

Abstract
6-Deoxyerythronolide B synthase (DEBS) is a modular polyketide synthase (PKS) responsible for the biosynthesis of 6-dEB (1), the parent aglycone of the broad spectrum macrolide antibiotic erythromycin. Individual DEBS modules, which contain the catalytic domains necessary for each step of polyketide chain elongation and chemical modification, can be deconstructed into constituent domains. To better understand the intrinsic stereospecificity of the ketoreductase (KR) domains, an in vitro reconstituted system has been developed involving combinations of ketosynthase (KS) – acyl transferase (AT) didomains with acyl-carrier protein (ACP) and KR domains from different DEBS modules. Incubations with (2S,3R)-2-methyl-3-hydroxypentanoic acid N-acetylcysteamine thioester (2) and methylmalonyl-CoA plus NADPH result in formation of a reduced, ACP-bound triketide that is converted to the corresponding triketide lactone 4 by either base- or enzyme-catalyzed hydrolysis/cyclization. A sensitive and robust GC-mass spectrometry technique has been developed to assign the stereochemistry of the resulting triketide lactones, on the basis of direct comparison with synthetic standards of each of the four possible diasteromers 4a–4d. Using the [KS][AT] didomains from either DEBS module 3 or module 6 in combination with KR domains from modules 2 or 6 gave in all cases exclusively (2R,3S,4R,5R)-3,5-dihydroxy-2,4-dimethyl-n-heptanoic acid-δ-lactone (4a). The same product was also generated by a chimeric module in which [KS3][AT3] was fused to [KR5][ACP5] and the DEBS thioesterase [TE] domain. Reductive quenching of the ACP-bound 2-methyl-3-ketoacyl triketide intermediate with sodium borohydride confirmed that in each case the triketide intermediate carried only an unepimerized D-2-methyl group. The results confirm the predicted stereospecificity of the individual KR domains, while revealing an unexpected configurational stability of the ACP-bound 2-methyl-3-ketoacyl thioester intermediate. The methodology should be applicable to the study of any combination of heterologous [KS][AT] and [KR] domains.
Modular polyketide synthases (PKSs) are multifunctional enzymes responsible for the biosynthesis of polyketides metabolites that exhibit a wide range of important biological properties, including antibacterial, antifungal, anticancer, and immunosuppressive activity.1 They catalyze repetitive Claisen-like condensations between methylmalonyl or malonyl thioester building blocks and the growing polyketide acyl thioesters. Using an assembly line of active sites, each module is responsible for one cycle of polyketide chain elongation and functional group modification before passing the resulting intermediate to the downstream module. Each module contains a set of three core domains: the β-ketoacyl-ACP synthase (KS), which accepts a polyketide chain from the appropriate upstream module and catalyzes the chain-building decarboxylative condensation between this intermediate and an ACP-bound methylmalonyl or malonyl chain extension unit; the acyl transferase (AT) domain, which loads the proper extender unit from the corresponding methylmalonyl or malonyl-CoA substrate onto the phosphopantetheine arm of the acyl-carrier protein (ACP), which in turn carries the growing polyketide intermediate from domain to domain. Additional domains that are found in varying combinations in most modules include a ketoreductase (KR), a dehydratase (DH) and an enoyl reductase (ER). Together these tailoring domains control the final oxidation state of the β-carbon of the growing polyketide acyl-ACP intermediate. A dedicated thioesterase (TE) domain, located at the C-terminus of the furthest downstream module, catalyzes release and concomitant cyclization of the mature, full-length macrocyclic polyketide.
The 6-deoxyerythronolide synthase (DEBS) from Saccharopolyspora erythraea, which is by far the most thoroughly studied modular PKS, is responsible for biosynthesis of 6-deoxyerythronolide (6-dEB, 1), the aglycone precursor of the broad spectrum antibiotic erythromycin A.2 The macrolide 6-dEB is assembled by three large polypeptides, denoted DEBS1, 2, and 3, each consisting of >3000 amino acids, that catalyze the formation of 6-dEB from a propionyl-CoA starter unit and six methylmalonyl-CoA extender units (Figure 1).
Figure 1.

Modular organization of 6-deoxyerythronolide B synthase (DEBS). The three polypeptides DEBS 1, 2 and 3 are denoted the yellow rectangles and modules by the solid lines. Each block represents a catalytic domain: KS, ketosynthase; AT, acyltransferase; ACP, acyl-carrier protein; KR, ketoreductase; DH, dehydratase; ER, enoyl reductase and TE, thioesterase. KR0 represents an inactive KR domain. The phosphopantetheinyl arm of the ACP domain is represented as a curly line.
