

Abstract
Stearoyl-acyl carrier protein desaturase (Δ9D) catalyzes the O2 and 2e- dependent desaturation of stearoyl-acyl carrier protein (18:0-ACP) to yield oleoyl-ACP (18:1-ACP). The 2e- are provided by essential interactions with reduced plant-type [2Fe-2S] ferredoxin (Fd). We have investigated the protein-protein interface involved in the Fd-Δ9D complex by use of chemical cross-linking, site-directed mutagenesis, steady-state kinetic approaches and molecular docking studies. Treatment of the different proteins with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and N-hydroxysuccinimide revealed that carboxylate residues from Fd and lysine residues from Δ9D contribute to the cross-linking. The single substitutions of K60A, K56A, and K230A on Δ9D decreased the kcat/KM for Fd by 4-, 22- and 2,400-fold, respectively, as compared to wt Δ9D and a K41A substitution. The double substitution K56A/K60A decreased the kcat/KM for Fd by 250-fold, while the triple mutation K56A/K60A/K230A decreased the kcat/KM for Fd by at least 700,000-fold. These results strongly implicate the triad of K56, K60 and K230 of Δ9D in the formation of a catalytic complex with Fd. Molecular docking studies indicate that electrostatic interactions between K56 and K60 and carboxylate groups on Fd may situate the [2Fe-2S] cluster of Fd near to W62, a surface residue that is structurally conserved in both ribonucleotide reductase and mycobacterial putative acyl-ACP desaturase DesA2. Owing to the considerably larger effects on catalysis, K230 appears to have other contributions to catalysis arising from its positioning in helix-7 and its close spatial location to the diiron center ligands E229 and H232. These results are considered in the light of the presently available models for Fd-mediated electron transfer in Δ9D and other protein-protein complexes.
Stearoyl-acyl carrier protein Δ9 desaturase (Δ9D)>1 catalyzes the O2- and NAD(P)H-dependent insertion of a cis double bond between carbons 9 and 10 of 18:0-ACP to yield 18:1-ACP. This soluble, homodimeric enzyme (Mr 84 000) contains a catalytically essential diiron cofactor in each subunit. The diiron center is maintained in the active site by endogenous ligands derived from two copies of the sequence motif (D/E)-X∼40EX2H separated by ∼100 amino acids (1). Related diiron binding motifs are found in other desaturases (2), the R2 component of ribonucleotide diphosphate reductase (3) and the bacterial hydrocarbon hydroxylases (4). The catalytic cycle for these enzymes consists of substrate binding, electron transfer to form a diferrous center, and O2 activation as essential prerequisites to substrate oxidation (5-7). In addition, ferritin (8) and rubrerythrin (9) have related diiron centers but differ in their catalytic cycles.
An acyl-ACP and a reduced plant-type [2Fe-2S] ferredoxin are both required for efficient Δ9D catalysis (10). ACP, a small, negatively charged protein, enhances the KM for the 18-carbon acyl chain relative to the comparable acyl-CoA (11), while kcat and kcat/KM are enhanced by hydrophobic contributions from the acyl chain (10). The addition of stoichiometric 18:0-ACP to sodium dithionite-reduced Δ9D (4e- Δ9D) changed the coordination geometry and redox-active orbitals of the diiron center (12) such that the reactivity with O2 might be enhanced (13) and, indeed, the addition O2 to the complex of 18:0-ACP and 4e- Δ9D yielded a quasi-stable μ-1,2 peroxo species (14, 15). However, this peroxo species ultimately decayed without formation of 18:1-ACP and without release of H2O2 (14), suggesting that key elements contributing to catalysis were missing from the simplified reaction system.
In contrast to the results described above with chemically reduced Δ9D, Fd is a catalytically competent electron donor for Δ9D during the conversion of 18:0-ACP to 18:1-ACP (16). For example, rapid mix of the Fd-reduced enzyme-substrate complex with aerobic buffer resulted in a burst phase (kburst = 95 s-1) of product formation accounting for ∼90% of the turnover expected from one subunit in the dimeric protein. This rapid phase was followed by a slower phase (klinear = 4.0 s-1) of product formation corresponding to the turnover expected from the second subunit. The reaction of the oxidized enzyme-substrate complex with excess reduced Fd resulted in a slower, linear rate (kods = 3.4 s-1) of product formation over ∼1.5 turnovers per Δ9D active site, implicating Fd-Δ9D complex formation as a distinct step of the single turnover reaction. However, as steady-state catalysis has kcat ≈ 0.5 s-1, product release appears to be the rate-limiting step in catalysis.
The significant role for Fd in catalytic desaturation was unexpected, as dithionite-reduced preparations of methane monooxygenase (17-20) and ribonucleotide reductase (21) were competent for methane oxidation and tyrosyl radical formation, respectively. Thus, unlike other enzymes in the diiron enzyme superfamily, Δ9D apparently requires electron transfer from the biological electron donor and this requirement may be further mediated by interactions with acyl-ACP.
This work examines potential sites of interaction between Fd and Δ9D by using chemical cross-linking and steady-state kinetic analysis. Figure 1 shows the spatial relationship of the lysine residues investigated during this work in the 3-dimensional structure of Δ9D. The results implicate participation of carboxylate residues from Fd and a triad of lysine residues on the surface of Δ9D in formation of the catalytic complex. Two of these lysine residues, K56 and K60, cluster near to W62, a surface tryptophan residue that has structurally conserved homologs in ribonucleotide reductase (22) and the putative acyl-ACP desaturase DesA2 from Mycobacterium tuberculosis (23). The third, K230, is hydrogen bonded to E233 along a conserved helix opposite to the diiron ligands E229 and H232. Mutagenesis of these three lysine residues to alanine essentially eliminates the ability of Δ9D to recognize Fd as an electron donor. In the discussion, we consider how protein-protein interactions involving these residues might contribute to Δ9D catalysis.
Figure 1.
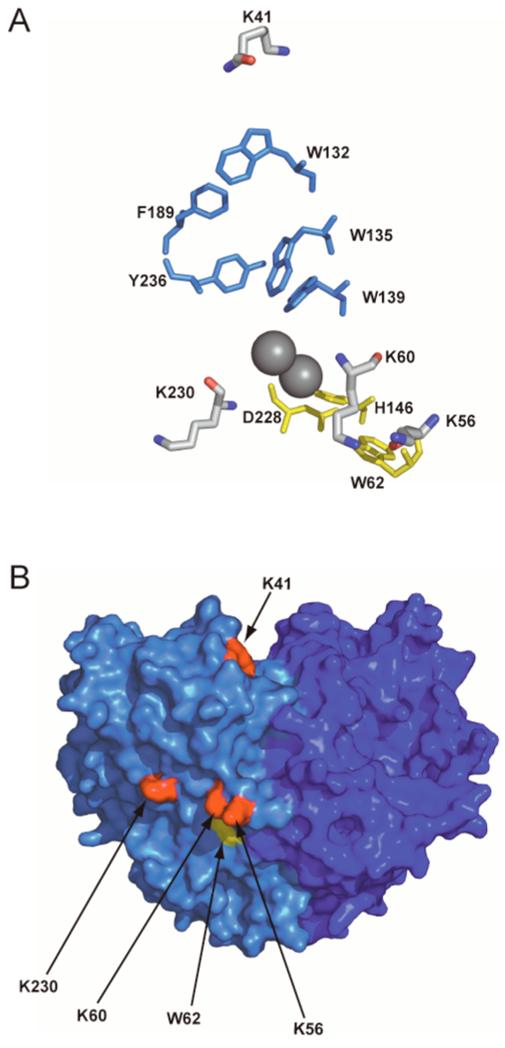
(A) Amino acids involved in putative electron transfer pathways to the diiron center (grey spheres) of Δ9D. One proposed pathway, consisting of W62, D228 and H146 (yellow), extends from the surface of Δ9D, and is close to K56, K60 and K230. The other proposed electron transfer pathway consists of W132, F189, Y236, W135 and W139 (blue) and is closest to K41. (B) Surface representation of the Δ9D dimer showing the relative location of the targeted surface lysines (red) in relation to W62 (yellow). The two subunits of Δ9D are shown in different shades of blue.
MATERIALS AND METHODS
Enzymes
Fd was expressed, purified, and characterized as previously described (24). The concentration of Δ9D active sites was determined using ε340 = 4200 M-1 cm-1 per diiron center, and cross-correlated by determination of protein concentration and total iron (1). For the preparations of Δ9D and each of the mutated isoforms of Δ9D studied in this work, these quantifications indicate greater than 95% occupation of the diiron active sites in the dimeric protein. Furthermore, the specific activity of the wt Δ9D preparations used in this work matched the range observed in other preparations of the enzyme (10). Recombinant Escherichia coli and spinach ACP were expressed, purified, phosphopantetheinylated, and acylated as previously described (10, 14). Acyl-chains were reductively cleaved, extracted, derivatized, and analyzed by GC-MS as previously described (10).
