

Abstract
Addition of a 5′ cap to RNA polymerase II transcripts, the first step of pre-mRNA processing in eukaryotes from yeasts to mammals, is catalyzed by the sequential action of RNA triphosphatase, guanylyltransferase, and (guanine-N-7)methyltransferase. The effects of knockdown of these capping enzymes in mammalian cells were investigated using T7 RNA polymerase-synthesized small interfering RNA and also a lentivirus-based inducible, short hairpin RNA system. Decreasing either guanylyltransferase or methyltransferase resulted in caspase-3 activation and elevated terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining characteristic of apoptosis. Induction of apoptosis was independent of p53 tumor suppressor but dependent on BAK or BAX. In addition, levels of the BH3 family member Bim increased, while Mcl-1 and Bik levels remained unchanged during apoptosis. In contrast to capping enzyme knockdown, apoptosis induced by cycloheximide inhibition of protein synthesis required BAK but not BAX. Both Bim and Mcl-1 levels decreased in cycloheximide-induced apoptosis while Bik levels were unchanged, suggesting that apoptosis in siRNA-treated cells is not a direct consequence of loss of mRNA translation. siRNA-treated BAK−/− BAX−/− double-knockout mouse embryonic fibroblasts failed to activate capase-3 or increase TUNEL staining but instead exhibited autophagy, as demonstrated by proteolytic processing of microtubule-associated protein 1 light chain 3 (LC3) and translocation of transfected green fluorescent protein-LC3 from the nucleus to punctate cytoplasmic structures.
A defining feature of eukaryotic gene expression is the addition of an m7GpppN cap to nascent pre-mRNAs shortly after initiation of synthesis (31). This 5′-terminal modification is catalyzed by the sequential action of RNA 5′ triphosphatase (RT), guanylyltransferase (GT), and (guanine-N-7)methyltransferase (MT) (8, 32). In mammalian and yeast cells, these enzymes bind to the phosphorylated C-terminal domain of the largest subunit of RNA polymerase II (Pol II), resulting in selective capping of Pol II transcripts. The cap enhances splicing, is retained on mature mRNAs, and is recognized by nuclear cap-binding complex for transport to the cytoplasm (9). Initiation of protein synthesis by ribosome binding near the capped end of mRNA is promoted by the cytoplasmic cap-binding initiation factor, eIF4F (7, 36). In addition to its roles in transcription, processing, transport, and translation, the cap stabilizes mRNA and is important for maintaining regulated cell growth and division.
Consistent with the multiple effects of the cap, mRNA capping is evolutionarily and functionally conserved. Yeast mutants defective for capping fail to grow but can be complemented by the corresponding mammalian enzymes, despite differences in gene organization, nucleotide sequence, and the catalytic mechanisms of RT (28, 39, 46). In Saccharomyces cerevisiae, the three steps in capping are catalyzed by separate proteins, while in metazoans, the RT and GT activities reside in N- and C-terminal domains, respectively, of a bifunctional capping enzyme (CE). RNA interference (RNAi) knockdown of CE in Caenorhabditis elegans was demonstrated previously to be embryo lethal (37), and deletion of the RT Cet1 or GT Ceg1 gene in S. cerevisiae also resulted in loss of viability (39). We analyzed human and mouse cells for effects of small interfering RNA (siRNA)-mediated knockdown of CE and MT in both transiently and stably transfected cell lines. Apoptosis, as detected by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay and caspase-3 activation, was induced in most of the lines studied, including p53-null H1299 human lung cancer cells and mouse embryonic fibroblasts (MEFs) missing either BAK or BAX. However, autophagy rather than apoptosis was induced in siRNA-treated MEFs that were null for both BAK and BAX.
MATERIALS AND METHODS
Constructs.
pEGFP-N3/hCE and pEGFP-N3/hMT were constructed by inserting human CE (hCE) and hMT coding sequences between XhoI and KpnI sites in pEGFP-N3 (Clontech, Mountain View, CA) to allow expression of green fluorescent protein (GFP) fused at the C terminus of hCE and hMT.
Cre was constructed into pEGFP-N3 between BglII and NotI sites, replacing the eGFP expression cassette. Catalytically inactive Cre mutants pEGFP-N3/Cre (Y324F) and pEGFP-N3/Cre (R173K) were made with the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA).
pEGFP-C1/LC3 was made by N. Mizushima and kindly provided by S. Jin.
In vitro screening of CE and MT siRNAs.
Sequences in the coding regions of hCE, hMT, and mouse CE (mCE) were synthesized in vitro by fusing a T7 promoter sequence at their 5′ ends. The sequences selected consisted of 21 nucleotides, started with G or GG to facilitate T7 RNA polymerase transcription, and showed no complementarity to any other genes in a BLAST search. Each siRNA is designated as CE or MT followed by the number corresponding to the nucleotide position in the coding region of CE or MT mRNA.
T7-synthesized RNAs were produced by using a T7-MEGAshortscript high-yield transcription kit (Ambion, Austin, TX). Antisense and sense transcripts were mixed, heated in buffer 2 (New England Biolabs, Ipswich, MA) at 95°C for 5 min, and slowly cooled to room temperature to allow RNA duplex formation.
TUNEL assays.
Cells were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA), and TUNEL assays were performed by using an in situ cell death detection kit, with fluorescein or tetramethylrhodamine red (Roche, Indianapolis, IN), all according to the manufacturers' protocols.
Western blots.
Rabbit polyclonal antibody made against gel-purified full-length recombinant hCE was purified by hCE-Sepharose affinity chromatography. Rabbit polyclonal antibody against glutathione S-transferase-MT was purified on protein A beads (GE Healthcare, Piscataway, NJ). Antibodies to cleaved caspase-3 (Cell Signaling, Danvers, MA), Bim (ΨProSci, Poway, CA), Mcl-1 (Rockland Immunochemicals, Gilbertsville, PA), Bik (Abcam, Cambridge, MA), and LC3 (MBL International, Woburn, MA) were used in Western blots. Cells were lysed with radioimmunoprecipitation assay buffer plus complete protease inhibitor cocktail (Roche), and protein concentrations were measured by the Bradford method. For Western blotting, 100 μg of total protein, unless otherwise specified in the figure legends, was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, blotted with the antibodies, and detected with SuperSignal West Pico substrate (Pierce Biotechnology, Rockford, IL).
pSico constructs.