One of the most powerful tools for the study of the mechanism and specificity of DEBS-catalyzed reactions has been the ability to express entire DEBS proteins or individual DEBS modules, alone or in combination, in heterologous hosts such as Escherichia coli or Streptomyces coelicolor.3 In vitro experiments with purified recombinant modules, most often carrying an appended TE domain fused to the C-terminus of the ACP domain, have allowed detailed studies of enzyme kinetics and mechanism, including determination of substrate specificity and tolerance.4 For example, DEBS module 3+TE has been shown to catalyze the conversion of (2S,3R)-2-methyl-3-hydroxypentanoic acid N-acetylcysteamine thioester (diketide-SNAC 2) and methylmalonyl-CoA to the triketide ketolactone 3 which is generated by TE-catalyzed release of the corresponding acyclic 2,4-dimethyl-3-keto-5-hydroxypentanoyl thioester from the ACP (Figure 2A).5 In similar fashion, DEBS module 2+TE, module 5+TE, and module 6+TE, each carrying an active KR domain, all convert diketide 2 and methylmalonyl-CoA to the reduced triketide 3-hydroxylactone 4a (Figure 2B).5 Site-directed mutagenesis of specific domains and swapping of entire domains between different DEBS modules as well as with heterologous PKS modules has further clarified the role of individual PKS domains, while allowing the generation of novel, rationally engineered polyketides.1,2
Figure 2.
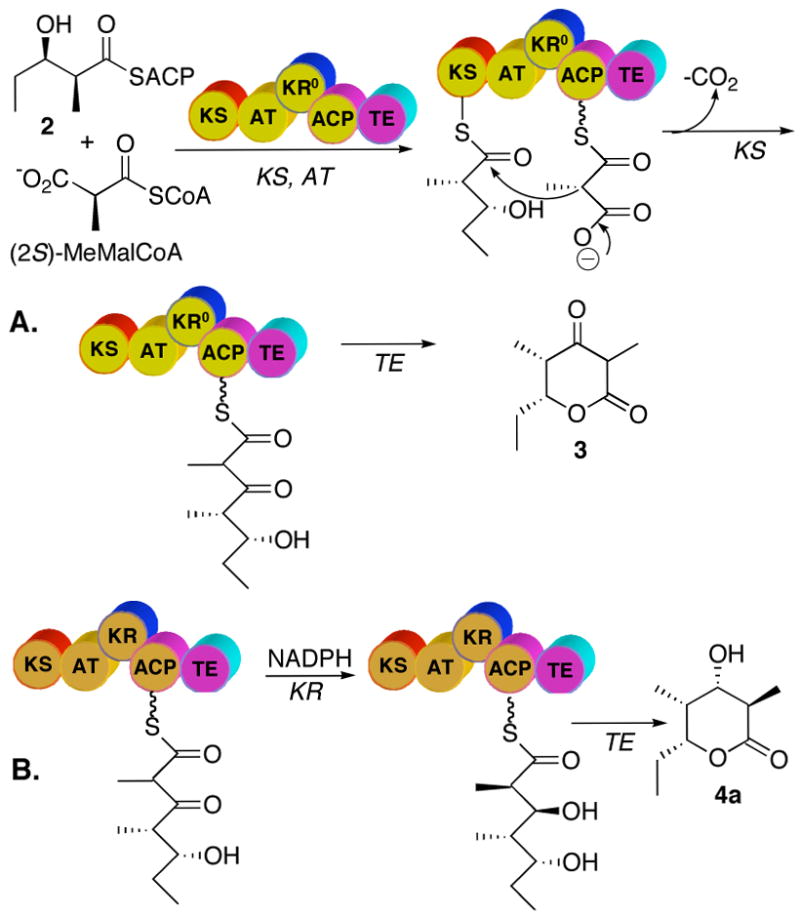
A. Sequence of reactions catalyzed by DEBS module 3+TE. The KS domain is first acylated by diketide-SNAc 2 and the AT domain primes the ACP domain with methylmalonyl-CoA. Following the KS-catalyzed condensation reaction, the TE domain catalyzes the cyclization and release of the resulting triketide ketolactone 3. B. Reaction catalyzed by DEBS module 2+TE (as well as by DEBS modules 5+TE and 6+TE). After the KS-catalyzed condensation reaction, the KR domain catalyzes the NADPH-dependent reduction of the 3-ketoacyl-ACP thioester, with release of the reduced triketide mediated by the TE domain to give the triketide lactone 4a.
More recently, limited proteolysis of DEBS proteins, in combination with high resolution structural studies, has facilitated the dissection and reconstitution of individual DEBS modules from their constituent domains.2a,6 Thus soluble active [KS][AT] didomains derived from DEBS modules 3 and 6 in combination with recombinant ACP domains have both been shown to catalyze the formation of triketide ketolactone 3 from diketide-SNAC 2 and methylmalonyl-CoA (Figure 3).7 The initially generated ACP-bound acyclic triketide can be released and lactonized either by base-catalyzed hydrolysis followed by acidification,7,8 or by addition of recombinant DEBS TE or the closely related PICS TE derived from the picromycin PKS.9 Importantly, addition of a recombinant KR domain, such as DEBS KR2, to the incubation mixtures results in formation of the corresponding reduced triketide lactone 4, which can readily be assayed by TLC-phosphorimaging.8 The stereochemistry of the 2-methyl and 3-hydroxyl substituents in samples of 4 resulting from combination of different [KS][AT] didomains with varied [KR] domains has not previously been determined.