Chemicals
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) was from Pierce (Rockford, IL). N-hydroxysuccinimide and buffers were from Sigma (Saint Louis, MO). HPLC grade acetic acid was from Fisher Scientific (Pittsburgh, PA). Methanol and acetonitrile (HPLC grade) were from Aldrich.
Cross-Linking Reactions
Δ9D and Fd were cross-linked in 25 mM HEPES, pH 7.4, containing 17 mM NaCl using a procedure based upon that of Sehgal, et al (25). Δ9D was added to a final concentration of 12.5 μM, Fd was added to a final concentration of 50 μM, and 18:0-ACP was added to a final concentration of 25 μM. The final reaction volume was 50 μL. A stock solution of EDC (50 mM in 25 mM HEPES, pH 7.4) was prepared ∼5 min prior to use and was added to a final concentration of 5 mM to a buffer solution containing either Fd or Δ9D. A stock solution of NHS (50 mM in 25 mM HEPES, pH 7.4) was then added to the EDC-treated protein to a final concentration of 10 mM. This reaction was incubated at room temperature for 5 min, BME (1.4 M in H2O) was added to a final concentration of 20 mM, and the protein solution was again incubated at room temperature. After 5 min, Fd, Δ9D, or pre-mixed Δ9D-18:0-ACP was added to the reaction and the reaction mixture was incubated for 2 h at 25°C with shaking. After this incubation period, hydroxylamine (100 or 300 mM in H2O) was added to the cross-linking reaction to a final concentration of 10 mM. Approximately 15-20 μL of the cross-linking reaction was diluted into 2X SDS sample buffer and analyzed by SDS-PAGE using BioRAD Tris-HCl Ready-Gels, Criterion gels (BioRad, Hercules, CA) or 10% Tris-tricine gels. The SDS-PAGE gels were stained with Coomassie Brilliant Blue and destained in 10% acetic acid/30% methanol.
Site-Directed Mutagenesis
All of the Δ9D variants carrying the different lysine to alanine single or multiple substitutions were prepared according to the QuikChange protocol (Stratagene, La Jolla, CA). The K41A mutation was generated with the primer 5′-ccccaaaagattgagatctttGCAtccctagacaattgggc -3′ and its reverse complement (the mutated codon is indicated in capital letters). The K56A mutation was generated with the primer 5′-gaacattctggttcatctGGCgccagttgagaaatgttggcaaccgc-3′ and its reverse complement. The K60A mutation was generated with the primer 5′- ctgaagccagttgagGCAtgttggcaaccgc -3′ and its reverse complement, and the double mutation, K56A/K60A, was prepared from the K60A background with the primer 5′-ctggttcatctGGCgccagttgagGCA-3′ and its reverse complement. The K230A mutation was constructed with the primer 5′-gtacaattgctgcagatgagAAGcgccatgagacagcctac-3′ and its reverse complement. The triple mutation K56A/K60A/K230A was generated using the same primer as for K230A with the gene containing the K56A/K60A mutations as template. Transformed clones were selected and sequenced by Big Dye DNA sequencing at the University of Wisconsin Biotechnology Center to identify individual clones containing the correct gene sequence.
Protein Expression
For protein expression, 1 μL of plasmid DNA (∼0.2 μg) was transformed into E. coli BL21(DE3) cells by electroporation and plated onto Luria-Bertani plates containing 50 μg mL-1 kanamycin. Kanamycin-resistant transformants (∼3 colonies) were used to inoculate 5 mL Luria-Bertani medium containing 50 μg mL-1 kanamycin and 1% (w/v) glucose. This culture was grown at 37°C with shaking (250 rpm). After the 5 mL culture reached an OD600 ∼0.2, 1 mL was used to inoculate 50 mL of Luria-Bertani medium containing 50 μg mL-1 kanamycin and 1% (w/v) glucose, and the culture was incubated at 37°C. After ∼4 h, this culture reached an OD600∼0.5 and 1 mL was used to inoculate each of six 2 L flasks containing 500 mL of Luria Bertani medium supplemented with 50 μg mL-1 kanamyacin and 1% (w/v) glucose. These cultures were incubated at 37°C, with shaking at 250 rpm, for ∼4 h or until they reached an OD600 of ∼0.5. The incubation temperature was reduced to 25°C and the cultures were induced by the addition of isopropyl-β-D-thiogalactopyranoside to a final concentration of 1 mM. At induction, each 500 mL culture was supplemented with 1 mL of 40 mM FeSO4 and 1 g of Casamino acids. After ∼12 h, the cells were harvested by centrifugation for 15 min at 4 400 g. The cell pellets were washed with 50 mM phosphate, pH 7.0, pelleted by centrifugation for 15 min at 39 000 g, and stored at -80°C. The growth procedure yielded ∼1.0 g L-1 of wet cell paste.
Protein Purification
The Δ9D variants bearing the different Lys mutations were purified from E. coli using a procedure similar to that previously described (26). Approximately 3 g of frozen cell paste was thawed in a small steel beaker (∼10 mL) and resuspended in 25 mM HEPES, pH 8.0. The cells were incubated on ice for 10 min after the addition of 25 mg each of lysozyme, RNase, and DNase to the cell suspension. The cell mixture was sonicated for a total of 1 min using 10-s pulses (Fisher Model 550 Sonic Dismembrator, 1/8-in horn, 45% maximum output). During sonication, the temperature of the cell suspension was maintained below 10°C by placing the beaker in an ice water bath containing NaCl. The suspension was diluted to 50 mL in 25 mM HEPES, pH 8.0 and centrifuged for 1 h at 39 000 g to remove cell debris. The supernatant was loaded onto a 30 mL Q-Sepharose column equilibrated in 25 mM HEPES, pH 8.0. The column was washed with 50 mL of 25 mM HEPES, pH 8.0 and the protein was eluted in 0.2-L linear gradient from 0 to 0.23 M NaCl in 25 mM HEPES, pH 8.0, at a linear flow rate of 7.6 cm h-1. Fractions were analyzed by SDS-PAGE and fractions containing Δ9D were pooled and concentrated by ultrafiltration (YM30 membrane, Amicon Inc., Beverly, MA). The concentrated sample was loaded onto a HiPrep 26/60 Sepharacyl S-100 column (Pharmacia LKB Biotechnology Inc., Piscataway NJ) equilibrated with 25 mM HEPES, pH 7.8, containing 50 mM NaCl and eluted at 6.8 cm h-1. Fractions containing Δ9D were identified by SDS-PAGE, pooled, and concentrated by ultrafiltration. Small aliquots of purified protein were frozen in liquid nitrogen and stored at -80°C.
Steady-State Evaluation of 18:1-ACP Formation
Enzyme reactions were performed in 25 mM HEPES buffer, pH 7.4, containing 17 mM NaCl. Assays contained varying concentrations of Fd, 40 μM of E. coli or spinach 18:0-ACP, 1.5 μM of ferredoxin reductase, and 0.02 or 0.04 nmol of Δ9D in a total volume of 500 μL. The reaction was initiated by the addition of NADPH to a final concentration of 400 μM. Aliquots were removed at timed intervals and quenched by rapid mixing with 200 μL of THF. Initial reaction velocities were determined using linear least-squares fitting from the time-dependence for appearance of the 18:1 products in reactions as determined by GC/EI-MS. The steady-state parameters kcat and KM were determined by nonlinear least squares fitting of the initial desaturation velocity and substrate concentration data to the Michaelis-Menten equation
| (1) |
using Kaleidagraph (Abelbeck Software, Reading, PA).
Mass Spectrometry
Cross-linking reactions were dialyzed in 1 L of 10 mM ammonium acetate, pH 5.0, for ∼16 h. The MALDI-MS analyses were obtained from either an α-cyano or a sinapinic acid matrix using a Bruker BIFLEX III spectrometer equipped with a 337 nm nitrogen laser.
Δ9D-Fd Docking Model
The protein-protein docking web server ClusPro (http://nrc.bu.edu/cluster/) was used to obtain 3D-models of the Fd- and ACP- complexes with Δ9D (27, 28). This server provides algorithms that filter docked conformations based on shape complementarity, desolvation and electrostatic energies. The PDB file 1AFR containing the coordinates for Δ9D was used as the static “receptor “ molecule and the files 1FXA containing the coordinates for [2Fe-2S] ferredoxin (29) and the coordinates for ACP2 were separately used as the moving “ligand” molecules. The top 60 docked conformations of Fd binding and the top 20 docked conformations of ACP from ZDOCK evaluations were examined in detail.
RESULTS
Covalent Cross-Linking of Fd and Δ9D
The combination of the zero-length cross-linker EDC and NHS can be used to define the source of the carboxyl and amine groups contributing to the cross-link (25). Figure 2 shows the reaction mechanism and sequence of additions used to perform cross-linking of Fd and Δ9D. In the first step of this sequence, carboxylate groups on protein P1 were reacted with EDC to form a reactive O-acylurea intermediate. By immediate reaction of the unstable O-acylurea intermediate with NHS (Figure 2A), a more stable N-hydroxysuccinimido ester derivative was formed from the activated carboxyl groups. After quenching of excess EDC by the addition of BME, the cross-linking reaction was initiated by addition of protein P2. Amide linkages were formed by displacement of NHS from P1 by an amine group from P2 (Figure 2B).
