

Abstract
Fifteen single-site charge-reversal mutations of yeast cytochrome c peroxidase (CcP) have been constructed in order to determine the effect of localized charge on the catalytic properties of the enzyme. The mutations are located on the front face of CcP, near the cytochrome c binding site identified in the crystallographic structure of the yeast cytochrome c/CcP complex (Pelletier and Kraut (1992) Science 258, 1748–1755). The mutants are characterized by absorption spectroscopy and hydrogen peroxide reactivity at both pH 6.0 and 7.5 and by steady-state kinetic studies using recombinant yeast iso-1 ferrocytochrome c(C102T) as substrate at pH 7.5. Some of the charge-reversal mutations cause detectable changes in the absorption spectrum, especially at pH 7.5, reflecting changes in the equilibrium between penta-and hexa-coordinate heme species in the enzyme. An increase in the amount of hexa-coordinate heme in the mutant enzymes correlates with an increase in the fraction of enzyme that does not react with hydrogen peroxide. Steady-state velocity measurements indicate that five of the fifteen mutations cause large increases in the Michaelis constant (R31E, D34K, D37K, E118K, and E290K). These data support the hypothesis that the cytochrome c/CcP complex observed in the crystal is the dominant catalytically active complex in solution.
Cytochrome c peroxidase, CcP1, is a detoxification enzyme localized between the inner and outer membranes of yeast mitochondria (1). CcP decreases toxic levels of hydrogen peroxide by catalyzing its reduction to water using ferrocytochrome c (2). The catalytic mechanism involves oxidation of the native enzyme by hydrogen peroxide to an enzyme intermediate called CcP Compound I, CcP-I. CcP-I contains two oxidized sites, an oxyferryl Fe(IV) heme group and a tryptophan π-cation radical located within van der Waals distance of the heme. Interaction with, and electron transfer from, ferrocytochrome c reduces CcP-I back to the native state via a second enzyme intermediate, CcP Compound II, CcP-II, completing the catalytic cycle. Since 1980, when the three-dimensional structure of CcP was first reported (3, 4), CcP has played an important role in elucidating the structural basis for heme protein reactivity, especially in the activation of hydrogen peroxide (5, 6), long-range electron transfer between heme proteins (7, 8), and protein-protein interactions (9, 10).
The nature of the cytochrome c/CcP interaction during catalysis is still under active investigation with a number of questions still unresolved, including the number of cytochrome c binding sites on CcP, the cytochrome c affinity at each binding site, the location of the binding sites, the dynamic nature of cytochrome c bound at each site, and the electron transfer activity of cytochrome c bound at the various sites (7, 11).
The first suggestion of multiple cytochrome c binding sites on the surface of CcP came from the steady-state velocity studies of Margoliash and co-workers in 1977 (12). Since that time a large number of studies have been published concerning the nature of the cytochrome c/CcP interaction and the potential locations of the cytochrome c binding sites on CcP (13–31). Northrup et al. published an influential study modeling the electrostatic interaction between the positively charged cytochrome c molecule and the negatively charged CcP using Brownian dynamics simulations (14). Northrup and colleagues identified three areas on the surface of CcP that had a high probability for formation of productive electron transfer complexes and these were located near Asp-34, Asp-148, and Asp-217 on the surface of CcP. An important milestone in the study of cytochrome c/CcP interactions was the determination of the crystallographic structures of two cytochrome c/CcP complexes by Pelletier and Kraut in 1992 (9). Pelletier and Kraut determined structures of 1:1 complexes between horse heart cytochrome c and CcP and between yeast iso-1 cytochrome c and CcP. They identified Asp-34 and Asp-290 as critical charged residues on CcP for formation of both complexes (9). A number of other studies using computer modeling (15–17) and chemical modification (18) have identified additional residues on CcP that may be important in cytochrome c binding. These include Asp-33, Asp-37, Asp-79, and residues between positions 221–224 and 290–294 as potential interaction sites.
Site-directed mutagenesis of CcP has been used to test some of the suggested locations for the cytochrome c binding sites (21–27). These studies consistently find that charge-neutralization or charge-reversal mutants of Asp-34, Asp-37, and Glu-290 inhibit cytochrome c binding while mutations at Glu-32, Glu-35, Asp-79, Asp-148, Asp-217, and Glu-291 have little effect on cytochrome c binding. The only experimental study that provides information on the location of the secondary binding site is that of Leesch et al. (24), who find that the K149E mutation increases formation of the 2:1 complex while having little effect of formation of the 1:1 complex.
The studies published to date are incomplete in that all potential cytochrome c binding sites may not have been identified by the theoretical and computer modeling studies and all of the potential binding sites have not been tested experimentally. We have initiated a systematic study to identify all of the negatively charged groups on CcP that may be involved in formation of both 1:1 and 2:1 yeast iso-1 cytochrome c/CcP complexes. We plan to mutate, individually, every aspartate and glutamate residue in CcP to a lysine residue. We will characterize the forty-five charge-reversal mutants in terms of their catalytic properties using yeast iso-1 ferrocytochrome c as substrate (32) and determine equilibrium constants for formation of both the 1:1 and 2:1 complexes using isothermal titration calorimetry (26, 27). If there is a single high-affinity binding site, the charge-reversal mutants that affect formation of the 1:1 complex will be clustered near the crystallographic binding site (9). On the other hand, if there are multiple binding sites with similar affinities for cytochrome c, then the locations of the charge-reversal mutations that affect formation of the 1:1 complex will be spread over the surface of CcP. Likewise, we will perform steady-state kinetic and calorimetric studies under conditions that facilitate formation of the 2:1 complex in order to determine which of the forty-five mutations affect binding of the second cytochrome and determine whether these mutations cluster in a single area or are distributed over the surface of CcP.
In this report, we present initial characterization of fifteen charge-reversal mutations on the front-face of CcP, the surface defined by the cytochrome c binding site in the crystal structure of the 1:1 complex (9). The mutants have been characterized by spectroscopic measurements, determination of their hydrogen peroxide reactivity, and steady-state kinetic studies at high ionic strength where only 1:1 complex formation between cytochrome c and CcP is observed. Five of these mutants show substantial increases in the Michaelis constant for yeast iso-1 ferrocytochrome c suggesting that these sites are involved in formation of the 1:1 complex, while ten of the front-face mutants have essentially no effect on the Michaelis constant indicating that these sites do not participate in binding of cytochrome c at high ionic strength.
MATERIALS AND METHODS
Starting Clones and Mutagenesis
The expression system for the recombinant CcP used in this study was provided by James Satterlee, Washington State University (33). The rCcP gene is inserted into the multiple cloning site of the Novagene vector pET24a(+) under control of the T7 promoter. The cloned gene has an identical sequence to that of mature baker’s yeast CcP with the exception of the methionine codon required for bacterial expression (34). The N-terminal methionine is removed from the recombinant CcP in this expression system so that there are no N-terminal modifications in the recombinant CcP compared to baker’s yeast CcP (35).
Gary Pielak, University of North Carolina kindly provided the plasmid pBTR(C102T) containing the gene for yeast iso-1 cytochrome c (36). The yeast iso-1 cytochrome c gene has been altered to replace cysteine-102 with a threonine residue (C102T) to prevent dimerization of the native protein via disulfide bond formation. The plasmid also contains the gene for yeast cytochrome c heme lyase to promote covalent attachment of the heme (37).
Mutations in CcP were created using Stratagene QuikChange mutagenesis kits and sequenced from 5′ to 3′ and from 3′ to 5′ to assure that, except for the intended mutation, the protein was identical to the published sequence.
Protein Expression and Purification
Recombinant rCcP, the charge-reversal mutants, and recombinant yeast iso-1 cytochrome c(C102T) were expressed in E. coli strain BL21(DE3) and isolated using published procedures (35–39).