Figure 3.
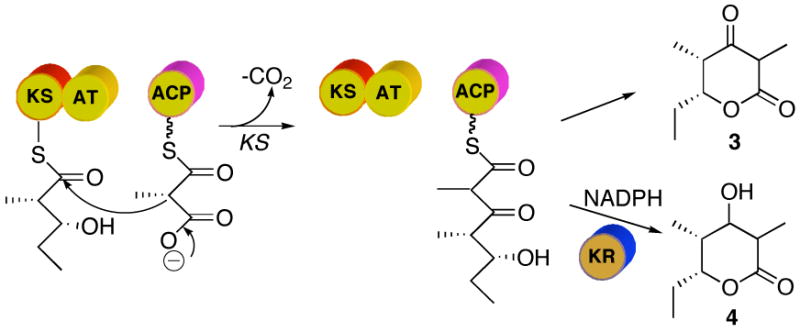
Reactions catalyzed by deconstructed DEBS module 3. After KS-catalyzed condensation, in the absence of an active KR domain the triketide ketolactone 3 is released by base- or TE-catalyzed hydrolysis. In the presence of an active KR domain such as DEBS KR2 and NADPH, the reduction of the ketone leads to formation of triketide lactone 4.
Reconstituted PKS systems have several attractive experimental features compared to intact PKS modules. Besides eliminating problems with incompatible domain boundaries, which have often interfered with earlier efforts to engineer catalytically efficient chimeric PKS modules from combinations of heterologous domains,10 the relative concentrations of the constituent domains can be independently varied. For example, it is often advantageous to use the ACP domain, which carries the extender unit and derived products, in stoichiometric excess over the catalytic [KS][AT] didomain. It is also possible to vary the nucleophilic substrate by using Sfp phosphopantetheinyl transferase11 to load malonyl-CoA analogs directly onto the ACP,7 thus bypassing the strict substrate specificity of the native AT domains. In like manner, we have determined the preference of individual [KS] domains for a variety of [ACP] domains carrying either the nucleophilic methylmalonyl or electrophilic polyketide acylthioester substrates.7
The macrolide aglycone 6-dEB (1) carries three L-hydroxy groups, at C-3, C-5, and C-11, and a single D-hydroxy substituent at C-13. The configuration of the methyl substituents shows a similar level of complexity, with D-methyl groups at C-2, C-4, and C-10, and L-methyls at C-6, C-8, and C-12 of 6-dEB (1). More than 40 years ago, Celmer pointed out the intriguing structural and stereochemical homology among a large number of 12-, 14-, and 16-membered macrolides, which could be summarized by a single stereochemical model.12 Since that time, the Celmer Model has provided a frame of reference for numerous investigations of the stereochemistry of macrolide biosynthesis. Although the modular organization of the DEBS PKS accounts in clear and elegant fashion for the observed pattern of substitution and oxidation in erythromycin and related macrolides, the detailed biochemical basis for the complex stereochemistry of the parent 6-dEB macrolide aglycone remains to be resolved.