Sequences for knocking down CE and MT expression were generated by pSicoOligomaker 1.5 (http://web.mit.edu/jacks-lab/protocols/pSico.html), a computer program that predicts the potential sequences for short hairpin RNAs (shRNAs) based on a published algorithm (27). Three CE sequences (CE612, CE699, and CE1521) and five MT sequences (MT73, MT1041, MT1086, MT1144, and MT1296) were selected and cloned into the pSico vector (40). All constructs were confirmed by sequencing.
Production of lentivirus.
293FT cells (6 × 106/10-cm culture dish; Invitrogen) were transfected with 3 μg of pSico construct together with three helper vectors (3.6 μg of pLP1, 2.5 μg of pLP2, and 2.9 μg of pLP/VSVG; Invitrogen) to produce lentivirus. Virus-containing culture medium was harvested 48 to 72 h posttransfection when more than 90% of the cells had detached from the dish. Virus particles were pelleted by centrifuging at 25,000 rpm for 90 min in a Beckman SW28 Ti rotor. Virus pellets were resuspended in 200 μl of Dulbecco's modified Eagle medium plus 10% fetal bovine serum and stored at −70°C.
Isolation of stable RNAi cell lines.
HeLa cells were infected overnight with 10 μl of the concentrated lentivirus suspension in the presence of 6 μg/ml of Polybrene. Cells were expanded by three serial passages after infection, and GFP-positive cells were isolated on a Coulter Epics Altra cell sorter. Collected cells were plated at a density of 5 × 103/10-cm dish to allow single colonies to form. Clones were examined by fluorescence microscopy, and those with bright, uniform GFP expression were selected, expanded, and used in RNAi experiments.
RESULTS
Knockdown of CE or MT by siRNA.
Twenty siRNAs synthesized by T7 RNA polymerase were individually transfected into HeLa or 293T cells together with pEGFP-N3/hCE or pEGFP-N3/mCE. Cells were monitored by measuring the decrease in levels of CE-GFP or MT-GFP. siRNA hCE332 strongly inhibited hCE-GFP expression (Fig. 1c versus a), and hCE614 and hCE1427 had a similar inhibitory effect (data not shown). Specificity was demonstrated using a 2-nucleotide (nt)-mismatch siRNA, hCE332m2 (Fig. 1b), and an ineffective siRNA, hCE120 (Fig. 1d); they showed little or almost no inhibitory effect on CE-GFP expression compared to the plasmid transfection control (Fig. 1a). The results were confirmed using mCE1558, which, like mCE332 and mCE754 (data not shown), strongly inhibited mCE-GFP expression (Fig. 1f). A negative-control 3-nt-mismatch siRNA, mCE1558m3, confirmed the specificity of the RNAi effect (Fig. 1e). In a similar screen, siRNAs hMT317 and hMT1085 effectively knocked down hMT (Fig. 1i and j), compared to cells transfected with the mismatch siRNA hMT317m2 (Fig. 1h) or pEGFP-N3/hMT only (Fig. 1g).
FIG. 1.
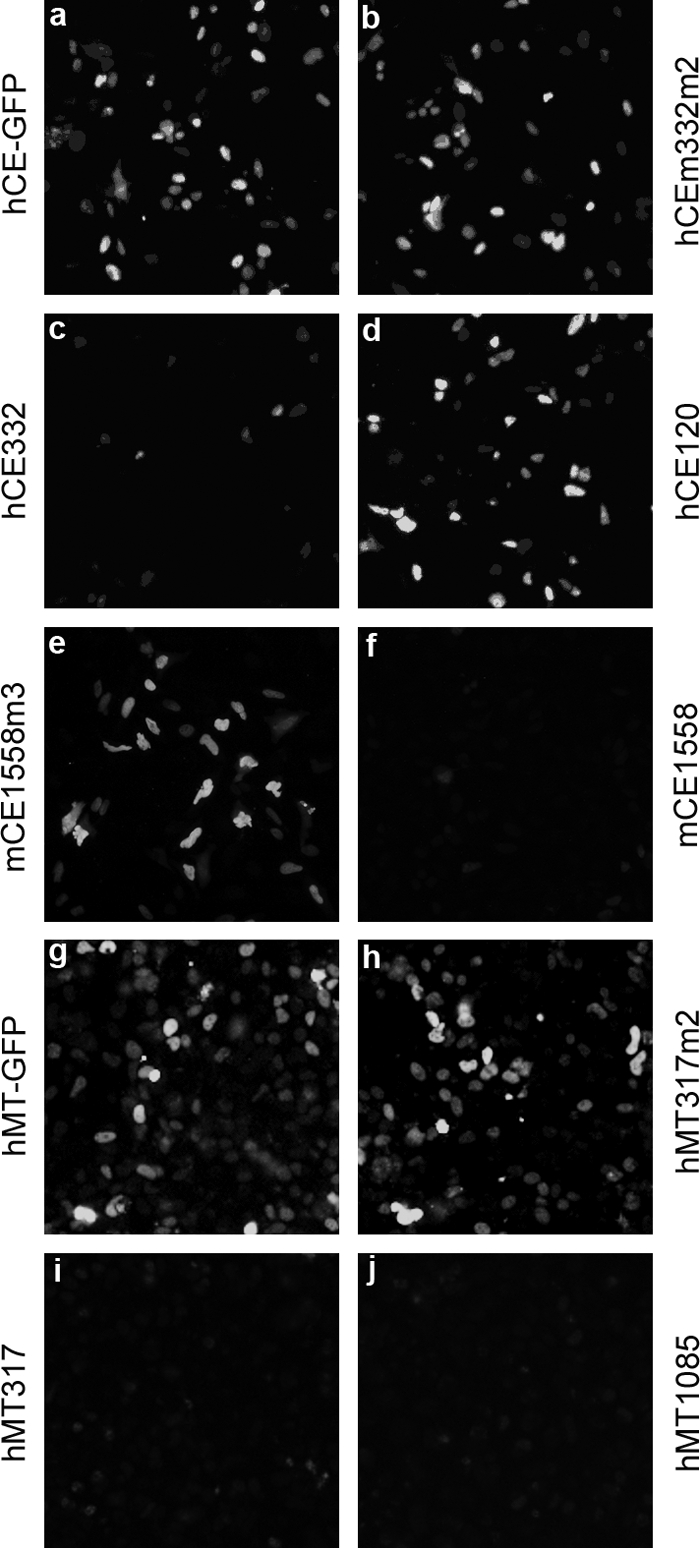
Screening for effective CE and MT siRNAs. HeLa cells were transfected with pEGFP-N3/hCE (a) along with siRNA hCE332m2 (b), hCE332 (c), and hCE120 (d); with pEGFP-N3/mCE plus mCE1558m3 (e) and mCE1558 (f); or with pEGFP-N3/hMT (g) plus hMT317m2 (h), hMT317 (i), and hMT1085 (j). In all cases, the amount of siRNAs and expression vectors was 0.32 μg. Cells were fixed 24 h posttransfection and examined by fluorescence microscopy.