Protein Concentration Determination
Protein concentrations were determined by absorbance measurements using either a Hewlett Packard Model 8452A diode array spectrophotometer or a Varian/Cary Model 3E spectrophotometer. Extinction coefficients and positions of the Soret maxima for the various proteins at pH 6.0 are: yCcP, 98 ± 3 mM−1 cm−1 at 408 nm; rCcP, 101 ± 3 mM−1 cm−1 at 408 nm; reduced cytochrome c(C102T), 150 ± 5 mM−1 cm−1 at 414 nm; and oxidized yeast cytochrome c(C102T), 118 ± 4 mM−1 cm−1 at 408 nm (29, 39).
Hydrogen Peroxide Concentration
Hydrogen peroxide was reagent grade 30% (v/v) purchased from Aldrich Chemical Company, Inc. The concentration of hydrogen peroxide stock solutions were determined by titration with cerium(IV) sulfate (40).
Hydrogen Peroxide Reactivity
A model SX.17MV stopped-flow spectroflorimeter, APL Ltd. Leatherhead, England was used to investigate the rate of reaction between the mutants and hydrogen peroxide in 0.100 M ionic strength potassium phosphate buffer, pH 6.0 and 7.5, 25°C. The reaction was investigated under pseudo first-order condition with hydrogen peroxide in excess. The enzyme was generally 1 μM and the reaction was monitored at 424 nm. The fraction of active enzyme was estimated by comparing the spectrum of the hydrogen peroxide-oxidized mutant to that of authentic yCcP Compound I.
Steady-State Kinetic Studies
Steady-state kinetic studies and activity measurements were performed at pH 7.5 in potassium phosphate buffers at 0.100 M ionic strength. Initial velocities were determined as a function of yeast iso-1 ferrocytochrome c(C102T) concentration (generally 1 to 100 μM) at constant hydrogen peroxide (200 μM). Initial velocities were determined by measuring the change in absorbance upon oxidation of recombinant yeast iso-1 ferrocytochrome c(C102T) at multiple wavelengths using a Hewlett Packard Model 8452A diode array spectrophotometer. Buffer, cytochrome c, and enzyme were thermally equilibrated at 25 °C in the spectrophotometer, initial absorbance readings made, then the reaction initiated by addition of the hydrogen peroxide. Five different wavelengths, generally chosen from 314, 362, 418, 448, 468, 478, 548, 564, and 574 nm depending upon the substrate concentration, were used to calculate the initial velocity at each set of experimental conditions using equation 1.
| (1) |
The symbols in equation 1 include the initial velocity, v0, the total enzyme concentration, e0, the change in absorbance with time, ΔA/Δt, and the difference in extinction coefficient, Δε, between oxidized and reduced cytochrome c. Samples of the substrate may contain small amounts of oxidized cytochrome c that can inhibit the reaction; fox is the fraction of oxidized cytochrome c in the substrate and is used to make small corrections to the initial velocity. The factor of 2 in the denominator converts cytochrome c turnover to enzyme turnover.
RESULTS
Locations of the Charge-Reversal Mutation Sites
The surface of CcP shown in Figure 1 contains the cytochrome c binding site identified using x-ray crystallography (9). We call this surface the ‘front-face’ of CcP. The cytochrome c binding site in Figure 1 is represented by the three residues shown in black, Asp-34, Ala-193, and Glu-290. Asp-34 and Glu-290 are within 4.2 and 4.4 Å of the cytochrome c surface residues, Lys-87 and Lys-73, respectively, and can potentially form salt-bridges through small adjustments of the side-chains (9). Ala-193 of CcP is in contact with a methyl group on the cytochrome c heme and is thought to be the primary electron transfer point between ferrocytochrome c and CcP during catalysis (9).
Figure 1.
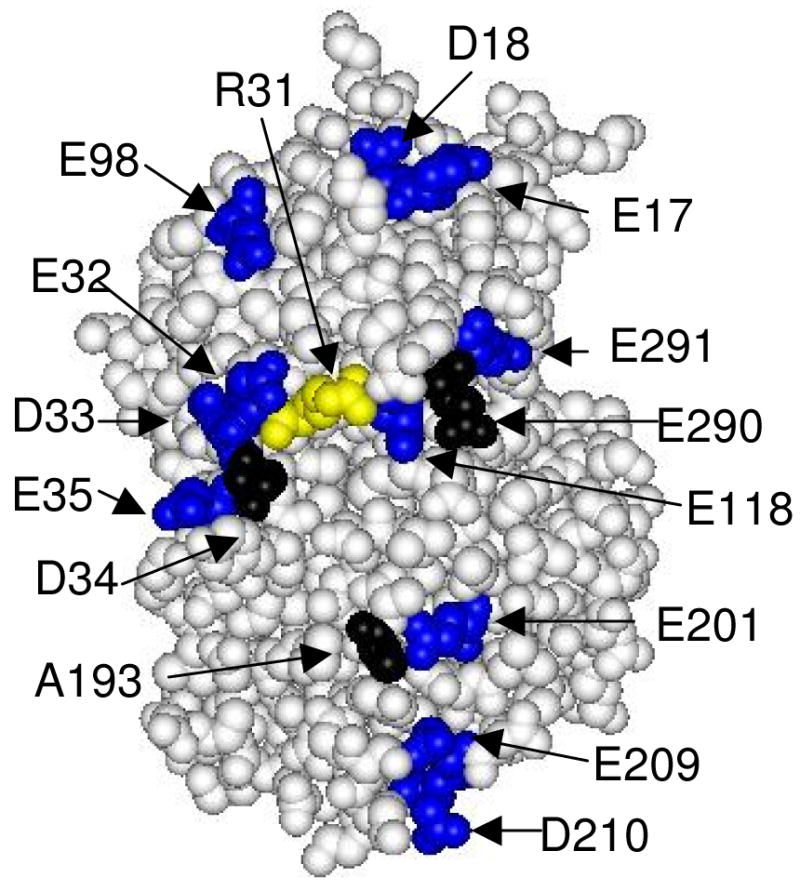
Space-filling model of the front face of CcP showing the location of residues that define the cytochrome c-binding site (black) and the location of residues mutated in this study (blue and yellow). Asp-34, Ala-193, and Glu-290 (black) define the yeast iso-1 cytochrome c binding site. The aspartate and glutamate residues on the front face of CcP, excluding Asp-34 and Glu-290, are shown in blue. The thirteen aspartate and glutamate residues shown in blue were individually mutated to lysine residues in this study. Arg-31 is shown in yellow and was mutated to a glutamate residue. The one-letter abbreviations and sequence numbers are used to identify the amino acid residues. Data from reference (9); Protein Data Bank (PDB) ID: 2PCC.
The locations of the mutation sites relative to the cytochrome c binding site can be somewhat misleading in Figure 1 due to the curvature of the CcP surface. Additional views of the mutation sites are shown in Figures S1-S3 in the Supporting Information accompanying this article. The figures in the Supporting Information include the bound cytochrome c and provide a perspective on the relationship between each of the mutation sites on CcP and the bound cytochrome c.
Spectroscopic Properties of the Charge-Reversal Mutants
Uv-visible absorption spectroscopy was used for the initial characterization of the mutants. Spectra were determined at pH 6.0, the center of the pH stability region for CcP, and at pH 7.5, the pH at which the steady-state and transient-state kinetic studies were performed. The spectra for all rCcP mutants at both pH 6.0 and 7.5 are shown in Figures S4 through S19 in the Supporting Information. Selected spectroscopic parameters, including the RZ value (purity number, the ratio of absorbance at the Soret maximum to the absorbance at the maximum of the protein band near 280 nm) at pH 6.0, the wavelength of the Soret maximum at both pH 6.0 and 7.5, and the ratio of the absorbance at the Soret maximum to that at 380 nm at both pH 6.0 and 7.5, are included in Table 1. Data for yCcP, CcP(MI) and rCcP are included in Table 1 for reference.
