Sepharose in a column (HiTrap NHS-activated HP 1 ml; Amersham Bioscience) and 5 mg of 3′-100/50 RNA fragment that had been synthesized in vitro by utilizing a commercial kit (RiboMAX large-scale RNA production systems; Promega, Madison, WI). The column was washed with 20 ml of buffer B containing 100 mM NaCl, and the bound proteins were eluted from the column by a step gradient of buffer B containing NaCl (100 to 1,000 mM). Each fraction was assayed by the UV cross-linking assay, and the fractions that showed maximum binding were combined and then concentrated by use of an Amicon Ultra-4 centrifugal filter device (4 ml, 1,000 normal molecular weight limit; Millipore, Bedford, MA).
Determination of internal amino acid sequence.
The purified fraction was further separated by two-dimensional electrophoresis with isoelectric focusing and sodium dodecyl sulfate-polyacrylamide electrophoresis (SDS-PAGE). The RNA-binding proteins in the combined and concentrated fractions were verified by Northwestern blotting analysis using 3′-100/50 RNA fragment as a radiolabeled probe. Thereafter, the spot corresponding to the 40-kDa one stained by Coomassie brilliant blue (CBB) was excised from a gel and subjected to internal amino acid sequence analysis by Shimadzu Biotech (Tsukuba, Ibaraki, Japan).
REMSA and UV cross-linking assay.
The RNA electromobility shift assay (REMSA) and UV cross-linking assay were carried out as described previously (45). The schemata of the probes used are shown in Fig. 3A. Extracted proteins, recombinant nucleophosmin (described below), or bovine serum albumin (BSA) was incubated at 25°C for 30 min with 5 × 104 cpm of radiolabeled RNA probes, and then the binding mixture was incubated with 1 μl of 1/100 diluted RNase cocktail (Ambion, Austin, TX) for a further 10 min at 37°C. The RNA-protein complex was subjected to 6% native PAGE in 0.5× Tris-borate-EDTA (TBE) buffer (Invitrogen, Carlsbad, CA) for REMSA. The gels were subsequently dried and autoradiographed.
FIG. 3.

Specific binding of chicken NPM to the 3′-100/50 element in the chicken ccn2 mRNA 3′-UTR. (A) Schematic representation of the radiolabeled probes utilized in this study. The 3′ half of the ccn2 mRNA, including the open reading frame (ORF) and the 3′-UTR, is illustrated at the top. The sense-strand RNA fragments of the chicken ccn2 mRNA 3′-UTR (3′-100/50 and 3′-50) were transcribed in vitro in the presence of [α-32P]UTP. (B) REMSA of the RNA fragments of ccn2 mRNA 3′-UTR. Two radiolabeled and folded RNA probes (3′-100/50 and 3′-50) corresponding to the ones shown in panel A were incubated with or without (−) 50 to 1,000 ng of recombinant chicken NPM or BSA. After RNase digestion, the mixtures were analyzed by electrophoresis through a 6% native polyacrylamide gel. (C) UV cross-linking assay. The radiolabeled and folded RNA probes were incubated with the protein and digested with RNase, as was performed in REMSA. Then, the mixtures were irradiated by UV on ice and analyzed by SDS-PAGE using a 12.5% gel. The positions of molecular mass standards (in kilodaltons) are shown at the left side of the panel. The arrow (labeled NPM) indicates the position corresponding to the molecular mass of the recombinant chicken NPM. (D) RNA immunoprecipitation analysis to confirm the specific binding of endogenous NPM in the nucleus to ccn2 mRNA. Total CEF RNA was mixed with CEF nuclear extract and was immunoprecipitated with the antibody indicated, and the presence of ccn2 or actin (control) mRNA in the immunocomplex was examined by RT-PCR after RNA extraction (Nuclear Extract). NPM, His, and Pre in the figure denote anti-chicken NPM antibody, antipolyhistidine antibody, and a preimmune IgG, respectively. Experiments were also repeated with recombinant NPM only, instead of CEF nuclear extract, which detected the interaction of NPM and ccn2 mRNA through the polyhistidine tag in the recombinant NPM as well. The RT-PCR analysis of input RNA is shown in lane Total. (−), negative-control antibody.
For the UV cross-linking assay, after RNase digestion, the protein-RNA complexes were put on ice and UV irradiated for 10 min by a UV cross-linker (Amersham Bioscience). Then, the samples were heated at 95°C for 5 min in an SDS sample buffer (Sigma Aldrich) in the presence of 5% 2-ME and separated by 12.5% or 4 to 20% gradient SDS-PAGE. The gels were subsequently dried and autoradiographed.
RNA immunoprecipitation assay.
Ten micrograms of total RNA was incubated with 2.5 μg of nuclear extract of CEFs or recombinant NPM at 30°C for 1 h in 250 μl of a reaction buffer (20 mM HEPES [pH 7.9], 20% glycerol, 50 mM KCl, 0.2 mM EDTA, 4.5 mM MgCl2, 0.5 mM ATP, 20 mM creatine phosphate, 2 units/mg RNAsin [Promega]), and then 200 ml of buffer N (50 mM Tris-HCl [pH 8], 100 mM NaCl, 0.05% NP-40, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol [DTT]) containing 200 ng of antibody or preimmune rabbit immunoglobulin G (IgG) was added, and the mixture was further incubated at 4°C for 16 h. Protein A-Sepharose beads (Amersham Bioscience) (20 μl) were washed twice with 750 μl of buffer K (100 mM Tris-HCl [pH 8], 12.5 mM EDTA, 150 mM NaCl, 1% SDS) and resuspended in the reaction mixture containing antibody-antigen-RNA complex. After incubation with shaking at 4°C for 1 h, the reaction mixture beads were washed three times with buffer K. Thereafter, the beads were suspended in 250 μl of buffer K, heated at 80°C for 10 min, and centrifuged. The RNA in the supernatant was purified by Isogen-LS (Nippon Gene, Tokyo, Japan) and subjected to reverse-transcription-mediated-PCR (RT-PCR) as described in our previous studies (44, 45).
Western blot analysis and Northwestern blot analysis.
Western blot analysis was carried out as described previously (45). The proteins in SDS sample buffer with 2-ME (Amersham Bioscience) (10 μg or protein amounts from the cells that conferred 18 μg of DNA) were heated, separated by 12.5% SDS-PAGE, and transferred to a polyvinylidene difluoride (PVDF) membrane (Hybond P; Amersham Bioscience). After the membrane was blocked with 5% skim milk, it was incubated with a 1/1,000 dilution of polyclonal anti-CCN2 antibodies (ABcam, Cambridge, United Kingdom), a 1/1,000 dilution of monoclonal anti-NPM antibody (American Research Products, Inc., Belmont, MA), a 1/2,500 dilution of monoclonal anti-α-tubulin antibody (Sigma Aldrich), or a 1/2,500 dilution of monoclonal anti-lamin B1 antibody (Zymed, South San Francisco, CA). Then, the membrane was incubated with a 1/20,000 dilution of peroxidase-conjugated goat anti-mouse IgG antibody (American Qualex, La Mirada, CA). Subsequently, the blot was visualized by use of an ECL Western blotting analysis system (Amersham Bioscience).
For Northwestern blotting, the protein was separated and transferred to a PVDF membrane, as performed in Western blotting. The membrane was stained with CBB prior to incubation with the probe. Then, the membrane was preincubated for 16 h at 4°C in blocking buffer consisting of 10 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 5% skim milk, 1 μg/ml yeast tRNA (Roche Applied Science, Penzberg, Germany), after which it was incubated for 2 h at 25°C with the radiolabeled probe (1 × 106 cpm/ml) in binding buffer comprising 10 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.25% skim milk, and 1 μg/ml yeast tRNA. Thereafter, the membrane was washed three times with wash buffer consisting of 10 mM Tris-HCl (pH 7.5), 50 mM KCl, 1 mM EDTA, and 1 mM DTT and was subjected to autoradiography.
RNA degradation analysis in vivo.
The cells in 3.5-cm and 10-cm tissue culture dishes were grown until subconfluent, and then 10 μg/ml of actinomycin D (Sigma Aldrich) was added to the cultures in order to arrest de novo RNA synthesis. In small interfering RNA (siRNA)-mediated knockdown experiments, CEFs were transfected with synthetic siRNAs as described elsewhere and subjected to actinomycin D treatment 24 h after transfection. After properly timed intervals, total cellular RNA was isolated and subjected to Northern blotting or real-time PCR analysis after reverse transcription in order to evaluate the ccn2 or gapdh mRNA level.
RNA preparation and Northern blot analysis.
Total cellular RNA was isolated by Isogen (Nippon Gene), according to the manufacturer's protocol. Northern blotting analysis was carried out as described previously (45). Total RNA was denatured by glyoxysal, separated on 1% agarose gels, and then blotted onto nylon membranes (Hybond N; Amersham Bioscience). After blotting, the membrane was fixed with 5% acetic acid and stained with 0.02% methylene blue to visualize rRNAs, hybridized, washed, and subsequently autoradiographed.
The probe for chicken ccn2 was prepared as described previously (45), and the probe for chicken npm is described below (see “Preparation of recombinant chicken NPM”). The plasmids were linearized by SpeI and transcribed in vitro by bacteriophage T7 RNA polymerase (Promega) in the presence of 50 μCi of [α-32P]UTP (3,000 Ci/mmol; Amersham Bioscience) for the preparation of radiolabeled antisense RNA, followed by RQ1 DNase (Promega) digestion and spin-column (ProbeQuant G-50; Amersham Bioscience) purification.
Real-time PCR analysis.
After being transcribed to cDNA as described elsewhere, the npm and ccn2 mRNA levels were quantitatively analyzed by a real-time PCR method with LightCycler (Roche). Quantification was performed by an intercalator methodology with SYBR green real-time PCR master mix (Toyobo, Osaka, Japan) as described previously (72). As a control, expression of the glyceraldehyde 3′-phosphate dehydrogenase (GAPDH) gene was also analyzed. The nucleotide sequences of the specific primers used in the amplification were 5′-GCA GAG AAG GAA TAT CAG TT-3′ (sense) and 5′-CTC AAA TCC ACC TAG TGA AAC-3′ (antisense) for npm, 5′-CAC CAA CGA TAA TGC TTT C3′ (sense) and 5′-ACT TAG CTC TGT ACG TCT TCA-3′ (antisense) for ccn2, and 5′-AGG CTG TGG GGA AAG TCA-3′ (sense) and 5′-GAC AAC CTG GTC CTC TGT GTA T-3′ (antisense) for gapdh. The integrity of all amplicons was confirmed by melting curve analysis.
Preparation of recombinant chicken NPM.