Figure 2.
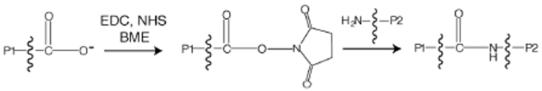
A summary of the sequence of reactions used for EDC-catalyzed cross-linking. (A) The carboxylate groups from one protein (P1) are activated with EDC and then reacted with NHS to yield a relatively stable succinimide-ester. (B) The NHS-activated carboxylate group on P1 then reacts with an amine group from protein P2 to generate a new peptide bond between proteins P1 and P2 upon displacement of the succinimide group.
Figure 3 shows the results from several control reactions performed in order to establish the behavior of the individual proteins in the presence of the cross-linking reagents. Lanes 2, 4 and 7 show the products of reactions containing untreated Fd (lane 2), Fd treated with BME-quenched EDC (lane 4) and EDC-activated Fd (lane 7). The products of these reactions were comparable based on SDS-PAGE analysis. Specifically, Fd appeared as an approximately equal distribution of two bands with apparent masses of ∼10 kDa and ∼22 kDa after treatment with BME at 25°C for 5 min. Further studies (not shown) revealed that the larger band was the exclusive product obtained from more vigorous treatment of Fd with BME at 90°C. The small change in the electrophoretic mobility of the 10 kDa Fd band in lanes 7 and 9 may be due to the modifications of carboxylic groups by EDC and NHS during the P1 reaction. Lanes 3 and 8 of Figure 3 show that reactions containing untreated Δ9D (lane 3) and Δ9D treated with BME-quenched EDC (lane 8) were also indistinguishable based on SDS-PAGE analysis. The single band with an apparent mass of 37 kDa apparently corresponded to the Δ9D subunit (Mr 42 000) obtained from denaturation of dimeric Δ9D by SDS. In contrast, SDS-PAGE analysis of EDC-activated Δ9D (lane 5) revealed two bands with apparent masses of ∼37 kDa and ∼75 kDa. The ∼37 kDa product corresponded to a single Δ9D subunit (Mr 42 000), while the ∼75 kDa band corresponded to a covalent cross-linking of two Δ9D subunits (Mr 84 000). Two possible inter-subunit salt bridges in Δ9D that might be activated for amide bond formation by the EDC and NHS treatment are K152 with E166 and K167 with E59 (i.e., less than 5 Å separation between Lys-NZ and Glu-CD). In addition, there are five possible intra-subunit salt bridges between lysine and glutamate residues (E38 and K41; E85 and K88; E182 and K261; E233 and K230; E245 and K242) and three possible intra-subunit salt bridges between lysine and aspartate residues (i.e., less than 5 Å separation between Lys-NZ and Asp-CG, D247 and K36; D257 and K261; D311 and K312).
Figure 3.
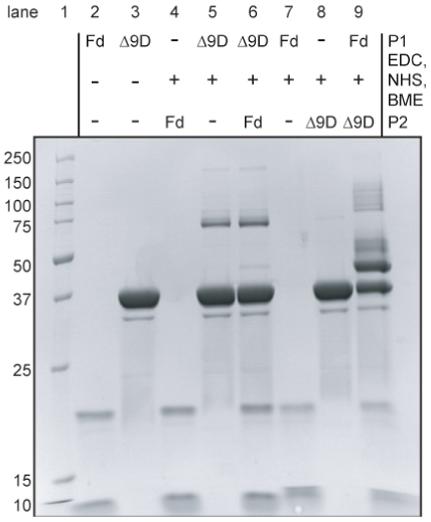
Coomassie-stained denaturing electrophoresis gel containing proteins obtained from the indicated cross-linking reactions. The identities of P1 and P2, as defined in Figure 2, are indicated. Lane 1 contains molecular mass standards.
Two reaction sequences were used to study the formation of a cross-linked Fd-Δ9D complex. In one reaction sequence, the carboxylate groups from Δ9D were first activated by treatment with EDC and NHS, the excess EDC was quenched by addition of BME, and then Fd was added. An SDS-PAGE analysis of this reaction sequence (lane 6 of Figure 3) revealed two bands with apparent masses of ∼37 kDa and ∼75 kDa, which is the same result obtained from reaction of Δ9D alone (lane 5 of Figure 3). In the other reaction sequence, the carboxylate groups from Fd were first activated by treatment with EDC and NHS, the excess EDC was quenched, and then Δ9D was added. SDS-PAGE analysis of this reaction sequence (lane 9 of Figure 3) revealed two prominent bands with apparent masses of ∼37 kDa and ∼45 kDa and a mixture of other minor, higher mass products. While the ∼37 kDa band represents the unreacted Δ9D subunit, the ∼45 kDa product was not observed from any of the other control reactions (lanes 2, 3, 5, 6 and 8 of Figure 3) and roughly corresponded to the combined mass of one Δ9D subunit (∼42 kDa) and one Fd (∼11 kDa). Thus only the activation of carboxylate residues on Fd led to cross-linking with Δ9D. It is also noted that activation of Fd bypassed the formation of the ∼75 kDa product assigned to a covalent linkage of the EDC-activated subunits of dimeric Δ9D.
MALDI-MS analyses were performed in order to more precisely determine the molecular masses of the polypeptides observed by SDS-PAGE. Table 1 shows the assignments made for the three prominent peaks of 10875, 41369, and 52219 m/z observed in the MALDI-MS spectrum. These masses closely corresponded to those expected for the singly protonated [M+H]+ Fd (m/z 10699), the Δ9D subunit (m/z 41646), and the cross-linking of Fd with the Δ9D subunit (m/z 52345). Several minor peaks were also observed, which likely represented the [M+2H]2+ ions of the Δ9D subunit (m/z 20823), the cross-linked complex of Δ9D and Fd (m/z 26173) and the [M+H]1+ ion from a single Δ9D subunit cross-linked to 2 Fd (m/z 63044).
Table 1.
MALDI-MS analysis of cross-linked products obtained from interactions of Fd and Δ9D
| Species a | Estimated mass(kDa)b | Observed peak m/z ratiosc | Charge stated | Calculated m/z ratiose |
|---|---|---|---|---|
| Fd | 10 | 10875 | [M+H]1+ | 10699 |
| Δ9D | 37 | 41369 | [M+H]1+ | 41646 |
| Δ9D | 20877 | [M+2H]2+ | 20823 | |
| Δ9D -Fd | 45 | 52219 | [M+H]1+ | 52345 |
| Δ9D -Fd | 26062 | [M+2H]2+ | 26173 | |
| Δ9D -Fd-Fd | 58 | 62325 | [M+H]1+ | 63044 |
| Δ9D-Δ9D | 75 | 83338 | [M+H]1+ | 83292 |
Fd, [2Fe-2S ferredoxin]; Δ9D, refers to a single subunit of the Δ9D dimer.
As determined from SDS-PAGE analysis.
Observed mass peak from MALDI-MS analysis performed as described in Materials and Methods.
Assigned charge state.
Calculated m/z from the observed mass peak and assigned charge state.
Mutagenesis of Δ9D and Kinetic Analysis
The results of Figure 3 and Table 1 demonstrated that amide linkages could be formed between Fd and Δ9D, but only in reactions where EDC was used to activate carboxylate groups from Fd. Thus Δ9D must provide lysine residues to the amide bond(s) formed in the cross-linking reactions. Figure 1B shows that K41, K56, K60, and K230 are positioned in close proximity to aromatic amino acids previously identified as possible electron transfer pathways for Δ9D (30) and other members of the diiron superfamily (22, 23).
In order to examine the potential role of these residues in formation of an Fd-Δ9D complex, they were mutated to alanine. The mutated Δ9D variants (along with wt Δ9D) were then characterized using steady-state kinetics by varying the concentration of Fd at fixed, saturating concentrations of 18:0-ACP. Figure 4 shows representative v versus [S] plots obtained from these characterizations, while Table 2 shows the kinetic parameters obtained from non-linear least squares fitting of the experimental results using equation 1. For wt Δ9D and the K41A mutation, both the kcat for formation of 18:1-ACP and the KM for Fd were comparable to the kinetic parameters previously determined for wt Δ9D (10).
Figure 4.
