

Abstract
Toxoplasma gondii, which causes toxoplasmic encephalitis and birth defects, contains an essential chloroplast-related organelle to which proteins are trafficked via the secretory system. This organelle, the apicoplast, is bounded by multiple membranes. In this report we identify a novel apicoplast-associated thioredoxin family protein, ATrx1, which is predominantly soluble or peripherally associated with membranes, and which localizes primarily to the outer compartments of the organelle. As such, it represents the first protein to be identified as residing in the apicoplast intermembrane spaces. ATrx1 lacks the apicoplast targeting sequences typical of luminal proteins. However, sequences near the N terminus are required for proper targeting of ATrx1, which is proteolytically processed from a larger precursor to multiple smaller forms. This protein reveals a population of vesicles, hitherto unrecognized as being highly abundant in the cell, which may serve to transport proteins to the apicoplast.
The apicoplast is a relict secondary chloroplast that resides within the cytosol of most apicomplexan parasites, including the intracellular pathogens Toxoplasma gondii and the malaria parasite Plasmodium. It is an essential organelle in these organisms, and since it does not have a counterpart in animal hosts, pathways known to function in the organelle have been considered as potential targets for prophylactic or therapeutic intervention for both toxoplasmosis and malaria (52). Indeed, specific antibiotics that target the transcription and translation systems of the apicoplast, including clindamycin and tetracycline, are known to be toxic to the parasites (9, 14, 23). Other small molecules inhibit key apicoplast metabolic pathways, such as isoprenoid biosynthesis (27) or fatty acid biosynthesis (22). Therefore, identification of additional essential molecules in the apicoplast could yield new drug targets.
The apicoplast was acquired by secondary endosymbiosis and is surrounded by four membranes (30); the inner two presumably correspond to the two original chloroplast membranes, while the outer two are thought to be derived from the algal plasma membrane and the endocytic vacuole. Thus, there are eight distinct locations within the apicoplast: one for each of the four membranes, three for the intermembrane spaces, and one for the lumen of the organelle. Little is known about proteins that reside in nonluminal compartments. Since the apicoplast genome has few genes other than those involved in genetic functions (53), the vast majority of apicoplast proteins are encoded by nuclear genes. Many proteins have been predicted to localize to the apicoplast lumen on the basis of a predicted apicoplast targeting sequence (17, 42), as described below.
To date, all proteins known to reside in the lumen of the apicoplast are synthesized with an N-terminal bipartite targeting sequence that routes them to the organelle. This sequence is composed of a signal peptide that diverts the nascent protein into the endoplasmic reticulum (ER). After cleavage of the signal sequence, the adjacent transit peptide that mediates trafficking to the apicoplast is revealed. Current data support a model of direct trafficking from the ER rather than passage through the Golgi apparatus (10, 28, 29, 48). The transit peptide is proteolytically removed after arrival in the apicoplast, yielding the mature protein residing in the organelle (10, 17, 49, 51, 54). The two membrane proteins described thus far in T. gondii lack such a bipartite targeting signal (28, 29), although one transporter in Plasmodium falciparum does have such a sequence (39). Interestingly, these two T. gondii apicoplast membrane proteins are not restricted to a single membrane but instead reside in multiple membranes within the apicoplast. No proteins localized to the intermembrane spaces have been described thus far.
In addition to proteins participating in the biosynthetic reactions mentioned above, several proteins involved in redox reactions have been demonstrated to reside in the apicoplast. These include ferredoxin and ferredoxin reductase (50), a superoxide dismutase, and a peroxiredoxin (40). Several enzymes found in the apicoplast are orthologues of chloroplast proteins that associate with, and are potentially regulated by, thioredoxin (Trx), including Clp protease, 1-deoxy-d-xylulose 5-reductoisomerase, and the protein translation factors EF-G and EF-Tu (1). However, no apicoplast Trx has been identified as yet. The Trx superfamily is a large set of proteins that contain domains that function biochemically by forming disulfide bonds with target molecules, resulting in conformational changes or rearrangement of disulfide bonds in the target. Although Trx proteins often have a single Trx domain, others contain multiple Trx domains and additional residues at the N and C termini. The cellular functions of Trx superfamily proteins include redox regulation, protein folding, intracellular signaling, and the response to oxidative stress (reviewed in reference 8). Trxs have been proposed as drug targets in cancer (41). Moreover, components of the redox cycle have been considered targets in malaria parasites and trypanosomatids (31).
In this report we describe a novel Trx family protein that is localized to the peripheral subcompartments of the apicoplast. We have therefore named this protein apicoplast Trx-like protein 1, or ATrx1. Several differently sized forms are present, as detected by a monoclonal antibody (MAb) directed against the protein or via expression of an epitope-tagged protein. Immunoelectron microscopy further shows that this molecule defines a novel set of highly abundant vesicles within the parasite.
MATERIALS AND METHODS
RH strain T. gondii was grown in human foreskin fibroblasts and transfected as previously described (28, 43). Version 4.3 of ToxoDB was used for analysis of T. gondii genomic, expressed sequence tag, and protein prediction models, as well as analysis of strain differences (20). The ATrx1 sequence from strain RH has been deposited in GenBank with accession number EU555314.
Production of the MAb 11G8 antibody.
To identify novel organellar proteins in T. gondii, MAbs were prepared against a partially purified fraction of organelles. The organellar fraction used for antibody production was essentially the “microneme-enriched” fraction as previously described (24). Briefly, extracellular parasites were washed into isotonic sucrose buffer SMDI (10 mM morpholinepropanesulfonic acid [pH 7.2], 250 mM sucrose, 2 mM dithiothreitol, Roche complete protease inhibitor cocktail) and disrupted in a French press. Unbroken parasites were removed by centrifugation, and the organellar pellet was obtained by centrifugation at 25,000 × g. The organellar pellet was resuspended in SMDI and loaded onto a 30% Percoll gradient as described previously (24). A 2-ml fraction located just above the rhoptry/dense granule band was collected, diluted, and pelleted to remove the Percoll. The fraction was used to immunize (200 μg/injection) a BALB/c mouse and after the fifth boost, the spleen was removed, and the fusion performed as described previously (21). The hybridoma supernatants were screened by immunofluorescence assay (IFA), and positive hybridomas were cloned and rescreened by IFA and Western blot analysis. The resulting hybridomas detected the apicoplast, mitochondrion, rhoptries, micronemes, and dense granules (not shown). One of the MAbs thus obtained, MAb 11G8, was used in the present study.
Cloning.
Genomic DNA was isolated from RH strain T. gondii. RNA was isolated by using TRIzol (Ambion) and treated with RNase-free DNase I. cDNA was synthesized by using a high-capacity cDNA reverse transcription kit from Ambion with the primers supplied and the conditions suggested by the manufacturer. Sequences of specific primers used are provided in supplemental material (see Table S1 in the supplemental material). To clone the full-length ATrx1 coding sequence, randomly primed cDNA was synthesized, and amplified by PCR using the primers (−)189s and (+)7695a (the numbers correspond to genomic nucleotide relative to the predicted start codon; the termination codon is at nucleotides [nt] 7358 to 7360). For expression of ATrx1 in T. gondii using its own promoter, the mRNA coding sequence was amplified from cDNA using primers XmaI(−)188s and AvrII(+)7357a, while the ATrx1 promoter was amplified from genomic DNA by using the primers HindIII(−)1535s and XmaI(−)195a. After cloning into pGEM-T Eeasy, the inserts were released with the indicated restriction enzymes and ligated into a T. gondii expression vector such that four hemagglutinin (HA) tags were added to the C terminus of the protein. This vector, which is derived from pHXGPRT (12), bears HXGPRT as the selectable marker. The ATrx1 coding sequence was checked by sequencing. The plasmid was transfected into T. gondii RHΔhgprt (12) expressing S+TACP-HcRed as an apicoplast luminal marker as described previously (28, 29). The plasmids ATrx1ΔN51-4HA and ATrx1(Δ50-210)-4HA were generated using from the ATrx1-4HA plasmid by mutagenesis using Pfu-Turbo (Strategene) plus the oligonucleotides Δ2-51s, Δ2-51a, Δ50-210s, and Δ50-210a, respectively, followed by DpnI to cleave the parent plasmid. The resulting plasmids were checked by sequencing the ATrx1 coding region. To prepare the plasmid ATrx1(1-225)-GFP, the ATrx1 promoter and coding sequence were ligated into a T. gondii expression vector such that green fluorescent protein (GFP) was added to the C terminus of the protein. The portion of ATrx1 encoding amino acids (aa) 226 to 742 was then removed by mutagenesis using the oligonucleotides Δ226-742s and Δ226-742a, and the resulting plasmid was checked by sequencing. For colocalization experiments, plasmids were transiently transfected into RHΔhxgprt cells or cell lines expressing the apicoplast membrane marker APT1 (28) or the ER marker FtsH(1-354) (29).