The RNAi effect was confirmed by quantitative PCR and Western blotting. CE and MT mRNA levels decreased 68% and 84% 2 days after siRNA transfection compared to 18S rRNA. At the protein level, CE in hCE332-transfected cells analyzed 1 to 4 days posttransfection decreased ca. 60% within 1 day (Fig. 2A, lanes 3 to 6) compared to untransfected cells (lane 1) and cells transfected with mismatch siRNA (lane 2). Since CE is highly conserved among mammalian species (e.g., hCE and mCE are 95% identical in amino acid sequence [46]), anti-hCE antibodies are difficult to elicit in rabbits, and the antibodies obtained are not very effective at detecting low levels of endogenous CE.
FIG. 2.
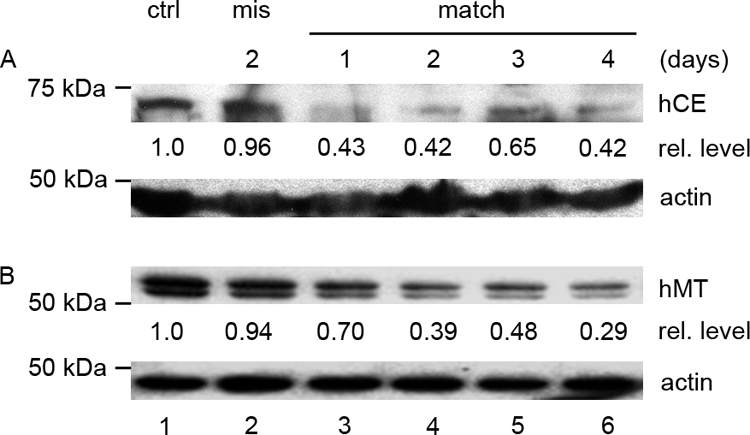
Decrease of CE and MT by siRNA treatment. HeLa cells (lane 1) in 10-cm plates were transfected with 8 μg of hCE322 siRNA (A) or hMT317 siRNA (B) (lanes 3 to 6) or mismatch siRNA CE332m2 (A) or hMT317m2 (B) (lane 2). Cells transfected with mismatch siRNA were collected at day 2, and the other cultures were harvested on the indicated days after transfection. Polyclonal antibodies against hCE or hMT were used to detect CE or MT measured with a charge-coupled device densitometer, and the levels of CE or MT relative to those in the untreated cells (ctrl, lane 1) are noted below the blots. In panel B, lysate containing 5 μg of total protein was loaded in each lane. The MT doublet in panel B results from a protease-sensitive site at lysine 445 and was eliminated by alanine substitution of K445 without affecting MT activity.
The level of endogenous MT was similarly decreased by transfection of MT siRNA (Fig. 2B, lanes 3 to 6) compared to untransfected cells (lane 1) or cells treated with mismatch siRNA (lane 2), demonstrating effective MT knockdown.
Levels of exogenously expressed hCE-GFP and MT-GFP were also found to decrease, as assayed by cotransfection of the expression vectors and corresponding siRNAs followed by blotting with antibodies against hCE, MT, or GFP (data not shown). The results are in agreement with the results shown in Fig. 1. Although not shown in all figures, an equal amount of total protein in each lysate was loaded, and equal levels of actin were detected by blotting (Fig. 2; also Fig. 7 and data not shown).
FIG. 7.

Activation of apoptosis is likely mediated through Bim but not by Mcl-1, Bik, or general protein synthesis shutdown. WT MEFs were treated with mCE1558 (lanes 6 to 8) and its mismatch (lanes 3 to 5) or with 10 μg/ml CHX (lanes 9 to 11) for the times shown and blotted for activated caspase-3 (a), Bim (b), Mcl-1 (c), and Bik (d). Actin (e) served as a loading control.
Induction of apoptosis by siRNA treatment of mammalian cells.
To test if knockdown of CE or MT affects viability, HeLa cells were transfected with hCE332, hCE614, hMT317, or hMT1085. Cell viability was markedly decreased based on MTT (methylthiazolyldiphenyl-tetrazolium bromide) staining (22) (data not shown), and there was a significant increase in TUNEL-positive cells (Fig. 3c, d, g, and h). In contrast, cultures that were treated with the mismatch siRNA hCE332m2 or hMT317m2 (Fig. 3b and f) or mock transfected (Fig. 3e) contained few apoptotic cells, like untreated cultures (Fig. 3a). These results indicate that the induction of apoptosis by siRNA treatment is highly specific.
FIG. 3.

Downregulation of CE or MT induces apoptosis. HeLa cells were transfected with hCE332m2 (b), hCE332 (c), hCE614 (d), hMT317m2 (f), hMT317 (g), or hMT1085 (h) and checked by TUNEL assay (fluorescein) 48 h later. Growing cells (a) and mock-transfected cells (e) were stained as controls. For Western blotting (i), HeLa cells were untreated (ctrl, lane 2), exposed to 1 μM STS for 2 days (lane 1), or treated with hCE614 (lanes 7 and 8), hMT1085 (lanes 9 and 10), and the corresponding mismatch siRNAs (lanes 3 to 6) and collected after 2 and 3 days. Caspase-3-activated fragment P17 was detected by cleaved caspase-3 (Asp175) antibody.
Activated caspase-3, one of the key executioners of apoptosis, is responsible for the cleavage of many important proteins in the death pathway (2, 6). Caspase-3 is activated by cleavage at Asp175, giving rise to the p19/p12 complex and finally the active p17/p12 products through slow autocatalysis or digestion by aspartate-specific cysteine protease-X (5). Caspase-3 activation was demonstrated by Western blot analysis at different times posttransfection. The active p17 form of caspase-3 was induced by hCE siRNA (Fig. 3i, lanes 7 and 8) and hMT siRNA (lanes 9 and 10) but not by the corresponding mismatch RNAs (lanes 3 and 4 and lanes 5 and 6). siRNA-induced cleavage of caspase-3 was similar to that obtained with staurosporine (STS), a potent protein kinase C inhibitor (lane 1).