Table 1.
Spectroscopic Properties for rCcP and Its Charge-Reversal Mutants.a
| pH 6.0 | pH 7.5 | |||||
|---|---|---|---|---|---|---|
| Mutant | λprotein (nm) | λSoret (nm) | RZb | ASoret/A380 | λSoret (nm) | ASoret/A380 |
| yCcPc | 282 | 408 | 1.28 ± 0.03 | 1.52 ± 0.04 | 409 | 1.60 ± 0.04 |
| CcP(MI)c | 282 | 408 | 1.28 ± 0.04 | 1.53 ± 0.02 | 410 | 1.67 ± 0.03 |
| rCcP | 282 | 408 | 1.31 | 1.54 | 409 | 1.62 |
| Negative Cluster Mutants | ||||||
| R31E | 281 | 407 | 1.40 | 1.53 | 410 | 1.78 |
| E32K | 282 | 409 | 1.29 | 1.58 | 409 | 1.62 |
| D33K | 282 | 409 | 1.29 | 1.60 | 410 | 1.69 |
| D34K | 282 | 408 | 1.29 | 1.57 | 408 | 1.63 |
| E35K | 282 | 408 | 1.30 | 1.58 | 408 | 1.64 |
| D37K | 283 | 411 | 1.34 | 2.23 | 414 | 2.59 |
| Mutants Near Ala-193 | ||||||
| E201K | 280 | 409 | 0.73 | 1.60 | 410 | 1.75 |
| E209K | 280 | 409 | 1.22 | 1.87 | 411 | 2.12 |
| D210K | 281 | 409 | 1.27 | 1.71 | 411 | 1.93 |
| Mutants Near Glu-290 | ||||||
| E118K | 279 | 409 | 1.12 | 1.62 | 410 | 1.76 |
| E290K | 281 | 407 | 1.26 | 1.57 | 410 | 1.84 |
| E291K | 281 | 408 | 1.23 | 1.53 | 411 | 1.87 |
| Top Front-Face Mutants | ||||||
| E17K | 280 | 408 | 1.22 | 1.61 | 411 | 2.00 |
| D18K | 281 | 408 | 1.28 | 1.60 | 410 | 1.74 |
| E98K | 281 | 408 | 1.31 | 1.55 | 409 | 1.62 |
Absorbance values were obtained at 1 nm intervals between 240 and 700 nm in 0.10 M ionic strength, potassium phosphate buffers.
RZ is the purity number, the ratio of the absorbance at the Soret maximum relative to the absorbance at the maximum of the protein band near 280 nm.
At pH 6.0, most of the charge-reversal mutants have spectra similar to that of penta-coordinate CcP but three mutants, D37K, E209K, and D210K, have spectra characteristic of mixtures of penta- and hexa-coordinate heme groups. Representative spectra for the two groups of mutants are shown in Figure 2 with the D34K mutant having a spectrum nearly identical to that of penta-coordinate CcP and the spectrum of the D37K mutant showing a significant contribution from hexa-coordinate heme.
Figure 2.

Spectra of the D34K and D37K mutants of rCcP at pH 6.0. The heme group in the D34K mutant is predominantly penta-coordinate, high-spin while the heme in the D37K mutant is a mixture of penta- and hexa-coordinate forms.
The most characteristic spectral feature of the penta-coordinate heme group in CcP is the high absorbance in the δ band near 380 nm, Figure 2. We have used the ratio of absorbancies at the Soret maximum to that at 380 nm (ASoret/A380) to monitor penta- and hexa-coordination, Table 1. Penta-coordinate yCcP, CcP(MI), and rCcP have ASoret/A380 values between 1.52 and 1.54. Twelve of the fifteen mutants have ASoret/A380 values ranging between 1.53 and 1.62. Three mutants show significant fractions of hexa-coordinate heme and have ASoret/A380 values between 1.71 and 2.23. At pH 7.5, the spectra of CcP and all of the mutants show an increased contribution from hexa-coordinate heme relative to that at pH 6.0, Table 1.
Steady-State Velocity Measurements
The dependence of the steady-state velocity on the yeast iso-1 ferrocytochrome c concentration was determined in 0.100 M ionic strength phosphate buffer, pH 7.5. Representative steady-state velocity plots are shown in Figure 3. Under these conditions, rCcP and thirteen of the fifteen mutants show simple Michaelis-Menten behavior, characterized by a Michaelis constant, KM, and a maximum velocity, Vmax/e0. The steady-state parameters are collected in Table 2.
Figure 3.

Steady-state velocities as a function of the recombinant yeast iso-1 ferrocytochrome c(C102T) concentration for rCcP (open circles) and two of its mutants, D33K (filled triangles) and D34K (filled circles). Experimental conditions: 0.100 M ionic strength potassium phosphate buffer, pH 7.5, 25°C, [H2O2] = 200 μM.
Table 2.
Steady-State Parameters for rCcP and Fifteen Charge-Reversal Mutants.a
| Major Phase | Minor Phase | |||
|---|---|---|---|---|
| Mutant | KM (μM) | Vmax/e0 (s−1) | KM (μM) | Vmax/e0 (s−1) |
| rCcPb | 2.1 ± 0.2 | 640 ± 20 | ||
| Negative Cluster Mutants | ||||
| R31E | >100 | >870 | 1.2 ± 0.4 | 30 ± 3 |
| E32K | 4.5 ± 0.5 | 750 ± 20 | ||
| D33K | 2.4 ± 0.3 | 680 ± 20 | ||
| D34K | >100 | >450 | 2.7 ± 0.8 | 20 ± 2 |
| E35K | 4.4 ± 0.4 | 760 ± 20 | ||
| D37K | 82 ± 12 | 41 ± 3 | ||
| Mutants Near Ala-193 | ||||
| E201K | 2.9 ± 0.9 | 2.8 ± 0.2 | ||
| E209K | 2.8 ± 0.5 | 200 ± 10 | ||
| D210K | 1.9 ± 0.3 | 470 ± 10 | ||
| Mutants Near Glu-290 | ||||
| E118K | 51 ± 8 | 230 ± 20 | ||
| E290K | 60 ± 11 | 140 ± 10 | ||
| E291K | 3.8 ± 0.4 | 520 ± 10 | ||
| Top Front-Face Mutants | ||||
| E17K | 2.3 ± 0.4 | 420 ± 20 | ||
| D18K | 2.0 ± 0.3 | 380 ± 10 | ||
| E98K | 3.1 ± 0.7 | 690 ± 40 | ||
Experimental Conditions: 0.100 M ionic strength, potassium phosphate buffer, pH 7.5, 25 °C, [H2O2] = 200 μM.
Data from reference (30).
Two of the mutants, R31E and D34K, show a biphasic dependence on the cytochrome c concentration with a minor phase characterized by Vmax/e0 and KM values and a major phase in which the velocity increases linearly up to the highest substrate concentrations used in the study. The major kinetic phase for the R31E and D34K mutants can only be characterized by giving lower limits for Vmax/e0 and KM values and these are included in Table 2. Based on the lower limits for the Vmax/e0 values for the major phases of the reaction, the minor phases contribute less than 5% to the maximum activity of R31E and D34K. Steady-state velocity plots for the R31E and D34K mutants are shown in Figure S20 of the Supporting Information.