ATrx1 transcripts were verified by random amplification of cDNA ends (RACE). The 3′ end of the cDNA was amplified by using the Second Generation 5′ 3′ RACE kit (Roche), using the anchor primer and the nested primers (+)4839s and (+)4978s. Similarly, to check the abundance of splice variants, oligo(dT)-primed cDNA was amplified by using (+)4978s and (+)7318a primers. The 5′ end of the transcript was mapped by RACE as described previously (28) using primers slightly downstream of the first peptides detected for band 3 (+)1156a for first-strand synthesis and (+)1053a and (+)1028a as nested primers for PCR.
Microscopy.
IFA was performed on intracellular parasites as previously described (10, 28). MAb 11G8, a mouse immunoglobulin G1 (IgG1), was used at a 1:500 dilution. The HA epitope tag was detected by using fluorescein isothiocyanate (FITC)-coupled rat anti-HA 3F10 MAb (3 μg/ml; Roche), Alexa 594-coupled mouse anti-HA MAb (10 μg/ml; Invitrogen), or rabbit anti-HA (10 μg/ml; Neomarkers) detected by anti-rabbit immunoglobulin coupled to Alexa 350 (4 μg/ml; Molecular Probes). Rabbit anti-IMC1 antiserum, a gift from Con Beckers (35), was used at a 1:2,000 dilution and detected with goat anti-rabbit IgG coupled to Alexa 680 (4 μg/ml; Molecular Probes). Rabbit anti-trypanosomal BiP antiserum (which cross-reacts with T. gondii BiP), a gift from Jay Bangs (2), was used at a 1:200 dilution; rabbit anti-T. gondii NTPase antiserum, a gift from Tim Stedmann and Keith Joiner (3), was used at a 1:200 dilution. Both antisera were detected with goat anti-rabbit IgG coupled to Texas Red (2 μg/ml; Southern Biotechnology). GFP was detected by using rabbit anti-GFP (0.5 μg/ml; Molecular Probes), followed by goat anti-rabbit IgG coupled to FITC (2 μg/ml; Southern Biotechnology). Slides were viewed on a Deltavision RT deconvolution microscope with an Olympus UPlan/Apo 100× 1.35NA objective. Images were deconvolved by using the conservative ratio method, other parameters were standard, and a single deconvolved plane is shown.
For immunoelectron microscopy, fibroblasts were infected with parasites expressing ATrx1-HA and grown overnight. Preparations were fixed, infiltrated, frozen, and sectioned by using published techniques (16). The sections were immunolabeled with rat anti-HA MAb 3F10 (2 μg/ml, in phosphate-buffered saline [PBS]-1% fish skin gelatin) and then with anti-mouse IgG antibodies, followed directly by protein A-10-nm gold, and visualized as described previously (28). To calculate the relative surface area of vesicles to the parasite cytoplasm, transmission electron micrographs of the sectioned infected cells were digitized by using photo scanner connected with a computer and imported as TIFF files. The digitized micrographs were pixel images that were transferred into vector graphics by tracing selected ATrx1-containing vesicles and the parasite surface area semiautomatically using the computer program Volocity 3.1 (ImproVision).
Protein fractionation, Western blotting, and immunoprecipitation.
To identify the protein(s) detected by MAb 11G8, the antibody was dimethylpimelimidate cross-linked to protein G-Sepharose, and proteins were isolated from cell lysates from 5 × 109 extracellular RH strain tachyzoites as described previously (21). The eluted proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and stained with Coomassie brilliant blue R-250. The stained bands were excised and rinsed in water, and proteins were subjected to tryptic digestion and mass spectrometry on a Thermo Electron LTQ linear ion trap mass spectrometer. The resulting collision-induced dissociation spectra were compared to the peptides predicted at ToxoDB (both from gene models and six-frame translations of the genome) to identify the corresponding proteins.
All extraction procedures were done on ice or at 4°C in the presence of 11 U of aprotinin/μl, 5 μg of leupeptin/ml, 1 μM pepstatin, 100 μM phenylmethylsulfonyl fluoride, and 1 mM EDTA unless otherwise noted. For carbonate and urea extractions, recently egressed parasites were suspended in nanopure water with protease inhibitors, sonicated for 5 min on ice and then transferred to a Dounce homogenizer and stroked 25 times. The sample was then adjusted to either 0.1 M Na2CO3 (as in reference 29) or 1× PBS for a final parasite concentration of 2 × 108/ml and then stroked 10 times every 5 min for 1 h. After removal of large debris by centrifugation at 5,000 × g for 5 min, membrane and soluble fractions were separated by centrifugation at 150,000 × g for 1 h. Soluble fractions were precipitated by 10% trichloroacetic acid. For urea extraction, cell pellets were suspended in ice-cold 6 M urea (108/ml) and processed as described above. For carbonate-, PBS-, and urea-treated samples, cell disruption (>99% lysis) was verified by microscopic analysis. Triton X-114 extraction was used to prepare integral membrane and soluble fractions from 2 × 108 RH parasites as described previously (21). Triton X-100 extraction was performed as described previously (29). The resulting fractions from these extractions were subjected to Western blot analysis with MAb 11G8 (1:5,000 or 1:1,000) or anti-HA (0.1 μg/ml; Covance) to detect endogenous or tagged ATrx1. Controls for soluble proteins were ROP1 (TG49 antiserum, 1:2,000) (45), Mic5 (6), and S+TACP-HcRed (detected by rabbit anti-HcRed [Becton Dickinson; 1:500]). Controls for membrane proteins were SAG1 (anti-SAG1, 1:30,000 [a gift from John Boothroyd]) or endogenous FtsH1 (affinity-purified anti-FtsH1, 1:250 [A. Karnataki, unpublished data]). Antibody binding was highlighted with goat anti-rabbit immunoglobulin coupled to IRDye 680 or goat anti-mouse coupled to immunoglobulin IRDye 800 as appropriate (100 ng/ml; LI-COR) and visualized by using an Odyssey infrared imaging system (LI-COR) except as noted.
In vivo labeling and immunoprecipitations were performed as previously described (10), except that samples were not denatured prior to immunoprecipitation. For pulse-chase analysis, parasites were labeled with [35S]methionine-cysteine (10), and ATrx1-HA was immunoprecipitated with anti-HA (8 μg/ml; Covance). Immunoprecipitates were separated by SDS-PAGE and transferred to nitrocellulose prior to phosphorimaging. The blots were subsequently probed with anti-HA or MAb 11G8.
RESULTS
Identification of an apicoplast Trx-like protein.
A panel of MAbs was raised against a T. gondii organellar fraction that contained apicoplasts, micronemes, rhoptries, dense granules, and mitochondria (4). Preliminary IFA revealed that one of the antibodies, MAb 11G8, stained a circular area located apical of the nucleus and distinct from the rhoptries. The antigen appeared to surround the DAPI (4′,6′-diamidino-2-phenylindole)-stained apicoplast DNA, suggesting that it resides at the membrane region of the apicoplast. When tested against a T. gondii cell line transfected with a red fluorescent apicoplast luminal marker (S+TACP-HcRed), the antibody stained an area surrounding the apicoplast lumen (Fig. 1A). Similarly, colocalization analysis with an HA-tagged version of the apicoplast membrane protein APT1 (28) showed that the MAb 11G8 antigen and APT1 colocalized in some parasites and partially overlapped in others (Fig. 1B). These data showed that that the protein recognized by this MAb was localized to the apicoplast and suggested it might be in or associated with one or more of the apicoplast membranes or reside in an intermembrane compartment.
FIG. 1.

Identification of ATrx1. (A) IFAs using MAb 11G8; S+TACP-HcRed (S+T-Red), a marker for the apicoplast lumen; and DAPI. Each panel is labeled according to the color shown in the merged image: green for MAb 11G8 (detected with FITC-coupled rabbit anti-mouse immunoglobulin), red for apicoplast lumen, and blue for DAPI. Bar, 2 μm. (B) IFA using MAb 11G8; APT1-HA, a marker for the apicoplast membranes; and DAPI. Each panel is labeled according to the color shown in the merged image: red for MAb 11G8 (detected with anti-mouse IgG1 coupled to Texas Red), green for apicoplast membranes (detected with FITC-coupled anti-HA antibody), and blue for DAPI. Bar, 2 μm. (C) Protein from 107 wild-type parasites was separated on a 10% acrylamide gel, transferred to nitrocellulose, and probed with MAb 11G8. Numbers on the right indicate the migration of molecular mass markers. (D) Immunopurification of MAb 11G8-reactive proteins, Coomassie blue-stained gel. Numbered bands from the immunopurified protein (IP) were subjected to mass spectrometric analysis. Std, molecular mass markers. (E) Diagram of ATrx1 and peptide coverage from mass spectrometry. Red indicates regions from bands 1 and 2 covered by detected peptides. Blue indicates where additional peptides were found in band 3. Details are shown in Fig. S1 in the supplemental material. No peptides were found for yellow regions. The location of the predicted signal anchor (SA) and putative TMD are indicated, as well as the thioredoxin domains. The predicted active site is marked with a star. Below the diagram, three deletion mutants are diagrammed, with dashed lines indicating sequences deleted near the N terminus.