Chicken npm cDNA, including the full-length open reading frame, was obtained by reverse-transcription-mediated-PCR (RT-PCR), utilizing total cellular RNA from CEFs. The nucleotide sequences of sense and antisense primers were 5′-CAT ATG GAG GAC AGC AGC GCC ATG GAC-3′ and 5′-CTC GAG AGT CTG TCT CCA CTG CCA G-3′, respectively. The amplicon was subcloned into pGEM-T Easy (Promega) by the TA-cloning method, which was also utilized for the preparation of the probe for Northern blotting. This plasmid was double digested with NdeI and XhoI, and the npm cDNA was isolated and subcloned into the corresponding site of pET21b(+) (EMD Biosciences, Madison, WI) in frame with the His6 tag at the C terminus. The resultant plasmid was designated pET21-cNPM. An Escherichia coli strain, RosettaBlue(DE3)pLysS (EMD Biosciences), was transformed with pET21-cNPM, and grown in Overnight Express instant TB (terrific broth) medium (EMD Biosciences) containing ampicillin (50 μg/ml), tetracycline (12.5 μg/ml), and kanamycin (34 μg/ml) at 37°C for 16 h. Thereafter, the cells were pelleted by centrifugation at 7,000 × g for 10 min, suspended in ice-cold binding buffer (20 mM phosphate [pH 7.4] containing 500 mM NaCl, 20 mM imidazole, 0.5% Tween 20, and 1 tablet/10 ml of Complete, Mini EDTA-free protease inhibitor cocktail tablets [Roche Applied Science]), and disrupted by sonication. The lysate was centrifuged at 12,000 × g for 10 min at 4°C, and the supernatant was filtered through a 0.45-μm filter. Chicken NPM-His6 fusion protein was purified with a HisTrap HP kit (Amersham Bioscience), according to the manufacturer's protocol. The filtered bacterial lysate was loaded onto the column equilibrated with binding buffer, and after being washed with binding buffer, the fusion protein was eluted with elution buffer (binding buffer containing 300 mM imidazole). The eluate was dialyzed and finally concentrated by an Amicon Ultra-4 centrifugal filter device. The quantity of purified recombinant chicken NPM-His6 fusion protein was determined by a bicinchoninic acid protein assay, and the quality was evaluated by CBB staining after SDS-PAGE and Western blotting with anti-chicken NPM or anti-His (C-terminal) antibody (Invitrogen; data not shown). The purified protein was aliquoted, supplemented with protease inhibitor cocktail (Sigma Aldrich), and stored at −80°C until used.
Preparation of firefly luciferase-ccn2 fusion gene constructs.
The reporter plasmids containing firefly luciferase-chicken ccn2 fusion genes on a pGL3L(+) backbone (34) were available from a previous study (45). In order to construct additional plasmids for preparation of the reporter mRNAs in vitro, the parental pGL3L(+) was double digested with HindIII and XbaI, and the isolated firefly luciferase gene was subcloned between the corresponding sites in pGEM4 (Promega). This plasmid was designated Luc(−). The plasmids referred to as 3′-Full, 3′-100/50, and 3′-50, which contained the full-length 3′-UTR of chicken ccn2 or ccn2 deletion mutations on a pGL3L(+) backbone (45), were double digested with XbaI and EcoRI, and each 3′-UTR fragment was subcloned into the corresponding sites in Luc(−). As a result, the 3′-UTR fragments were located at the 3′ end of the firefly luciferase gene in these three resultant chimeric fusion constructs, which were designated Luc-3′-Full, Luc-3′-100/50, and Luc-3′-50. Proper construction of all plasmids was confirmed by nucleotide sequencing (58) and restriction enzymatic digestion.
DNA transfection and luciferase assay.
Twenty-four hours prior to transfection, 2 × 105 cells were seeded into each well in 35-mm tissue culture dishes. Cationic liposome-mediated DNA transfection was carried out with 1 μg of each pGL derivative, which is described in the previous paragraph under “Preparation of firefly luciferase-ccn2 fusion gene constructs,” in combination with 0.5 μg of pRL-TK, according to the manufacturer's optimized methodology (FuGENE6; Roche, Indianapolis, IN). Forty-eight hours after transfection, the cells were lysed in 500 μl of a passive lysis buffer (Promega), and the cell lysate was directly used for the luciferase assay.
The dual luciferase assay system (Promega) was applied for the sequential measurement of firefly (reporter) and Renilla luciferase (transfection efficiency standard) activities with specific substrates. Quantification of both luciferase activities and calculation of relative ratios were done as described previously (34).
IVDA.
An in vitro RNA degradation assay (IVDA) was carried out as described previously (43) with slight modifications. Luc-3′-Full, Luc-3′-100/50, and Luc-3′-50 were linearized by SpeI, transcribed, capped, and polyadenylated in vitro in the presence of [α-32P]UTP by using a commercial kit (mMESSAGE mMACHINE T7 Ultra; Ambion) according to the manufacturer's protocol. Schemata of the transcripts are shown in Fig. 5A. Fifty thousand cpm of each radiolabeled RNA was incubated with 10 μg of cytoplasmic extract in IVDA reaction buffer consisting of 10 mM Tris-HCl (pH 7.5) containing 100 mM potassium acetate, 2 mM magnesium acetate, 2 mM DTT, 10 mM creatine phosphate, 1 mM ATP, 0.4 mM GTP, and 0.1 mM spermine. After the appropriate incubation at 37°C, the RNA was purified by use of Isogen LS (Nippon Gene) and subjected to 6% PAGE in the presence of 6 M urea in 1× TBE buffer, followed by autoradiography. The optical density of each signal in the autoradiograph was quantified by using a commercial computer software, Quantity One (PDI Inc., New York, NY). In certain experiments, recombinant chicken NPM (0.1 or 0.5 μg) or the eluate from the RNA affinity column (1 μg) was added together with the cytosolic extract.
FIG. 5.

Effect of NPM on the degradation of target RNA with or without a chicken ccn2 3′-UTR segment in a reconstituted system with CEF cytosolic extract in vitro. (A) Schematic representation of the structures of the RNAs utilized in this study. The 3′ half of the ccn2 mRNA including the 3′ half of the open reading frame (ORF) and the 3′-UTR are illustrated at the top, and the full-length and deletion mutant fusion mRNAs are schematically displayed. The full-length ccn2 3′-UTR and two short segments of ccn2 3′-UTR were connected in the sense orientation to the 3′ end of firefly luciferase genes (F. Luciferase) derived from pGL3L(+). These three fusion RNAs (Luc-2′-Full, Luc-3′-100/50, and Luc-3′-50) and firefly luciferase-only RNA [Luc(−) as a negative control] were capped (hatched box) and polyadenylated (AAA) in the presence of [α-32P]UTP by using a commercial kit (see Materials and Methods). The results of in vivo expression of the same mRNAs are also summarized. Arrows pointing down and right indicate lower and equivalent gene expression levels relative to that of Luc(−) in chicken cells (44). (B) IVDA of the fusion RNAs. The radiolabeled RNAs shown in panel A were incubated with cytoplasmic extracts (10 μg) from CEFs in the absence (−) or presence of recombinant chicken NPM (0.5 μg) or eluate from the chicken ccn2 mRNA affinity column (1 μg). After timed intervals, RNA was purified and subjected to urea-denatured 6% PAGE. The graphs at the bottom of the panel show the relative remaining values and approximate fitted degradation curves of the RNA standardized against each RNA at time zero. The values and curves for the negative control (closed ovals and solid line), NPM (closed rectangles and dotted line), and eluate (closed triangles and broken line) are depicted. The values are shown as percentages on semilogarithmic graphs (value at time zero = 100). Statistically significant differences from the values for the control without NPM or eluate are indicated by asterisks as follows: *, P < 0.05; **, P < 0.001.
Synthesis and application of siRNA against chicken npm1.
Two sets of siRNA duplexes targeted to chicken npm1 mRNA were synthesized by iGene Therapeutics (Tsukuba, Japan). The nucleotide sequences of the sense strands of the two double-stranded RNAs, si546 and si207, were 5′-UGA UGA GGA GGA GAU UAA AAC ACC A-3′ and 5′-CGA AGG CAA CCC AAC UAA AGU AGU A-3′, respectively. For the knockdown experiments, 50 nM of each siRNA was transfected into CEFs with the aid of a cationic liposomal transfection reagent (RNAiFect; Qiagen, Hilden, Germany). The cells were harvested at 48 h after transfection, and then total RNAs were extracted and subjected to reverse transcription by avian myeloblastosis virus reverse transcriptase (Takara, Tokyo, Japan), followed by real-time PCR analysis for quantification.
Statistical analysis.
Unless otherwise specified, all experiments were repeated at least twice, and similar results were obtained in the repeated experiments. Statistical analysis was carried out by repeated measure analysis of variance, Bonferroni/Dunn, or paired t test. Data were expressed as the means ± standard deviations.
RESULTS
Purification and identification of the protein(s) binding to the 3′-UTR of ccn2 mRNA.
Recently, we found that a 40-kDa trans-factor protein bound specifically to an RNA cis element (3′-100/50) in the chicken ccn2 3′-UTR, which posttranscriptionally regulated gene expression during the course of chondrocyte differentiation (Fig. 1A) (45). First of all, we examined whether the previous findings at the mRNA level correlated with the resultant production of CCN2 protein during chondrocytic differentiation. Using CEF, LS, and US cells representing immature mesenchymal cells, proliferating chondrocytes, and hypertrophic chondrocytes, Western blotting analysis was performed with proteins from an equal number of cells. As expected, increased production of CCN2 along with chondrocytic differentiation was clearly observed (Fig. 1B). Thereafter, in order to identify this protein, we employed the experimental strategy summarized in Fig. 1D. In the first step of purification, a nuclear extract of CEFs was applied to a heparin-Sepharose column equilibrated with buffer B. Fractions that were eluted from the column were assayed for protein content and binding to the 3′-100/50 fragment, the minimal repressive cis element in the 3′-UTR of chicken ccn2 mRNA (45), by conducting a UV cross-linking assay. Fractions that were eluted at 200 to 600 mM NaCl concentrations showed maximal binding to the radiolabeled 3′-100/50 fragment (data not shown), so these fractions were combined, pooled, and subjected to further purification. In the next step, the pooled fractions (2 mg of protein) were subjected to RNA affinity chromatography. The pooled fractions were loaded onto the 3′-100/50 RNA affinity column, and after excessive washing of the column, the bound protein was eluted by a linear step gradient of 100 to 1,000 mM NaCl in buffer B. The results of the UV cross-linking assay (Fig. 2A) conducted on the resulting fractions revealed that the fractions eluting at 200 to 600 mM NaCl contained the protein bound to the RNA, whereas no positive signal was observed in the flowthrough fraction, suggesting successful purification by the RNA chromatography. Therefore, these fractions were combined and pooled. For further verification of the binding between the proteins and the 3′-100/50 RNA fragment, Northwestern blotting was carried out (Fig. 2B). Five micrograms of the eluted protein was subjected to 4 to 20% SDS-PAGE and stained with CBB (Fig. 2B). A major band corresponding to an approximate molecular mass of 40 kDa and several minor bands were observed. Thereafter, the membrane was incubated with radiolabeled 3′-100/50 probe (Fig. 2B, NW lanes). A 40-kDa protein, which corresponded to the major band seen by CBB staining, strongly bound to the probe, whereas another major band was also observed, one corresponding to an approximate molecular mass of 90 kDa. Other minor bands stained with CBB showed no binding to the probe. Furthermore, molecular mass markers did not bind to the probe, indicating that the bands were probe-specific ones. Since the 40-kDa protein corresponded to the one we had determined to be a putative trans factor of ccn2 mRNA, this protein was subjected to protein sequencing for identification.