The KR domains of DEBS modules 1, 2, 5, and 6 all use the 4-si (4-proS) hydride of the NADPH cofactor, identical to the known stereochemical course for fatty acid synthase KR domains.13 We have also demonstrated that the stereochemistry of ketone reduction is an intrinsic property of the PKS KR domain, with the configuration of the resulting hydroxyl group independent of either modular context or substrate structure.14 Both Reid15a and Caffrey15b have recently independently compared the sequences of some 200 modular KR domains belonging to the superfamily of short-chain dehydrogenases/reductases (SDRs). Consistent with the known structures of several members of this family, an active site triad of Ser, Tyr, and Lys residues is strictly conserved among all the KR domains analyzed. An interesting correlation was also noted between the observed formation of D-hydroxy groups and the presence of a conserved Asp found within a highly conserved LDD motif in such D-specific reductases. Notably, this conserved Asp is not present in the KR2, KR5, and KR6 domains responsible for generating L-3-hydroxyacyl-ACP intermediates that serve as precursors of the C-3, C-5, and C-11 hydroxyl groups found in 6-dEB (1). Intriguingly, this conserved Asp residue is also found in KR domains of cryptic stereochemical specificity that are paired with a dehydratase (DH) domain, such as the KR domains of DEBS module 4 and of PICS module 2. Although the stereospecificity of DEBS KR4 remains to be determined, we have established that PICS KR2 indeed generates a D-hydroxy group, as predicted by the bioinformatic analysis.16 On the other hand, attempts to use site-directed mutagenesis to modify the intrinsic stereospecificity of DEBS KR domains have achieved only limited success, suggesting that the characteristic Asp and other conserved residues are not the unique determinants of the intrinsic stereospecificity of 3-ketoacyl thioester reduction.17
The factors controlling the stereochemistry of methyl substitution in 6-dEB (1) and other polyketides are even less well understood. Except for the C-6 L-methyl, whose configuration is the consequence of the ER4-mediated reduction of the unsaturated 2-methyl pentaketide-ACP4 intermediate, the configuration of the remaining methyl-bearing carbons must be fixed either immediately before or following the KS-catalyzed chain elongation reaction. All 6 AT domains of DEBS have been shown to utilize exclusively (2S)-methylmalonyl-CoA,18 with the generation of methyl-bearing centers with D-configuration resulting in net inversion of configuration and retention of the original C-2 proton of the methylmalonyl unit.19 By contrast, generation of centers with L-methyl groups (C-8 and C-12 of 6-dEB) results in net loss of the original C-2 proton of the (2S)-methylmalonyl chain extension unit.19a The epimerization itself must occur either immediately preceding or just following the KS-catalyzed condensation reaction (Scheme 1). According to Pathway A, the KS domain would first epimerize the (2S)-methylmalonyl-SACP substrate to the corresponding (2R)-methylmalonyl-SACP, which would then undergo inversion in the course of KS-catalyzed decarboxylative condensation with the acyl-SACP substrate donated by the proximal upstream module. In Pathway A, control of methyl stereochemistry would be vested exclusively in the KS domain, with the KR domain simply reducing the resultant [L-methyl]-2-methyl-3-ketoacyl-SACP intermediate. Alternatively, in Pathway B, the KS domain first catalyzes the decarboxylative condensation of the (2S)-methylmalonyl-SACP with inversion of configuration to generate the [D-methyl]-(2R)-2-methyl-3-ketoacyl-SACP intermediate.20 For those modules with an associated epimerase activity, this ketoacyl thioester then would undergo epimerization followed by diastereoselective reduction mediated by the KR domain, (for example the KR of DEBS module 1), thereby fixing the configuration at both C-2 and C-3 of the 2-methyl-3-hydroxyacyl-SACP chain elongation intermediate.
Scheme 1.
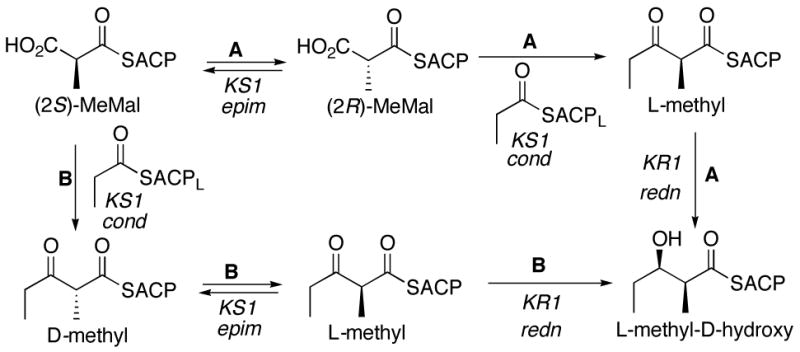
Possible pathways for the observed epimerization mediated by DEBS module 1.
A serious obstacle to experimental analysis of these complex mechanistic and stereochemical issues is that the initially generated 2-methyl-3-ketoacyl thioesters are expected to be configurationally labile, undergoing rapid, non-enzyme catalyzed epimerization if not immediately trapped in situ by reduction or chain extension. With this in mind, we have used reconstituted PKS modules to test systematically varied combinations of [KS][AT] didomains and recombinant KR domains, based on rigorous determination of the stereochemistry of the resultant triketide lactones (Figure 4). The results shed light on both the timing of epimerization and the diastereoselectivity of the individual ketoreductase domains. We also report the unexpected configurational stability of ACP-bound 2-methyl-3-ketoacyl polyketides.
Figure 4.
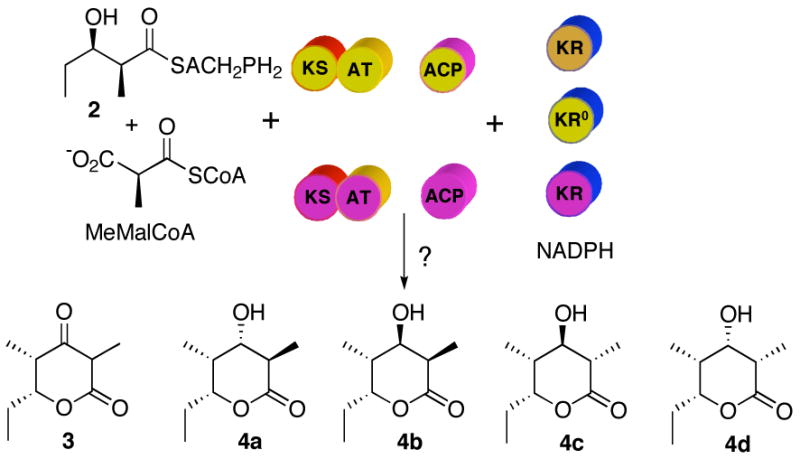
Reconstituted DEBS modules. Incubation of different combinations of [KS][AT], [ACP] and [KR] domains from different DEBS modules (color code according to Figure 1) with the appropriate substrates will result in the formation of triketide lactones whose stereochemistry at C-2 and C-3 can be determined by GC-MS comparison with authentic standards.