Immunoblot analysis of T. gondii lysates revealed multiple bands using MAb 11G8 (Fig. 1C). Three closely spaced species were detected around 60 kDa—at 57, 62, and 65 kDa. Bands at about 70 and 85 kDa were also seen. To identify the proteins detected by MAb 11G8, the antibody was used to immunoaffinity purify proteins from T. gondii for mass spectrometry. Five bands were detected on a Coomassie blue-stained gel (Fig. 1D). These included a doublet at around 60 kDa (bands 1 and 2) and a fainter band at 85 kDa (band 3). These correspond to the bands seen on immunoblots, although the deliberate loading of large amounts of protein reduced the resolution of the 60-kDa region (see Fig. 1D). Two large species were detected near the top of the Coomassie blue-stained gel (bands 4 and 5), which were not typically seen on MAb 11G8 immunoblots. The regions containing the protein bands were removed from the gel, with slow-migrating bands 4 and 5 extracted together, and then all four samples were subjected to tryptic digestion and tandem mass spectroscopy. The peptide data were screened against the T. gondii databases (both the predicted protein database and the conceptual translation of the nucleotide database). All four samples showed numerous peptides that matched gene model 583.m00655, a protein with Trx domains (see Fig. S1 in the supplemental material for detailed peptide coverage). Band 3 also revealed numerous peptides that matched T. gondii gene model 39.m00367, a hypothetical protein with no conserved domains. This protein was not investigated further since matching peptides were only present in band 3. Bands 4 plus 5 had multiple peptides matching gene model 583.m00655, as well as peptides matching to model 50.m05651, a surface protein related to PspC. This last protein was also found upon immunoaffinity purification and mass spectrometry of an unrelated rhoptry protein (data not shown), suggesting that it is a nonspecific contaminant. Since all bands contained protein corresponding to the gene model 583.m00655, we propose that this is the MAb 11G8 target antigen. Based on the protein's location around the apicoplast and the presence of a Trx domain, we named it apicoplast Trx family protein 1 (ATrx1).
The coding sequence of ATrx1 contains two Trx-fold domains (InterPro IPR012335) (reviewed in reference 8); only the second of these includes the conserved active site WCPPC. ATrx1 is most similar overall to nucleoredoxin proteins from vertebrates, and the individual Trx domains belong to the TryX subfamily of Trx domains (see alignment in Fig. S2 in the supplemental material). TryX domains are found on tryparedoxins, which are well known in trypanosomatids (34, 44), and are also present on certain Trx family proteins in higher eukaryotes. A second gene in T. gondii is also annotated as encoding a nucleoredoxin (42.m00038), but the predicted protein is unlikely to be redundant to ATrx1 because it has only a single Trx domain, and its homology with ATrx1 is limited to that domain.
Verification of the coding sequence.
Gene model 583.m00655 has 10 exons and encodes a protein of 742 aa with a predicted size of 81.8 kDa (see Fig. S2 in the supplemental material). This size correlated well with band 3 (∼85 kDa), being substantially larger than the more abundant proteins present in bands 1 and 2. Indeed, peptides from bands 1 and 2 both mapped exclusively in the C-terminal 58 kDa of the protein, with very high coverage of that region (82 and 80%, respectively; see Fig. S1 in the supplemental material). Peptides from bands 4 and 5 also mapped to the C-terminal region of the protein, although coverage was not as definitive. In contrast, peptides from band 3 extended further N terminally by about 113 aa (Fig. 1E). In our analyses, no peptides were identified that mapped to the N-terminal 112 aa of the protein, but an additional peptide commencing at aa 85 within exon 2 is seen on ToxoDB (data provided by the Einstein Biodefense Proteomics Center, http://toro.aecom.yu.edu/biodefense/). No peptides were found corresponding to exon 1 or to conceptual translations of predicted introns. Whether the 85-kDa form represents the primary translation product or a form processed near the amino terminus (e.g., after the putative signal anchor) cannot be determined from the current data. The mass spectrometry analysis did not reveal any obvious reasons for the difference in sizes of the smaller isoforms, but it is likely that the gel bands were not sufficiently separated to rule out cross-contamination. The molecular differences between the smaller isoforms could reflect heterogeneity of proteolytic cleavage or a distinct posttranslational modification such as phosphorylation or glycosylation.
To verify that the first exon was indeed present in the mRNA, we conducted 5′ RACE using cDNA prepared from strain RH parasites. Primers were positioned slightly downstream of the regions encoding the first peptides detected in band 3. Sequencing of the cloned 5′ RACE products showed that transcripts begin approximately 346 nt upstream of the start codon. This is 33 nt upstream of the predicted transcription start site at nt 2432437 of chromosome IX in strain ME49 annotated on ToxoDB. The termination codon and a 3′ untranslated region of 740 nt were defined by 3′ RACE, matching the gene model. We also amplified the entire predicted open reading frame from randomly primed cDNA. Two independently amplified cDNAs were sequenced; except for a single synonymous substitution, the sequences matched that of gene model 583.m00655 of T. gondii strain GT1, which belongs to the same lineage as RH, the strain used here (20). Interestingly, one cDNA sequence was 48 nt shorter than the other. This reflected a different splice acceptor site being used for exon 8. To determine which of these two variants was dominant in the mRNA population, a PCR assay was designed such that a 586-nt or a 538-nt product would result, depending on the splice junction. The larger product was dominant, while the smaller product was barely detectable (not shown). Consequently, the longer coding sequence was used to construct the expression plasmid discussed below. The 16 aa absent from the protein encoded by the smaller splice variant lie within a conserved region of the second, presumed functional Trx domain (see Fig. S2 in the supplemental material, blue-shaded residues), so it is unlikely that the resulting smaller protein would have Trx activity.
Confirmation of gene identification.
To verify that the identified gene corresponds to the antigen detected by MAb 11G8, we expressed an epitope-tagged version of ATrx1 in T. gondii. A clone was constructed encoding four HA tags (molecular mass of 4.9 kDa) at the C terminus of ATrx1. The tagged sequence was cloned into a promoter-less T. gondii expression vector with approximately 1.5 kb of ATrx1 5′ flanking sequence, so that expression was driven by the ATrx1 promoter. After isolation of a clonal, stable transfectant, immunoblot and immunofluorescence analyses were performed. When transfectants expressing ATrx1-HA were examined, anti-HA detected four major bands at approximately 65, 70, 72, and 92 kDa and a faint band at ∼78 kDa, all of which were detected by antibody MAb 11G8 (Fig. 2A). The size difference between the wild-type and HA-tagged bands is consistent with the 5-kDa peptide encoded by the four HA tags. The presence of these different forms following expression of the tagged cDNA indicates that they likely result from processing or other posttranslational modifications rather than alternative splicing. The amount of MAb 11G8 immunoreactive protein resulting from the transfected ATrx1 coding sequence appears to be similar to that of the endogenous protein (Fig. 2A), even though the transfected gene lacks introns and has a heterologous 3′ UTR. However, we did observe relatively more of the larger HA-tagged isoform versus the smaller isoforms compared to the untagged form. Similar patterns were seen on anti-HA blots of reducing and nonreducing gels (Fig. 2B), except that a smeared band at ∼200 kDa, similar to that seen in Fig. 1D, was occasionally observed in nonreduced samples. No differences were seen between extracellular parasites and those that were mechanically released from host cells when analyzed with MAb 11G8 or anti-HA (not shown).
FIG. 2.

Western blot, showing multiple bands with MAb 11G8 and anti-HA. (A) Lysates of parasites expressing ATrx1-HA or control parasites not expressing an HA-tagged protein were separated on 7.5% acrylamide gels and analyzed by immunoblot analysis using anti-HA and MAb 11G8. Note the additional, slightly larger bands detected in ATrx1-HA transfectants which comigrate with those detected by anti-HA. The same pattern was seen in an independent transfectant. The migration of molecular mass markers is indicated. Arrows mark the bands described in the text. (B) Reducing (+BME, β-mercaptoethanol) and nonreducing (−BME) gels show similar patterns. Samples were separated on 10% acrylamide gels and analyzed by immunoblot analysis using anti-HA. The cell lines used were ATrx1-HA transfectants and the parental line RH (control). The migration of molecular mass markers is indicated.