Activation of apoptosis by CE knockdown is p53 independent.
The tumor suppressor p53 mediates cellular responses to various kinds of stress, dependent on or independent of transcription activation (19, 21, 30). To assess if p53 is also involved in apoptosis induced by the loss of mRNA capping activity, the p53-null human non-small-cell lung cancer cell line H1299 (ATCC CRL-5803) was transfected with CE or MT siRNA. Induction of apoptosis detected by TUNEL assay was similar to that observed in HeLa cells (data not shown). Analysis of caspase-3 by Western blotting showed that, as in STS-treated cells (Fig. 4a and b, lane 1), downregulation of either CE (Fig. 4a) or MT (Fig. 4b) resulted in formation of the caspase-3 activation products p19 and p17, characteristic of apoptosis (lanes 6 to 8), while mismatch RNAs had no effect (lanes 3 to 5). The results indicate that the induction of apoptosis by CE or MT depletion is p53 independent.
FIG. 4.

Activation of caspase-3 is independent of p53. H1299 cells were untreated (ctrl, lane 2) or treated with 1 μM STS for 2 days (lane 1) or with hCE614 (a), hMT1085 (b), and the corresponding mismatch siRNAs and collected on the indicated days. Activated caspase-3 P19 and P17 fragments were detected by cleaved caspase-3 (Asp175) antibody.
Apoptosis induction is dependent on both BAK and BAX.
Apoptosis can be induced through the intrinsic and extrinsic major pathways. In the extrinsic pathway, binding of an extracellular ligand to a cell surface receptor (e.g., FasL/Fas) triggers recruitment of cytosolic adaptor proteins (e.g., FADD) and activates downstream caspases (13). In the intrinsic pathway, release of cytochrome c from mitochondria is the key triggering event. Efflux of cytochrome c is tightly regulated by pro- and antiapoptotic factors, notably, Bcl-2 family proteins. Among the Bcl-2 family members, activation of BAX and BAK promotes cytochrome c release, while Bcl-2 and Bcl-XL inhibit this process (29). Release of cytochrome c from mitochondria into the cytosol initiates a cascade of caspase activations leading to rapid and irreversible cell death.
To test for activation of the intrinsic pathway, we compared the mCE RNAi effect in wild-type (WT) MEFs with that in BAK−/−, BAX−/−, or BAK−/− BAX−/− double-knockout (DKO) MEFs. Cells were transfected with mCE siRNA and checked for the induction of apoptosis by TUNEL staining 24 h later. Simian virus 40 (SV40)-immortalized and spontaneously immortalized WT MEFs both showed a striking increase in TUNEL staining when CE was knocked down by mCE1558 transfection (Fig. 5b and e), while cells treated with the mismatch RNA mCE1558m3 remained at background levels (Fig. 5a and d). The SV40-immortalized cultures contained more TUNEL-stained cells than spontaneously immortalized MEFs, a result also seen for caspase-3 activation (Fig. 5c and f). In BAK−/− and BAX−/− MEFs, immortalized spontaneously and by SV40 transformation, respectively, mCE1558 treatment also resulted in significant TUNEL staining, with comparatively more in BAX−/− cells (Fig. 5k versus h). The higher level of TUNEL staining in BAX−/− cells versus greater caspase-3 activation in BAK−/− cells suggests that SV40 immortalization did not have an important effect on apoptosis. DKO MEFs had the same background level of TUNEL staining as with mismatch siRNA treatment (Fig. 5n and m), indicating that induction of apoptosis by downregulation of mCE requires either BAK or, more effectively, BAX (Fig. 5i versus l).
FIG. 5.

Induction of apoptosis is dependent on BAK or BAX. WT MEFs, immortalized by SV40 or spontaneously, and BAK−/−, BAX−/−, or DKO MEFs were transfected with the siRNA mCE1558 or the mismatch siRNA mCE1558m3 and checked for induction of apoptosis by TUNEL assay (left and middle) and Western blotting (right). For TUNEL assay, cells were stained by tetramethylrhodamine red 24 h posttransfection, and cells treated with 0.1 μM STS for 8 h served as positive controls. For Western blotting, cells treated with 0.1 μM STS (right, lanes 1 and 2), mCE1558 siRNA (lanes 6 to 8), or mismatch (lanes 3 to 5) were collected on different days and analyzed for caspase-3 activation.
MEFs transfected with mCE1558 siRNA were also analyzed for caspase-3 activation by Western blotting after 24, 48, and 72 h. Knockdown of mCE by siRNA transfection of SV40-immortalized WT MEFs resulted in increasing levels of activated caspase-3 (Fig. 5c, lanes 6 to 8), compared to a higher level after 1 day of STS treatment (Fig. 5c, lanes 1 and 2). Mismatch siRNA-treated cells showed no caspase-3 cleavage (Fig. 5c, lanes 3 to 5), indicating that the siRNA-induced apoptosis was authentic and specific. To rule out a possible impact of SV40, which was used to establish the cell line, we also tested a spontaneously immortalized WT MEF cell line. The results showed increasing accumulation of activated caspase-3 during the first two days and a decrease on the third day posttransfection, presumably due to the loss of dead cells (Fig. 5f, lanes 6 to 8). Mismatch RNA treated cells again showed no detectable level of activated caspase-3 (Fig. 5f, lanes 3 to 5). The extent of apoptosis in the spontaneously immortalized MEFs was generally less than in SV40-immortalized MEFs, like STS-induced apoptosis (Fig. 5f versus c, lanes 1 and 2).
When CE was knocked down in either BAK−/− or BAX−/− MEFs, a low level of activated caspase-3 was detected (Fig. 5i and l, lanes 6 to 8), while mismatch siRNA treatment showed no activation (Fig. 5i and l, lanes 3 to 5). Apoptosis in MEFs bearing a single deletion, notably BAX null cells, was weaker than that in WT cells, consistent with the response to STS (Fig. 5i and l, lanes 1 and 2). Thus, deletion of either BAK or BAX severely decreased but did not completely eliminate the capacity of the mutant MEFs to undergo apoptosis in response to mCE siRNA. However, in DKO cells treated with mCE siRNA or STS, apoptosis measured by caspase-3 activation was not detectable even in somewhat overexposed blots (Fig. 5o, lanes 1 and 2 and lanes 6 to 8), as also observed with mismatch siRNA transfection (Fig. 5o, lanes 3 to 5).