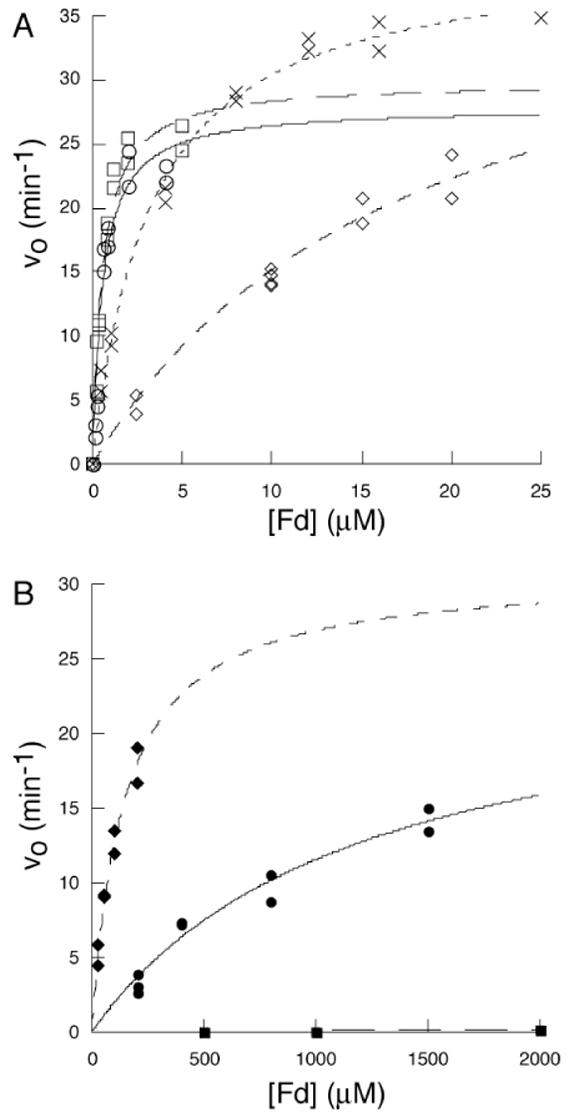
The dependence of vo for 18:1-ACP formation on the concentration of Fd. (A) wt Δ9D, (○); K41A Δ9D, (□); K60A Δ9D, (x); K56A Δ9D, (◇). (B) K56A/K60A Δ9D, (◆); K230A Δ9D, (●); and K56A/K60A/K230A Δ9D (■). The lines are the results of non-linear least squares fitting of the experimental data using equation 1.
Table 2.
Kinetic parameters for the Δ9D-catalyzed conversion of 18:0-ACP to 18:1-ACP measured at varying concentrations of Fd
| Δ9D isoform | kcat (min-1)a | KM (mM) | kcat/KM (mM-1min-1) | R b |
|---|---|---|---|---|
| Wild-type | 28 (1.8) | 0.56 (0.11) | 50 (7.6) | 0.981 |
| K41A | 30 (1.4) | 0.54 (0.082) | 55 (6.5) | 0.986 |
| K56A | 42 (1.6) | 18 (1.7) | 2.4 (0.15) | 0.992 |
| K60A | 40 (0.95) | 3.2 (0.27) | 12 (0.84) | 0.997 |
| K56A/K60A | 30 (4) | 140 (30) | 0.2(0.02) | 0.979 |
| K230A | 24 (3.8) | 1,194 (327) | 0.02 (0.006) | 0.988 |
| K56A/K60A/K230Ac | NDd | ND | 0.00007 (0.000008) | 0.957 |
The kcat- and KM-values were obtained from nonlinear least-squares fitting to equation 1. The kcat-values are reported per diiron center of D9D.
Correlation coefficient of the non-linear least squares fitting.
The product formation data were analyzed by linear least squares fitting as described in Materials and Methods using the first order assumption d [18:1-ACP] / d t = vo [Fd].
Not determined.
Steady-state kinetic analysis revealed that the K56A and K60A variants had kcat-values equivalent to wt Δ9D and the K41A Δ9D; however, the KM-values determined for these two variants were altered slightly to values ∼30-fold and ∼6-fold greater, respectively, than the KM-value determined for wt Δ9D. Based on these relatively modest changes in KM, the doubly mutated K56A/K60A Δ9D was also generated and it yielded a kcat of ∼30 min-1 (again similar to wt Δ9D) along with a KM of ∼140 μM (an ∼250-fold increase in KM as compared to wt Δ9D). Surprisingly, when K230A Δ9D was examined, a KM of ∼1,200 μM was obtained, representing an ∼2,200-fold decrease in the apparent affinity for Fd. However, the K230A Δ9D also had a kcat value of ∼25 min-1, which was only slightly diminished relative to wt Δ9D.
To further probe the role of these lysines in Fd binding to Δ9D, the triply mutated K56A/K60A/K230A Δ9D was constructed. This isoform retained the catalytic diiron centers as judged by iron analysis and optical spectroscopy, yet catalytic activity was nearly eliminated, even at Fd concentrations up to 2 mM (solid squares Figure 4B). Under the assumption of low affinity for Fd, the detected rate of product formation corresponded to the first order rate, kcat/KM in the accessible concentration range for Fd. Table 2 shows that K56A/K60A/K230A Δ9D has a catalytic efficiency decreased by at least 700,000-fold relative to Δ9D.
Cross-Linking Reactions of Mutated Δ9D
The large increases in the apparent KM-values observed for K230A Δ9D and the doubly and triply mutated variants suggested that weaker binding affinities might also correspond to decreased efficiency of the EDC and NHS reactions. Surprisingly, however, similar yields of the ∼45 kDa product assigned to the cross-linking of Fd and Δ9D were observed in all reactions, including complexes of Fd and K56A/K60A/K230A Δ9D, where the apparent affinity for Fd should have decreased by greater than 104 according to steady-state kinetic analysis.
Figure 5 and Table 3 summarize the results of the cross-linking of Fd and Δ9D in the presence of 18:0-ACP, and resolves this apparent inconsistency. Figure 5 shows an SDS-PAGE analysis of cross-linking reactions between EDC-activated Fd and the pre-formed complexes of either Δ9D (Figure 5, P2 = Δ9D) or K56A/K60A Δ9D (Figure 5, P2 = K56A/K60A Δ9D, abbreviated as Δ9D* in the figure) with 18:0-ACP (Figure 5, S) and other control reactions. Table 3 summarizes the results from comparable cross-linking reactions performed with the various mutated forms of Δ9D. Under the conditions used in these experiments, Δ9D was fully saturated with 18:0-ACP [KD1= 13 nM, KD2 = 170 nM (31)]. Lane 4 is a control that shows Δ9D and 18:0-ACP are separated by the denaturing electrophoresis. This lane also shows that 18:0-ACP (apparent mass of ∼12 kDa) is partially converted into the holo-ACP dimer (apparent mass of ∼20 kDa) during the electrophoresis. Moreover, lane 5 is a control that shows EDC-activated Fd does not cross-link with 18:0-ACP.
Figure 5.
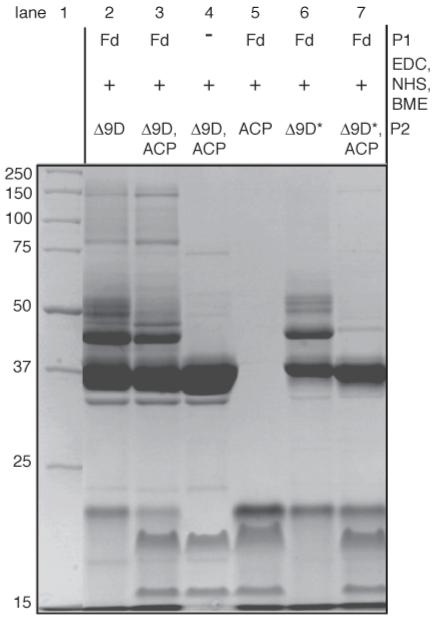
Coomassie-stained denaturing electrophoresis gel showing the influence of 18:0-ACP (abbreviated as ACP to fit the figure) on the cross-linking of Fd to either wt Δ9D (P2 = Δ9D) or K56A/K60A Δ9D (P2 = K56A/K60A Δ9D, abbreviated as Δ9D* to fit in the figure). Lane 1 contains molecular mass standards. Cross-linking of wt Δ9D in the absence (lane 2) or the presence (lane 3) of 18:0-ACP appears similar. Lane 4 shows Δ9D and 18:0-ACP in the presence of EDC, NHS and BME. Cross-linking of K56A/K60A Δ9D is affected by the presence of 18:0-ACP (compare lanes 6 and 7). Activated Fd does not react with 18:0-ACP (lane 5). 18:0-ACP (∼16 kDa) is partially converted to holo-ACP (∼20 kDa) during the electrophoresis.
Table 3.
Results of cross-linking EDC-activated Fd to complexes of Δ9D isoforms with 18:0-ACP
| Δ9D isoforma | 18:0-ACPb | Cross-linking with EDC-Fd observed |
|---|---|---|
| wt | - | + |
| K56A | - | + |
| K60A | - | + |
| K230A | - | + |
| K56A/K60A | - | + |
| K56A/K60A/K230A | - | + |
| wt | + | + |
| K56A | + | + |
| K60A | + | + |
| K230A | + | + |
| K56A/K60A | + | - |
| K56A/K60A/K230A | + | - |
Isoforms complexed with 18:0-ACP as indicated and then mixed with EDC-activated Fd as described in Materials and Methods.