Anti-HA and MAb 11G8 showed similar if not identical patterns on IFA (Fig. 3A). The staining with anti-HA was around the luminal marker, S+TACP-HcRed, but some cells showed only circumplastid staining (as in the bottom set of panels), while other cells additionally showed apparent vesicles or tubules near the apicoplast (as in top set of panels). For the two other T. gondii apicoplast membrane proteins analyzed thus far (APT1 and FtsH1), such staining patterns correlated with the plastid and cell division cycle (28, 29). We therefore assessed the plastid and cell division cycle of a number of cells by the shape and location of the plastid, the nucleus, and the formation of inner membrane complex as described previously (28, 46) (Fig. 3B). As with APT1 and FtsH1, ATrx1-HA showed a predominantly circumplastid staining in stage 1 cells, although some also showed additional staining in the ER. Stage 1 cells, which have round plastids that are separated from the nucleus, are in interphase and are highly abundant in an asynchronous population. As the plastid began to elongate and move closer to the nucleus (stage 2), more vesicular and/or tubular staining was seen. The presence of apparent tubules and vesicles continued during stages 3 and 4, in which the plastid was enlarging and elongating, while the inner membrane complex of the daughter cell was forming. Once the plastid had divided (stage 5) and the nucleus had divided (stage 6), ATrx1 resumed a circumplastid location in some cells, with some cells also showing ER staining. Parasites within the same vacuole showed similar ATrx1-HA distribution. This cell cycle-associated staining pattern is similar to that seen for FtsH1 and APT1, except that the tubule-like staining for ATrx1 extends more prominently into the later stages (5-6). Examples of circumplastid staining, plastid-plus-tubule staining, and plastid-plus-ER staining are shown at the right of Fig. 3B.
FIG. 3.

Epitope-tagged ATrx1 localizes to the apicoplast. (A) IFA of transfectants expressing ATrx1-HA. The bottom set of panels shows a circumplastid localization of both the HA-tagged protein and the MAb 11G8 antigen, while the upper set shows more additional extended tubules. Each panel is labeled according to the color shown in the merged images, and the outline of the parasite within the vacuole is marked as a gray line on the merged image. Bar, 2 μm. (B) Localization of ATrx1-HA is modulated during the cell cycle. Cells in different vacuoles were staged according to the shape and location of the apicoplast, the shape and division of the nucleus, and the formation of the inner membrane complex (46) as follows: stage 1, round apicoplast is away from nucleus; stage 2, apicoplasts begin to elongate and move closer to the nucleus; stage 3, the apicoplast elongates further and extends across the apical face of the nucleus; stage 4, the daughter cell IMC is visible (as revealed by anti-IMC1) and the apicoplast is V-shaped; stage 5, the apicoplast has divided; and stage 6, nuclear division is complete. The proportion of vacuoles with different distribution patterns for ATrx1 during the plastid and cell division cycle is shown graphically. The patterns were tabulated as circumplastid (CP, yellow shading), plastid plus tubule (P+T, red shading) and plastid plus ER (P+ER, blue shading). Next to the key, examples of the staining patterns categorized are shown. In these images, ATrx1-HA is green and DAPI is blue. In the CP and P+T panels, the apicoplast luminal marker (S+TACP-HcRed) is red; in the P+ER panel, the ER (as revealed by anti-BiP) is red. The nucleus is marked with an “N”. Note the perinuclear staining of ATrx1 in the plastid plus ER panel (green).
The N-terminal region of ATrx1 functions in apicoplast localization.
All proteins targeted to the apicoplast lumen that have been studied thus far bear an N-terminal extension containing the essential information for trafficking, including a signal peptide and transit sequence. This extension is typically removed by proteolytic processing. ATrx1 protein has a predicted signal anchor sequence (13), from aa 37 to 50. This could direct the protein into the endomembrane system for trafficking to the apicoplast. Neither the sequence preceding nor that following the predicted signal anchor resemble a transit peptide, since they are not enriched for basic amino acids, a property shown to be critical for directing proteins to the apicoplast lumen in P. falciparum (17) and T. gondii (47).
We therefore tested whether the region upstream of the first peptides detected in band 1 contained information important for trafficking of ATrx1 (Fig. 4). Two deletion constructs were made from ATrx1-HA, as diagrammed in Fig. 1E. The first, ATrx1ΔN51-HA lacked the first 51 aa, which includes the predicted signal anchor domain, whereas the second, ATrx1Δ50-210-HA, lacked the region immediately downstream of the signal anchor. ATrx1ΔN51-HA showed a dispersed, punctate pattern having minimal colocalization with the ER marker BiP and did not colocalize with the dense granule marker NTPase (3) or the apicoplast luminal marker S+TACP-HcRed (Fig. 4A to C). The modest level of apparent ER staining could reflect the import of a minor fraction of the mutant protein into the ER, association with the external face of the ER, or simply cytosolic protein very close to the ER. In any case, the protein did not localize to the plastid. Much of the second mutant protein, ATrx1Δ50-210-HA, which contains the putative signal sequence colocalized with the ER marker BiP (Fig. 4D, note particularly the perinuclear staining characteristic of the ER), and some was detected outside the parasite in the parasitophorous vacuole (Fig. 4D to F, arrows). ATrx1(Δ50-210)-HA did not colocalize with the dense granule marker NTPase within the cell but did colocalize with a deposit of NTPase within the vacuole (Fig. 4E, arrow) These data suggest that the mutant protein might be deposited in the residual body upon daughter cell formation. Although the protein appeared to be entering the secretory system, it was not diverted from the ER to the apicoplast (Fig. 4F). When aa 1 to 225 of ATrx1 were placed at the N terminus of GFP, the protein was directed to the region of the apicoplast, near but not colocalizing with the luminal marker (Fig. 4G). In most cells, it colocalized with the apicoplast membrane marker APT1 (Fig. 4H), although in some cells it showed less complete overlap (not shown). Our previous studies have shown that GFP alone lacks organelle targeting information when expressed in T. gondii (11). These data show that the N-terminal “extension” of ATrx1 is both required and sufficient for localization to the apicoplast.
FIG. 4.

ATrx1 deletion analysis. Expression plasmids encoding ATrx1ΔN51-HA (A to C) ATrx1(Δ50-210)-HA (D to F), or ATrx(1-225)-GFP (G and H) were transfected either into T. gondii expressing S+TACP-HcRed (C, F, and G) or V5-APT1 (H) or into wild-type cells (A, B, D, and E). Cells were processed for IFA as follows. ER was visualized with rabbit anti-BiP, dense granules (DG) with anti-NTPase, the apicoplast lumen with S+TACP-HcRed, and the apicoplast membranes with V5-APT1 (as detected by anti-V5). GFP was detected with anti-GFP. Secondary antibodies were chosen to allow specificity and avoid spectral overlap. All samples were also stained with DAPI, and differential interference contrast (DIC) images of the vacuoles are shown.
ATrx1 undergoes processing.
Signal and transit domains of apicoplast luminal proteins are cleaved upon import into the ER and the lumen of the apicoplast, respectively (49), but the mature form of the protein and the mature-plus-transit-peptide forms of proteins are visible on immunoblots. In pulse-chase experiments, the abundance of the former increases at the expense of the latter (10, 49). We performed pulse-chase analysis of ATrx1-HA metabolically labeled with [35S]methionine-cysteine, using anti-HA for immunoprecipitation (anti-HA produced a much stronger signal than MAb 11G8). After SDS-PAGE, the samples were transferred to nitrocellulose, and the samples were subjected to both phosphorimaging and immunoblot analysis with anti-HA. The latter revealed the expected pattern as seen in Fig. 5A, and all immunodetected bands were seen in at least one time point of the pulse-chase analysis. These bands were not observed after immunoprecipitation of cells that did not express the tagged protein (Fig. 5B, lane c). After a 30-min pulse, the radiolabeled 92-kDa species was easily observed, as was the fainter 78-kDa species, and they slowly declined in abundance over the 3-h chase period (Fig. 5A). The smaller isoforms were not visible after the pulse, but with a 1-h chase, a band appeared at ∼65 kDa that persisted through 3 h. An additional radiolabeled band migrating at ∼72 kDa was also seen. Since the relative intensity of the bands at the 3-hour chase did not yet reflect the steady-state abundance of the ATrx1 isoforms, we also conducted a 3-h labeling, followed by an 8-h chase and an 18-h chase (Fig. 5B, note that these samples were separated on a different percentage gel). By 8 h (approximately one cell cycle), the relative intensity of the bands was very similar to the steady-state abundance of ATrx1 isoforms. Interestingly, the 65-kDa band was seen in the 3-h pulse sample, but not in either the 8- or 18-h chase samples. Furthermore, the 72-kDa band was seen in the 3-h pulse-labeled sample, while the 70-kDa band was not. However, both were easily seen at the 18-h time point. These data suggest that the 92-kDa molecule is the precursor of the other species, which likely result from multiple posttranslational modifications of ATrx1, including N-terminal proteolytic cleavage.
FIG. 5.

Pulse-chase analysis. (A) ATrx1-HA intracellular parasites were labeled for 30 min with [35S]methionine-cysteine, and chased for the indicated times. Samples were subjected to immunoprecipitation with anti-HA and separated on an 8 to 16% acrylamide gel. Radiolabeled proteins were revealed by phosphorimaging, and the same blot was probed with rabbit anti-HA antibody. The migration of molecular mass markers is indicated on the left. Double-headed arrows mark the largest and smallest ATrx1 isoforms detected by phosphorimaging and immunoblotting. (B) ATrx1-HA intracellular parasites were labeled for 3 h with [35S]methionine-cysteine and chased for the indicated times. Samples were analyzed as described above, except that a 10% acrylamide gel was used. As a control to show that higher-molecular-mass bands seen with ATrx1-HA are specific, parasites expressing APT1-HA were labeled for 3 h and immunoprecipitated with anti-HA (lane c). The lane shown was from the same exposure of the same gel as the ATrx1-HA samples. APT1-HA is not visible because it migrates at ∼40 kDa.