Rate of Reaction between H2O2 and the Charge-Reversal Mutants
The mutant enzymes were further characterized by determining their rate of reaction with H2O2 using stopped-flow techniques at both pH 6.0 and 7.5. At pH 7.5, seven of the mutants gave monophasic kinetics with H2O2 while eight of the mutants gave biphasic kinetics, with the slow phase of the reaction at least 10 times slower than the fast phase. The rates of monophasic reaction and the fastest phase of the biphasic reactions are linearly dependent upon the H2O2 concentration and are due to the bimolecular reaction between enzyme and H2O2 to form Compound I. The bimolecular rate constant is defined as k1. Values of k1 vary between 36 ± 5 μM−1s−1 and 51 ± 6 μM−1s−1 for E291K and E290K, respectively, averaging 45 ± 4 μM−1s−1 for the fifteen mutants. The average value of k1 for the mutant enzymes is identical to the rate constant of 45 ± 3 μM−1s−1 determined for yCcP at pH 7.5 (41, 44). The results of the stopped-flow studies at pH 7.5 are summarized in Table 3. Kinetic data for the H2O2 reaction at pH 6.0 are given in Table S1 of the Supporting Information, along with a discussion of the slow phase of the H2O2 reaction.
Table 3.
Kinetic Parameters for the Reaction of rCcP and Its Charge-Reversal Mutants with H2O2.a
| Fast Phase | Slow Phase | Inactiveb | ||||
|---|---|---|---|---|---|---|
| Mutant | % Enzyme | k1 (μM−1s−1) | % Enzyme | k2 (μM−1s−1) | k3 (s−1) | % Enzyme |
| yCcPc | 96 | 45 ± 3 | 0 | - | - | 4 |
| CcP(MI)c | 76 | 47 ± 4 | 12 | - | 11 ± 7 | 12 |
| rCcP | 82 | 48 ± 2 | 3 | 1.3 ± 0.1 | - | 15 |
| Negative Cluster Mutants | ||||||
| R31E | 59 | 44 ± 3 | 0 | - | - | 41 |
| E32K | 98 | 43 ± 6 | 2 | 2.1 ± 0.3 | - | 0 |
| D33K | 94 | 41 ± 7 | 0 | - | - | 6 |
| D34K | 94 | 49 ± 4 | 0 | - | - | 6 |
| E35K | 89 | 44 ± 6 | 3 | 2.2 ± 0.3 | - | 8 |
| D37K | 10 | 37 ± 6 | 2 | - | 6 ± 3 | 88 |
| Mutants Near Ala-193 | ||||||
| E201K | 42 | 43 ± 7 | 0 | - | - | 58 |
| E209K | 27 | 40 ± 12 | 0 | - | - | 73 |
| D210K | 58 | 39 ± 11 | 0 | - | - | 42 |
| Mutants Near Glu-290 | ||||||
| E118K | 57 | 44 ± 8 | 0 | - | - | 43 |
| E290K | 61 | 51 ± 6 | 8 | 2.5 ± 0.4 | - | 31 |
| E291K | 40 | 36 ± 5 | 7 | 1.8 ± 0.3 | - | 53 |
| Top Front-Face Mutants | ||||||
| E17K | 31 | 38 ± 8 | 10 | 2.0 ± 0.3 | - | 59 |
| D18K | 68 | 43 ± 14 | 7 | 2.8 ± 1.5 | - | 25 |
| E98K | 93 | 42 ± 6 | 5 | - | 22 ± 7 | 2 |
Experimental conditions: 0.100 M ionic strength, potassium phosphate buffers, pH 7.5, 25°C.
Percent inactive enzyme estimated from the increase in the absorbance at 424 nm in the presence of a stoichiometric excess of hydrogen peroxide relative to that for yeast CcP.
Estimation of the Fraction of H2O2-Reactive Enzyme for the Charge-Reversal Mutants
During the stopped-flow studies of the reaction between the charge-reversal mutants and H2O2 it became apparent that the absorbance changes at 424 nm for some of the mutants were much smaller than those for wild-type CcP. This led us to determine the absolute spectrum of the H2O2-oxidized form of the mutant enzymes. Some of the mutants, such as E32K, react with H2O2 producing Compound I spectra essentially identical to that of yCcP Compound I, Figure 4, while other mutants, such as D37K, showed relatively small changes in the absorption spectrum upon the addition of a slight excess of H2O2, Figure 5.
Figure 4.

Spectra of the E32K mutant of rCcP in the absence (thin line) and presence (thick line) of a slight stoichiometric excess of hydrogen peroxide at pH 7.5. Spectrum of yCcP Compound I (dashed line).
Figure 5.

Spectra of the D37K mutant of rCcP in the absence (thin line) and presence (thick line) of a slight stoichiometric excess of hydrogen peroxide at pH 7.5. Spectrum of yCcP Compound I (dashed line).
The most likely reason for the small absorbance changes for the H2O2 reaction in some of the mutants is that these mutants do not react stoichiometrically with H2O2. An estimate of the fraction of H2O2-reactive enzyme for all of the mutants was made based on the assumption that the spectrum of Compound I for the mutant enzymes is identical to that of yCcP Compound I. This is a reasonable assumption since the heme ligation for Compound I of the mutants should be identical to that of yCcP Compound I. The fraction of H2O2-reactive mutant was estimated based on the observed absorbance change at 424 nm in the presence of a slight excess of H2O2 relative to the absorbance change at 424 nm calculated for the complete conversion of the mutant enzyme to the equivalent of yCcP Compound I, Figures 4 and 5. Based on these estimates, the fraction of H2O2-reactive enzyme varies from 100% for E32K to 12% for D37K at pH 7.5. These estimates, converted to percent of inactive enzyme, are included in the last column in Table 3. The results of equivalent estimates at pH 6 are given in Table S1 of the Supporting Information. A discussion of potential errors in estimating the fraction of inactive enzyme by this procedure is included in the Supporting Information.
DISCUSSION
Variation in KM for the Charge-Reversal Mutants
We are most interested in those mutants showing an altered Michaelis constant since a review of the literature shows that the KM values for the cytochrome c dependence of the steady-state kinetic velocities are within experimental error of the equilibrium dissociation constants for the cytochrome c/CcP complex when carried out at identical conditions of ionic strength (11, 30). In this study, KM has been used to assess the effects of charge-reversal mutations on the front-face of CcP on the interaction of rCcP with yeast iso-1 cytochrome c(C102T). Figure 6 shows a bar graph of the KM values for the charge-reversal mutants as a function of the primary sequence position within CcP. It is obvious that five of the mutations, R31E, D34K, D37K, E118K, and E290K, have very large effects on the binding of cytochrome c with KM values that are more that 24 times larger than that of wild-type rCcP, Table 2. The properties of these five mutants will be discussed below. The KM values for the remaining ten charge-reversal mutants are within a factor of ~2 of that for wild-type rCcP, varying between 1.5 and 4.5 μM, and these mutations are considered to have minimal effect on the interaction between CcP and yeast cytochrome c.
Figure 6.

Michaelis constants, KM, for the interaction of yeast iso-1 ferrocytochrome c with rCcP and with fifteen charge-reversal mutants of CcP. The KM value for rCcP is given in the left-most position followed by the KM values of the mutants listed in order of the primary sequence position of the charge-reversal mutations. The KM values for R31E and D34K are lower limits, while error bars are included for all other KM values. Experimental conditions: 0.100 M ionic strength potassium phosphate buffer, pH 7.5, 25°C, [H2O2] = 200 μM.