Results
Synthesis of triketide ketolactone 3 and triketide 3-hydroxylactones 4a–4d
The requisite synthetic reference standards of the triketide ketolactone 3 and the four 2-methyl-3-hydroxy diastereomers of the reduced triketide lactones 4a–4d were prepared using a common synthetic scheme based on the well-established Evans methodology for acyclic diastereoselection.14,21 The diastereomeric 2-methyl-3-ketoacyloxazolidinone derivatives 6 and 10 were each prepared as previously described from the corresponding (4R)- and (4S)-4-benzyl-3-propyl-2-oxazolidinones via the intermediate 3-ketooxazolidinones 5 and 9, respectively (Schemes 2 and 3). Stereoselective reduction of 6 and 10 gave the corresponding acyclic syn- or anti-2-methyl-3-hydroxy derivatives, which were readily converted to the desired triketide lactones 4a–4d by treatment with lithium hydroperoxide.21c Although hydrolysis of 11 unexpectedly gave a mixture of the predicted lactone 4c mixed with diastereomer 4b, the two compounds could be easily distinguished by NMR and were readily resolved by GC-MS analysis. The configuration of each newly generated stereocenter was unambiguously confirmed by detailed 1H NMR analysis and comparison where relevant with literature data for the same compounds prepared by alternative methods.14,22 For the synthesis of triketide ketolactone 3, attempted direct cyclization of 3-keto-oxazolidinones 6 or 10 gave low yields of impure product. The desired 3 could be generated cleanly by Dess-Martin oxidation of the mixture of triketide 3-hydroxylactones 4b and 4c (Scheme 4). The 3-keto form of 3 is in rapid equilibrium with the enol tautomer, with the latter being the predominant form in water, as previously reported.23 Detailed synthetic protocols and spectroscopic data can be found in the Supporting Information.
Scheme 2.

Synthesis of triketide lactones 4a and 4b. a. i. Bu2BOTf, i-Pr2NEt, propionaldehyde, CH2Cl2, −78 °C, 60%; ii, SO3Pyr, Et3N, DMSO, 97%; b. Sn(OTf)2, Et3N, propionaldehyde, CH2Cl2, −78 °C, 61%; c. Na(OAc)3BH, AcOH, 25 °C, 89%; d. LiOOH, 0 °C, 68%; e. DIBAL-H, THF, −78 °C, 86%; f. LiOOH, 0 °C, 51%.
Scheme 3.

Synthesis of triketide lactones 4c and 4d. a. i. Bu2BOTf, Et3N, propionaldehyde, CH2Cl2, −78 °C, 54%; ii, SO3Pyr, Et3N, DMSO, 93%; b. TiCl4, i-Pr2NEt, propionaldehyde, CH2Cl2, −78 °C, 80%; c. Zn(BH4)2, CH2Cl2, −78 °C, 99%; d. LiOOH, 0 °C, 75%; e. Na(OAc)3BH, AcOH, 25 °C, 29%; f. LiOOH, 0 °C, 20%.
Scheme 4.
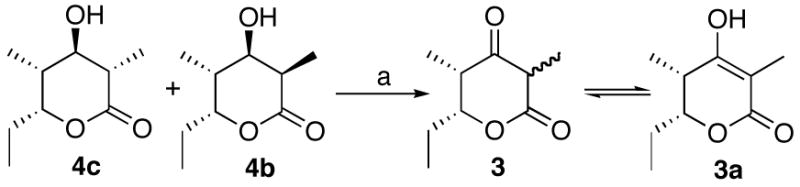
Synthesis of triketide ketolactone 3. a. Dess-Martin periodinane, CH2Cl2, rt, 38%.