Fractionation properties of ATrx1.
IFA of ATrx1 indicated that the protein was predominantly localized in a circumplastid pattern, as was previously observed for two integral membrane proteins (28, 29). Since the apicoplast has multiple intermembrane spaces, as well as multiple membranes, the protein could be localized to either nonmembrane or membrane compartments. Analysis of ATrx1 with programs that predict transmembrane domains (TMDs) yielded divergent results, with TMHMM predicting no TMDs, ConPredII predicting one (at aa 262 to 288) and TmPred and TopPred predicting two TMDs (the putative signal anchor region and aa 262 to 288) (Fig. 1E and see Fig. S2 in the supplemental material). The signal anchor region is clearly missing from the smaller isoforms, while the putative TMD region is present in all forms, as evidenced by the mass spectrometry data.
We assessed whether any or all isoforms of ATrx1-HA were strongly associated with membranes. When hypotonically lysed transfectants were extracted with PBS, ATrx1 was present in both supernatant and pellet fractions (Fig. 6A). In contrast, almost all of the integral membrane protein FtsH1 was in the pellet, and most of the apicoplast luminal protein S+TACP-HcRed was in the supernatant (the mature form is shown in all panels). A similar pattern was seen after treatment with 0.1 M carbonate (pH 11) (Fig. 6B) or 6 M urea (data not shown), both of which extract soluble and peripherally associated membrane proteins—only FtsH1 was predominantly in the membrane fraction. The presence of somewhat more ATrx1 than the soluble marker in the pellet fractions may result from a specific membrane interaction of a portion of ATrx1 or from some nonspecific precipitation. The fractionation pattern of the 92-kDa band was similar to the 65 to 72-kDa bands; thus, there was no evidence for preferential membrane association of the species most likely to contain the signal anchor domain. The nonionic detergent Triton X-100 released ATrx1-HA and the marker proteins to the supernatant, indicating that none of them are forming insoluble aggregates. Wild-type ATrx1 detected by MAb 11G8 had the same carbonate and Triton X-100 extraction profile as the tagged protein (Fig. 6C, in this experiment Mic5 served as the soluble marker). Another test of integral association to membranes is Triton X-114 extraction, in which integral membrane proteins partition to the detergent phase, while soluble and peripheral membrane proteins partition to the aqueous phase. When T. gondii were extracted with Triton X-114, the protein detected by MAb 11G8 was found in the aqueous fraction and not in the detergent fraction (Fig. 6D). Some proteolysis was evident in the Triton X-114 treated samples, which may account for the difficulty in detecting the larger isoform of ATrx1. In this experiment, ROP1 served as the soluble marker, while SAG1 was the membrane marker. Taken together, the evidence argues that ATrx1 is not an integral membrane but rather is either soluble or peripherally associated with the membrane.
FIG. 6.
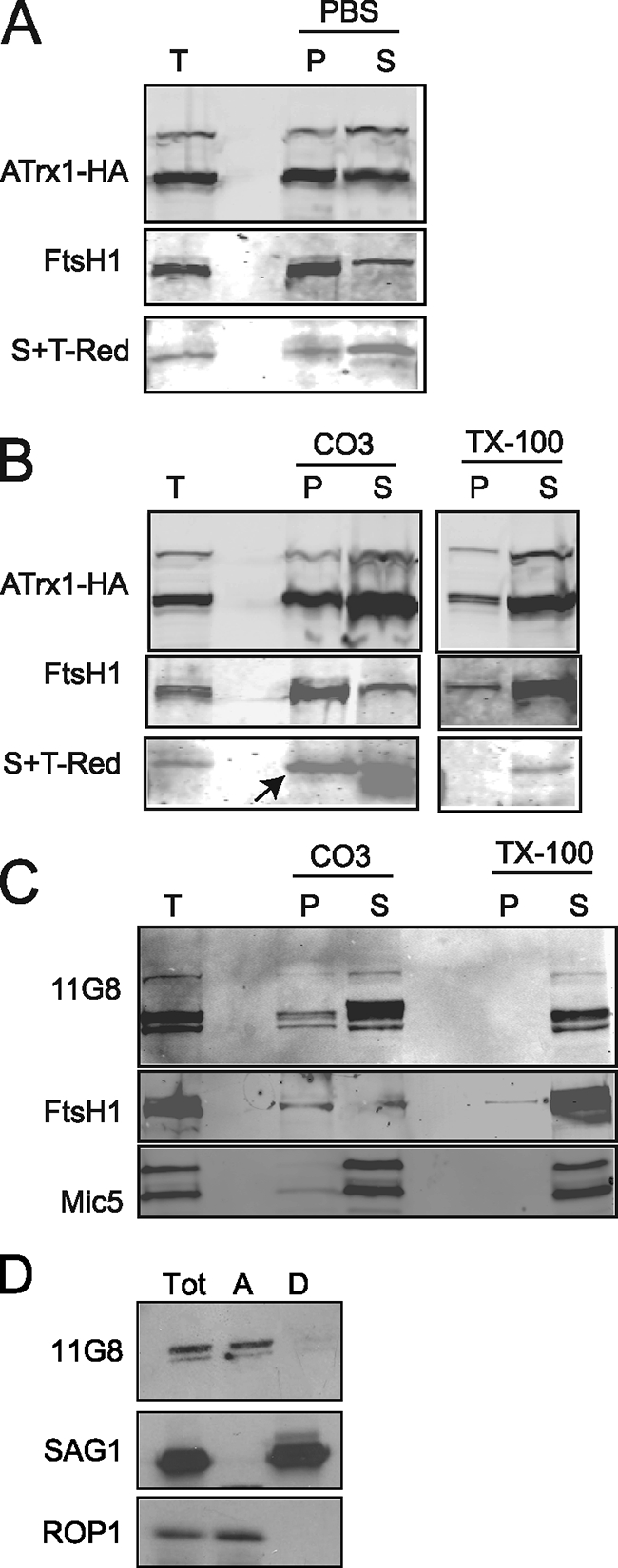
Fractionation behavior of ATrx1. Immunoblot analysis was performed after various extractions. S+TACP-HcRed, Mic5, and ROP1 served as soluble protein markers, and FtsH1 and SAG1 served as membrane protein markers. (A) Transfectants expressing ATrx1-HA and the apicoplast luminal marker S+TACP-HcRed were Dounce homogenized in PBS, and large debris was removed by centrifugation. After high-speed centrifugation, the pellet (P) and supernatant (S) were analyzed by immunoblotting with anti-HA, anti-FtsH1, and anti-HcRed. The band corresponding to the mature form of S+TACP-HcRed is shown. (B) Transfectants expressing ATrx1-HA and S+TACP-HcRed were extracted with 0.1 M carbonate (pH 11) or 2% Triton X-100. After centrifugation to separate the soluble fraction (S) from insoluble pellet (P), both fractions were analyzed by immunoblotting with anti-HA, anti-FtsH1, and anti-HcRed. The band corresponding to the mature form of S+TACP-HcRed is shown (arrow); the lower band on this image is a low-molecular-mass protein that cross-reacts with anti-FtsH1. (C) T. gondii transfected with an irrelevant plasmid were extracted with 0.1 M carbonate (pH 11) or 2% Triton X-100. After centrifugation to separate the soluble fraction (S) from insoluble pellet (P), both fractions were analyzed by immunoblotting with MAb 11G8, anti-FtsH1, and anti-Mic5. (D) Wild-type parasites were extracted with Triton-X114 at 37°C, and the detergent (lane D) and aqueous (lane A) protein fractions were analyzed by immunoblotting with MAb 11G8 (1:1,000) followed by goat anti-mouse antibody coupled to horseradish peroxidase (1:1,000; Sigma). Detection was performed by using the ECL system (Pierce).
ATrx1 is localized to peripheral compartments of the apicoplast and marks an abundant class of vesicles.
The MAb 11G8 was found to be unsuitable for immunoelectron microscopy, so we analyzed the location of ATrx1 using the transfectants expressing ATrx1-HA. ATrx1 was abundant in the membrane region of the apicoplast (Fig. 7). Like the apicoplast membrane proteins that have been characterized to date, Apt1 and FtsH1, ATrx1 was associated with multiple peripheral subcompartments of the apicoplast. However, given the close spacing of the membranes, it is not possible to determine whether the molecule is predominantly in the intermembrane spaces or molecularly associated with membranes. Some gold particles were associated with or adjacent to electron-dense region observed in the lumen of most apicoplasts (Fig. 7C). However, in several of these cases, a sliver of membrane appeared nearby, suggesting potential membrane association. We did not see such apparently internal labeling of the apicoplast when analyzing APT1-HA (28) or V5-FtsH1 (29), although the intensity of labeling was lower. Rarely, as in Fig. 8E, the apicoplast showed only weak labeling.
FIG. 7.