Effect of D34K and E290K on Binding
The large increase in KM for both D34K and E290K, Table 2, is consistent with the crystal structure of the 1:1 yeast iso-1 cytochrome c/CcP complex (9), which shows potential interactions between Asp-34 and Glu-290 with lysines 87 and 73 on cytochrome c, respectively. Previous studies have also shown that charge-neutralization or charge-reversal mutations at positions 34 and 290 decrease the affinity for binding cytochrome c (24–27). Steady-state kinetic studies with D34N and E290N mutants show 4- to 18-fold increases in the KM values for yeast iso-1 ferrocytochrome c, depending upon ionic strength (25) while calorimetry studies with these same mutants find a 4-fold decrease in binding affinity for horse cytochrome c in 50 mM ionic strength buffers, pH 6.0 (26). Photoinitiated electron transfer kinetic studies between E290K and zinc-substituted horse cytochrome c indicate that the affinity for horse cytochrome c is reduced 20-fold in 18 mM ionic strength buffer, pH 7.0 (24). All of these data are consistent with the involvement of both Asp-34 and Glu-290 in binding cytochrome c to form a 1:1 complex.
Effect of E118K on Binding
The E118K mutation increased KM for yeast cytochrome c by a factor of 24, Table 2. Although Glu-118 is not in direct contact with the bound cytochrome (see Figure S3, Supporting Information), Glu-118 is near Glu-290 and located on the surface of CcP between the two protein molecules. The carboxylate side-chain of Glu-118 is partially buried with the OE2 oxygen hydrogen-bonded to the peptide nitrogen of Leu-289 in the interior of CcP. Mutation of Glu-118 to lysine will disrupt the hydrogen bonding to Leu-289 and most likely cause the positively charged lysine side-chain to rotate toward the surface of CcP. This would increase the electrostatic repulsion between CcP and cytochrome c weakening the binding as observed. Another possible contribution to the weakening of binding could be the disruption of the Glu-118/Leu-289 hydrogen bond, altering the conformation of the polypeptide backbone from Leu-289 through the C-terminal residue, Leu -294, including Glu-290. This could affect the Glu-290/Lys-73 interaction between CcP and cytochrome c.
Effect of D37K on Binding
Previous studies have shown that the D37K mutation decreases the affinity for both horse and yeast cytochrome c, with decreases ranging from 5- to 67-fold depending upon experimental conditions (21–23). Our steady-state data are consistent with previous studies, showing that the D37K mutation has a ~40-fold decrease in affinity for yeast cytochrome c at pH 7.5, 100 mM ionic strength. However, Asp-37 is not involved in binding cytochrome c in the crystal structures of the horse and yeast cytochrome c/CcP complexes (9). We do not believe that the influence of Asp-37 is the result of a through-space electrostatic repulsion of cytochrome binding at the crystallographic site unless there is a major reorientation of the lysine side-chain in the mutant with respect to the aspartate side-chain in wild-type CcP. This is based on the observation that Glu-32, Asp-33, and Glu-35 are closer to the bound cytochrome in the crystal structure than is Asp-37 and the E32K, D33K, and E35K mutations have no significant effect on KM, Table 2. It is also difficult to visualize an orientation of cytochrome c that would allow the cytochrome to simultaneously interact with Asp-37, Asp-34, and Glu-290 without also involving Glu-32, Asp-34, and Glu-35.
Another consideration that must be taken into account is the localized nature of the charge reversal mutation effects. D34K causes a 48-fold decrease in cytochrome c affinity while mutations at both adjacent residues, D33K and E35K, have almost no effect. This is also seen with E290K and E291K. The former mutation causes a 30-fold decrease in binding affinity while the latter has essentially no effect. Assuming that the D37K mutation causes a localized effect on cytochrome c binding leads to the conclusion that cytochrome c can bind in a different orientation from that shown in the crystal structure of the 1:1 complex, one that involves direct interaction between Asp-37 and a positively charged residue on cytochrome c. Alternatively, Asp-37 could be part of a second binding site that is adjacent to and/or overlaps the crystallographic site as suggested by Nocek et al. (7). Nocek et al. suggest that the second binding site is essentially that proposed by Poulos and Kraut (15) involving residues Asp-34, Asp-37, Asp-79, Gln-86, Asn-87 and Asp-217. Asp-37 is at the boundary between the crystallographic site and the Poulos/Kraut site and could affect cytochrome c binding at both sites. On the other hand, the data of Corin et al. (21, 22) would seem to eliminate Asp-79 and Asp-217 from the second site.
A recent NMR study (31) of the CcP/yeast iso-1 cytochrome c complex demonstrates that the bound cytochrome has considerable mobility, but generally corroborates the crystallographic structure, indicating that cytochrome c resides at the crystallographic site about 70% of the time and in much more dynamic encounter complexes about 30% of the time. The encounter complexes encompass much of the CcP surface as first postulated by Northrup et al. (14). Using covalently attached paramagnetic spin labels, Volkov et al. (31) show that encounter complexes exist near residues 38, 200, and 288 on the surface of CcP and but not at residues 137 and 263. The spin label probe at residue 38 should detect cytochrome c binding near Asp-37.
There is also the possibility that the D37K mutation decreases the affinity for cytochrome c by altering the surface conformation of CcP. The carboxylate side-chain of Asp-37 is rotated toward the interior of CcP with the OD2 oxygen atom hydrogen bonding to nitrogen NE2 of His-181 (4). His-181 is part of a hydrogen-bonding network that connects residues in the distal heme pocket with those in the proximal heme pocket (43). Disruption of the hydrogen-bonding network by the lysine for aspartate substitution could account for the increased concentration of hexa-coordinate heme in this mutant, Table 1 and Figure 2, and, perhaps for an altered surface conformation of the cytochrome c binding site. It is likely that the lysine for aspartate substitution would rotate the lysine side-chain toward the surface of CcP, bringing it closer to cytochrome c bound at the crystallographic site, increasing the electrostatic repulsion of bound cytochrome c, and decreasing the affinity at the crystallographic site.
Effect of R31E on Binding
The fifth mutation with a large effect on the Michaelis constant is the R31E mutation. Arg-31 is a positively charged residue that precedes the string of negatively-charged residues, Glu-32 through Asp-37. We decided to mutate Arg-31 to a glutamate in anticipation that it might increase the affinity of CcP for cytochrome c. Much to our surprise, the R31E mutation had a profound effect on the steady-state kinetics, generating a biphasic Michaelis-Menten plot, Figure S20 in the Supporting Information, with the major phase having a KM value so large that only a lower limit of 100 μM could be established. The steady-state data suggests that the R31E mutation decreases the binding affinity for cytochrome c by more than a factor of 50. This is difficult to understand based on simple electrostatic considerations and something more profound may be happening in the R31E mutant.
Within the yeast cytochrome c/CcP complex (9), the side chain of Arg-31 is hydrogen bonded to the C-terminal carboxylate of Leu-294 and is within 4.7 Å of the ε-amino group of Lys-86 in cytochrome c. The effect of the R31E mutation appears to be the result of opposite effects, strengthening the interaction with Lys-86 but disrupting the hydrogen bond with Leu-294. Disruption of the R31E/Leu-294 interaction apparently has the more profound effect, perhaps by changing the conformation of the C-terminus and altering the Glu-290/Lys-73 interaction as was proposed for the effects of the E118K mutation above. Both Arg-31 and Glu-118 are involved in hydrogen bonding to the last few residues at the C-terminus of CcP and certainly play some role in stabilizing the conformation of the C-terminus. The C-terminus includes Glu-290, which can form a salt bridge with Lys-73 of cytochrome c in the 1:1 complex (9).