Isoforms complexed with 18:0-ACP as indicated and then mixed with EDC-activated Fd as described in Materials and Methods.
Cross-linking products detected using SDS-PAGE as shown in Figure 5, lanes 6 and 7 for reactions containing K56A/K60A Δ9D without and with 18:0-ACP.
Lanes 2 and 3 of Figure 5 show that EDC-activated Fd can be cross-linked to Δ9D to give the ∼45 kDa complex in both the absence (lane 2) and in the presence of 18:0-ACP (lane 3). This result confirms that binding of 18:0-ACP and Fd are not mutually exclusive. Thus a ternary complex is a feasible intermediate in Δ9D catalysis.
Lane 6 of Figure 5 shows that EDC-activated Fd can also be efficiently cross-linked to K56A/K60A Δ9D in the absence of 18:0-ACP, even as the KM-value of this Δ9D isoform was increased by ∼250-fold (Table 2). Moreover, Table 3 indicates that EDC-activated Fd was also efficiently cross-linked to the triply mutated Δ9D. These results reveal the presence of an alternative Fd binding site on Δ9D after the proposed Fd binding site was eliminated by mutagenesis. Indeed, Table 3 shows that cross-linking was obtained among all combinations of the mutated Δ9D and Fd in the absence of 18:0-ACP.
Lane 7 of Figure 5 and Table 3 show that the cross-linking of Fd to either K56A/K60A Δ9D or K56A/K60A/K230A Δ9D was eliminated in the presence of 18:0-ACP. As mutations of both K56 and K60 were required to prevent the cross-linking in the presence of 18:0-ACP, these two residues may have a distinct role in complex formation relative to the contributions of K230. The elimination of the cross-linking between Fd and the doubly and triply mutated Δ9D in the presence of 18:0-ACP is also notable in that it defines an overall specificity for protein-protein interactions, as no alternative lysine residues on the surface of Δ9D were capable of yielding cross-linking with Fd. Therefore, we conclude that Fd has one functionally relevant binding site on the Δ9D surface near to K56 and K60, and that Fd can adventitiously cross-link to the 18:0-ACP binding site in the mutated Δ9D or in the absence of 18:0-ACP.
Docking Results
ClusPro (27, 28) was used to investigate complexes of Fd (PDB file 1FXA) as the moving molecule and with Δ9D (PDB file 1AFR) as the static molecule. Figure 6A shows the locations of two favored regions for Fd binding on the Δ9D surface. From a total of 60 different conformations examined in detail, 17 were located near to our proposed Fd binding site, including K56, K60, K230 (red surface) and W62 (yellow surface). The green backbone traces represent an ensemble of 6 of these conformations at the Fd binding site of one subunit of Δ9D. Multiple conformations of Fd were also located near to the opening of the proposed 18:0-ACP binding channel, including the three closely spaced lysine residues K261, K262 and K322 (violet surface). The red backbone traces represent an ensemble of 6 Fd adventitiously bound at the 18:0-ACP binding site. The other Fd conformations examined were singletons whose positions were distributed nearly all over the surface of Δ9D. It is notable, however, that none of these latter conformations contained Fd interacting with the surface of Δ9D nearest to K41, which is likely due to steric factors. Thus the docking experiments also support the conclusion that two different binding sites for Fd exist on the surface of Δ9D, with one binding site directed toward a catalytically relevant electron transfer site and the other apparently overlapping with the high affinity binding site for 18:0-ACP.
Figure 6.
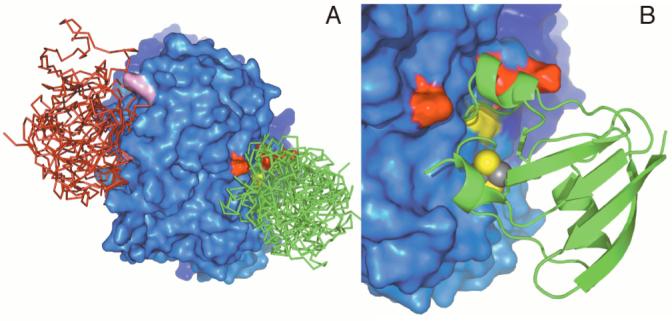
Predicted sites for protein-protein complex formation between Δ9D and Fd. This orientation allows a view of only one subunit of the Δ9D dimer. (A) Surface representation of Δ9D showing the lysine residues important in Fd binding (red), W62 (yellow), and lysine residues in the ACP binding site (violet). Fd molecules docked at the proposed electron transfer site are shown as green backbone trace; Fd molecules docked in the ACP binding site are shown as red backbone trace. (B) A close-up view of a representative Δ9D-Fd conformation with coloring of the residues as in (A). In this conformation, the [2Fe-2S] cluster of Fd is ∼11 Å from W62.
The set of 17 conformations at the proposed Fd binding site had individual conformations that placed the [2Fe-2S] cluster either near to or far from the surface of Δ9D. For the entire set of 17, the average distance from C41-SG (ligand of the [2Fe-2S] cluster of Fd) to W62-N of Δ9D was ∼20 ± 10 Å. However, by selecting the 7 binding conformations that had the [2Fe-2S] cluster oriented toward the Δ9D surface, the average distance shortened to 14 ± 4 Å. This distance compares favorably to the proposed optimal electron tunneling distance between two redox cofactors of ∼14 Å (32). Figure 6B shows one representative conformation from this subset of Fd-Δ9D conformations.
A ClusPro docking of 18:0-ACP to Δ9D was also undertaken. From a total of 20 configurations examined, 18 clustered at the 18:0-ACP binding site defined here by possible electrostatic contributions of K262, K262 and K322. Surprisingly, the 18:0-ACP configurations did not cluster at the proposed Fd binding site, which again may be due to steric considerations. Figure 7A shows one of the representative conformations for 18:0-ACP binding, along with Fd bound at its proposed functional site to make a ternary complex with Δ9D. The ACP binding conformation places Ser36 (the position of covalent attachment of phosphopantetheine and acyl chain) near to the entry to the substrate channel leading to the diiron active site.
Figure 7.
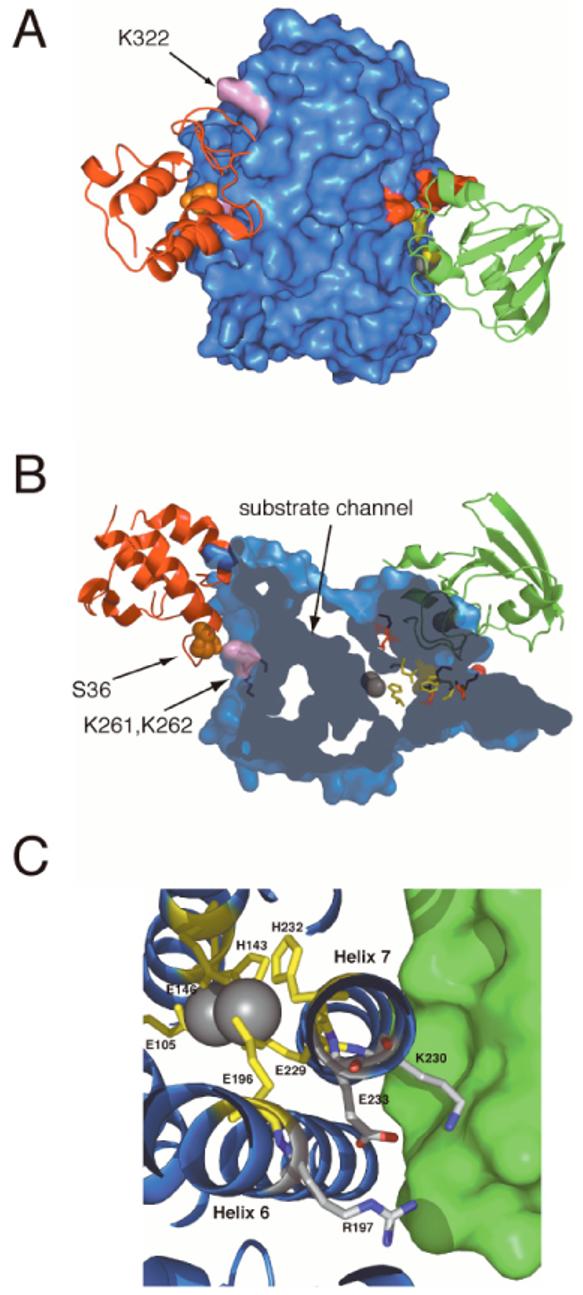
A model for the tertiary complex of Δ9D. This orientation allows a view of only one subunit of the Δ9D dimer. The Fd and ACP binding sites are separated by ∼50 Å across subunits of the Δ9D dimer. (A) Surface representation of Δ9D with Fd (green) and ACP (red) shown in ribbon representation. Lysine residues involved in forming the Fd binding site are shown in red, W62, D228 and H146 are shown in yellow, and lysine residues in the ACP binding site are shown in pink. The S36 residue of ACP (site of phosphopantetheinylation and acylation) is shown as orange spheres. B) A clipped view representation of Δ9D revealing the position of the channel leading to the active site diiron center (grey spheres). The Fd and ACP are in the same binding sites as show in (A). (C) A close-up view of the region of Δ9D near K230 in the model for the complex of Fd and Δ9D. K230 and E233 are found on the solvent-exposed face of helix-7, which also provides the diiron ligands E229 and H232. E233 is hydrogen bonded to both K230 and R197. It is proposed that these interactions may be perturbed by Fd binding and thus alter the active site in order to facilitate catalysis.