Activation of apoptosis is likely mediated through Bim and not Mcl-1, Bik, or general protein synthesis shutdown.
To determine if apoptosis induced by the loss of mRNA capping is the direct consequence of global protein synthesis shutdown, MEFs were treated with 10 μg/ml of cycloheximide (CHX) to block protein synthesis and then checked for caspase-3 activation. In WT and BAX−/− MEFs, CHX treatment induced caspase-3 activation within 4 h (Fig. 6a, b, and d), while BAK−/− and DKO MEFs showed no caspase-3 cleavage even after 24 h (Fig. 6c and e). These results indicate that apoptosis induced by a block in protein synthesis is specifically BAK dependent (34), in contrast to death induced by depletion of CE, which is dependent on either BAX or BAK. Thus, apoptosis induced by siRNA knockdown of CE involves a signaling pathway(s) in addition to those activated by loss of protein synthesis.
FIG. 6.

Apoptosis induced by CHX is BAK dependent. WT MEFs (a and b) or MEFs with deletion of BAK (c), BAX (d), or both (DKO) (e) were treated with 10 μg/ml CHX for the times shown and blotted for activated caspase-3.
MEF WT cells were used to assess the involvement of BH3 family members in the induction of apoptosis. Among these family members, NOXA and PUMA have been shown to be dependent on p53 transactivation (23, 26), in contrast to the p53-independent apoptosis induction in this study, making NOXA and PUMA unlikely as participants in CE knockdown-induced apoptosis. Bid is also unlikely to be involved, since it is thought to be activated through death receptors that reside on the cell surface (16).
Other BH3 family members, i.e., Mcl-1, Bik, and Bim, were assayed in parallel with caspase-3 activation in MEF WT cells transfected with mCE1558 siRNA or its mismatch or treated with CHX. Transfection resulted in the activation of caspase-3 (Fig. 7a, lanes 6 to 8), as also seen in response to STS treatment (Fig. 7a, lane 2) and CHX treatment (Fig. 7a, lanes 9 to 11) but not by mismatch siRNA transfection (Fig. 7a, lanes 3 to 5). Bim was found to increase with both match and mismatch siRNA treatment, but the increase was considerately greater with match siRNA (Fig. 7b, lanes 6 to 8 versus 3 to 5), while the levels of Mcl-1 and Bik did not change during the 72 h study (Fig. 7c and d, lanes 6 to 8). However, Bim and Mcl-1 were found to decrease sharply, Bim within 4 h, when translation was blocked by CHX (Fig. 7b and c, lanes 9 to 11), while Bik remained relatively unaffected (Fig. 7d, lanes 9 to 11). These results confirmed that apoptosis induced by siRNA knockdown of CE adopts a different signaling pathway(s) than those activated by loss of protein synthesis.
Induction of autophagy in DKO cells.
DKO MEFs failed to undergo apoptosis when CE was knocked down by RNAi, and we hypothesized that these cells may respond by activating an alternative death pathway(s), as cultures treated with mCE1558 siRNA showed significantly lower growth rates, as assayed by MTT staining 4 days after transfection, compared to normal, mock-transfected, or mismatch siRNA-transfected cells (see Fig. S1 in the supplemental material). Autophagy is an evolutionarily conserved, stress-induced alternative pathway that occurs by degradation of cytosolic proteins and organelles within the autophagosome, a double membrane vesicle formed during autophagy (14). Cell survival for a limited time is facilitated by autophagy during nutrient or growth factor depletion. A commonly used marker for detecting autophagy is microtubule-associated protein 1 light chain 3 (LC3), a homolog of the yeast Apg8p protein associated with autophagosome membranes and essential for autophagy in yeast (14). LC3 is posttranslationally modified by removal of its C terminus to produce LC3-I, with phosphatidylethanolamine lipidation giving rise to LC3-II (14, 15). LC3-I (∼18 kDa) is cytosolic, while LC3-II (∼16 kDa) is membrane bound and enriched in autophagosomes (1, 14, 20).
Growing cultures of DKO MEFs at low density contained no detectable levels of LC3-II (Fig. 8a, lane 1), while in cells placed under conditions of ischemia (25), which served as a positive control, there was almost complete conversion of LC3-I to LC3-II after 1 day (lanes 2 to 3). Cells that were treated with CE siRNA also converted most of the LC3-I to LC3-II by 24 h posttransfection (Fig. 8a, lanes 6 to 7). By contrast, mismatch siRNA-treated cells had little LC3 conversion on day 1 (Fig. 8a, lane 4) but an increase on day 2 (lane 5), possibly due to overgrowth of the culture, as reported previously for the MEF line NIH 3T3 (44). DKO cells treated with 10 μg/ml of CHX for 3 days had a decrease in LC3-I and LC3-II but no conversion, suggesting that induction of autophagy by CE siRNA is not due to protein synthesis inhibition (see Fig. S2 in the supplemental material). Interestingly, DKO cells treated with CHX for 4 days stopped growing (see Fig. S1 in the supplemental material) and died gradually during a 2-week treatment; however, the cells that remained rapidly resumed growth once CHX was removed (data not shown).
FIG. 8.

Conversion of LC3 and translocation of GFP-LC3 in DKO MEFs. Autophagy was induced in growing DKO MEFs (a, lane 1) by ischemia treatment (lanes 2 and 3) or by transfection of siRNA mCE1558 (lanes 6 and 7) or the corresponding mismatch RNA (lanes 4 and 5) and assayed for conversion of LC3-I to LC3-II, both detected by anti-LC3 monoclonal antibody. DKO cells transfected with GFP-LC3 expression vector (b) were subjected to ischemia treatment (c) or transfected with CE siRNA (e) or mismatch siRNA (d) and analyzed by fluorescence microscopy 24 h later.