Mass spectrometric analysis of triketide ketolactone 3 and triketide lactones 4a–4d
For routine analysis of enzymatically generated triketide lactones, we wanted a method that would be simple, sensitive, and robust. Although the individual diastereomers of lactones 4a–4d can be readily distinguished by 1H NMR, such analysis normally requires a minimum of 1–10 μmol of purified material. By contrast, GC-MS analysis is at least 3–4 orders of magnitude more sensitive and requires minimal manipulation of the sample prior to analysis. Indeed, the triketide ketolactone 3 and the four triketide lactone diastereomers 4a–4d were readily resolved by capillary GC and detected by chemical ionization (CI)-MS (Table 1). The sensitivity and resolution were further improved by analysis of the corresponding trimethylsilyl ether derivatives 4a–TMS-4d–TMS, which were conveniently generated by treatment of the concentrated EtOAc extract of the enzymatic incubation mixture with N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) containing 1 % (v/v) TMS-imidazole (Scheme 5, Figure 5, and Table 1). In this manner, as little as 1 nmol of each TMS-lactone could readily be detected. For analysis of enzymatic incubation mixtures, the detection sensitivity was enhanced with concomitant elimination of signals from non-enzymatic side-products by the use of extracted ion monitoring (XIM) of the parent [M+H]+ (m/z 245). Alternatively, the TMS-lactones 4a–TMS-4d–TMS could be analyzed by 70 eV electron impact (EI) MS, monitoring the m/z 171 base peak corresponding to the [M−TMS]+ species.
Table 1.
GC-CI-MS retention times for synthetic triketide ketolactone 3 and triketide lactones 4a–4d, as well as the corresponding TMS derivatives 4a–TMS-4d–TMS.
| Lactone | Retention time (min:s) | TMS-lactone | Retention time (min:s) |
|---|---|---|---|
| 3 | 6:09 | ||
| 4a | 6:43 | 4a–TMS | 6:55 |
| 4b | 6:50 | 4b–TMS | 7:02 |
| 4c | 6:33 | 4c–TMS | 6:49 |
| 4d | 6:52 | 4d–TMS | 7:17 |
Scheme 5.
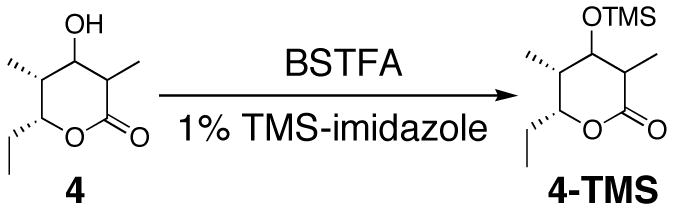
Derivatization of triketide lactones 4a–4d.
Figure 5.
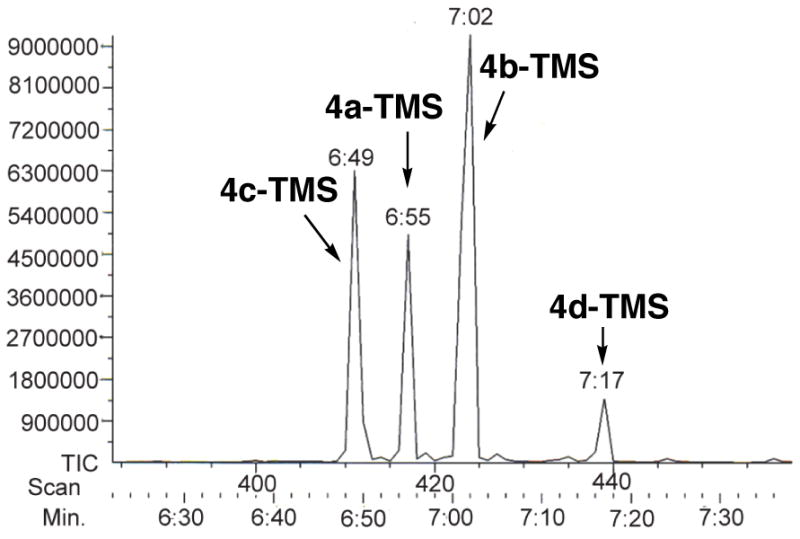
GC-CI-MS analysis of the 4 TMS-derivatized diastereomers of (4R,5R)-3,5-dihydroxy-2,4-dimethyl-n-heptanoic acid-δ-lactones (4a–TMS-4d–TMS).