DISCUSSION
Electron Transfer Pathways in Δ9D and Related Enzymes
Examination of the 3-dimensional structure showed that Fd has 21 carboxylate groups, including many on the surface near to the [2Fe-2S] cluster. Likewise, each subunit of Δ9D has 24 lysine residues. In combination, this number of functional groups suggests potentially many interactions that might be subjected to chemical cross-linking and belies the utility of cross-linking alone to reveal molecular details of protein-protein interactions.
The crystal structure of Δ9D (30) revealed two possible pathways for electron transfer extending from the surface of the protein to the diiron center (see Figure 1). One pathway includes the amino acids W62, D228 and H146, and extends from W62 at the surface loop region between helix-1 and helix-2 to the internal diiron ligand H146 (Figure 1). Both Escherichia coli ribonucleotide reductase (22) and the putative acyl-ACP desaturase DesA2 from Mycobacterium tuberculosis (23) have this triad of structurally homologous residues, but methane monooxygenase has R146 in this position as a substitute for W62 (33). In the E. coli enzyme, W48 is hydrogen-bonded to D237, which is hydrogen-bonded to the diiron ligand H118. W48 forms a cation radical during tyrosyl radical formation, and mutagenesis of W48 profoundly changes the outcome of single-turnover catalysis (34-36). Δ9D also has K230 in close proximity to K56 and K60. These three lysine residues are 7.5 Å, 14 Å and 7.3 Å, respectively, from W62. The combination of results presented here strongly implicates a role for these surface residues in Δ9D catalysis.
The Δ9D structure also showed that K41, the other surface lysine investigated in this work, is in close proximity to a proposed electron transfer pathway (30) including the amino acids W132, W135, W139, F189 and Y236 (Figure 1). These residues are completely buried and extend along the interior of the four-helix bundle toward diiron center ligand E143. These residues are mostly conserved in M. tuberculosis DesA2 (where the closest structurally comparable residues are W96, W99, W103, L152 and F196 respectively), less so in ribonucleotide reductase (F18, W107, W111, L197 and T245), and completely diverged in methane monooxygenase (M53 or Q133, Y137, Q140, T253 and Q205 and G250). Furthermore, the lack of effects upon mutation of K41 (Table 2) suggests that the proposed electron path involving W132 is not used in Δ9D catalysis.
Cross-Linking Implicates Carboxylate Residues from Fd and Lysine Residues from Δ9D
EDC-catalyzed cross-linking with SDS analysis showed that a distinct product with an apparent mass of ∼45 kDa was formed only in reactions containing EDC-activated Fd and Δ9D (Figure 2B, lane 9). Analysis by MALDI-MS confirmed that this cross-linked product consisted of Fd and a single subunit of dimeric Δ9D (Table 1). The reaction conditions (Figure 2) established that the covalent cross-link was generated between carboxylate groups from Fd and lysine(s) from Δ9D.
Catalytic Consequences of Lysine Mutations
The K41A mutation gave no change in either kcat or kcat/KM as compared to wt Δ9D. Therefore, no other lysine residues were studied in this region of Δ9D. In contrast, the apparent KM values of the K56A, K60A and K230A isoforms for Fd binding were substantially altered (Table 2), even as the kcat values were not significantly changed as compared to wt Δ9D. Notably, the doubly mutated K60A/K56A Δ9D had a ∼250-fold reduction in the apparent KM but no significant reduction in kcat. Furthermore, the single mutation K230 caused a ∼2,200-fold reduction in kcat/KM, which was also primarily associated with a decrease in the apparent KM for Fd. Substitution of all three lysine residues effectively abolished the ability of wt Δ9D to recognize Fd as electron partner (Table 2), although owing to the sensitivity of the assay for 18:1 production, the triply mutated protein did indeed retain detectable ability to catalyze double bond insertion. These results strongly implicate K56, K60 and K230 in one or more aspects of Δ9D catalysis, but also suggest that these surface residues are not essential for the chemical steps of catalysis.
Effect of 18:0-ACP on Fd Cross-linking
Cross-linking reactions undertaken using the catalytically-relevant ternary complex of Δ9D, Fd and 18:0-ACP (16) suggest that Fd can adventitiously cross-link to the 18:0-ACP binding site of the K56A/K60A and K56A/K60A/K230A mutated Δ9D in the absence of 18:0-ACP. In contrast, cross-linking of Fd to the doubly and triply mutated Δ9D was eliminated in the presence of 18:0-ACP (which likely blocks access to K261, K262 and K322), showing that no other lysine groups from Δ9D are involved in complex formation. Since ACP and Fd are small acidic proteins (∼9 kDa with pI 3.8 and ∼11 kDa with pI 3.9, respectively), it is reasonable that both may interact with Δ9D by carboxylate-lysine charge pairs. The originally proposed ACP binding region consists of a depression near to the opening of an extended channel leading to the diiron center (30). This depression is on the opposite side of the Δ9D monomer from the proposed Fd binding site encompassing K56, K60, W62 and K230 (see Figures 6A, 7A and 7B). As 18:0-ACP exhibits nanomolar binding affinities (31), its presence would likely eliminate adventitious Fd binding, which is apparently several orders of magnitude weaker [Table 2 and (31)].
Further consideration of the results of Figure 5 and Table 3 reveals that K56 and K60 are essential for the formation of a cross-linked complex between Fd and Δ9D, supporting a role for these lysine residues in formation of an electrostatic complex at the proposed electron transfer site during catalysis. Since the presence of K230 in the doubly mutated Δ9D did not lead to the formation of the diagnostic ∼45 kDa cross-linked adduct (Table 3), the strong contribution of this lysine residue to catalysis (Table 2) may arise from a different role than electrostatic complex formation.
Other Studies of Fd-Protein Interactions
EDC-catalyzed cross-linking of the [2Fe-2S] Fd domain of the reductase and hydroxylase components of methane monooxygenase yielded a cross-link between acidic residues from the Fd domain (E56 and E91) and the N-terminus of the hydroxylase (37). The Fd domain has 19 carboxylate residues, while the hydroxylase has 122 lysine residues. Whether the Fd domain has the same binding interactions as the intact reductase has not been determined. However, in a related study (38), no significant line broadening of the 1H 15N HSQC cross-peaks assigned to Fd domain residues E56 and E91 occurred upon addition of the hydroxylase.
Ferredoxins from several other enzyme complexes have been cross-linked to their physiological redox partners through acidic residues (39, 40). For example, an acidic residue from Fd has been shown to cross-link to Fd:NADP+ reductase (41) and to photosystem I in reactions containing EDC (39). The binding interactions and electron transfer reaction between Fd and Fd:NADP+ reductase from Anabaena have been studied extensively (42-45), and these results show that electrostatic interactions are are largely mediated by acidic residues from Fd (46) and basic residues from Fd:NADP+ reductase. These interactions are important for “steering” the two proteins into the correct orientation for electron transfer (47). In the crystal structure of the Fd-Fd:NADP+ complex (45), a salt bridge between E94 from Fd and K75 from Fd:NADP+ reductase was observed at the protein-protein interface. Site-directed mutagenesis of K75 from Fd:NADP+ reductase increased the KM for Fd binding by at least 50-fold, while no effect was observed on kcat (43), suggesting an electrostatic role for K75 in formation of the Fd-Fd:NADP+ complex. A similar magnitude of increase in KM for Fd binding and no change in kcat was observed with the K60A mutation of Δ9D, further supporting the role this lysine residue in electrostatic contributions to the orientation of Fd binding.
Δ9D Tertiary Complex
It is reasonable to assume that a functional Fd binding site on Δ9D will place the [2Fe-2S] cluster and diiron cofactor in close proximity in order to facilitate electron transfer during multiple turnover catalysis. While two of the cysteine ligands to the [2Fe-2S] cluster in Fd are solvent exposed (29), all of the ligands to the diiron center of Δ9D are buried within a four-helix bundle (30). Thus electron transfer from Fd to the diiron center of Δ9D is presumably facilitated by alignment of the two protein surfaces through amino acid contacts. Our results also indicate that 18:0-ACP binding precedes electron transfer (16). Indeed, many of the models calculated for the docking of 18:0-ACP with Δ9D predict interactions of ACP with K261, K262 and K322. These lysine residues are on the opposite face of the Δ9D subunit from the lysines proposed to interact with Fd in the electron transfer reactions (∼ 38 Å apart). Furthermore, the proposed 18:0-ACP and Fd binding sites are separated by ∼50 Å between subunits and so are unlikely to spatially overlap. Figure 7A provides a model for the ternary complex of Fd and 18:0-ACP with one subunit of the Δ9D dimer.