GFP-LC3 in transfected cells translocates from the nucleus to the cytoplasm during autophagy and accumulates in punctate structures (17, 18), a response we used to detect autophagy in siRNA-treated cells. DKO MEFs were first transfected with pEGFP-C1/LC3, allowed to recover overnight, and then transfected with CE siRNAs or subjected to ischemia treatment. GFP-LC3 in growing MEFs was uniformly distributed (Fig. 8b). Cells expressing GFP-LC3 and placed under conditions of ischemia for 24 h showed typical nuclear exclusion and cytoplasmic aggregates (Fig. 8c). The same pattern of GFP-LC3 nuclear exclusion and aggregation in the cytosol occurred in CE siRNA-treated cells (Fig. 8e) but not in cells treated with mismatch siRNA (Fig. 8d). These results confirmed that CE and MT are of critical importance in maintaining cell growth. Their depletion resulted in loss of viability, either by apoptosis in WT, BAK−/−, and BAX−/− MEFs or by autophagy in DKO MEFs.
Production of lentiviruses harboring CE or MT shRNAs.
CE and MT siRNA treatment resulted in cell death, but the effect was transient and dependent on transfection efficiency, which can vary greatly in different cell lines and with transfection reagents. An inducible lentivirus-based RNAi system was recently described (40), and we used this method to create stable cell lines expressing inducible CE or MT shRNAs.
HeLa cell cultures were infected with the eight lentivirus strains containing eight shRNA sequences plus four mismatch controls and selected by cell sorting for positive transduction. shRNA expression was induced by transfecting cells with vectors expressing Cre. More than 85% of the cells transfected with pEGFP-N3/Cre in each of the eight cell lines stopped growing and appeared to die within 2 to 3 days posttransfection, while cells harboring mismatch shRNA sequences grew normally after Cre treatment. The shRNA effect in all eight lentivirus-containing cell lines was greater than that with the siRNA transfections, confirming that both CE and MT are required for mammalian cell viability (data not shown). To rule out the unlikely possibility that cell death was caused by transfection or by overexpression of Cre, cells with match or mismatch shRNA were mock transfected or transfected with vector expressing mutant inactive Cre. They showed no indication of induced cell death (data not shown).
Induction of apoptosis in lentivirus-based inducible RNAi system.
To investigate if cells also die by apoptosis in this lentivirus RNAi system, TUNEL assays were performed. Two cell lines, containing inducible shCE612 and shMT1296, were either transfected with pEGFP-N3/Cre or mock transfected and were examined 36 h later. Cells expressing shCE612 or shMT1296 showed extensive TUNEL staining (Fig. 9b and d), while in mock-transfected cultures there were very few stained cells (Fig. 9a and c).
FIG. 9.

Induction of apoptosis in the inducible lentivirus RNAi system. HeLa cells containing stably integrated lentivirus RNAi against CE or MT were mock transfected (a and c) or transfected with Cre (b and d) and TUNEL stained for apoptosis. For Western blotting, HeLa cells harboring lentivirus shCE612 (e, lanes 2 and 3 and 7 to 9), shCE612m3 (e, lanes 4 to 6), shCE1521 (e, lanes 10 to 12), shMT1086 (f, lanes 2 and 3 and 7 to 9), shMT1086m3 (f, lanes 4 to 6), and shMT1296 (f, lanes 11 to 13) were transfected with Cre (e, lanes 4 to 12; f, lanes 4 to 9 and 11 to 13) or Cre-inactive mutants R173K (e, lanes 2 and 3) or Y324F (f, lanes 2 and 3). On the indicated day, activated caspase-3 was detected by Western blotting. STS-induced caspase-3 cleavage (lane 1) served as a positive control.
Activation of caspase-3 was also assayed in the lentivirus-based RNAi-inducible cells. RNAi induction by transfection of Cre vector in cell lines expressing shCE612 or shCE1521 resulted in caspase-3 cleavage. Similar to results obtained with STS treatment (Fig. 9e, lane 1), there was strong activation of caspase-3 (Fig. 9e, lanes 7 to 9 and 10 to 12). By contrast, there was no activation in cells expressing the 3-nt-mismatch siRNA shCE612m3 or shCE612 with a Cre-inactive mutant, R173K (42) (Fig. 9e, lanes 4 to 6 and lanes 2 and 3, respectively). These results indicate specificity of the RNAi induction of apoptosis and rule out the possibility that the apoptosis observed in two shRNA-expressing cell lines was triggered by Cre overexpression.
For MT RNAi, cells expressing shMT1086 or shMT1296 were transfected with Cre-expressing vector and assayed by Western blotting for caspase-3 cleavage. From day 1 to day 3 after induction, cells expressing shMT1086 or shMT1296 contained cleaved caspase-3 (Fig. 9f, lanes 7 to 9 and 11 to 13), as also seen with STS treatment (lane 1). Cells harboring the 3-nt-mismatch siRNA shMT1086m3 or the match siRNA shMT1086 and treated with another active-site Cre mutant, Y324F, showed no detectable caspase-3 activation (Fig. 9f, lanes 4 to 6 and lanes 2 and 3, respectively). Taken together, the results demonstrate that both CE and MT are essential for mammalian cell viability. Depletion of either by two different siRNA methods resulted in caspase-3 activation and apoptosis.
DISCUSSION
We used transient transfection and stable, inducible-RNAi strategies to test if mRNA capping is essential for homeostasis and viability in mammalian cells. We found that siRNA knockdown of CE or MT in mouse and human cells resulted in decreased growth followed by induction of apoptosis, as shown by caspase-3 activation and TUNEL staining. The results demonstrated that the requirement for mRNA capping is evolutionarily conserved from lower eukaryotes, including S. cerevisiae (10, 33, 38) and C. elegans (37), to mammals.
The tumor suppressor p53 plays a critical role in response to cellular stress and mediates either cell cycle arrest or apoptosis, depending on cell type and the nature of the stress (12). p53 can activate transcription of many genes, including those involved in regulating apoptosis (41), and can induce apoptosis in response to the loss of RNA or protein synthesis (3, 19). However, in our experiments, CE or MT knockdown in p53−/− human H1299 cells resulted in cleavage of caspase-3, indicative of apoptosis. This result indicates that cell death induced by depletion of RNA CE and MT is independent of p53.
Bcl-2 family members play important roles in regulating apoptosis (4, 45). We tested for dependence on BAK and BAX, the two key proapoptotic factors in this family. In response to a cell death signal, BAK and BAX change conformation, oligomerize, and form pores in the outer mitochondrial membrane, leading to the efflux of cytochrome c and activation of downstream caspases, including caspase-3 (24, 35). Decreasing levels of mRNA CE and MT induced apoptosis in BAK−/− and in BAX−/− MEFs but not in MEFs missing both proapoptotic proteins.