Relation to Other Studies
The data presented here are consistent with other studies that support the hypothesis that there is a unique catalytically-competent cytochrome c/CcP complex and this complex has a structure similar to, or identical to, the structures determined by Pelletier and Kraut (9) for the yeast iso-1 cytochrome c/CcP and horse heart cytochrome c/CcP complexes in the crystalline state. This hypothesis is supported by the cytochrome c/CcP cross-linking studies of Poulos and co-workers (28, 45, 46) as well as those of Nakani and colleagues (29, 30). Pappa and Poulos (45) and Papa et al. (46) used site-directed mutagenesis to engineer specific cysteine residues into both yeast cytochrome c and CcP to covalently attach cytochrome c to the Pelletier/Kraut binding site through a CcP Cys-290/cytochrome c Cys-73 disulfide bond. The covalent complex is electron transfer competent with the covalently bound ferrocytochrome c rapidly reducing the Trp-191 radical when CcP Compound I is formed. Equally important is the observation that the covalent complex has a very slow turnover rate for exogenous ferrocytochrome c indicating that if a second cytochrome c binding site existed, its catalytic activity is low. Nakani et al. (29) also synthesized the CcP Cys-290/cytochrome c Cys-73 covalent complex and showed that the residual activity of the covalent complex during steady-state turnover of exogenous ferrocytochrome c is due to small amounts of rCcP(E290C) that co-purified with the covalent complex and that the covalent complex is completely inactive toward exogenous ferrocytochrome c, eliminating the possibility of a second catalytically-active cytochrome c binding site on CcP. Guo et al. (28) synthesized a second covalent complex in which cytochrome c is bound at the Pelletier/Kraut binding site through a CcP Cys-197/cytochrome c Cys-81 disulfide bond. This covalent complex has rapid electron transfer from the covalently bound ferrocytochrome c to the Trp-191 radical in CcP Compound I and also has a very slow rate of electron turnover of exogenous ferrocytochrome c. Guo et al. (28) obtained a high-resolution x-ray structure of the covalent complex and demonstrated that it closely resembles the Pelletier/Kraut structure (9). As in the Pelletier/Kraut structure, there are no direct salt bridges between charged residues in CcP and cytochrome c at the interface between the two proteins in the covalent complex but there are a number of water-mediated interactions between polar groups on CcP and cytochrome c (28).
Crane and co-workers (47–49) have provided some of the most definitive evidence that the 1:1 CcP/cytochrome c complexes observed in the crystalline state are the electron transfer-active species. Crane and co-workers have crystallized several complexes between zinc-porphyrin CcP (ZnCcP) and cytochrome c and have shown that the quenching rate of the zinc porphyrin triplet by the Fe(III) cytochrome c within the crystal is essentially the same as that in solution (47–48). This provides strong support for the idea that the 1:1 complexes observed in the crystalline state are in fact the complexes involved in electron transfer between cytochrome c and CcP.
Another important aspect of the work of Crane and colleagues is that they have provided crystal structures for six additional 1:1 complexes of various modified forms of CcP and cytochrome c (47, 48) to go along with the original yeast and horse cytochrome complexes with wild-type CcP provided by Pelletier and Kraut (9). These complexes include the yeast and horse cytochrome complexes with ZnCcP (47) and four Phe-82 mutants of yeast cytochrome c binding to ZnCcP (48). In the crystal structures of all eight complexes published to date, cytochrome c binds to the same general surface region of CcP but with some variation as originally found for the yeast and horse cytochrome/CcP structures (9, 48). Kang and Crane (48) have also described a low resolution crystal structure of a ninth complex, that between a K72S/F82Y double mutant of yeast cytochrome c and ZnCcP, in which the cytochrome binds in a substantially different region on the surface of ZnCcP and shows no electron transfer activity in the crystal. Interestingly, none of the cytochrome c molecules in any of the nine complexes is near Asp-37 in CcP and the inhibition of binding of cytochrome c by the D37K mutant still remains an enigma.
Variation in Vmax/e0
At the beginning of these studies, we had anticipated that the charge-reversal mutations would modulate the binding affinity of cytochrome c to CcP but that cytochrome c would bind to the same site and have essentially the same maximum velocity as wild-type enzyme. The observation of the large variation in the maximum velocity was unexpected. The maximum velocities actually have a larger variation than the Michaelis constants, varying >300-fold, from 2.8 s−1 for E201K to >870 s−1 for R31E, Table 2. A major part of the variation in Vmax/e0 is due to the presence of H2O2-unreactive forms in the mutant enzymes, Table 3. If the Vmax/e0 values are corrected for the fraction of H2O2-reactive enzyme, there is much less variation in the maximum turnover rates.
The fraction of H2O2-inactive enzyme correlates with the amount of hexa-coordinate heme as monitored by the ASoret/A380 ratio, Tables 1 and 3. The observation that charge-reversal mutations on the surface of CcP caused changes in heme ligation in the interior of the protein was another unanticipated finding although not central to the major objective of this study. We hypothesize that the charge-reversal mutation affects the apparent pKA for the alkaline transition in CcP (42, 50) by modulating the binding of hydroxide ion to the heme iron, the predominant hexa-coordinate heme species observed in the CcP mutants at pH 6 and 7.5, Table 1. A discussion of the effects of the charge-reversal mutations on heme ligation and the consequent changes in H2O2 reactivity is included in the Supporting Information accompanying this article.
CONCLUSIONS
Five of the fifteen (R31E, D34K, D37K, E118K, and E290K) charge-reversal mutants of CcP investigated in this study have significantly decreased affinity for yeast iso-1 ferrocytochrome c as monitored by the Michaelis constant in steady-state kinetic studies. Three of the five (D34K, D37K, and E290K) have previously been shown to have decrease affinity for cytochrome c (21–27) while two (R31E and E118K) have been shown to decrease the binding affinity for the first time. Three of the mutation sites that affect cytochrome c binding, D34K, E118K, and E290K, are within the cytochrome c binding domain identified in the crystal structure of the yeast iso-1 cytochrome c/CcP complex (9) while two are on the periphery, R31E, and D37K. The R31E mutation most likely alters the cytochrome c affinity by changing the conformation of the C-terminus of CcP, the region that includes Glu-290. The effect of Asp-37 on the binding of cytochrome c is not completely understood. It is unlikely that the effect is due to direct electrostatic repulsion of cytochrome c bound at the Pelletier/Kraut site (9). The D37K mutation could alter the conformation of CcP near Asp-34 and Asp-35, indirectly weakening the binding of cytochrome c, or Asp-37 could be part of a second binding site of the left-hand side of CcP, not yet observed in any of the crystal structures of the various cytochrome c/CcP complexes (9, 47, 48).
A second important result of these studies is that ten of the fifteen mutation sites on the front face of CcP, Figure 1, do not affect cytochrome c binding. Previously, three of these ten sites (E32K, E35K, and E291K) have been shown not to affect cytochrome binding (24, 25). This study adds seven more sites that can be eliminated as potential cytochrome c binding sites for formation of 1:1 complexes.
Supplementary Material
Figures describing the localization of mutation sites relative to the cytochrome c-binding site on CcP, spectra of all mutants at pH 6.0 and 7.5, hydrogen peroxide reactivity at pH 6.0, and steady-state velocity plots for selected charge-reversal mutants are available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Professor James Satterlee, Washington State University, for providing the plasmid containing the gene for rCcP and Professor Gary Pielak, University of North Carolina, for providing the plasmid containing the yeast iso-1 cytochrome c(C102T) and heme lyase genes.
Footnotes
This work was supported in part by a grant from the National Institutes of Health (R15 GM59740).