Fine localization of ATrx1 to subcompartments of the apicoplast. Infected fibroblasts were processed for immunogold labeling to reveal the fine distribution of ATrx1-HA. Panels A to C show apicoplasts containing ATrx1-gold particles. Panel A shows evidence of a membrane-bound association of the protein (arrow). Panel B reveals the presence of gold particles on multiple membranes of the apicoplast (triple arrows). Panel C illustrates the protein on a more internal membrane of the organelle (arrow). Bars: 200 nm in panels A and C; 100 nm in panel B.
FIG. 8.

Ultrastructural detection of ATrx1 associated with the apicoplast and small cytoplasmic vesicles. Panels A and B show gold particles on the apicoplast (a) and on vesicles (v) surrounding the apicoplast. Arrows pinpoint intimate contact between both structures, a finding suggestive of fusion or fission events. Panels C and D reveal the presence of Atrx1 frequently associated with the ER and occasionally on the Golgi complex (Go, arrows). Panel E shows that the labeled vesicles are very abundant and morphologically distinct from parasite secretory organelles such as dense granules (dg), micronemes (m), and rhoptries (r). ATrx1 is observed predominantly at the limiting membrane of the vesicles, as illustrated in panels F and G. Bars: 200 nm in panels A to D; 500 nm in panel E; 100 nm in panels F and G.
ATrx1-HA also defined a set of highly abundant vesicles (Fig. 8A and E). The vesicles were typically spherical, 135 ± 10 nm in diameter, and with similar electron density as the material present in the lumen of the apicoplast (Fig. 8F and G). By monitoring the surface area of the vesicles relative to the parasite cytoplasm, we have calculated that these vesicles comprise ca. 3% of the total parasite surface area seen on a series of random sections. These vesicles were more frequently observed than several other characterized organelles of T. gondii, such as the dense granules. Several images showed them to be very close to or merging with the apicoplast (Fig. 8A and B). Most of the gold particles lay very close to the vesicular membrane, indicating that ATrx1 is predominantly membrane associated in the vesicles. About 25% of the 10-nm gold particles were more than two gold particles' width away from the vesicle membrane; this may indicate that a modest proportion is luminal or that the section was tangential. This distribution is very different from that seen upon immunogold staining of dense granule proteins, which are not membrane associated until after secretion. These show gold particles distributed throughout the organelle (33, 37).
The origin of the vesicles was addressed by examining the distribution of immunogold particles over the Golgi complex and ER. Dispersed immunogold labeling was frequently observed in cortical ER and perinuclear ER (Fig. 8C), but in a few sections it was also seen associated with or adjacent to the parasite cis-Golgi complex (Fig. 8D). This labeling was clearly less abundant than that observed on vesicles. Thus, here, as with previous studies (10, 28, 29, 48), the evidence does not support sorting of apicoplast proteins in the Golgi complex.
DISCUSSION
We report here the identification of an apicoplast-localized Trx family protein, ATrx1. The majority of apicoplast ATrx1-HA was detected in the peripheral compartments of the plastid by immunoelectron microscopy. A smaller fraction was seen apparently within the lumen of the organelle, but often traces of membranes were visible near the gold particles. As with apicoplast integral membrane proteins APT1 and FtsH1, ATrx1-HA was associated with vesicles. At the level of fluorescence microscopy, the vesicles appear as tubules and dots, which are more noticeable during the time of plastid enlargement and division. The ATrx1-HA bearing vesicles were more easily detected upon scans of T. gondii sections than were those revealed with antibodies to APT1-HA or V5-FtsH1, possibly as a result of higher expression or a longer period of expression within the cell cycle (or both). The lower immunogold labeling intensity of APT1-HA (28) and V5-FtsH1 (29) likely left some vesicles undetected. Since similar vesicles were observed with all three proteins, the appearance of such vesicles is not a response to the overproduction of a specific protein. Indeed, these vesicles are observed on normal embedding, but without the gold marker they can be erroneously considered as casual secretory vesicles, immature dense granules, or small lipid droplets (I. Coppens, unpublished data). As revealed by ATrx1-HA, the vesicles are surprisingly abundant, being more prevalent in random sections than dense granules. Future double-labeling studies will determine whether the same vesicles bear ATrx1, APT1, and FtsH1. Apicoplast luminal proteins have not been detected in vesicles as yet, although these vesicles are moderately electron dense and hence carry some type of internal cargo. While we believe it is likely that these vesicles are transport intermediates, it is also possible that they could fulfill some other, perhaps metabolic, functions.
Although immunoelectron microscopy suggests a membrane association for ATrx1 in the vesicles and potential association in the apicoplast, the molecular means of that association remains unclear. Biochemical fractionation indicates that rather than being an integral membrane protein, the preponderance of ATrx1 is instead either soluble or peripherally associated with the membrane. Given these biochemical properties, ATrx1 represents the first demonstrated intermembrane space protein of the apicoplast.
Previous studies have shown that at least some apicoplast integral membrane proteins lack the bipartite targeting sequence characteristic of soluble luminal proteins (28, 29, 39). Even though most ATrx1 is not integral to membranes, the protein also lacks the canonical targeting sequences. Nonetheless, analysis of deletion mutants and a fusion protein showed that information in the N-terminal extension of ATrx1 (which is absent from the fully processed form of the protein) is required and sufficient for trafficking to the apicoplast. Further, this region is functionally distinct from luminal targeting sequences, since it directs a soluble reporter, GFP, to the outer compartments of the apicoplast and not to the lumen of the organelle. Bioinformatic detection of the bipartite targeting sequence has been utilized to predict the apicoplast proteome in P. falciparum (17) and is generally used to evaluate protein localization to the apicoplast of T. gondii in silico. However, these results show that definition of the apicoplast proteome solely by informatic detection of a bipartite sequence will leave some gaps.
Because of its association with vesicle membranes and the peripheral compartments of the apicoplast, as well as its lack of a bipartite targeting sequence, it is tempting to speculate that ATrx1 traffics similarly to a membrane protein. Indeed, it may be transiently membrane integral, perhaps at the time of being packed into vesicles. The N-terminal sequence that is processed from ATrx1 could mediate trafficking, either by virtue of its own motifs or by interacting with a membrane protein. Alternatively, this presequence could function to keep ATrx1 as a zymogen until it arrives at the correct cellular location and is processed to its mature form(s), conceptually similar to the processing of apicoplast luminal proteins. The processing of ATrx1-HA appears to be complex, involving multiple events which include N-terminal cleavage, possibly at more than one site, and potentially additional modifications such as phosphorylation or glycosylation. These events could occur at different sites en route to and/or within the apicoplast. Bands detected by MAb 11G8 all have cognates which are detected by anti-HA, suggesting that ATrx1 is not proteolytically cleaved at the C terminus (the HA tag is at the C terminus). Interestingly, we have recently observed that both the N and the C termini of the apicoplast membrane protein FtsH1 are processed (A. Karnataki et al., unpublished data). Together, these data suggest that the protein processing associated with the apicoplast is more complex than previously suspected.
In addition to multiple “orphan” Trxs, many Trxs cluster into three subfamilies on the basis of sequence similarity: Trxs, glutaredoxins, and nucleoredoxins. ATrx1 belongs to the NRX subfamily of Trxs, which is characterized by the tryparedoxin variant of the Trx domain. Indeed, at the sequence level, ATrx1 is most similar to nucleoredoxin, the prototype NRX protein. Nucleoredoxin was initially reported to be nuclear (32), and more recently cytosolic (18), perhaps shuttling between the two compartments. It appears to function in signal transduction, particularly in the WNT pathway, presumably by altering the oxidation state of other signaling molecules in the pathway (18, 25). In contrast, tryparedoxin functions in the trypanothione redox pathway of trypanosomatids, which is not present in apicomplexans. Clearly, the cellular function of ATrx1 is distinct, albeit not yet known. Furthermore, the various isoforms of ATrx1 may perform similar or different cellular functions. Although we have not confirmed activity of the protein, the active-site motif is present in the second of the two Trx domains. It is not unusual for members of the Trx superfamily to have multiple Trx domains, some of which lack the redox active site (19).
The reductant for nucleoredoxin proteins is not known. Trxs can be reduced through the action of ferredoxin Trx reductases or NADPH Trx reductases, which exist as multiple isozymes in plants (38). No reductase of either type has been identified in the apicoplast as yet, although multiple Trx reductases are evident from the genome sequence of T. gondii.