A direct consequence of disruption of capping would be a decrease in mRNA levels, as we demonstrated by quantitative PCR, leading to loss of protein synthesis. However, shutdown of translation by CHX treatment of BAX−/−, BAK−/−, or DKO MEFs led to caspase-3 activation and cell death only in BAX−/− cells, i.e., only in cells containing BAK. This finding resembles the BAK-dependent apoptosis induced following inhibition of protein synthesis by the bacterial toxin MazF (34). However, apoptosis induced by siRNA depletion of CE or MT apparently was not directly due to a block in protein synthesis, since the induction of cell death was mediated through either BAK or BAX. Thus, disruption of mRNA capping can activate BAK and/or BAX and induce apoptosis before global protein synthesis shutdown, possibly as a consequence of mRNA surveillance.
Our data suggest that the elevation of Bim plays a role in activation of apoptosis in CE-depleted cells. Inhibition of mRNA capping would be expected to affect most if not all Pol II transcripts, including nuclear snRNAs (11, 43), rather than only a small subset of mRNAs, through decreases in levels of transcripts and splicing. Changes in mRNA and protein levels upon disruption of mRNA capping depend not only on the half-lives of pre-existing mRNAs but also on protein turnover, and it is likely that loss of essential mRNAs and proteins with the least stability would lead to activation of cell death signals. We assessed the changes in several Bcl-2 family members during apoptosis and found that Bim significantly increased during apoptosis induced by the loss of CE. The opposite effect occurred in response to apoptosis induced by CHX inhibition of protein synthesis, and the level of Bim was drastically decreased. Mcl-1 levels were not affected by the knockdown of CE but decreased in parallel with global synthesis shutdown by CHX treatment. Another Bcl-2 family member, Bik, remained little changed in both treatments. Taken together, the data demonstrate a distinct difference in the mechanisms of apoptosis induced by downregulation of CE and by CHX-mediated loss of protein synthesis. A comparison of global changes in mRNA and/or protein levels may shed light on which short-half-life mRNA(s) and high-turnover protein(s) contribute most to apoptosis induction.
An intriguing issue is the fate of cells unable to undergo apoptosis if CE or MT is depleted. Our results show that cells die by autophagy in response to knockdown of CE if the apoptotic pathway is unavailable, as in DKO MEFs, while protein synthesis inhibition by CHX in the same cells failed to induce autophagy. This result emphasizes the functional and evolutionary importance of mRNA capping in maintaining cellular homeostasis.
Supplementary Material
Acknowledgments
We thank Céline Gélinas for MEF lines and helpful comments on the manuscript and Eileen White for assistance with ischemia treatment of MEFs.
Footnotes
Published ahead of print on 4 August 2008.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Asanuma, K., I. Tanida, I. Shirato, T. Ueno, H. Takahara, T. Nishitani, E. Kominami, and Y. Tomino. 2003. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J. 171165-1167. [DOI] [PubMed] [Google Scholar]
- 2.Boatright, K. M., and G. S. Salvesen. 2003. Mechanisms of caspase activation. Curr. Opin. Cell Biol. 15725-731. [DOI] [PubMed] [Google Scholar]
- 3.Caelles, C., A. Helmberg, and M. Karin. 1994. p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature 370220-223. [DOI] [PubMed] [Google Scholar]
- 4.Danial, N. N. 2007. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin. Cancer Res. 137254-7263. [DOI] [PubMed] [Google Scholar]
- 5.Fernandes-Alnemri, T., R. C. Armstrong, J. Krebs, S. M. Srinivasula, L. Wang, F. Bullrich, L. C. Fritz, J. A. Trapani, K. J. Tomaselli, G. Litwack, and E. S. Alnemri. 1996. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc. Natl. Acad. Sci. USA 937464-7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandes-Alnemri, T., G. Litwack, and E. S. Alnemri. 1994. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J. Biol. Chem. 26930761-30764. [PubMed] [Google Scholar]
- 7.Filipowicz, W., Y. Furuichi, J. M. Sierra, S. Muthukrishnan, A. J. Shatkin, and S. Ochoa. 1976. A protein binding the methylated 5′-terminal sequence, m7GpppN, of eukaryotic messenger RNA. Proc. Natl. Acad. Sci. USA 731559-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furuichi, Y., and A. J. Shatkin. 2000. Viral and cellular mRNA capping: past and prospects. Adv. Virus Res. 55135-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamm, J., and I. W. Mattaj. 1990. Monomethylated cap structures facilitate RNA export from the nucleus. Cell 63109-118. [DOI] [PubMed] [Google Scholar]
- 10.Ho, C. K., B. Schwer, and S. Shuman. 1998. Genetic, physical, and functional interactions between the triphosphatase and guanylyltransferase components of the yeast mRNA capping apparatus. Mol. Cell. Biol. 185189-5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobson, M. R., and T. Pederson. 1998. A 7-methylguanosine cap commits U3 and U8 small nuclear RNAs to the nucleolar localization pathway. Nucleic Acids Res. 26756-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin, S., and A. J. Levine. 2001. The p53 functional circuit. J. Cell Sci. 1144139-4140. [DOI] [PubMed] [Google Scholar]
- 13.Jin, Z., and W. S. El-Deiry. 2005. Overview of cell death signaling pathways. Cancer Biol. Ther. 4139-163. [DOI] [PubMed] [Google Scholar]
- 14.Kabeya, Y., N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi, and T. Yoshimori. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 195720-5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kabeya, Y., N. Mizushima, A. Yamamoto, S. Oshitani-Okamoto, Y. Ohsumi, and T. Yoshimori. 2004. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 1172805-2812. [DOI] [PubMed] [Google Scholar]
- 16.Luo, X., I. Budihardjo, H. Zou, C. Slaughter, and X. Wang. 1998. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94481-490. [DOI] [PubMed] [Google Scholar]