Stereochemical assignment of the triketide lactone formed by [KS6][AT6], [ACP6], and [KR6]
DEBS module 6+TE has been shown to catalyze conversion of diketide 2 and methylmalonyl-CoA in the presence of NADPH to triketide lactone 4a as the exclusive product.5 We have recently reported that incubation of the reconstituted mixture of recombinant [KS6][AT6] didomain, [ACP6] and [KR6] with the same substrates resulted in formation of triketide lactone 4 of unspecified stereochemistry at C-2 and C-3, as determined by TLC-phosphorimaging.8 To validate the sensitivity and accuracy of the mass spectrometric analysis and to establish the stereochemistry of the resulting triketide lactone, the recombinant [KS6][AT6], [ACP6] and [KR6] fragments from DEBS module 6 were incubated for 1.5 h with diketide-SNAC 2, methylmalonyl-CoA, and NADPH, followed by base-catalyzed hydrolysis of the ACP-bound product and acidification. Following treatment of the crude EtOAc extract with BSTFA containing 1% (v/v) TMS-imidazole, GC-CI-MS analysis with XIM detection of the m/z 245 components revealed the presence of a single peak, ret. time 6:55 min, corresponding to TMS-lactone 4a–TMS (Figure 6). GC-CI-MS analysis of the underivatized extract also confirmed the formation of the single triketide lactone stereoisomer 4a, ret. time 6:43 min, accompanied as previously reported by the unreduced triketide ketolactone 3. Preincubation of [KS6][AT6] with diketide-SNAC 2 for 1 h to allow complete acylation of the active Cys of the KS,8 followed by addition of [ACP6] and [KR6] along with methylmalonyl-CoA and NADPH gave identical results. Decreasing the concentration of [KR6] had no effect on the stereochemical purity of the product nor, most interestingly, did delaying the addition of [KR6] and NADPH by 20 or 45 min following the addition of [ACP6] and methylmalonyl-CoA. In all cases, the exclusive triketide lactone product formed was diastereomer 4a, under conditions that could easily have detected as little as 2–3% of any of the other 3 diastereomers, 4b–4d.
Figure 6.
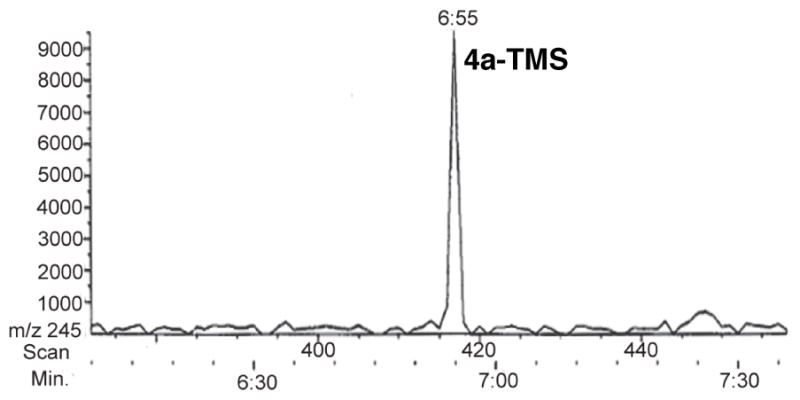
GC-CI-MS (XIC at m/z 245) of the derivatized triketide lactone 4a–TMS formed by incubation of [KS6][AT6], [ACP6], and [KR6] with diketide-SNAC 2, methylmalonyl-CoA, and NADPH.
Incubation of heterologous combinations of [KS][AT], [ACP], and [KR] domains
Having established the sensitivity and accuracy of the GC-CI-MS analysis of the diastereomers of triketide lactone 4, we carried out a series of incubations using the [KS][AT] didomains from DEBS modules 3 or DEBS module 6 in combination with either DEBS [KR2] or [KR6], both of which had been shown to generate the triketide lactone product 4, with reduction of the 3-ketotriketide intermediate by KR2 being more efficient than KR6-catalyzed reduction.8 In each case, we used an ACP domain derived from the same module as the [KS][AT] didomain, since we had previously established that each KS domain has a significant preference for its cognate ACP domain.7 Notably, the KS3 domain is from DEBS module 3 whose normal tetraketide product eventually carries an epimerized L-methyl group, corresponding to the C-8 methyl of 6-dEB (1). KS6 on the other hand generates a product with a D-methyl group, as found at C-2 of 6-dEB (1) produced by the complete DEBS, or C-2 of 4a, produced by DEBS module 6+TE. As summarized in Table 2, all 4 combinations of [KS][AT] and [KR] domains gave exclusively triketide lactone isomer 4a. In each case, the [KS][AT] didomain was pre-incubated with diketide-SNAC 2 for 1 h, followed by addition of the [ACP] and [KR] proteins along with the remaining substrates, methylmalonyl-CoA and NADPH. Addition of the [ACP] and [KR] domains together or with an interval of 45 min between introduction of [ACP] and addition of the [KR] domain all gave triketide lactone 4a of unchanged stereochemical purity. The 45 min delay did result in a small increase in the proportion of unreduced triketide ketolactone 3 in the product mixture due to competing buffer-catalyzed release of the ACP-bound acyclic 3-ketotriketide, as previously reported. The use of DEBS TE to catalyze the enzymatic release of the triketide lactone in place of the usual base-catalyzed hydrolysis/acid cyclization protocol also had no effect on the observed stereochemical purity of the resultant triketide lactone product 4a, thereby ruling out any influence of the base treatment on the configurational integrity of the product.
Table 2.