FIG. 1.
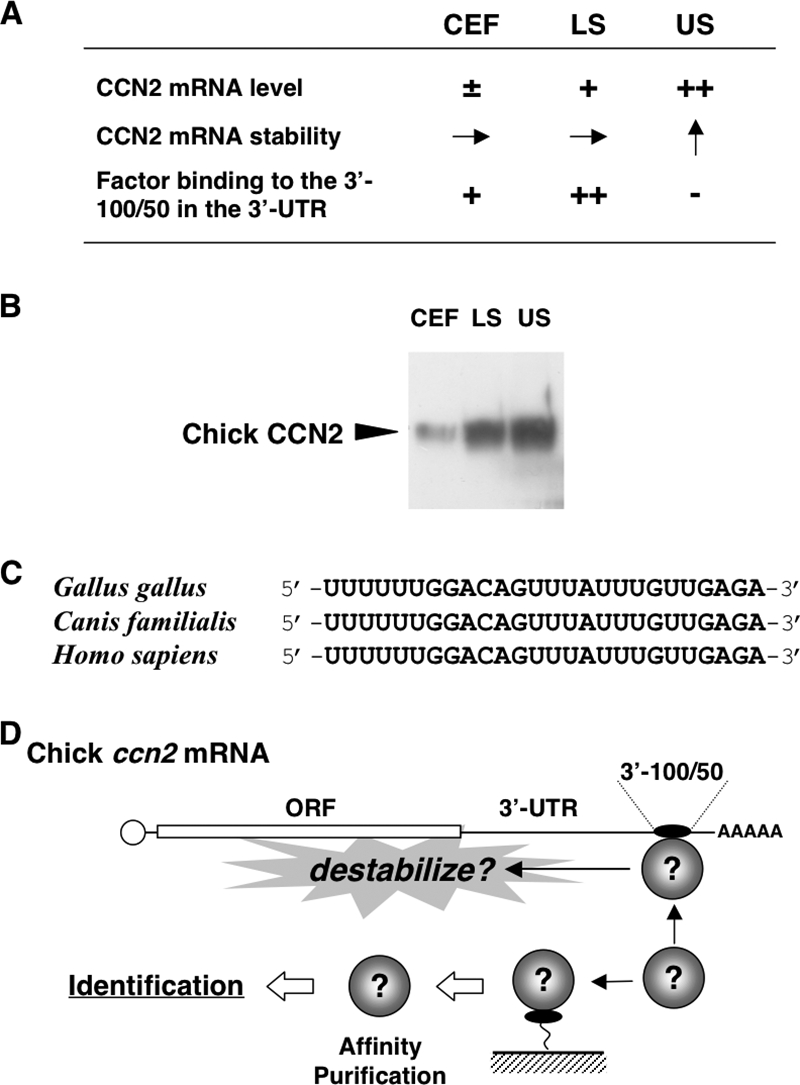
(A) Chicken in vitro model of endochondral ossification and posttranscriptional regulatory outcome of ccn2 expression therein. CEF, LS, and US denote chicken embryonic fibroblasts representing undifferentiated mesenchymal cells, lower sternum cartilage cells representing immature/proliferating chondrocytes, and upper sternum cartilage cells representing hypertrophic chondrocytes, respectively. −, ±, +, and ++ indicate approximate levels of each mRNA or protein in each cell; → and ↑ indicate equivalent and increased mRNA stability, respectively. Note that ccn2 mRNA stability is increased in US cells (arrow pointing up), where binding of a 40-kDa protein to the 3′-100/50 posttranscriptional regulatory element is nearly absent (−). (B) Western blotting analysis of the CCN2 proteins in CEF, LS, and US cells. Total cell lysates comprising 18 μg of DNA were analyzed. (C) A core nucleotide sequence conserved among mammalian species in the 3′-100/50 region and the corresponding sequence in the ccn2 mRNA 3′-UTR. (D) Experimental strategy for the identification of the 40-kDa protein. As summarized in panel A, the protein (illustrated as spheres) binds specifically to the 3′-100/50 RNA fragment in the 3′-UTR, which has been supposed to destabilize ccn2 mRNA. On the basis of this specific RNA-protein interaction, affinity purification methodology was employed, as illustrated. ORF, open reading frame.
FIG. 2.

Purification and identification of chicken ccn2 mRNA 3′-UTR-binding protein by affinity chromatography. (A) UV cross-linking assay of eluate from a chicken ccn2 mRNA 3′-UTR affinity column. NHS-activated Sepharose in a column was covalently conjugated to the 3′-100/50 fragment of chicken ccn2 mRNA 3′-UTR, which had been transcribed in vitro (see Materials and Methods). The nuclear proteins from CEFs that bound to the RNA were eluted by a linear step gradient of NaCl concentrations (100 to 1,000 mM) and concentrated. The proteins in the column flowthrough (FT) and fractions that bound to the 3′-100/50 RNA fragment were examined by conducting a UV cross-linking assay with SDS-PAGE in a 4 to 20% gradient gel. The positions of molecular mass standards (in kilodaltons) (Rainbow markers; Amersham Bioscience) are shown at the left side of the panel. (B) Northwestern blotting. The molecular mass standards (Marker) and 5 μg of eluted proteins (Extract) from the combined positive fractions (obtained with 200 to 600 mM NaCl in panel A) were subjected to SDS-PAGE in a 4 to 20% gradient gel and blotted onto a PVDF membrane. The membrane was first stained with CBB and then incubated with radiolabeled 3′-100/50 probe for Northwestern analysis (NW). The prominent bands bound by the probe are indicated by arrows at the right side of the panel. (C) Identification of the purified protein as nucleophosmin. A tryptic digestion product of the 40-kDa protein was subjected to Edman degradation, and the internal peptide sequences obtained were analyzed by the BLAST program of the National Center of Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov). Amino acid sequences from five fractions (fractions [Fr.] 14, 16, 18, 20, and 22; see the figure in supplemental material for details) purified through a high-pressure liquid chromatograph column were found to be identical to those of chicken NPM (GenBank accession number NM_205267), as indicated by the fraction numbers and underlined sequences. The boldface superscript numbers to the right of the sequences represent residue numbers of NPM counting from the initiation methionine.
Tryptic digestion products of the approximately 40-kDa protein excised from the SDS-polyacrylamide gel were separated by passage through a reverse-phase high-pressure liquid chromatograph column, and 11 specific peaks, which were not found in the blank control gel, were selected (see figure in supplemental material). These peaks were applied to an amino acid sequencer, and the results were subjected to a BLAST (National Center for Biotechnology Information; http://www.ncbi.nlm.nih.gov) amino acid homology search. Of the sequences of the proteins in these peaks, five (Fig. 2C) showed complete homology with the sequence of chicken NPM/B23 (GenBank accession number NM_205267), a protein known to shuttle between the nucleus and cytoplasm (3).
NPM binds to the repressive cis element of the 3′-UTR of chicken ccn2 mRNA.
The ability of recombinant chicken NPM (with His6 at its C-terminal region) to bind to the 3′-UTR of chicken ccn2 mRNA was determined by REMSA (Fig. 3B) and the UV cross-linking assay (Fig. 3C). In these experiments, two radiolabeled RNA probes were prepared (Fig. 3A) on the basis of our recent study (45). One was the 3′-100/50 probe, the minimal element in the 3′-UTR of chicken ccn2 mRNA for RNA destabilization through binding to the 40-kDa putative trans factor. The other was 3′-50, which was not capable of binding to the putative trans factor. In REMSA (Fig. 3B), the recombinant chicken NPM bound to 3′-100/50, resulting in a shift of the position of the probe after electrophoresis. The binding was enhanced by increasing the concentration of NPM. In contrast, no shifted band was observed after incubation of BSA with 3′-100/50. Furthermore, the results of the UV cross-linking assay (Fig. 3C) clearly indicated the binding between NPM and 3′-100/50. By incubating NPM with 3′-100/50, the 40-kDa band was observed, and its density increased with an increase in the concentration of NPM used. Also in this case, no specific band was observed when BSA was incubated with 3′-100/50. As expected, no signal was observed with the negative-control probe, 3′-50 (Fig. 3B). Finally, to further ascertain the interaction of NPM and the ccn2 mRNA, we also carried out an RNA immunoprecipitation analysis. As shown in Fig. 3D, an anti-NPM antibody successfully coimmunoprecipitated a ccn2 mRNA segment with native NPM in the nuclear extract, further indicating the interaction between NPM and the ccn2 mRNA. These results together demonstrate the specific binding of NPM to 3′-100/50, thus strongly suggesting that NPM is a trans factor of ccn2 mRNA.
Relevance of the ccn2 mRNA stability with subcellular distribution of NPM during differentiation of chondrocytes.
Since the 40-kDa trans-factor protein was anticipated to be an RNA-destabilizing regulator during chondrocyte differentiation, the intracellular fate of the ccn2 mRNA and the intracellular behavior of NPM were compared between proliferative LS and hypertrophic US chondrocytes. As also described earlier, the half-life (t1/2) of the ccn2 mRNA was prolonged by more than twofold in US cells (20.4% remaining after 2 h of actinomycin D treatment) compared to that in LS cells (3.8% remaining under the same condition) (Fig. 4A), which was also consistent with the results at the protein level (Fig. 1B). Using the same conditions, we next analyzed NPM in chondrocytes. Since NPM is known to be a shuttle protein moving between the nucleus and cytoplasm, we were quite interested in the intracellular distribution and the total content of NPM in these cells. Therefore, we carried out Western blotting to determine the intracellular location of the trans factor (Fig. 4B). By analyzing the proteins from an equal number of cells, total NPM was found to be lower in US cells than in LS cells. Of more interest, NPM was distributed dominantly in the cytoplasm in LS cells, whereas most of the protein in the US cells had accumulated in the nuclei, with quite low amounts in the cytoplasm. The results of Western blotting analysis for lamin B1 (a nuclear protein) and α-tubulin (a cytoplasmic protein) confirmed the quality and quantity of each protein fraction (Fig. 4C). These findings indicate that cytoplasmic NPM can be an RNA-binding destabilizer that regulates ccn2 expression during chondrocyte differentiation.
FIG. 4.