- 17.Martinet, W., G. R. De Meyer, L. Andries, A. G. Herman, and M. M. Kockx. 2006. Detection of autophagy in tissue by standard immunohistochemistry: possibilities and limitations. Autophagy 255-57. [DOI] [PubMed] [Google Scholar]
- 18.Martinet, W., G. R. De Meyer, L. Andries, A. G. Herman, and M. M. Kockx. 2006. In situ detection of starvation-induced autophagy. J. Histochem. Cytochem. 5485-96. [DOI] [PubMed] [Google Scholar]
- 19.Mihara, M., S. Erster, A. Zaika, O. Petrenko, T. Chittenden, P. Pancoska, and U. M. Moll. 2003. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 11577-590. [DOI] [PubMed] [Google Scholar]
- 20.Mizushima, N., A. Yamamoto, M. Hatano, Y. Kobayashi, Y. Kabeya, K. Suzuki, T. Tokuhisa, Y. Ohsumi, and T. Yoshimori. 2001. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152657-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moll, U. M., S. Wolff, D. Speidel, and W. Deppert. 2005. Transcription-independent pro-apoptotic functions of p53. Curr. Opin. Cell Biol. 17631-636. [DOI] [PubMed] [Google Scholar]
- 22.Mosmann, T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 6555-63. [DOI] [PubMed] [Google Scholar]
- 23.Nakano, K., and K. H. Vousden. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7683-694. [DOI] [PubMed] [Google Scholar]
- 24.Narita, M., S. Shimizu, T. Ito, T. Chittenden, R. J. Lutz, H. Matsuda, and Y. Tsujimoto. 1998. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 9514681-14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson, D. A., T.-T. Tan, A. B. Rabson, D. Anderson, K. Degenhardt, and E. White. 2004. Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes Dev. 182095-2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oda, E., R. Ohki, H. Murasawa, J. Nemoto, T. Shibue, T. Yamashita, T. Tokino, T. Taniguchi, and N. Tanaka. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2881053-1058. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds, A., D. Leake, Q. Boese, S. Scaringe, W. S. Marshall, and A. Khvorova. 2004. Rational siRNA design for RNA interference. Nat. Biotechnol. 22326-330. [DOI] [PubMed] [Google Scholar]
- 28.Saha, N., B. Schwer, and S. Shuman. 1999. Characterization of human, Schizosaccharomyces pombe, and Candida albicans mRNA cap methyltransferases and complete replacement of the yeast capping apparatus by mammalian enzymes. J. Biol. Chem. 27416553-16562. [DOI] [PubMed] [Google Scholar]
- 29.Schafer, Z. T., and S. Kornbluth. 2006. The apoptosome: physiological, developmental, and pathological modes of regulation. Dev. Cell 10549-561. [DOI] [PubMed] [Google Scholar]
- 30.Schuler, M., and D. R. Green. 2005. Transcription, apoptosis and p53: catch-22. Trends Genet. 21182-187. [DOI] [PubMed] [Google Scholar]
- 31.Shatkin, A. J. 1976. Capping of eucaryotic mRNAs. Cell 9645-653. [DOI] [PubMed] [Google Scholar]
- 32.Shatkin, A. J., and J. L. Manley. 2000. The ends of the affair: capping and polyadenylation. Nat. Struct. Biol. 7838-842. [DOI] [PubMed] [Google Scholar]
- 33.Shibagaki, Y., N. Itoh, H. Yamada, S. Nagata, and K. Mizumoto. 1992. mRNA capping enzyme. Isolation and characterization of the gene encoding mRNA guanylyltransferase subunit from Saccharomyces cerevisiae. J. Biol. Chem. 2679521-9528. [PubMed] [Google Scholar]
- 34.Shimazu, T., K. Degenhardt, E. K. A. Nur, J. Zhang, T. Yoshida, Y. Zhang, R. Mathew, E. White, and M. Inouye. 2007. NBK/BIK antagonizes MCL-1 and BCL-XL and activates BAK-mediated apoptosis in response to protein synthesis inhibition. Genes Dev. 21929-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimizu, S., and Y. Tsujimoto. 2000. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc. Natl. Acad. Sci. USA 97577-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sonenberg, N., H. Trachsel, S. Hecht, and A. J. Shatkin. 1980. Differential stimulation of capped mRNA translation in vitro by cap binding protein. Nature 285331-333. [DOI] [PubMed] [Google Scholar]
- 37.Srinivasan, P., F. Piano, and A. J. Shatkin. 2003. mRNA capping enzyme requirement for Caenorhabditis elegans viability. J. Biol. Chem. 27814168-14173. [DOI] [PubMed] [Google Scholar]
- 38.Takase, Y., T. Takagi, P. B. Komarnitsky, and S. Buratowski. 2000. The essential interaction between yeast mRNA capping enzyme subunits is not required for triphosphatase function in vivo. Mol. Cell. Biol. 209307-9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsukamoto, T., Y. Shibagaki, S. Imajoh-Ohmi, T. Murakoshi, M. Suzuki, A. Nakamura, H. Gotoh, and K. Mizumoto. 1997. Isolation and characterization of the yeast mRNA capping enzyme beta subunit gene encoding RNA 5′-triphosphatase, which is essential for cell viability. Biochem. Biophys. Res. Commun. 239116-122. [DOI] [PubMed] [Google Scholar]
- 40.Ventura, A., A. Meissner, C. P. Dillon, M. McManus, P. A. Sharp, L. Van Parijs, R. Jaenisch, and T. Jacks. 2004. Cre-lox-regulated conditional RNA interference from transgenes. Proc. Natl. Acad. Sci. USA 10110380-10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vousden, K. H., and X. Lu. 2002. Live or let die: the cell's response to p53. Nat. Rev. Cancer 2594-604. [DOI] [PubMed] [Google Scholar]
- 42.Wierzbicki, A., M. Kendall, K. Abremski, and R. Hoess. 1987. A mutational analysis of the bacteriophage P1 recombinase Cre. J. Mol. Biol. 195785-794. [DOI] [PubMed] [Google Scholar]
- 43.Worch, R., A. Niedzwiecka, J. Stepinski, C. Mazza, M. Jankowska-Anyszka, E. Darzynkiewicz, S. Cusack, and R. Stolarski. 2005. Specificity of recognition of mRNA 5′ cap by human nuclear cap-binding complex. RNA 111355-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu, H., J. M. Yang, S. Jin, H. Zhang, and W. N. Hait. 2006. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res. 663015-3023. [DOI] [PubMed] [Google Scholar]
- 45.Youle, R. J., and A. Strasser. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 947-59. [DOI] [PubMed] [Google Scholar]
- 46.Yue, Z., E. Maldonado, R. Pillutla, H. Cho, D. Reinberg, and A. J. Shatkin. 1997. Mammalian capping enzyme complements mutant Saccharomyces cerevisiae lacking mRNA guanylyltransferase and selectively binds the elongating form of RNA polymerase II. Proc. Natl. Acad. Sci. USA 9412898-12903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.