Figure 7B shows a more detailed view of the docked position of ACP and the extent of the substrate-binding channel. This channel proceeds from the surface toward the diiron center and the region of helix-7 containing K230 and E233. It has been previously demonstrated that 18:0-ACP binding causes structural changes in the active site of sodium dithionite-reduced Δ9D. However, these changes were not sufficient to permit O2-dependent desaturation, but instead led to the formation of a quasi-stable unreactive peroxo intermediate (14, 15). One possibility accounting for the lack of reactivity of the sodium dithionite-reduced Δ9D might be that Fd binding is required to promote further reactivity. Thus occupation of substrate channel by an 18:0 acyl chain could lead to conformation changes that optimize the Fd binding site for efficient electron transfer, promote of ligand rearrangements that lead to O2 activation (12), or other aspects. Alternatively, Fd binding may transmit information into the active site that optimally aligns the acyl chain to allow catalysis to proceed.
Recent X-ray studies of methane monooxygenase revealed conformational changes in helix E in the presence of a relatively large alternative product, 6-bromohexan-1-ol (48). Similar conformational changes may occur upon formation of the more extensive interactions predicted by the binding of 18:0-ACP with Δ9D (Figure 7B). Upon formation of the 18:0-ACP and Δ9D complex, it is also feasible that any rearrangements caused by binding could be transmitted to the region of helix-7 including K230. Figure 7C shows that K230 is within hydrogen-bonding distance of E233, the latter of which also forms an ion pair with R197. R197 is located in helix-6, which is structurally homologous to helix E of methane monooxygenase. K230 and E233 are adjacent to the diiron ligands E229 and H232, while R197 is adjacent to the diiron ligand E196. By making contacts with K230, binding of Fd may alter this configuration of residues situated near to or in the active site.
Implications for the Δ9D Mechanism
These studies have provided new insight into protein-protein interactions required for Δ9D catalysis. Our proposed location of the Fd binding site has been supported by biochemical, catalytic and computational approaches. This preferred binding site involves surface complementarity and electrostatic interactions between Fd and Δ9D. The site also appears to encompass a set of surface residues from Δ9D that may lead to conformational changes from binding of either 18:0-ACP or Fd. The presence of only one Fd binding site implies that two molecules of Fd must successively bind at this position in order to provide the 2e- required for catalysis. The exact nature and timing of molecular events encompassed by the reaction cycle are presently not known, but a number of intriguing possibilities can be considered in light of this model.
ACKNOWLEDGEMENTS
We thank Dr. Julie Mitchell (University of Wisconsin) for helpful discussions on modeling the Δ9D-Fd complex.
Footnotes
This work was supported by the National Institutes of Health GM-50853 to B.G.F.
- ACP
- acyl carrier protein
- 18:0-ACP
- ACP with stearic acid covalently attached to ACP through a phosphopantetheine thioester bond
- 18:1-ACP
- ACP with oleic acid covalently attached to ACP through a phosphopantetheine thioester bond
- Δ9D
- 18:0-ACP Δ9 desaturase
- resting Δ9D
- as-isolated form of Δ9D containing all ferric sites
- 4e- Δ9D
- chemically reduced form of Δ9D containing all ferrous sites
- Fd
- Anabaena 7120 vegetative [2Fe-2S] ferredoxin
- GC
- gas chromatography
- MS
- mass spectrometry
- EDC
- 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- NHS
- N-hydroxysuccinimide
- BME
- β-mercaptoethanol.
REFERENCES
- 1.Fox BG, Shanklin J, Somerville C, Münck E. Stearoyl-acyl carrier protein Δ9 desaturase from Ricinus communis is a diiron-oxo protein. Proc. Natl. Acad. Sci. U.S.A. 1993;90:2486–2490. doi: 10.1073/pnas.90.6.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shanklin J, Cahoon EB. Desaturation and related modifications of fatty acids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998;49:611–641. doi: 10.1146/annurev.arplant.49.1.611. [DOI] [PubMed] [Google Scholar]
- 3.Nordlund P, Eklund H. Di-iron-carboxylate proteins. Curr. Opin. Struct. Biol. 1995;5:758–766. doi: 10.1016/0959-440x(95)80008-5. [DOI] [PubMed] [Google Scholar]
- 4.Leahy JG, Batchelor PJ, Morcomb SM. Evolution of the soluble diiron monooxygenases. FEMS Microbiol. Rev. 2003;27:449–479. doi: 10.1016/S0168-6445(03)00023-8. [DOI] [PubMed] [Google Scholar]
- 5.Wallar BJ, Lipscomb JD. Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem. Rev. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 6.Fox BG. In: Comprehensive biological catalysis. Sinnott M, editor. Academic Press; London, U. K.: 1997. pp. 261–348. [Google Scholar]
- 7.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Muller J, Lippard SJ. Dioxygen activation and methane hydroxylation by soluble methane monooxygenase: A tale of two irons and three proteins. Angew. Chem. Int. Ed. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 8.Thiel EC. Ferritin: dynamic management of biological iron and oxygen chemistry. Acc. Chem. Res. 2005;38:167–175. doi: 10.1021/ar0302336. [DOI] [PubMed] [Google Scholar]
- 9.deMare F, Kurtz DM, Jr., Nordlund P. The structure of Desulfovibrio vulgaris rubrerythrin reveals a unique combination of rubredoxin-like FeS4 and ferritin-like diiron domains. Nat. Struct. Biol. 1996;3:539–546. doi: 10.1038/nsb0696-539. [DOI] [PubMed] [Google Scholar]
- 10.Haas JA, Fox BG. Role of hydrophobic partitioning in substrate selectivity and turnover of the Ricinus communis stearoyl acyl carrier portein Δ9 desaturase. Biochemistry. 1999;38:12833–12840. doi: 10.1021/bi991318a. [DOI] [PubMed] [Google Scholar]
- 11.McKeon TA, Stumpf PK. Purfication and characterization of the stearoyl-acyl carrier protein desaturase and the acyl-acyl carrier protein thioesterase from maturing seeds of safflower. J. Biol. Chem. 1982;257:12141–12147. [PubMed] [Google Scholar]
- 12.Yang Y, Broadwater JA, Pulver SC, Fox BG, Solomon EI. Circular dichroism and magnetic circular dichroism studies of the reduced binuclear non-heme iron site of stearoyl-ACP Δ9 desaturase: Substrate binding and comparison to ribonucleotide reductase. J. Am. Chem. Soc. 1999;121:2770–2783. [Google Scholar]
- 13.Skulan AJ, Brunold TC, Baldwin J, Saleh L, Bollinger JM, Jr., Solomon EI. Natur of the peroxo intermediate of the W48F/D84E ribonucleotide reductase variant: Implications for O2 activation by binuclear non-heme iron enzymes. J. am. Chem. Soc. 2004;126:8842–8855. doi: 10.1021/ja049106a. [DOI] [PubMed] [Google Scholar]
- 14.Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Peroxodiferric intermediate of stearoyl-acyl carrier protein Δ9 desaturase: Oxidase reactivity during single turnover and implications for the mechanism of desaturation. Biochemistry. 1998;37:14664–14671. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- 15.Broadwater JA, Achim C, Münck E, Fox BG. Mössbauer studies of the formation and reactivity of a quasi-stable peroxo intermediate of stearoyl-acyl carrier protein Δ9-desaturase. Biochemistry. 1999;38:12197–12204. doi: 10.1021/bi9914199. [DOI] [PubMed] [Google Scholar]
- 16.Lyle KS, Haas JA, Fox BG. Rapid-mix and chemical quench studies of ferredoxin-reduced stearoyl-acyl carrier protein desaturase. Biochemistry. 2003;42:5857–5866. doi: 10.1021/bi030020o. [DOI] [PubMed] [Google Scholar]
- 17.Fox BG, Froland WA, Dege JE, Lipscomb JD. Methane monooxygenase from Methylosinus trichosporium OB3b purification and properties of a three-component system with high specific activity from a type II methanotroph. J. Biol. Chem. 1989;264:10023–10033. [PubMed] [Google Scholar]