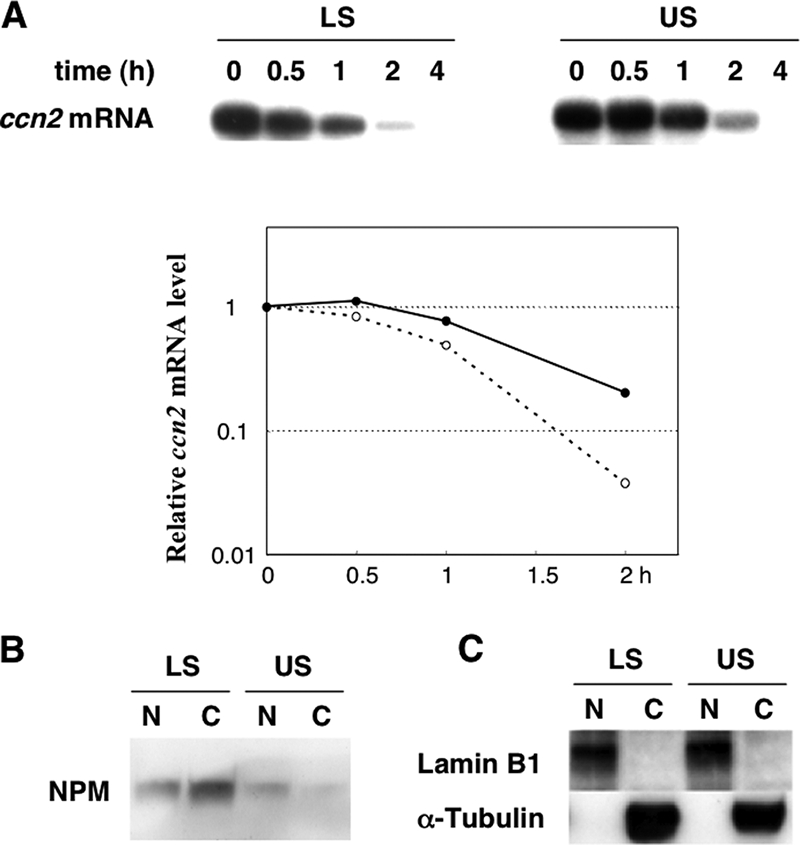
Relationship between intracellular ccn2 mRNA stability and subcellular distribution of NPM in chondrocytes. (A) Degradation profiles of ccn2 mRNA in LS and US cells. RNA synthesis in LS or US cells was arrested by actinomycin D, and a time-course study of the fate of the remaining ccn2 mRNA was analyzed by Northern blotting analysis (top panels). The level of 28S rRNA remained unchanged through the experimental time course (data not shown). The numbers (hours) indicate the time periods after transcriptional arrest was initiated. The signal intensities of the autoradiograms were quantitatively analyzed and plotted compared to the values at time zero (bottom panel). The broken line with open circles and solid line with closed circles denote the data obtained with LS and US cells, respectively. (B) Nucleocytoplasmic distribution of NPM in LS and US cells. Proteins from the nuclear extract (N) or cytoplasmic extract (C) from cells comprising the same amount of DNA were subjected to 12.5% SDS-PAGE and blotted onto a PVDF membrane. The blot was then incubated with anti-NPM antibody and then with secondary antibodies for visualization of signals, as described in Materials and Methods. (C) Successful fractionation confirmed by Western blotting of each fraction (10 μg protein) with anti-lamin B1 (nuclear marker) or anti-α-tubulin (a cytoplasmic marker) antibody.
In vitro reconstruction of the 5′-100/50-mediated posttranscriptional regulation by recombinant NPM.
Specific binding of NPM to 3′-100/50, the repressive cis element, and its negative correlation with ccn2 mRNA stability suggest that NPM may regulate the level of ccn2 mRNA by acting as an RNA destabilizer. In order to investigate this point, we established an IVDA system. As shown in Fig. 5A, four chimeric fusion probes [Luc(−), Luc-3′-Full, Luc-3′-100/50, and Luc-3′-50], in which the full-length 3′-UTR of ccn2 mRNA and deletion mutants of 3′-UTR of ccn2 mRNA were conjugated to the end of the open reading frame of firefly luciferase, were prepared to mimic RNA decay in vitro, according to previous studies (43, 63). It has been widely known that regulated mRNA degradation occurs in processing bodies (P bodies) in the cytosol (51). The IVDA was carried out by using 10 μg of the cytosolic proteins of CEFs in the presence or absence of 0.5 μg of recombinant chicken NPM or 1 μg of the eluate from the chicken ccn2 mRNA affinity column (Fig. 5B). When incubated with the cytosolic extract from CEFs alone, not only Luc-3′-Full but also Luc-3′-100/50 underwent rapid degradation (t1/2 = 0.5 h), whereas degradation of Luc(−) and Luc-3′-50 was relatively slower (t1/2 = 1 h) (Fig. 5B), suggesting that this in vitro system could mimic the events that occurred in vivo (Fig. 5A). Furthermore, in the presence of the recombinant chicken NPM, the stability of Luc-3′-Full and Luc-3′-100/50 was drastically decreased (t1/2 = 0.2 h), whereas that of Luc(−) and Luc-3′-50 was only modestly decreased (t1/2 = 0.7 h). Thus, NPM was shown to be associated with both ccn2-specific and nonspecific RNA degradation events in CEFs. Consistent with these findings, results with the eluate from the RNA affinity column, which contained endogenous NPM, were almost the same as those with recombinant chicken NPM. Therefore, these data indicate that NPM is the trans-regulatory factor of chicken ccn2 mRNA acting via RNA destabilization.
Destabilization effect of NPM on ccn2 mRNA is more specific and robust in chondrocytes.
Next, IVDA was conducted by using cytosolic extracts from both LS and US cells (Fig. 6) as well as from CEFs. Each of the three radiolabeled probes (shown in Fig. 5A) was incubated with 10 μg of cytosolic protein from LS or US cells. With LS proteins, the half-life of Luc-3′-50 was as long as that of Luc(−) (t1/2 = 0.7 h), and the stability of Luc-3′-Full and Luc-3′-100/50 was decreased (t1/2 = 0.25 h). Interestingly, the addition of NPM to Luc-3′-Full or Luc-3′-100/50 in the LS cytosolic extract accelerated mRNA decay far more drastically (Fig. 6) (t1/2 < 0.1 h) than that in the CEF extract (Fig. 5B), whereas the effect of NPM was minimal on Luc(−) and Luc-3′-50 (t1/2 = 0.52 h). These findings indicate that NPM acts as a more specific and efficient regulator of ccn2 mRNA degradation in the cytosolic extract of chondrocytes. Similar increased potential and functional specificity of NPM were also observed in the case of the US cell cytosolic extract. However, all of the RNA probes were more stable in the presence of the US cell extract than in the presence of the LS cell extract, and the half-lives of Luc(−) and Luc-3′-50 were longer than those of Luc-3′-Full and Luc-3′-100/50 [t1/2 = 1.6 h for Luc(−), t1/2 = 0.9 h for Luc-3′-Full, t1/2 = 0.55 h for Luc-3′-100/50, and t1/2 = 1.4 h for Luc-3′-50]. The addition of NPM to the US cell extract also resulted in drastically quick degradation of Luc-3′-Full and Luc-3′-100/50 (t1/2 < 0.1 h), whereas the nonspecific effect on Luc(−) and Luc-3′-50 was quite small (t1/2 = 1 h for Luc(−) and t1/2 = 0.8 h for Luc-3′-50). These findings indicate that NPM obtains the ability to exert highly specific action to regulate ccn2 mRNA stability in chondrocytes, which is supported by the composition of the cytosolic molecules that are specific to chondrocytes.
FIG. 6.

Robust effect of NPM on selective degradation of target RNA in a reconstituted system with chondrocyte cytosolic extract in vitro. The radiolabeled RNAs shown in Fig. 5A were incubated with cytoplasmic extracts (10 μg) from LS or US cells in the absence (−) or presence of recombinant chicken NPM (0.5 μg). After timed intervals, the RNA was purified and subjected to urea-denaturing 6% PAGE. In the graphs at the bottom of the panel, the relative remaining amounts and fitted degradation curves of RNA standardized against each RNA at time zero (negative control [closed ovals and solid line] and NPM [closed rectangles and dotted line]) are shown on a semilogarithmic graph (value at time zero = 100). Statistically significant differences between the two groups are indicated by asterisks as follows: *, P < 0.05; **, P < 0.001. A clear-cut and selective effect of NPM on the degradation of the 3′-100/50-containing RNA was observed.
Modulation of the intracellular distribution of NPM by growth factors.
In addition to CCN2, several growth factors, such as TGF-β (26, 49), BMPs (14, 18, 42, 78), and PDGF (27), have been reported to play important roles in growth and differentiation of chondrocytes. Moreover, these growth factors not only stimulate the expression of ccn2 (46) but also alter the stability of ccn2 mRNA in vivo (45). In fact, these growth factors repressed the degradation of ccn2 mRNA in LS cells, whereas conversely, they accelerated it in US cells. Therefore, suspecting the involvement of NPM in the regulation of ccn2 by such growth factors, we investigated npm gene expression and the subcellular distribution of NPM protein in LS and US cells stimulated by these growth factors. Northern blotting (Fig. 7A) revealed that all of the growth factors enhanced the expression of npm mRNA in LS cells. However, the level of NPM protein was found to be rather decreased by growth factor treatment, as revealed by Western blotting analysis (Fig. 7B). These data suggest a possible regulation of NPM production at a translation level. More importantly, Western blotting also revealed that stimulation of LS cells by all of the growth factors resulted in remarkable decreases in cytoplasmic NPM (Fig. 7B). This effect was most prominent by stimulation with TGF-β and BMP 2 and was modest with PDGF and CCN2. To our surprise, the effect of the growth factors on the US cells was the opposite. In US cells, NPM was found mostly in the nuclei without stimulation. However, stimulation by any of the growth factors resulted in drastic accumulation of NPM in the cytoplasm but also in an increased amount of NPM in the nuclei. The results of Western blotting analyses of marker proteins confirmed the quality and quantity of each protein fraction. The different redistribution patterns of NPM that resulted from stimulation by the growth factors are consistent with the differentiation-dependent effects of these growth factors on the resultant ccn2 gene expression between LS and US cells (45), suggesting the role of NPM as an RNA destabilizer acting in the cytoplasm in the ccn2 regulation by the growth factors.
FIG. 7.

Effects of growth factors on the expression and subcellular distribution of NPM in LS and US chondrocytes. (A) Northern blot analysis of LS and US cells stimulated with growth factors. LS and US cells were stimulated with TGF-β (10 ng/ml), BMP 2 (200 ng/ml), PDGF (10 ng/ml), or CCN2 (30 ng/ml) for 24 h in the absence of FBS or not stimulated (−) as a negative control, and total RNA was then extracted, denatured by glyoxysal, separated on 1% agarose gel, and blotted onto a nylon membrane. Prior to hybridization with the npm probe, the membrane was stained by methylene blue (28S) in order to show the same amount of total RNA. The signals were quantified, standardized against the level of 28S RNA, and presented as relative values versus control (1.00) at the bottom of the panel. (B) Subcellular fractionation and Western blot analysis. LS and US cells were stimulated with growth factors or not stimulated as described above for panel A, and 10-μg amounts of proteins from the nuclear extract (N) or cytoplasmic extract (C) were subjected to 12.5% SDS-PAGE and blotted onto a PVDF membrane. The blot was incubated with anti-NPM, anti-lamin B1 (nuclear control), or anti-α-tubulin (cytosolic control) antibody. The NPM signals from the nuclei (N) and cytosol (C) were standardized against the signals of lamin B1 (NPM/L) and α-tubulin (NPM/T), respectively. Normalized signals relative to the control (1.00) are displayed.