In chloroplasts, Trxs reduce the disulfide groups of other proteins, stimulating a variety of processes ranging from photosynthesis to protein synthesis in the organelle (7). Several molecules that require activation by or typically associate with Trx are known to be localized to the apicoplast: e.g., a Trx-dependent peroxidase (40), acetyl coenzyme A carboxylase (26), superoxide dismutase (40), and phosphoglycerate kinase (15). However, these enzymes have been proposed to be reduced directly by ferredoxin (50). Furthermore, the enzymes are thought to reside in the apicoplast lumen, whereas most ATrx1 is associated with peripheral apicoplast compartments and vesicles that likely represent transport intermediates. However, proteomic analysis has shown that the preponderance of Trx targets in the photosynthetic cyanobacterium Synechocystis are membrane proteins, many of which are localized to chloroplasts in plants (36). Interestingly, one of the targets is the FtsH protease, which we have also identified as an apicoplast membrane protein in T. gondii (29). Other possible functions of ATrx1, by analogy with other Trxs, include protection against oxidative stresses or redox-related signaling. Candidate ATrx1 orthologues exist in Eimeria tenella (www.GeneDB.org) and Babesia bovis (BBOV_II002190), although the latter lacks the typical active-site cysteines (see Fig. S2 in the supplemental material). (5). Interestingly, orthologues are apparently absent in other plastid-containing apicomplexans sequenced thus far (Plasmodium and Theileria). Whether the functions of ATrx1 are fulfilled by another, as-yet-unknown apicoplast-localized Trx in Plasmodium and Theileria, or whether its phylogenetic distribution reflects differences in apicoplast metabolism remains to be seen.
Supplementary Material
Acknowledgments
We thank Yuko Ogata at the SBRI Global Health Biotechnology Center for mass spectrometry, Marc Pypaert of the Yale Center for Cell and Molecular Imaging for excellent advice and assistance with immunoelectron microscopy, John Boothroyd for support for the initial preparation of MAbs, and Pashmi Vaney for assistance with cloning and parasite culture. Antibodies were kindly provided by Jay Bangs, John Boothroyd, Con Beckers, Vern Carruthers, Keith Joiner, and Tim Stedmann. Preliminary genomic and/or cDNA sequence data for T. gondii was accessed via http://ToxoDB.org and http://www.tigr.org/tdb/t_gondii/. T. gondii genomic data were provided by The Institute for Genomic Research (supported by the NIH grant AI05093) and by the Sanger Center (supported by The Wellcome Trust).
This study was supported in part by NIH R01 AI50506 (to M.P.), AI21423 (to John Boothroyd), AI064616 (to P.J.B.), and AI060767 (to I.C.).
Footnotes
Published ahead of print on 27 June 2008.
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1.Balmer, Y., A. Koller, G. del Val, W. Manieri, P. Schurmann, and B. B. Buchanan. 2003. Proteomics gives insight into the regulatory function of chloroplast thioredoxins. Proc. Natl. Acad. Sci. USA 100370-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bangs, J. D., L. Uyetake, M. J. Brickman, A. E. Balber, and J. C. Boothroyd. 1993. Molecular cloning and cellular localization of a BiP homologue in Trypanosoma brucei. Divergent ER retention signals in a lower eukaryote. J. Cell Sci. 1051101-1113. [DOI] [PubMed] [Google Scholar]
- 3.Bermudes, D., K. R. Peck, M. A. Afifi, C. J. Beckers, and K. A. Joiner. 1994. Tandemly repeated genes encode nucleoside triphosphate hydrolase isoforms secreted into the parasitophorous vacuole of Toxoplasma gondii. J. Biol. Chem. 26929252-29260. [PubMed] [Google Scholar]
- 4.Bradley, P. J., C. Ward, S. J. Cheng, D. L. Alexander, S. Coller, G. H. Coombs, J. D. Dunn, D. J. Ferguson, S. J. Sanderson, J. M. Wastling, and J. C. Boothroyd. 2005. Proteomic analysis of rhoptry organelles reveals many novel constituents for host-parasite interactions in Toxoplasma gondii. J. Biol. Chem. 403425-34258. [DOI] [PubMed] [Google Scholar]
- 5.Brayton, K. A., A. O. Lau, D. R. Herndon, L. Hannick, L. S. Kappmeyer, S. J. Berens, S. L. Bidwell, W. C. Brown, J. Crabtree, D. Fadrosh, T. Feldblum, H. A. Forberger, B. J. Haas, J. M. Howell, H. Khouri, H. Koo, D. J. Mann, J. Norimine, I. T. Paulsen, D. Radune, Q. Ren, R. K. Smith, Jr., C. E. Suarez, O. White, J. R. Wortman, D. P. Knowles, Jr., T. F. McElwain, and V. M. Nene. 2007. Genome sequence of Babesia bovis and comparative analysis of apicomplexan hemoprotozoa. PLoS Pathog. 31401-1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brydges, S. D., G. D. Sherman, S. Nockemann, A. Loyens, W. Daubener, J. F. Dubremetz, and V. B. Carruthers. 2000. Molecular characterization of TgMIC5, a proteolytically processed antigen secreted from the micronemes of Toxoplasma gondii. Mol. Biochem. Parasitol. 11151-66. [DOI] [PubMed] [Google Scholar]
- 7.Buchanan, B. B., and Y. Balmer. 2005. Redox regulation: a broadening horizon. Annu. Rev. Plant Biol. 56187-220. [DOI] [PubMed] [Google Scholar]
- 8.Carvalho, A. P., P. A. Fernandes, and M. J. Ramos. 2006. Similarities and differences in the thioredoxin superfamily. Prog. Biophys. Mol. Biol. 91229-248. [DOI] [PubMed] [Google Scholar]
- 9.Dahl, E. L., J. L. Shock, B. R. Shenai, J. Gut, J. L. DeRisi, and P. J. Rosenthal. 2006. Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob. Agents Chemother. 503124-3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeRocher, A., B. Gilbert, J. E. Feagin, and M. Parsons. 2005. Dissection of brefeldin A-sensitive and -insensitive steps in apicoplast protein targeting. J. Cell Sci. 118565-574. [DOI] [PubMed] [Google Scholar]
- 11.DeRocher, A., C. B. Hagen, J. E. Froehlich, J. E. Feagin, and M. Parsons. 2000. Analysis of targeting sequences demonstrates that trafficking to the Toxoplasma gondii plastid branches off the secretory system. J. Cell Sci. 1133969-3977. [DOI] [PubMed] [Google Scholar]
- 12.Donald, R. G., and D. S. Roos. 1998. Gene knockouts and allelic replacements in Toxoplasma gondii: HXGPRT as a selectable marker for hit-and-run mutagenesis. Mol. Biochem. Parasitol. 91295-305. [DOI] [PubMed] [Google Scholar]
- 13.Emanuelsson, O., S. Brunak, H. G. von, and H. Nielsen. 2007. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2953-971. [DOI] [PubMed] [Google Scholar]
- 14.Fichera, M. E., and D. S. Roos. 1997. A plastid organelle as a drug target in apicomplexan parasites. Nature 390407-409. [DOI] [PubMed] [Google Scholar]
- 15.Fleige, T., K. Fischer, D. J. Ferguson, U. Gross, and W. Bohne. 2007. Carbohydrate metabolism in the Toxoplasma gondii apicoplast: localization of three glycolytic isoenzymes, the single pyruvate dehydrogenase complex and a plastid phosphate translocator. Eukaryot. Cell 6984-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Folsch, H., M. Pypaert, P. Schu, and I. Mellman. 2001. Distribution and function of AP-1 clathrin adaptor complexes in polarized epithelial cells. J. Cell Biol. 152595-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foth, B. J., S. A. Ralph, C. J. Tonkin, N. S. Struck, M. Fraunholz, D. S. Roos, A. F. Cowman, and G. I. McFadden. 2003. Dissecting apicoplast targeting in the malaria parasite Plasmodium falciparum. Science 299705-708. [DOI] [PubMed] [Google Scholar]
- 18.Funato, Y., T. Michiue, M. Asashima, and H. Miki. 2006. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat. Cell Biol. 8501-508. [DOI] [PubMed] [Google Scholar]
- 19.Funato, Y., and H. Miki. 2007. Nucleoredoxin, a novel thioredoxin family member involved in cell growth and differentiation. Antioxid. Redox. Signal. 91035-1057. [DOI] [PubMed] [Google Scholar]
- 20.Gajria, B., A. Bahl, J. Brestelli, J. Dommer, S. Fischer, X. Gao, M. Heiges, J. Iodice, J. C. Kissinger, A. J. Mackey, D. F. Pinney, D. S. Roos, C. J. Stoeckert, Jr., H. Wang, and B. P. Brunk. 2007. ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic Acids Res. 36D553-D556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert, L. A., S. Ravindran, J. M. Turetzky, J. C. Boothroyd, and P. J. Bradley. 2007. Toxoplasma gondii targets a protein phosphatase 2C to the nuclei of infected host cells. Eukaryot. Cell 673-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodman, C. D., and G. I. McFadden. 2007. Fatty acid biosynthesis as a drug target in apicomplexan parasites. Curr. Drug Targets 815-30. [DOI] [PubMed] [Google Scholar]