Abbreviations: Mutations in the amino acid sequences of either CcP or cytochrome c are indicated by using the one letter code for the amino acid residue in the wild-type protein, followed by the residue number and the one letter code for the amino acid residue in the mutant protein, i.e., C102T represents a mutant in which a threonine residue replaces the cysteine residue at position 102 of the wild-type protein; CcP, generic abbreviation for cytochrome c peroxidase whatever the source; yCcP, authentic yeast cytochrome c peroxidase isolated from bakers’ yeast, Saccharomyces cervisiae; rCcP, recombinant CcP expressed in E. coli with an identical amino acid sequence as yCcP; CcP(MI), recombinant CcP expressed in E. coli with four amino acid variations compared to yCcP, a Met-Ile N-terminal extension and mutations T53I and D152G; ZnCcP, CcP in which zinc porphyrin replaces the heme; CcP-I, CcP Compound I, the first intermediate observed in the catalytic cycle produced upon oxidation of CcP with hydrogen peroxide; CcP-II, CcP Compound II, the second intermediate observed in the catalytic cycle produced by one-electron reduction of CcP-I; RZ, reinheitzahl or purity number, the ratio of the absorbance at the Soret maximum to the absorbance at the protein band maximum near 280 nm.
References
- 1.Yonetani T, Ohnisi T. Cytochrome c peroxidase, a mitochondrial enzyme of yeast. J Biol Chem. 1966;241:2983–2984. [PubMed] [Google Scholar]
- 2.Yonetani T. Studies on cytochrome c peroxidase IV. A comparison of peroxide-induced complexes of horseradish and cytochrome c peroxidases. J Biol Chem. 1966;241:2562–2571. [PubMed] [Google Scholar]
- 3.Poulos TL, Freer ST, Alden RA, Edwards SL, Skogland U, Takio K, Eriksson B, Xuong N, Yonetani T, Kraut J. The crystal structure of cytochrome c peroxidase. J Biol Chem. 1980;255:575–580. [PubMed] [Google Scholar]
- 4.Finzel BC, Poulos TL, Kraut J. Crystal structure of yeast cytochrome c peroxidase refined at 1.7-Å resolution. J Biol Chem. 1984;259:13027–13036. [PubMed] [Google Scholar]
- 5.Poulos TL, Kraut J. The stereochemistry of peroxidase catalysis. J Biol Chem. 1980;255:8199–8205. [PubMed] [Google Scholar]
- 6.Erman JE, Vitello LB, Miller MA, Shaw A, Brown KA, Kraut J. Histidine 52 is a critical residue for rapid formation of cytochrome c peroxidase compound I. Biochemistry. 1993;32:9798–9806. doi: 10.1021/bi00088a035. [DOI] [PubMed] [Google Scholar]
- 7.Nocek JM, Zhou JS, De Forest S, Priyadarshi S, Beratan DN, Onuchic JN, Hoffman BM. Theory and practice of electron transfer within protein-protein complexes: application to the multidomain binding of cytochrome c by cytochrome c peroxidase. Chem Rev. 1996;96:2459–2489. doi: 10.1021/cr9500444. [DOI] [PubMed] [Google Scholar]
- 8.Bendal DS, editor. Protein electron transfer. Bios Scientific Publishers; Oxford: 1996. [Google Scholar]
- 9.Pelletier H, Kraut J. Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science. 1992;258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 10.Mathews FS, Mauk AG, Moore GR. Protein-protein complexes formed by electron transfer proteins. In: Kleanthous C, editor. Protein-protein recognition. Oxford University Press; Oxford: 2000. pp. 60–101. [Google Scholar]
- 11.Erman JE, Vitello LB. Yeast cytochrome c peroxidase: mechanistic studies via protein engineering. Biochim Biophys Acta. 2002;1597:193–220. doi: 10.1016/s0167-4838(02)00317-5. [DOI] [PubMed] [Google Scholar]
- 12.Kang CH, Ferguson-Miller S, Margoliash E. Steady state kinetics and binding of eukaryotic cytochromes c with yeast cytochrome c peroxidase. J Biol Chem. 1977;252:919–926. [PubMed] [Google Scholar]
- 13.Kang DS, Erman JE. The cytochrome c peroxidase-catalyzed oxidation of ferrocytochrome c by hydrogen peroxide. Steady state kinetic mechanism. J Biol Chem. 1982;257:12775–12779. [PubMed] [Google Scholar]
- 14.Northrup SH, Boles JO, Reynolds JCL. Brownian dynamics of cytochrome c and cytochrome c peroxidase association. Science. 1988;241:67–70. doi: 10.1126/science.2838904. [DOI] [PubMed] [Google Scholar]
- 15.Poulos TL, Kraut J. A hypothetical model of the cytochrome c peroxidase · cytochrome c electron transfer complex. J Biol Chem. 1980;255:10322–10330. [PubMed] [Google Scholar]
- 16.Poulos TL, Finzel BC. Heme enzyme structure and function. Peptide and Protein Reviews. 1984;4:115–171. [Google Scholar]
- 17.Lum VR, Brayer GD, Louie GV, Smith M, Mauk AG. Computer modeling of yeast iso-1 cytochrome c-yeast cytochrome c peroxidase complexes. Prot Struct Fold Design. 1987;2:143–150. [Google Scholar]
- 18.Bechtold R, Bosshard HR. Structure of an electron transfer complex. II. Chemical modification of carboxyl groups of cytochrome c peroxidase in presence and absence of cytochrome c. J Biol Chem. 1985;260:5191–5200. [PubMed] [Google Scholar]
- 19.Stemp EDA, Hoffman BM. Cytochrome c peroxidase binds two molecules of cytochrome c: evidence for a low-affinity, electron-transfer-active site on cytochrome c peroxidase. Biochemistry. 1993;32:10848–10865. doi: 10.1021/bi00091a041. [DOI] [PubMed] [Google Scholar]
- 20.Mauk MR, Ferrer JC, Mauk AG. Proton linkage in formation of the cytochrome c-cytochrome c peroxidase complex: electrostatic properties of the high- and low-affinity cytochrome c binding sites on the peroxidase. Biochemistry. 1994;33:12609–12614. doi: 10.1021/bi00208a011. [DOI] [PubMed] [Google Scholar]
- 21.Corin AF, McLendon G, Zhang Q, Hake RA, Falvo J, Lu KS, Ciccarelli RB, Holzschu D. Effects of surface amino acid replacements in cytochrome c peroxidase on complex formation with cytochrome c. Biochemistry. 1991;30:11585–11595. doi: 10.1021/bi00113a014. [DOI] [PubMed] [Google Scholar]
- 22.Corin AF, Hake RA, McLendon G, Hazzard JT, Tollin T. Effects of surface amino acid replacements in cytochrome c peroxidase on intracomplex electron transfer from cytochrome c. Biochemistry. 1993;32:2756–2762. doi: 10.1021/bi00062a004. [DOI] [PubMed] [Google Scholar]
- 23.Zhou JS, Tran ST, McLendon G, Hoffman BM. Photoinduced electron transfer between cytochrome c peroxidase (D37K) and Zn-substituted cytochrome c: probing the two-domain binding and reactivity of the peroxidase. J Amer Chem Soc. 1997;119:269–277. [Google Scholar]
- 24.Leesch VW, Bujons J, Mauk AG, Hoffman BM. Cytochrome c peroxidase-cytochrome c complex: locating the second binding domain on cytochrome c peroxidase with site-directed mutagenesis. Biochemistry. 2000;39:10132–10139. doi: 10.1021/bi000760m. [DOI] [PubMed] [Google Scholar]
- 25.Miller MA. A complete mechanism for steady-state oxidation of yeast cytochrome c by cytochrome c peroxidase. Biochemistry. 1996;35:15791–15799. doi: 10.1021/bi961488c. [DOI] [PubMed] [Google Scholar]