Reproduction of the effect of growth factors on ccn2 mRNA stability in IVDA, which was saturable by excess NPM.
As stated above (see “Modulation of the intracellular distribution of NPM by growth factors”), certain factors had the opposite effect on the stabilization of ccn2 mRNA between LS and US cells. In the present study, IVDA (Fig. 8), in which Luc-3′-100/50 was incubated with cytoplasmic proteins, yielded the same results, which again indicates the reliability of IVDA as an in vitro assay system. In LS cells, the RNA was more stable in the presence of cytosolic proteins from cells stimulated with TGF-β (t1/2 = 1.7 h), BMP 2 (t1/2 = 1.2 h), PDGF (t1/2 = 0.6 h), or CCN2 (t1/2 = 0.9 h), than in the presence of control cell extract (t1/2 = 0.25 h). However, of importance, the RNA stabilization effect of the growth factors was totally abolished by the addition of 0.1 μg NPM. Indeed, in the presence of excess NPM, ccn2 mRNA decayed in the growth factor-treated cell lysates as rapidly as in the control cell lysate. On the other hand, in US cells, stimulation by the growth factors rather decreased the RNA stability (t1/2 = 0.6 h for the control, t1/2 = 0.15 h for TGF-β, t1/2 = 0.35 h for BMB 2, t1/2 = 0.15 h for PDGF, and t1/2 = 0.2 h for CCN2). More importantly, the addition of NPM resulted in rapid and comparable degradation of the RNA in all cell extracts. These results further confirm that NPM plays an important role in the regulated degradation of ccn2 mRNA and that NPM is a ccn2-specific RNA-destabilizing molecule, and this effect can be precisely controlled by its subcellular redistribution during chondrocytic differentiation.
FIG. 8.

Differentiation-dependent regulation of the stability of the target RNA containing 3′-100/50 by growth factors via NPM in vitro. LS and US cells were stimulated with TGF-β (10 ng/ml), BMP 2 (200 ng/ml), PDGF (10 ng/ml), or CCN2 (30 ng/ml) for 24 h in the absence of FBS or left alone (−) as a negative control, and the cytoplasmic extract was obtained. Radiolabeled RNA (Luc-3′-100/50) was incubated with the cytoplasmic extracts (10 μg) in the absence (−) or presence of recombinant chicken NPM (0.1 μg). After timed intervals, the RNA was purified and subjected to urea-denaturing 6% PAGE. In the graphs at the bottom of the panel, the relative remaining amounts and fitted degradation curves of RNA standardized against each RNA at time zero (negative control [closed ovals and solid line] and NPM [closed rectangles and dotted line]) are shown on semilogarithmic graphs (value at time zero = 100). Note the opposite effects of growth factors on the reporter RNA stability between the two, both of which were abolished when excess NPM was added. Statistically significant differences in the absence of NPM between the values of growth factor-treated groups and the control group are indicated by asterisks as follows: *, P < 0.05.
Effect of npm silencing on ccn2 gene expression by siRNAs in vivo.
Finally, in order to confirm the requirement of NPM in the regulation of ccn2 in living cells, we employed an RNA interference-mediated knockdown strategy with CEFs. Two independent siRNAs were synthesized. One siRNA duplex, designated si546, was predicted to exert a maximal knockdown effect against the npm mRNA, whereas the other one, si207, was predicted to be less effective in silico. Consistent with this prediction, transfection of si546 into CEFs showed striking gene silencing effect on npm, while a modest repressive effect was observed with si207 (Fig. 9). The expression level of gapdh was not significantly altered by the siRNAs (Fig. 10), ruling out the possibility of off-target effects, and thus, it was utilized as an internal control to compute the relative gene expression levels. Also, the resultant NPM protein levels produced by the transfectants corresponded well to the mRNA levels observed above (Fig. 9B). Next, under the same conditions, the steady-state ccn2 mRNA level was evaluated. Exactly as expected, strong or modest silencing of npm resulted in a remarkable or subtle increase in the steady-state ccn2 mRNA level. These results clearly indicate that NPM is acting as a specific negative regulator of ccn2 gene expression in vivo, confirming the utility and reliability of our in vitro system.
FIG. 9.
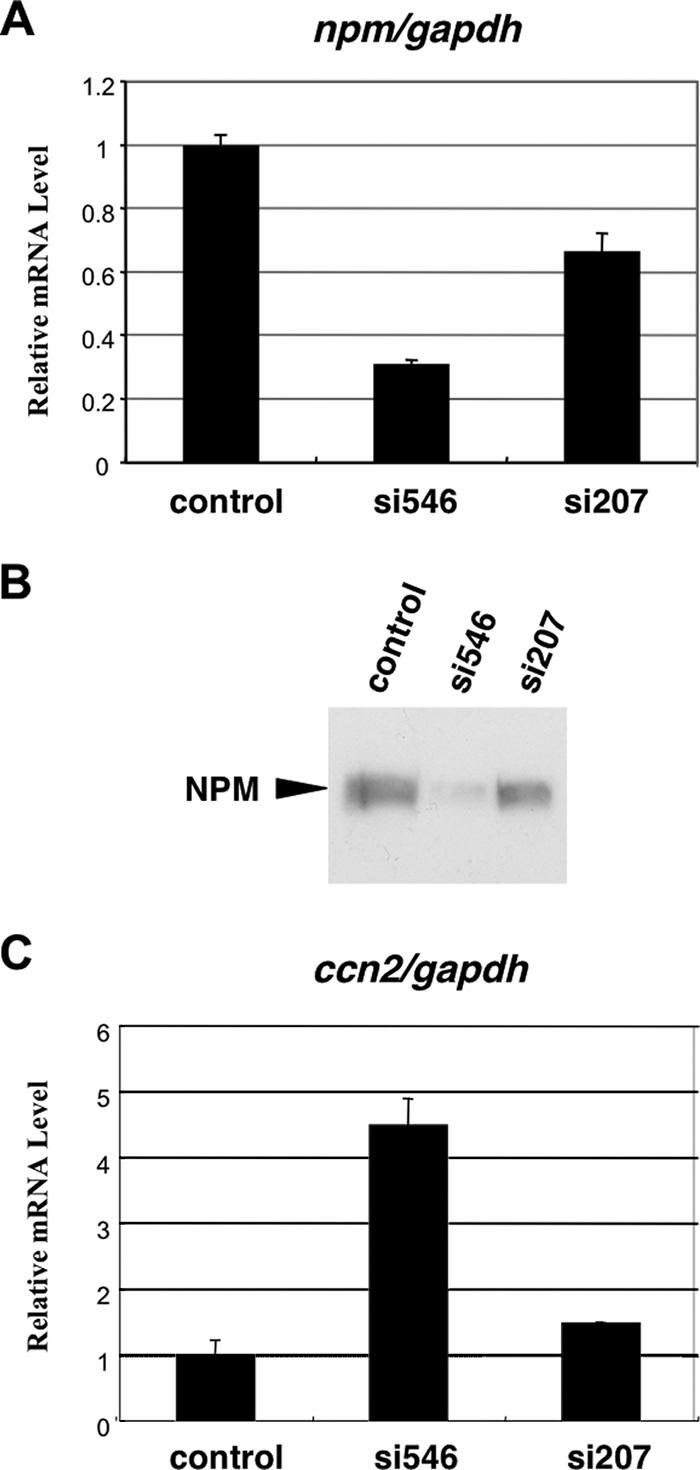
RNA interference-mediated knockdown of npm and its effect on ccn2 gene expression. (A) Steady-state mRNA levels of npm in CEFs at 48 h after the transfection of siRNAs as evaluated by real-time RT-PCR analysis. Two different siRNAs targeted to npm were synthesized and applied. The gapdh mRNA level, which was not significantly altered by those siRNAs (data not shown), was used as a standard. The mRNA levels are represented as relative values against that obtained with a control RNA duplex. (B) Western blotting analysis of NPM in CEFs transfected with the corresponding siRNAs under the same conditions as those shown in panel A. The same amount of total proteins (10 μg) was loaded for each sample. (C) Effects of npm knockdown by the siRNAs on the steady-state mRNA levels of ccn2 in CEFs. The ccn2 mRNA levels were standardized against gapdh mRNA levels and are shown as relative values against those with a control RNA duplex.
FIG. 10.
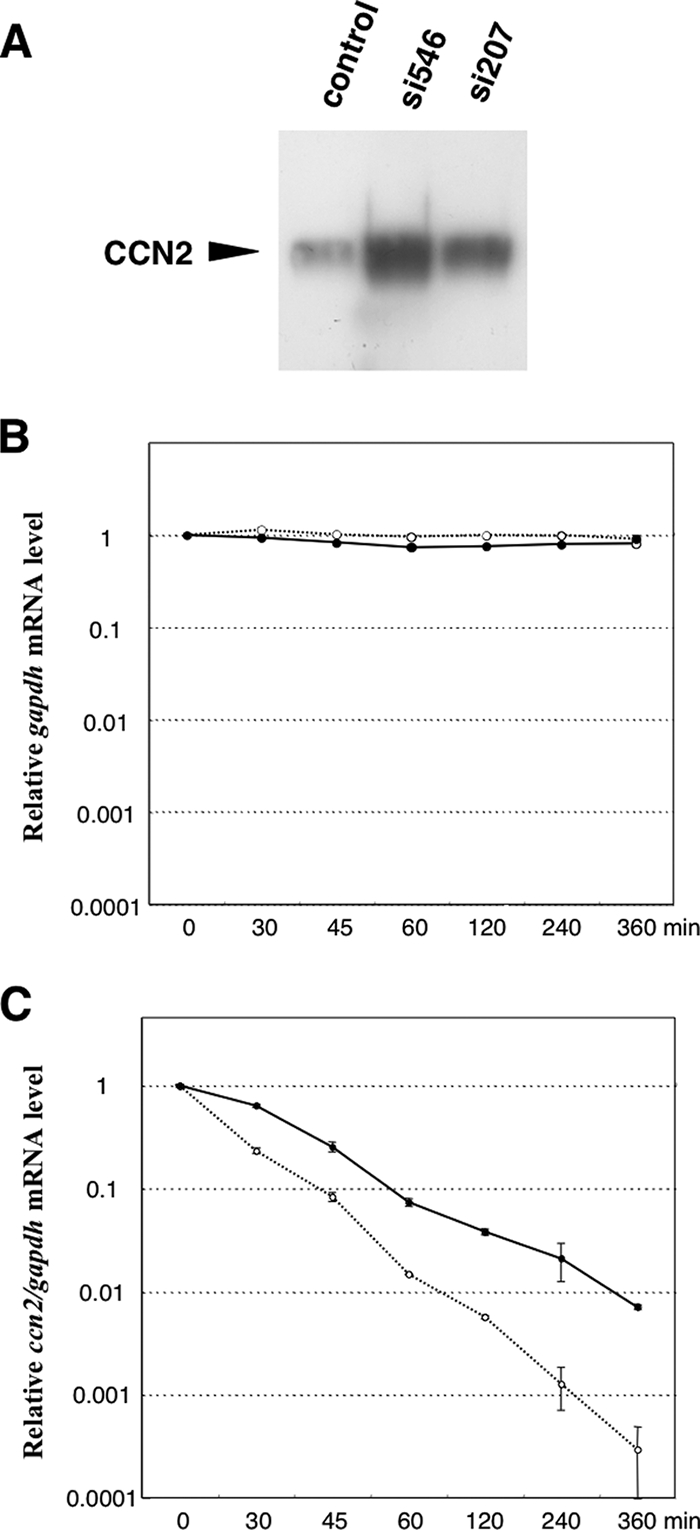
Outcome and mechanism of enhanced ccn2 gene expression by npm gene silencing. (A) Cellular CCN2 levels in CEFs as evaluated by Western blotting after the introduction of the siRNAs targeting npm. The same set of siRNAs was employed as in Fig. 9. The same amount of total proteins (10 μg) was loaded for each sample. (B) No significant effect of npm gene silencing on the degradation of gapdh mRNA in vivo. Nascent mRNA synthesis was arrested by actinomycin D treatment, and the fate of the mRNA in CEFs with a control RNA duplex (solid line) or si546 (dotted line) was pursued by real-time RT-PCR analysis. Relative values were computed against those at time zero. (C) Effect of npm gene silencing on the degradation of ccn2 mRNA in CEFs transfected with a control RNA duplex (solid line) or si546 (dotted line). Standardized values of ccn2 mRNA levels (ccn2/gapdh) were further normalized against those at time zero and presented.