- 18.Froland WA, Andersson KK, Lee S-K, Liu K, Lipscomb JD. Methane monooxygnease component B and reductase alter the regioselectivity of the hydroxylase component-catalyzed reactions: A novel role for protein-protein interactions in an oxygenase mechanism. J. Biol. Chem. 1992;267:17588–17597. [PubMed] [Google Scholar]
- 19.Lee S-K, Nesheim JC, Lipscomb JD. Transient intermediates of the methane monooxygenase catalytic cycle. J. Biol. Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- 20.Valentine AM, Stahl SS, Lippard SJ. Mechanistic studies of the reaction of reduced methane monooxygenase hydroxylase with dioxygen and substrates. J. Am. Chem. Soc. 1999;121:3876–3887. [Google Scholar]
- 21.Lynch JB, Juarez-Garcia C, Münck E, Que LJ. Mössbauer and EPR studies of the binuclear iron center in ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 1989;264:8091–8096. [PubMed] [Google Scholar]
- 22.Nordlund P, Eklund H. Structure and function of the Escherichia coli ribonucleotide reductase protein R2. J. Mol. Biol. 1993;232:123–164. doi: 10.1006/jmbi.1993.1374. [DOI] [PubMed] [Google Scholar]
- 23.Dyer DH, Lyle KS, Rayment I, Fox BG. X-ray structure of putative acyl-ACP desaturase DesA2 from Mycobacterium tuberculosis H37Rv. Prot. Sci. 2005;14:1508–17. doi: 10.1110/ps.041288005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng H, Westler WM, Xia B, Oh B, Markley JL. Protein expression, selective isotopic labeling, and analysis of hyperfine-shifted NMR signals of Anabaena 7120 vegetative [2Fe-2S] ferredoxin. Arch. Biochem. Biophys. 1995;316:619–634. doi: 10.1006/abbi.1995.1082. [DOI] [PubMed] [Google Scholar]
- 25.Sehgal D, Vijay IK. A method for the high efficiency of water-soluble carbodiimide-mediated amidation. Anal. Biochem. 1994;218:87–91. doi: 10.1006/abio.1994.1144. [DOI] [PubMed] [Google Scholar]
- 26.Hoffman BJ, Broadwater JA, Johnson P, Harper J, Fox BG, Kenealy WR. Lactose fed-batch overexpression of recombinant metalloproteins in Escherichia coli BL21(DE3): Process control yielding high levels of metal-incorporated, soluble protein. Protein Expression Purif. 1995;6:646–654. doi: 10.1006/prep.1995.1085. [DOI] [PubMed] [Google Scholar]
- 27.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: A fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32:W96–9. doi: 10.1093/nar/gkh354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: An automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 29.Rypniewski WR, Breiter DR, Benning MM, Wesenberg G, Oh B-H, Markley JL, Rayment I, Holden HM. Crystallization and structure determination to 2.5-Å resolution of the oxidized [2Fe-2S] ferredoxin isolated from Anabaena 7120. Biochemistry. 1991;30:4126–4131. doi: 10.1021/bi00231a003. [DOI] [PubMed] [Google Scholar]
- 30.Lindqvist Y, Huang W, Schneider G, Shanklin J. Crystal structure of stearoyl-acyl carrier protein Δ9 desaturase from castor seed and its relationship to other diiron proteins. EMBO J. 1996;15:4081–4092. [PMC free article] [PubMed] [Google Scholar]
- 31.Haas JA, Fox BG. Fluorescence anisotropy studies of enzyme-substrate complex formation in stearoyl-ACP desaturase. Biochemistry. 2002;41:14472–14481. doi: 10.1021/bi020340s. [DOI] [PubMed] [Google Scholar]
- 32.Page CC, Moser CC, Chen X, Dutton LP. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 33.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature. 1993;366:537–43. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 34.Baldwin J, Krebs K, Ley BA, Edmondson DE, Huynh BH, Bollinger JM. Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 1. Evidence for a transient tryptophan radical. J. Am. Chem. Soc. 2000;122:12195–12206. [Google Scholar]
- 35.Krebs C, Chen S, Baldwin J, Ley BA, Patel U, Edmondson DE, Huynh BH, Bollinger JM., Jr. Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 2. Evidence for and consequences of blocked electron transfer in the W48F variant. J. Am. Chem. Soc. 2000;122:12207–12219. [Google Scholar]
- 36.Bollinger JM, Jr., Tong WH, Ravi N, Huynh BH, Edmondson DE, Stubbe J. Mechanism of assembly of the tyrosyl radical-diiron(III) cofactor of E. coli ribonucleotide reductase. 3. Kinetics of the limiting Fe2+ reaction by optical, EPR, and Möessbauer spectroscopies. J. Am. Chem. Soc. 1994;116:8024–8032. [Google Scholar]
- 37.Kopp DA, Berg EA, Costello CE, Lippard SJ. Structural features of covalently cross-linked hydroxylase and reductase proteins of soluble methane monooxygenase as revealed by mass spectrometric analysis. J. Biol. Chem. 2003;278:20939–20945. doi: 10.1074/jbc.M301581200. [DOI] [PubMed] [Google Scholar]
- 38.Muller J, Lugovskoy AA, Wagner G, Lippard SJ. NMR structure of the [2Fe-2S] ferredoxin domain from soluble methane monooxygenase reductase and interaction with its hydroxylase. Biochemistry. 2002;41:42–51. doi: 10.1021/bi015668k. [DOI] [PubMed] [Google Scholar]
- 39.Lelong C, Setif P, Lagoutte B, Bottin H. Identification of the amino acids involved in the functional interaction between photosystem I and ferredoxin from Synechocystis sp. PCC 6803 by chemical cross-linking. J. Biol. Chem. 1994;269:10034–10039. [PubMed] [Google Scholar]
- 40.Golinelli MP, Gagnon J, Meyer J. Specific interaction of the [2Fe-2S] ferredoxin from Clostridium pasteurianum with the nitrogenase MoFe protein. Biochemistry. 1997;36:11797–11803. doi: 10.1021/bi970528p. [DOI] [PubMed] [Google Scholar]
- 41.Zanetti G, Morelli D, Ronchi S, Negri A, Aliverti A, Curti B. Structural studies on the interaction between ferredoxin and ferredoxin-NADP+ reductase. Biochemistry. 1988;27:3753–3759. [Google Scholar]
- 42.Nogues I, Martinez-Julvez M, Navarro JA, Hervas M, Armenteros L, Angel de la Rosa M, Brodie TB, Hurley JK, Tollin G, Gomez-Moreno C, Medina M. Role of hydrophobic interactions in the flavodoxin mediated electron transfer from photosystem I to ferredoxin-NADP+ reductase in Anabaena PCC 7119. Biochemistry. 2003;42:2036–2045. doi: 10.1021/bi0270541. [DOI] [PubMed] [Google Scholar]
- 43.Martinez-Julvez M, Medina M, Hurley JK, Hafezi R, Brodie TB, Tollin G, Gomez-Moreno C. Lys75 of Anabaena ferredoxin-NADP+ reductase is a critical residue for binding ferredoxin and flavodoxin during electron transfer. Biochemistry. 1998;37:13604–13613. doi: 10.1021/bi9807411. [DOI] [PubMed] [Google Scholar]
- 44.Hurley JK, Weber-Main AM, Stankovich MT, Benning MM, Thoden JB, Vanhooke JL, Holden HM, Chae YK, Xia B, Cheng H, Markley JL, Martinez-Julvez M, Gomez-Moreno C, Schmeits JL, Tollin G. Structure-function relationships in anabaena ferredoxin: Correlations between x-ray crystal structures, reduction potentials, and rate constants of electron transfer to ferredoxin:NADP+ reductase for site-specific ferredoxin mutants. Biochemistry. 1997;36:11100–11117. doi: 10.1021/bi9709001. [DOI] [PubMed] [Google Scholar]
- 45.Morales R, Charon M-H, Kachalova G, Serre L, Medina M, Gomez-Moreno C, Frey M. A redox-dependent interaction between two electron-transfer partners involved in photosynthesis. EMBO Reports. 2000;1:271–276. doi: 10.1093/embo-reports/kvd057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hurley JKM, Martinez-Julvez M, Brodie TB, Medina M, Gomez-Moreno C, Tollin G. Structure-function relationships in anabaena ferredoxin/ferredoxin-NADP+ reductase electron transfer: Insights from site-directed mutagenesis, transient absorption spectroscopy, and x-ray crystallography. Biochim. Biophys. Acta. 2002;1554:5–21. doi: 10.1016/s0005-2728(02)00188-3. R. [DOI] [PubMed] [Google Scholar]
- 47.De Pascalis AR, Jelesarov I, Ackermann F, Koppenol WH, Hirasawa M, Knaff DB, Bosshard HR. Binding of ferredoxin to ferredoxin:NADP+ reductase: The role of carboxyl groups, electrostatic surface potential, and molecular dipole moment. Prot. Sci. 1993;2:1126–1135. doi: 10.1002/pro.5560020707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sazinsky MH, Lippard SJ. Product bound structures of the soluble methane monooxygenase hydroxylase from methylococcus capsulatus (bath): Protein motion in the alpha-subunit. J. Am. Chem. Soc. 2005;127:5814–5825. doi: 10.1021/ja044099b. [DOI] [PubMed] [Google Scholar]