- 23.Goodman, C. D., V. Su, and G. I. McFadden. 2007. The effects of antibacterials on the malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 152181-191. [DOI] [PubMed] [Google Scholar]
- 24.Hehl, A. B., C. Lekutis, M. E. Grigg, P. J. Bradley, J. F. Dubremetz, E. Ortega-Barria, and J. C. Boothroyd. 2000. Toxoplasma gondii homologue of plasmodium apical membrane antigen 1 is involved in invasion of host cells. Infect. Immun. 687078-7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirota, K., M. Matsui, M. Murata, Y. Takashima, F. S. Cheng, T. Itoh, K. Fukuda, and J. Yodoi. 2000. Nucleoredoxin, glutaredoxin, and thioredoxin differentially regulate NF-κB, AP-1, and CREB activation in HEK293 cells. Biochem. Biophys. Res. Commun. 274177-182. [DOI] [PubMed] [Google Scholar]
- 26.Jelenska, J., M. J. Crawford, O. S. Harb, E. Zuther, R. Haselkorn, D. S. Roos, and P. Gornicki. 2001. Subcellular localization of acetyl-CoA carboxylase in the apicomplexan parasite Toxoplasma gondii. Proc. Natl. Acad. Sci. USA 982723-2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jomaa, H., J. Wiesner, S. Sanderbrand, B. Altincicek, C. Weidemeyer, M. Hintz, I. Türbachova, M. Eberl, J. Zeidler, H. K. Lichtenthaler, D. Soldati, and E. Beck. 1999. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 2851573-1576. [DOI] [PubMed] [Google Scholar]
- 28.Karnataki, A., A. DeRocher, I. Coppens, C. Nash, J. E. Feagin, and M. Parsons. 2007. Cell cycle-regulated vesicular trafficking of Toxoplasma APT1, a protein localized to multiple apicoplast membranes. Mol. Microbiol. 631653-1668. [DOI] [PubMed] [Google Scholar]
- 29.Karnataki, A., A. E. Derocher, I. Coppens, J. E. Feagin, and M. Parsons. 2007. A membrane protease is targeted to the relict plastid of Toxoplasma via an internal signal sequence. Traffic 81543-1553. [DOI] [PubMed] [Google Scholar]
- 30.Kohler, S., C. F. Delwiche, P. W. Denny, L. G. Tilney, P. Webster, R. J. Wilson, J. D. Palmer, and D. S. Roos. 1997. A plastid of probable green algal origin in Apicomplexan parasites. Science 2751485-1489. [DOI] [PubMed] [Google Scholar]
- 31.Krauth-Siegel, R. L., S. K. Meiering, and H. Schmidt. 2003. The parasite-specific trypanothione metabolism of Trypanosoma and Leishmania. Biol. Chem. 384539-549. [DOI] [PubMed] [Google Scholar]
- 32.Kurooka, H., K. Kato, S. Minoguchi, Y. Takahashi, J. Ikeda, S. Habu, N. Osawa, A. M. Buchberg, K. Moriwaki, H. Shisa, and T. Honjo. 1997. Cloning and characterization of the nucleoredoxin gene that encodes a novel nuclear protein related to thioredoxin. Genomics 39331-339. [DOI] [PubMed] [Google Scholar]
- 33.Lecordier, L., C. Mercier, L. D. Sibley, and M. F. Cesbron-Delauw. 1999. Transmembrane insertion of the Toxoplasma gondii GRA5 protein occurs after soluble secretion into the host cell. Mol. Biol. Cell 101277-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ludemann, H., M. Dormeyer, C. Sticherling, D. Stallmann, H. Follmann, and R. L. Krauth-Siegel. 1998. Trypanosoma brucei tryparedoxin, a thioredoxin-like protein in African trypanosomes. FEBS Lett. 431381-385. [DOI] [PubMed] [Google Scholar]
- 35.Mann, T., and C. Beckers. 2001. Characterization of the subpellicular network, a filamentous membrane skeletal component in the parasite Toxoplasma gondii. Mol. Biochem. Parasitol. 115257-268. [DOI] [PubMed] [Google Scholar]
- 36.Mata-Cabana, A., F. J. Florencio, and M. Lindahl. 2007. Membrane proteins from the cyanobacterium Synechocystis sp. PCC 6803 interacting with thioredoxin. Proteomics 73953-3963. [DOI] [PubMed] [Google Scholar]
- 37.Mercier, C., M. F. Cesbron-Delauw, and L. D. Sibley. 1998. The amphipathic alpha helices of the Toxoplasma protein GRA2 mediate post-secretory membrane association. J. Cell Sci. 111(Pt. 15)2171-2180. [DOI] [PubMed] [Google Scholar]
- 38.Meyer, Y., W. Siala, T. Bashandy, C. Riondet, F. Vignols, and J. P. Reichheld. 2007. Glutaredoxins and thioredoxins in plants. Biochim. Biophys. Acta 1783589-600. [DOI] [PubMed] [Google Scholar]
- 39.Mullin, K. A., L. Lim, S. A. Ralph, T. P. Spurck, E. Handman, and G. I. McFadden. 2006. Membrane transporters in the relict plastid of malaria parasites. Proc. Natl. Acad. Sci. USA 1039572-9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pino, P., B. J. Foth, L. Y. Kwok, L. Sheiner, R. Schepers, T. Soldati, and D. Soldati-Favre. 2007. Dual targeting of antioxidant and metabolic enzymes to the mitochondrion and the apicoplast of Toxoplasma gondii. PLoS Pathog. 3e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powis, G., and D. L. Kirkpatrick. 2007. Thioredoxin signaling as a target for cancer therapy. Curr. Opin. Pharmacol. 7392-397. [DOI] [PubMed] [Google Scholar]
- 42.Ralph, S. A., G. G. van Dooren, R. F. Waller, M. J. Crawford, M. J. Fraunholz, B. J. Foth, C. J. Tonkin, D. S. Roos, and G. I. McFadden. 2004. Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat. Rev. Microbiol. 2203-216. [DOI] [PubMed] [Google Scholar]
- 43.Roos, D. S., R. G. Donald, N. S. Morrisette, and A. L. Moulton. 1994. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 4527-63. [DOI] [PubMed] [Google Scholar]
- 44.Schlecker, T., A. Schmidt, N. Dirdjaja, F. Voncken, C. Clayton, and R. L. Krauth-Siegel. 2005. Substrate specificity, localization, and essential role of the glutathione peroxidase-type tryparedoxin peroxidases in Trypanosoma brucei. J. Biol. Chem. 28014385-14394. [DOI] [PubMed] [Google Scholar]
- 45.Schwartzman, J. D. 1986. Inhibition of a penetration-enhancing factor of Toxoplasma gondii by monoclonal antibodies specific for rhoptries. Infect. Immun. 51760-764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Striepen, B., M. J. Crawford, M. K. Shaw, L. G. Tilney, F. Seeber, and D. S. Roos. 2000. The plastid of Toxoplasma gondii is divided by association with the centrosomes. J. Cell Biol. 1511423-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tonkin, C. J., D. S. Roos, and G. I. McFadden. 2006. N-terminal positively charged amino acids, but not their exact position, are important for apicoplast transit peptide fidelity in Toxoplasma gondii. Mol. Biochem. Parasitol. 150192-200. [DOI] [PubMed] [Google Scholar]
- 48.Tonkin, C. J., N. S. Struck, K. A. Mullin, L. M. Stimmler, and G. I. McFadden. 2006. Evidence for Golgi-independent transport from the early secretory pathway to the plastid in malaria parasites. Mol. Microbiol. 61614-630. [DOI] [PubMed] [Google Scholar]
- 49.van Dooren, G. G., V. Su, M. C. D'Ombrain, and G. I. McFadden. 2002. Processing of an apicoplast leader sequence in Plasmodium falciparum and the identification of a putative leader cleavage enzyme. J. Biol. Chem. 27723612-23619. [DOI] [PubMed] [Google Scholar]
- 50.Vollmer, M., N. Thomsen, S. Wiek, and F. Seeber. 2001. Apicomplexan parasites possess distinct nuclear encoded but apicoplast-localized plant-type ferredoxin-NADP+-reductase and ferredoxin. J. Biol. Chem. 2765483-5490. [DOI] [PubMed] [Google Scholar]
- 51.Waller, R. F., P. J. Keeling, R. G. K. Donald, B. Striepen, E. Handman, N. Lang-Unnasch, A. F. Cowman, G. S. Besra, D. S. Roos, and G. McFadden. 1998. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 9512352-12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wiesner, J., and F. Seeber. 2005. The plastid-derived organelle of protozoan human parasites as a target of established and emerging drugs. Expert Opin. Ther. Targets 923-44. [DOI] [PubMed] [Google Scholar]
- 53.Wilson, R. J., P. W. Denny, P. R. Preiser, K. Rangachari, K. Roberts, A. Roy, A. Whyte, M. Strath, D. J. Moore, P. W. Moore, and D. H. Williamson. 1996. Complete gene map of the plastid-like DNA of the malaria parasite Plasmodium falciparum. J. Mol. Biol. 261155-172. [DOI] [PubMed] [Google Scholar]
- 54.Yung, S., T. R. Unnasch, and N. Lang-Unnasch. 2001. Analysis of apicoplast targeting and transit peptide processing in Toxoplasma gondii by deletional and insertional mutagenesis. Mol. Biochem. Parasitol. 11811-21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.