Increased production of CCN2 by npm silencing and the mechanism of action.
Subsequently, the effects of npm silencing on CCN2 production were analyzed by Western blotting analysis in order to estimate the biological significance of the upregulated ccn2 expression. As clearly seen in Fig. 10, CCN2 production levels showed changes comparable to those in mRNA levels (Fig. 9), indicating that NPM acts as a regulator of CCN2 production. Then, to further examine whether NPM regulated ccn2 expression by altering the stability of ccn2 mRNA in vivo, which was clearly indicated by the in vitro data, mRNA degradation analysis was carried out in vivo. After the pretreatment by the siRNAs for 24 h, nascent mRNA synthesis was arrested by actinomycin D, and the fate of remaining mRNAs was chased following a time course. As a result, degradation of ccn2 mRNA was found to be remarkably slower in si546-treated cells than in the cells with control siRNA. Additionally, the degradation profile of another mRNA, gapdh, was not altered by si546. These data represent two critical aspects concerning target specificity; one is the specificity of the siRNAs to npm mRNA, and the other is the specificity of NPM to ccn2 mRNA. Collectively, in vivo knockdown of npm resulted in a prolonged half-life of ccn2 mRNA, whereas in vitro addition of NPM caused accelerated degradation of ccn2 mRNA. Thus, the consistency of in vivo and in vitro data strongly indicate that our IVDA system indeed reflects the biological reality taking place in living cells.
DISCUSSION
Our previous study (44) revealed that the 3′-UTR of chicken ccn2 mRNA contained a repressive cis element of gene expression, as does that of mammalian species (30-32, 34-36). Furthermore, our recent study (45) indicated that the stability of chicken ccn2 mRNA is regulated in a differentiation stage-dependent manner in chondrocytes and that the stability of chicken ccn2 mRNA negatively correlates with the interaction between a putative 40-kDa trans factor and the repressive cis element of the 3′-UTR of ccn2 mRNA. Following these studies, we sought to identify the putative 40-kDa trans-factor by employing an RNA affinity chromatography technique with a 3′-100/50 fragment of ccn2 mRNA 3′-UTR. The results of a UV cross-linking assay of the 3′-100/50 fragment with the purified proteins (Fig. 2A) showed a few bands representing their specific binding. Also, upon Northwestern blotting (Fig. 2B), two specific bands were observed. One was approximately 40 kDa in molecular mass, and the other was approximately 90 kDa. On the basis of our previous study (45), we chose and further investigated the 40-kDa protein in the present study. However, the 90-kDa protein may not be disregarded, since it also bound to the probe rather more strongly than the 40-kDa protein did (Fig. 2B). Therefore, in the future, we will investigate the properties of this protein as well.
The protein was processed by an endopeptidase, and purified fragments were subjected to Edman analysis. The five internal peptide sequences obtained (Fig. 2C) indicated that NPM (40, 41, 50) was the 40-kDa binding protein for the 3′-100/50 fragment. NPM has been given other names, such as B23 (7, 8), NO38 (60, 61), and numatrin (16). NPM was first found to be located mainly in a granular component of nuclei (56, 74) and to play a role in the assembly of preribosomal particles (60). Later, it was shown to shuttle between the nucleus and cytoplasm (3). Furthermore, a number of recent reports revealed that NPM interacts with proteins, such as the transcriptional factor YY1 (24), Rev protein of human immunodeficiency virus type 1 (15), and tumor suppressor p53 (10), and with single- or double-stranded DNA (9, 10, 76, 77) and RNA (3, 70, 73). Since these findings suggested multifunctional roles of NPM as a nucleocytoplasmic shuttle protein, we investigated whether or not NPM is a trans factor critical for the posttranscriptional regulation of ccn2 mRNA.
First, in order to confirm the binding of NPM to the 3′-UTR of ccn2 mRNA, we prepared two radiolabeled probes, 3′-100/50 and 3′-50 (Fig. 3A). The former probe was the target RNA segment, whereas the latter was a negative control. Indeed, REMSA and the UV cross-linking assay (Fig. 3B and C) indicated the specific binding of recombinant chicken NPM to 3′-100/50. Moreover, this specific interaction of NPM and the 3′-100/50 probe was further confirmed by RNA immunoprecipitation analysis. With this protein, we could eventually reconstitute the 3′-100/50-mediated regulation of ccn2 expression in vitro, which was actually observed during chondrocyte differentiation in vivo.
Prior to establishing an IVDA system, we comparatively evaluated the gene expression and subcellular distribution of NPM in LS and US cells. Northern blotting analysis showed comparable ccn2 gene expression levels in LS and US cells (data not shown). However, interestingly, in US cells, most of the NPM protein had accumulated in the nuclei, and its distribution in the cytoplasm was very low (Fig. 4B). Considering the increased ccn2 mRNA stability in US cells (Fig. 4A), NPM was thus suspected to be the critical determinant to accelerate the selective degradation of ccn2 mRNA in the cytoplasm. Involvement of the shuttling property of NPM in ccn2 regulation was also indicated herein.
For IVDA, four chimeric reporter RNAs were prepared, and their stability was assessed in cytosolic extract from CEFs. Unexpectedly, the stabilities of all of the reporter RNAs were significantly decreased by the addition of NPM to the CEF extract. Several earlier studies (21, 22, 59) showed that NPM possesses an intrinsic RNase activity that preferentially cleaves single-stranded poly(A), poly(U), and poly(C). Hence, the decreased RNA stability in the presence of NPM might involve the effect of the nonspecific RNase activity of NPM in part. However, the degree of decreased stability caused by NPM was greater for the mRNAs with the binding target (Luc′-3′-Full and Luc-3′-100/50) than for those of the controls [Luc(−) and Luc-3′-50]. These results indicate the sequence-specific enhancing effect of NPM on RNA degradation. Therefore, together with the results of the RNA-binding analysis, we showed that NPM is an mRNA destabilizer that binds to the repressive cis element of 3′-UTR of ccn2 mRNA and accelerates the degradation with significant specificity in CEF extracts. Furthermore, this NPM function as a negative posttranscriptional regulator of ccn2 expression indicated in vitro was clearly confirmed in vivo through an siRNA-mediated gene silencing approach.
As stated above, the effect of NPM on mRNA degradation did not appear to be highly specific in CEF extracts. Nevertheless, subsequent analysis with chondrocyte cytosolic extracts revealed a quite interesting aspect of NPM function. Surprisingly, IVDA with cytosolic proteins from LS and US cells (Fig. 6) showed much stronger and more specific effects of NPM on the stability of ccn2 mRNA than the effects obtained with cytosolic proteins from CEFs. The molecular background of this cell-type-dependent enhancement of functional specificity is unknown. However, since CCN2 plays a critical role in both the growth and differentiation of chondrocytes, precise regulation of ccn2 mRNA would be especially required in this particular type of cells. Therefore, the enhanced regulatory potential of NPM in LS and US cells suggests that the NPM-mediated posttranscriptional regulation is one of the precise control systems that have been developed specifically in chondrocytes.
TGF-β, BMP 2, PDGF, and CCN2 are known to promote the differentiation of growing cartilage cells. Therefore, upon stimulation, these factors increase ccn2 expression in LS cells, whereas they rather decrease it in US cells by driving the cells to terminal hypertrophy (45). It is of interest that although stimulation with these factors hardly changed the expression level of the npm gene in LS and US cells (Fig. 7A), the cellular distribution of NPM protein was drastically changed depending on the differentiation stage of the chondrocytes (Fig. 7B). In LS cells, incubation with the growth factors resulted in the accumulation of NPM in the nuclei. In contrast, in growth factor-treated US cells, NPM was observed in the cytosolic fraction, in which it was hardly observed without stimulation; thus, opposite results were obtained with LS and US cells. It is noteworthy that the results of IVDA (Fig. 8) indicated that the stimulation by growth factors stabilized the target RNA in LS cell extract, whereas it rather destabilized the target in US cell extracts, which is in agreement with our previous study (45). Here again, the higher the NPM content in the cytosol, the more fragile was the ccn2 mRNA. Total ablation of such an effect of these growth factors by excess NPM in vitro indicates that NPM acts as a major regulator of ccn2 mRNA stability upon growth factor stimulation. These results further indicate that the stability of ccn2 mRNA in chondrocytes was critically controlled by the intracellular distribution of shuttling NPM.
In a number of previous studies, important physiological or pathological roles of CCN2 were reported, such as roles in cell growth and differentiation in development (46, 47, 71), angiogenesis (2, 64, 68), and wound healing (23). In addition, CCN2 is known to play a critical role in chondrocytes during endochondral ossification (46-48, 65, 69), with its gene expression being precisely controlled during differentiation. Indeed, expression of the ccn2 gene is controlled not only at the transcriptional level (11-13, 19) but also by posttranscriptional events (30-32, 34-36, 44, 45). In the present study, we first identified the posttranscriptional regulatory protein as NPM binding to the repressive cis element to enhance RNA degradation with pronounced efficacy in chondrocytes. Moreover, we demonstrated that the intracellular distribution of NPM was a critical parameter of ccn2 regulation during the differentiation of chondrocytes. To our knowledge, no study concerning the role of NPM in skeletal development has previously appeared in the literature; thus, our present findings represent the first report of a novel NPM-mediated gene regulation system critical for proper chondrogenesis and endochondral ossification. Generally, mRNAs are regulated posttranscriptionally by multiple complexes composed of an RNA cis element and trans-factor protein(s). Hence, our next goal is to uncover the precise regulatory mechanism of degradation of ccn2 mRNA by the interaction among 3′-UTR, NPM, and any other factor(s) that needs to be identified. Further investigation is now ongoing.