- 26.Erman JE, Kresheck GK, Vitello LB, Miller MA. Cytochrome c/cytochrome c peroxidase complex: Effect of binding-site mutations on the thermodynamics of complex formation. Biochemistry. 1997;36:4054–4060. doi: 10.1021/bi962632x. [DOI] [PubMed] [Google Scholar]
- 27.Pielak GJ, Wang X. Interactions between yeast iso-1-cytochrome c and its peroxidase. Biochemistry. 2001;40:422–428. doi: 10.1021/bi002124u. [DOI] [PubMed] [Google Scholar]
- 28.Guo M, Bhaskar B, Huiying L, Barrows TP, Poulos TL. Crystal structure and characterization of a cytochrome c peroxidase-cytochrome c site-specific cross-link. Proc Nat’l Acad Sci USA. 2004;101:5940–5945. doi: 10.1073/pnas.0306708101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakani S, Viriyakul T, Mitchell R, Vitello LB, Erman JE. Characterization of a covalently-linked yeast cytochrome c/cytochrome c peroxidase complex: Evidence for a single, catalytically-active cytochrome c binding site on cytochrome c peroxidase. Biochemistry. 2006;45:9887–9893. doi: 10.1021/bi060586n. [DOI] [PubMed] [Google Scholar]
- 30.Nakani S, Vitello LB, Erman JE. Characterization of four covalently-linked yeast cytochrome c/cytochrome c peroxidase complexes: Evidence for electrostatic interaction between bound cytochrome c molecules. Biochemistry. 2006;45:14371–14378. doi: 10.1021/bi061662p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volkov AN, Worrall JAR, Holtzmann E, Ubbink M. Solution structure and dynamics of the complex between cytochrome c and cytochrome c peroxidase determined by paramagnetic NMR. Proc Nat’l Acad Sci USA. 2006;103:18945–18950. doi: 10.1073/pnas.0603551103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matthis AL, Erman JE. Cytochrome c peroxidase-catalyzed oxidation of yeast iso-1 ferrocytochrome c by hydrogen peroxide. Ionic strength dependence of the steady-state parameters. Biochemistry. 1995;34:9991–9999. doi: 10.1021/bi00031a021. [DOI] [PubMed] [Google Scholar]
- 33.Savenkova MI, Satterlee JD, Erman JE, Siems WF, Helms GL. Expression, purification, characterization, and NMR studies of highly deuterated recombinant cytochrome c peroxidase. Biochemistry. 2001;40:12123–12131. doi: 10.1021/bi0111000. [DOI] [PubMed] [Google Scholar]
- 34.Takio K, Titani K, Ericsson LH, Yonetani T. Primary structure of yeast cytochrome c peroxidase II. The complete amino acid sequence. Arch Biochem Biophys. 1980;203:615–6299. doi: 10.1016/0003-9861(80)90219-2. [DOI] [PubMed] [Google Scholar]
- 35.Teske JG, Savenkova MI, Mauro JM, Erman JE, Satterlee JD. Yeast cytochrome c peroxidase expression in Escherichia coli and rapid isolation of various highly purified holoenzymes. Protein Expression Purif. 2000;19:139–147. doi: 10.1006/prep.2000.1220. [DOI] [PubMed] [Google Scholar]
- 36.Morar AS, Kakouras D, Young GB, Boyd J, Pielak GJ. Expression of 15N-labeled eukaryotic cytochrome c in Escherichia coli. J Biol Inorg Chem. 1999;4:220–222. doi: 10.1007/s007750050307. [DOI] [PubMed] [Google Scholar]
- 37.Pollock WBR, Rosell FI, Twitchett MB, Dumont ME, Mauk AG. Bacterial expression of a mitochondrial cytochrome c. Trimethylation of Lys72 in yeast iso-1-cytochrome c and the alkaline conformational transition. Biochemistry. 1998;37:6124–6131. doi: 10.1021/bi972188d. [DOI] [PubMed] [Google Scholar]
- 38.Fishel LA, Villafranca JE, Mauro JM, Kraut J. Yeast cytochrome c peroxidase: mutagenesis and expression in Escherichia coli show tryptophan-51 is not the radical site in compound I. Biochemistry. 1987;26:351–360. doi: 10.1021/bi00376a004. [DOI] [PubMed] [Google Scholar]
- 39.Vitello LB, Huang M, Erman JE. pH-dependent spectral and kinetic properties of cytochrome c peroxidase: Comparison of freshly isolated and stored enzyme. Biochemistry. 1990;29:4283–4288. doi: 10.1021/bi00470a004. [DOI] [PubMed] [Google Scholar]
- 40.Kolthoff IM, Belcher R. Volumetric Analysis. Vol. 3. Interscience; New York: 1957. Hydrogen peroxide; pp. 75–76. [Google Scholar]
- 41.Loo S, Erman JE. A kinetic study of the reaction between cytochrome c peroxidase and hydrogen peroxide. Dependence on pH and ionic strength. Biochemistry. 1975;14:3467–3470. doi: 10.1021/bi00686a027. [DOI] [PubMed] [Google Scholar]
- 42.Vitello LB, Erman JE, Miller MA, Mauro JM, Kraut J. Effect of asp-235 → asn substitution on the absorption spectrum and hydrogen peroxide reactivity of cytochrome c peroxidase. Biochemistry. 1992;31:11524–11535. doi: 10.1021/bi00161a034. [DOI] [PubMed] [Google Scholar]
- 43.Miller MA, Hazzard JT, Mauro JM, Edwards SL, Simons PC, Tollin G, Kraut J. Site-directed mutagenesis of yeast cytochrome c peroxidase shows histidine 181 is not required for oxidation of ferrocytochrome c. Biochemistry. 1988;27:9081–9088. doi: 10.1021/bi00426a003. [DOI] [PubMed] [Google Scholar]
- 44.Vitello LB, Erman JE, Mauro JM, Kraut J. Characterization of the hydrogen peroxide - enzyme reaction for two cytochrome c peroxidase mutants. Biochim, Biophys Acta. 1990;1038:90–97. doi: 10.1016/0167-4838(90)90015-8. [DOI] [PubMed] [Google Scholar]
- 45.Pappa HS, Poulos TL. Site-specific cross-linking as a method for studying intramolecular electron transfer. Biochemistry. 1995;34:6573–6580. doi: 10.1021/bi00020a001. [DOI] [PubMed] [Google Scholar]
- 46.Pappa HS, Tajbaksh S, Saunders AJ, Pielak GJ, Poulos TL. Probing the cytochrome c peroxidase-cytochrome c electron transfer reaction using site specific cross-linking. Biochemistry. 1996;35:4837–4845. doi: 10.1021/bi952935b. [DOI] [PubMed] [Google Scholar]
- 47.Kang SA, Marjavaara PJ, Crane BR. Electron transfer between cytochrome c and cytochrome c peroxidase in single crystals. J Amer Chem Soc. 2004;126:10836–10837. doi: 10.1021/ja049230u. [DOI] [PubMed] [Google Scholar]
- 48.Kang SA, Crane BR. Effects of interface mutations on association modes and electron-transfer rates between protein. Proc Nat’l Acad Sci USA. 2005;102:15465–15470. doi: 10.1073/pnas.0505176102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang SA, Hoke KR, Crane BR. Solvent isotope effects on interfacial protein electron transfer in crystals and electrode films. J Amer Chem Soc. 2006;128:2346–3355. doi: 10.1021/ja0557482. [DOI] [PubMed] [Google Scholar]
- 50.Dhaliwal BK, Erman JE. A kinetic study of the alkaline transitions in cytochrome c peroxidase. Biochim Biophys Acta. 1985;827:174–182. doi: 10.1016/0167-4838(85)90087-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures describing the localization of mutation sites relative to the cytochrome c-binding site on CcP, spectra of all mutants at pH 6.0 and 7.5, hydrogen peroxide reactivity at pH 6.0, and steady-state velocity plots for selected charge-reversal mutants are available free of charge via the Internet at http://pubs.acs.org.