Supplementary Material
Acknowledgments
This work was supported by the programs Grants-in-Aid for Scientific Research (grant S to M.T.) and (grant C to S. Kubota) and Grants-in-Aid for Exploratory Research (to M.T.) of the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and by a grant from the Foundation of Sanyo Broadcasting (to S. Kubota).
We thank Takako Hattori, Takashi Nishida, Masanao Minato, Tsuyoshi Yanagita, Kazumi Kawata, Chisa Kuroda, Kyouji Nakao, and Toshihiro Ogawara for helpful suggestions; Kazumi Ohyama for technical assistance; and Yuki Nonami for secretarial assistance.
Footnotes
Published ahead of print on 4 August 2008.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Almendral, J. M., D. Sommer, H. Macdonald-Bravo, J. Burckhardt, J. Perera, and R. Bravo. 1988. Complexity of the early genetic response to growth factors in mouse fibroblasts. Mol. Cell. Biol. 82140-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babic, A. M., C. C. Chen, and L. F. Lau. 1999. Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin αvβ3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol. Cell. Biol. 192958-2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borer, R. A., C. F. Lehner, H. M. Eppenberger, and E. A. Nigg. 1989. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 56379-390. [DOI] [PubMed] [Google Scholar]
- 4.Bork, P. 1993. The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 327125-130. [DOI] [PubMed] [Google Scholar]
- 5.Bradham, D. M., A. Igarashi, R. L. Potter, and G. R. Grotendorst. 1991. Connective tissue growth factor: a cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J. Cell Biol. 1141285-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brigstock, D. R. 1999. The connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family. Endocr. Rev. 20189-206. [DOI] [PubMed] [Google Scholar]
- 7.Chan, P. K., M. Aldrich, R. G. Cook, and H. Busch. 1986. Amino acid sequence of protein B23 phosphorylation site. J. Biol. Chem. 2611868-1872. [PubMed] [Google Scholar]
- 8.Chan, P. K., W. Y. Chan, B. Y. Yung, R. G. Cook, M. B. Aldrich, D. Ku, I. L. Goldknopf, and H. Busch. 1986. Amino acid sequence of a specific antigenic peptide of protein B23. J. Biol. Chem. 26114335-14341. [PubMed] [Google Scholar]
- 9.Chen, H., B. Li, and J. L. Workman. 1994. A histone-binding protein, nucleoplasmin, stimulates transcription factor binding to nucleosomes and factor-induced nucleosome disassembly. EMBO J. 13380-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colombo, E., J. C. Marine, D. Danovi, B. Falini, and P. G. Pelicci. 2002. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 4529-533. [DOI] [PubMed] [Google Scholar]
- 11.Eguchi, T., S. Kubota, S. Kondo, T. Shimo, T. Hattori, T. Nakanishi, T. Kuboki, H. Yatani, and M. Takigawa. 2001. Regulatory mechanism of human connective tissue growth factor (CTGF/Hcs24) gene expression in a human chondrocytic cell line, HCS-2/8. J. Biochem. (Tokyo) 13079-87. [DOI] [PubMed] [Google Scholar]
- 12.Eguchi, T., S. Kubota, S. Kondo, T. Kuboki, H. Yatani, and M. Takigawa. 2002. A novel cis-element that enhances connective tissue growth factor gene expression in chondrocytic cells. Biochem. Biophys. Res. Commun. 295445-451. [DOI] [PubMed] [Google Scholar]
- 13.Eguchi, T., S. Kubota, K. Kawata, Y. Mukudai, T. Ohgawara, K. Miyazono, K. Nakao, S. Kondo, and M. Takigawa. 2007. Different transcriptional strategies for ccn2/ctgf gene induction between human chondrocytic and breast cancer cell lines. Biochimie 89278-288. [DOI] [PubMed] [Google Scholar]
- 14.Enomoto-Iwamoto, M., M. Iwamoto, Y. Mukudai, Y. Kawakami, T. Nohno, Y. Higuchi, S. Takemoto, H. Ohuchi, S. Noji, and K. Kurisu. 1998. Bone morphogenetic protein signaling is required for maintenance of differentiated phenotype, control of proliferation, and hypertrophy in chondrocytes. J. Cell Biol. 140409-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fankhauser, C., E. Izaurralde, Y. Adachi, P. Wingfield, and U. K. Laemmli. 1991. Specific complex of human immunodeficiency virus type 1 Rev and nucleolar B23 proteins: dissociation by the Rev response element. Mol. Cell. Biol. 112567-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feuerstein, N., P. K. Chan, and J. J. Mond. 1988. Identification of numatrin, the nuclear matrix protein associated with induction of mitogenesis, as the nucleolar protein B23. Implication for the role of the nucleolus in early transduction of mitogenic signals. J. Biol. Chem. 26310608-10612. [PubMed] [Google Scholar]
- 17.Frazier, K., S. Williams, D. Kothapalli, H. Klapper, and G. R. Grotendorst. 1996. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J. Investig. Dermatol. 107404-411. [DOI] [PubMed] [Google Scholar]
- 18.Gitelman, S. E., M. S. Kobrin, J. Q. Ye, A. R. Lopez, A. Lee, and R. Derynck. 1994. Recombinant Vgr-1/BMP-6-expressing tumors induce fibrosis and endochondral bone formation in vivo. J. Cell Biol. 1261595-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grotendorst, G. R. 1997. Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 8171-179. [DOI] [PubMed] [Google Scholar]
- 20.Hashimoto, Y., N. Shindo-Okada, M. Tani, Y. Nagamachi, K. Takeuchi, T. Shiroishi, H. Toma, and J. Yokota. 1998. Expression of the Elm1 gene, a novel gene of the CCN (connective tissue growth factor, Cyr61/Cef10, and neuroblastoma overexpressed gene) family, suppresses in vivo tumor growth and metastasis of K-1735 murine melanoma cells. J. Exp. Med. 187289-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herrera, J. E., R. Savkur, and M. O. Olson. 1995. The ribonuclease activity of nucleolar protein B23. Nucleic Acids Res. 233974-3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hingorani, K., A. Szebeni, and M. O. Olson. 2000. Mapping the functional domains of nucleolar protein B23. J. Biol. Chem. 27524451-24457. [DOI] [PubMed] [Google Scholar]
- 23.Igarashi, A., H. Okochi, D. M. Bradham, and G. R. Grotendorst. 1993. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol. Biol. Cell 4637-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inouye, C. J., and E. Seto. 1994. Relief of YY1-induced transcriptional repression by protein-protein interaction with the nucleolar phosphoprotein B23. J. Biol. Chem. 2696506-6510. [PubMed] [Google Scholar]
- 25.Joliot, V., C. Martinerie, G. Dambrine, G. Plassiart, M. Brisac, J. Crochet, and B. Perbal. 1992. Proviral rearrangements and overexpression of a new cellular gene (nov) in myeloblastosis-associated virus type 1-induced nephroblastomas. Mol. Cell. Biol. 1210-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kato, Y., M. Iwamoto, T. Koike, F. Suzuki, and Y. Takano. 1988. Terminal differentiation and calcification in rabbit chondrocyte cultures grown in centrifuge tubes: regulation by transforming growth factor beta and serum factors. Proc. Natl. Acad. Sci. USA 859552-9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kieswetter, K., Z. Schwartz, M. Alderete, D. D. Dean, and B. D. Boyan. 1997. Platelet derived growth factor stimulates chondrocyte proliferation but prevents endochondral maturation. Endocr. J. 6257-264. [DOI] [PubMed] [Google Scholar]
- 28.Kikuchi, K., T. Kadono, H. Ihn, S. Sato, A. Igarashi, H. Nakagawa, K. Tamaki, and K. Takehara. 1995. Growth regulation in scleroderma fibroblasts: increased response to transforming growth factor-beta 1. J. Investig. Dermatol. 105128-132. [DOI] [PubMed] [Google Scholar]
- 29.Kireeva, M. L., B. V. Latinkic, T. V. Kolesnikova, C. C. Chen, G. P. Yang, A. S. Abler, and L. F. Lau. 1997. Cyr61 and Fisp12 are both ECM-associated signaling molecules: activities, metabolism, and localization during development. Exp. Cell Res. 23363-77. [DOI] [PubMed] [Google Scholar]
- 30.Kondo, S., S. Kubota, T. Eguchi, T. Hattori, T. Nakanishi, T. Sugahara, and M. Takigawa. 2000. Characterization of a mouse ctgf 3′-UTR segment that mediates repressive regulation of gene expression. Biochem. Biophys. Res. Commun. 278119-124. [DOI] [PubMed] [Google Scholar]
- 31.Kondo, S., S. Kubota, T. Shimo, T. Nishida, G. Yosimichi, T. Eguchi, T. Sugahara, and M. Takigawa. 2002. Connective tissue growth factor increased by hypoxia may initiate angiogenesis in collaboration with matrix metalloproteinases. Carcinogenesis 23769-776. [DOI] [PubMed] [Google Scholar]
- 32.Kondo, S., S. Kubota, Y. Mukudai, N. Moritani, T. Nishida, H. Matsushita, S. Matsumoto, T. Sugahara, and M. Takigawa. 2006. Hypoxic regulation of stability of connective tissue growth factor/CCN2 mRNA by 3′-untranslated region interacting with a cellular protein in human chondrosarcoma cells. Oncogene 251099-1110. [DOI] [PubMed] [Google Scholar]
- 33.Kothapalli, D., K. S. Frazier, A. Welply, P. R. Segarini, and G. R. Grotendorst. 1997. Transforming growth factor beta induces anchorage-independent growth of NRK fibroblasts via a connective tissue growth factor-dependent signaling pathway. Cell Growth Differ. 861-68. [PubMed] [Google Scholar]
- 34.Kubota, S., T. Hattori, T. Nakanishi, and M. Takigawa. 1999. Involvement of cis-acting repressive element(s) in the 3′-untranslated region of human connective tissue growth factor gene. FEBS Lett. 45084-88. [DOI] [PubMed] [Google Scholar]
- 35.Kubota, S., S. Kondo, T. Eguchi, T. Hattori, T. Nakanishi, R. J. Pomerantz, and M. Takigawa. 2000. Identification of an RNA element that confers post-transcriptional repression of connective tissue growth factor/hypertrophic chondrocyte specific 24 (ctgf/hcs24) gene: similarities to retroviral RNA-protein interactions. Oncogene 194773-4786. [DOI] [PubMed] [Google Scholar]
- 36.Kubota, S., Y. Mukudai, N. H. Moritani, K. Nakao, K. Kawata, and M. Takigawa. 2005. Translational repression by the cis-acting element of structure-anchored repression (CAESAR) of human ctgf/ccn2 mRNA. FEBS Lett. 5793751-3758. [DOI] [PubMed] [Google Scholar]
- 37.Lau, L. F., and D. Nathans. 1985. Identification of a set of genes expressed during the G0/G1 transition of cultured mouse cells. EMBO J. 43145-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lau, L. F., and S. C. Lam. 1999. The CCN family of angiogenic regulators: the integrin connection. Exp. Cell Res. 24844-57. [DOI] [PubMed] [Google Scholar]
- 39.Leask, A., A. Holmes, C. M. Black, and D. J. Abraham. 2003. Connective tissue growth factor gene regulation. Requirements for its induction by transforming growth factor-beta 2 in fibroblasts. J. Biol. Chem. 27813008-13015. [DOI] [PubMed] [Google Scholar]
- 40.Maridor, G., W. Krek, and E. A. Nigg. 1990. Structure and developmental expression of chicken nucleolin and NO38: coordinate expression of two abundant non-ribosomal nucleolar proteins. Biochim. Biophys. Acta 1049126-133. [DOI] [PubMed] [Google Scholar]
- 41.Maridor, G., and E. A. Nigg. 1990. cDNA sequences of chicken nucleolin/C23 and NO38/B23, two major nucleolar proteins. Nucleic Acids Res. 181286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minina, E., H. M. Wenzel, C. Kreschel, S. Karp, W. Gaffield, A. P. McMahon, and A. Vortkamp. 2001. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 1284523-4534. [DOI] [PubMed] [Google Scholar]
- 43.Moallem, E., R. Kilav, J. Silver, and T. Naveh-Many. 1998. RNA-protein binding and post-transcriptional regulation of parathyroid hormone gene expression by calcium and phosphate. J. Biol. Chem. 2735253-5259. [DOI] [PubMed] [Google Scholar]
- 44.Mukudai, Y., S. Kubota, and M. Takigawa. 2003. Conserved repressive regulation of connective tissue growth factor/hypertrophic chondrocyte-specific gene 24 (ctgf/hcs24) enabled by different elements and factors among vertebrate species. Biol. Chem. 3841-9. [DOI] [PubMed] [Google Scholar]
- 45.Mukudai, Y., S. Kubota, T. Eguchi, S. Kondo, K. Nakao, and M. Takigawa. 2005. Regulation of chicken ccn2 gene by interaction between RNA cis-element and putative trans-factor during differentiation of chondrocytes. J. Biol. Chem. 2803166-3177. [DOI] [PubMed] [Google Scholar]
- 46.Nakanishi, T., Y. Kimura, T. Tamura, H. Ichikawa, Y. Yamaai, T. Sugimoto, and M. Takigawa. 1997. Cloning of a mRNA preferentially expressed in chondrocytes by differential display-PCR from a human chondrocytic cell line that is identical with connective tissue growth factor (CTGF) mRNA. Biochem. Biophys. Res. Commun. 234206-210. [DOI] [PubMed] [Google Scholar]
- 47.Nakanishi, T., T. Nishida, T. Shimo, K. Kobayashi, T. Kubo, T. Tamatani, K. Tezuka, and M. Takigawa. 2000. Effects of CTGF/Hcs24, a product of a hypertrophic chondrocyte-specific gene, on the proliferation and differentiation of chondrocytes in culture. Endocrinology 141264-273. [DOI] [PubMed] [Google Scholar]
- 48.Nishida, T., T. Nakanishi, M. Asano, T. Shimo, and M. Takigawa. 2000. Effects of CTGF/Hcs24, a hypertrophic chondrocyte-specific gene product, on the proliferation and differentiation of osteoblastic cells in vitro. J. Cell. Physiol. 184197-206. [DOI] [PubMed] [Google Scholar]
- 49.Noda, M., and J. J. Camilliere. 1989. In vivo stimulation of bone formation by transforming growth factor-beta. Endocrinology 1242991-2994. [DOI] [PubMed] [Google Scholar]
- 50.Orrick, L. R., M. O. Olson, and H. Busch. 1973. Comparison of nucleolar proteins of normal rat liver and Novikoff hepatoma ascites cells by two-dimensional polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. USA 701316-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parker, R., and U. Sheth. 2007. P bodies and the control of mRNA translation and degradation. Mol. Cell 25635-646. [DOI] [PubMed] [Google Scholar]
- 52.Pennica, D., T. A. Swanson, J. W. Welsh, M. A. Roy, D. A. Lawrence, J. Lee, J. Brush, L. A. Taneyhill, B. Deuel, M. Lew, C. Watanabe, R. L. Cohen, M. F. Melhem, G. G. Finley, P. Quirke, A. D. Goddard, K. J. Hillan, A. L. Gurney, D. Botstein, and A. J. Levine. 1998. WISP genes are members of the connective tissue growth factor family that are up-regulated in wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc. Natl. Acad. Sci. USA 9514717-14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perbal, B. 2004. CCN proteins: multifunctional signalling regulators. Lancet 36362-64. [DOI] [PubMed] [Google Scholar]
- 54.Perbal, B., and M. Takigawa (ed.). 2005. CCN proteins—a new family of cell growth and differentiation regulators. Imperial College Press, London, United Kingdom.
- 55.Phanish, M. K., N. A. Wahab, P. Colville-Nash, B. M. Hendry, and M. E. Dockrell. 2006. The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem. J. 393601-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prestayko, A. W., G. R. Klomp, D. J. Schmoll, and H. Busch. 1974. Comparison of proteins of ribosomal subunits and nucleolar preribosomal particles from Novikoff hepatoma ascites cells by two-dimensional polyacrylamide gel electrophoresis. Biochemistry 131945-1951. [DOI] [PubMed] [Google Scholar]
- 57.Ryseck, R. P., H. Macdonald-Bravo, M. G. Mattei, and R. Bravo. 1991. Structure, mapping, and expression of fisp-12, a growth factor-inducible gene encoding a secreted cysteine-rich protein. Cell Growth Differ. 2225-233. [PubMed] [Google Scholar]
- 58.Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 745463-5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savkur, R. S., and M. O. Olson. 1998. Preferential cleavage in pre-ribosomal RNA by protein B23 endoribonuclease. Nucleic Acids Res. 264508-4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmidt-Zachmann, M. S., B. Hugle-Dorr, and W. W. Franke. 1987. A constitutive nucleolar protein identified as a member of the nucleoplasmin family. EMBO J. 61881-1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmidt-Zachmann, M. S., and W. W. Franke. 1988. DNA cloning and amino acid sequence determination of a major constituent protein of mammalian nucleoli. Correspondence of the nucleoplasmin-related protein NO38 to mammalian protein B23. Chromosoma 96417-426. [DOI] [PubMed] [Google Scholar]
- 62.Sela-Brown, A., J. Silver, G. Brewer, and T. Naveh-Many. 2000. Identification of AUF1 as a parathyroid hormone mRNA 3′-untranslated region-binding protein that determines parathyroid hormone mRNA stability. J. Biol. Chem. 2757424-7429. [DOI] [PubMed] [Google Scholar]
- 63.Sengupta, T. K., S. Bandyopadhyay, D. J. Fernandes, and E. K. Spicer. 2004. Identification of nucleolin as an AU-rich element binding protein involved in bcl-2 mRNA stabilization. J. Biol. Chem. 27910855-10863. [DOI] [PubMed] [Google Scholar]
- 64.Shimo, T., T. Nakanishi, T. Nishida, M. Asano, M. Kanyama, T. Kuboki, T. Tamatani, K. Tezuka, M. Takemura, T. Matsumura, and M. Takigawa. 1999. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J. Biochem. (Tokyo) 126137-145. [DOI] [PubMed] [Google Scholar]
- 65.Shimo, T., E. Koyama, H. Sugito, C. Wu, S. Shimo, and M. Pacifici. 2005. Retinoid signaling regulates CTGF expression in hypertrophic chondrocytes with differential involvement of MAP kinases. J. Bone Miner. Res. 20867-877. [DOI] [PubMed] [Google Scholar]
- 66.Simmons, D. L., D. B. Levy, Y. Yannoni, and R. L. Erikson. 1989. Identification of a phorbol ester-repressible v-src-inducible gene. Proc. Natl. Acad. Sci. USA 861178-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith, P. K., R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson, and D. C. Klenk. 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem. 15076-85. [DOI] [PubMed] [Google Scholar]
- 68.Takigawa, M. 2003. CTGF/Hcs24 as a multifunctional growth factor for fibroblasts, chondrocytes and vascular endothelial cells. Drug News Persp. 1611-21. [DOI] [PubMed] [Google Scholar]
- 69.Takigawa, M., T. Nakanishi, S. Kubota, and T. Nishida. 2003. Role of CTGF/Hcs24/ecogenin in skeletal growth control. J. Cell. Physiol. 194256-266. [DOI] [PubMed] [Google Scholar]
- 70.Wang, D., A. Baumann, A. Szebeni, and M. O. Olson. 1994. The nucleic acid binding activity of nucleolar protein B23.1 resides in its carboxyl-terminal end. J. Biol. Chem. 26930994-30998. [PubMed] [Google Scholar]
- 71.Yamaai, T., T. Nakanishi, M. Asano, K. Nawachi, G. Yoshimichi, K. Ohyama, T. Komori, T. Sugimoto, and M. Takigawa. 2005. Gene expression of connective tissue growth factor (CTGF/CCN2) in calcifying tissues of normal and cbfa1-null mutant mice in late stage of embryonic development. J. Bone Miner. Metab. 23280-288. [DOI] [PubMed] [Google Scholar]
- 72.Yanagita, T., S. Kubota, K. Kawata, H. Kawaki, S. Kondo, T. Takano-Yamamoto, S. Tanaka, and M. Takigawa. 2007. Expression and physiological role of CCN4/Wnt-induced secreted protein 1 mRNA splicing variants in chondrocytes. FEBS J. 2741655-1665. [DOI] [PubMed] [Google Scholar]
- 73.Yang, C., D. A. Maiguel, and F. Carrier. 2002. Identification of nucleolin and nucleophosmin as genotoxic stress-responsive RNA-binding proteins. Nucleic Acids Res. 302251-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yung, B. Y., H. Busch, and P. K. Chan. 1986. Effects of luzopeptins on protein B23 translocation and ribosomal RNA synthesis in HeLa cells. Cancer Res. 46922-925. [PubMed] [Google Scholar]
- 75.Zhang, R., L. Averboukh, W. Zhu, H. Zhang, H. Jo, P. J. Dempsey, R. J. Coffey, A. B. Pardee, and P. Liang. 1998. Identification of rCop-1, a new member of the CCN protein family, as a negative regulator for cell transformation. Mol. Cell. Biol. 186131-6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zirwes, R. F., A. P. Kouzmenko, J. M. Peters, W. W. Franke, and M. S. Schmidt-Zachmann. 1997. Topogenesis of a nucleolar protein: determination of molecular segments directing nucleolar association. Mol. Biol. Cell 8231-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zirwes, R. F., M. S. Schmidt-Zachmann, and W. W. Franke. 1997. Identification of a small, very acidic constitutive nucleolar protein (NO29) as a member of the nucleoplasmin family. Proc. Natl. Acad. Sci. USA 9411387-11392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zou, H., R. Wieser, J. Massague, and L. Niswander. 1997. Distinct roles of type I bone morphogenetic protein receptors in the formation and differentiation of cartilage. Genes Dev. 112191-2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.