Identification of triketide lactone isolated after incubation of [KS][AT], [ACP] and [KR] domains from different DEBS modules.
| [KS][AT] didomain | [ACP] domain | [KR] domain | Triketide lactone |
|---|---|---|---|
| [KS6][AT6] | [ACP6] | [KR6] | 4a |
| [KS6][AT6] | [ACP6] | [KR2] | 4a |
| [KS3][AT3] | [ACP3] | [KR6] | 4a |
| [KS3][AT3] | [ACP3] | [KR2] | 4a |
Stereochemistry of the triketide lactone formed by the hybrid module [KS3][AT3][KR5][ACP5][TE]
We have previously reported the construction of a chimeric module engineered by fusing the [KS3][AT3] chain elongation didomain to the reductive [KR5][ACP5] didomain, along with an appended DEBS TE domain.7 Incubation of this [KS3][AT3][KR5][ACP5][TE] with diketide 2 and methylmalonyl-CoA in the presence of NADPH led to a mixture of triketide ketolactone 3 and the reduced triketide lactone 4 of unspecified configuration. GC-CI-MS analysis has now established that this chimeric module produces exclusively triketide lactone 4a, with no trace of diastereomers 4b–4d (Scheme 6). The formation of triketide lactone 4a by the hybrid module is completely consistent with the results obtained with the reconstituted modules, as well as the exclusive formation of 4a by the native DEBS module 5+TE itself.5
Scheme 6.
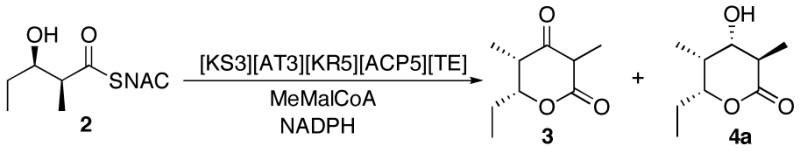
Stereochemistry of triketide lactone formation catalyzed by hybrid module [KS3][AT3][KR5][ACP5][TE].
Reduction of the ACP-bound 3-ketotriketide by sodium borohydride
The formation of triketide lactone 4a carrying an L-hydroxy group at C-3 as a result of incubations with either KR2 or KR6 is consistent with the known stereochemistry of 3-ketoacyl thioester reduction that is catalyzed by each ketoreductase domain.5 Furthermore, the D-methyl stereochemistry of 4a is consistent with the established stereospecificity of DEBS modules 2 and 6. On the other hand, the exclusive formation of lactone 4a with an unepimerized D-2-methyl group is unexpected for the KS3 domain of DEBS module 3. These results also do not distinguish whether either KS3 or KS6 generates exclusively the D-2-methyl-3-ketoacyl triketide product, or whether the KR domain selectively reduces only the D-2-methyl diastereomer that may be present in an epimeric mixture of chain-elongation intermediates. Notably, allowing the initially-generated ACP-bound 3-ketoacyl triketide to incubate for an additional 45 min prior to introduction of ketoreductase had no detectable effect on the diastereomeric purity of the resulting triketide lactone 4a. We therefore sought to trap the ACP-bound 2-methyl-3-ketoacyl triketide by in situ reduction with NaBH4, in order to establish directly the configuration of the C-2 methyl group.
The α-methyl group of a β-ketoacyl thioester or oxoester normally undergoes rapid epimerization even at neutral pH. Indeed, we observed that room temperature incubation of the model β-ketoester, ethyl 2-methylacetoacetate, in 100 mM sodium phosphate in D2O, pD 7.2, showed a t1/2 for proton exchange of 4.7 min, as monitored directly by 1H NMR. Were the ACP-bound 2-methyl-3-ketoacyl thioesters to behave similarly, an interval of 20 to 45 min before addition of the KR domain to the incubation mixture would correspond to 4–9 proton-exchange half-lives, resulting in effectively complete epimerization of the 2-methyl substituent.
Incubation of [KS3][AT3] with diketide 2 for 1 h, followed by the addition of [ACP3] and methylmalonyl-CoA and incubation for a further 45 min, with termination by basic hydrolysis led to the formation of triketide ketolactone 3, as expected. Reductive quenching of a parallel enzymatic reaction after 45 min incubation by addition of an 80-fold excess of NaBH4, followed by base hydrolysis (Figure 7), gave a ~ 1:1.6 mixture of triketide lactones 4a and 4b, as established by the standard GC-CI-MS analysis of the corresponding TMS-derivatives (Figure 8). Neither 4c nor 4d could be detected in the product mixture. The acyclic ketone had thus been reduced non-stereospecifically by NaBH4, as expected, but the methyl group was clearly only in the D configuration. When NaBH4 was used to reductively quench an incubation mixture containing [KS6][AT6] and [ACP6] a 1.3:1 mixture of 4a and 4b was obtained, establishing that both KS3 and KS6 generate the identical (2R,4R,5R)-2,4-dimethyl-3-keto-5-hydroxypentanoyl-ACP product from diketide 2 and methylmalonyl-CoA.
Figure 7.
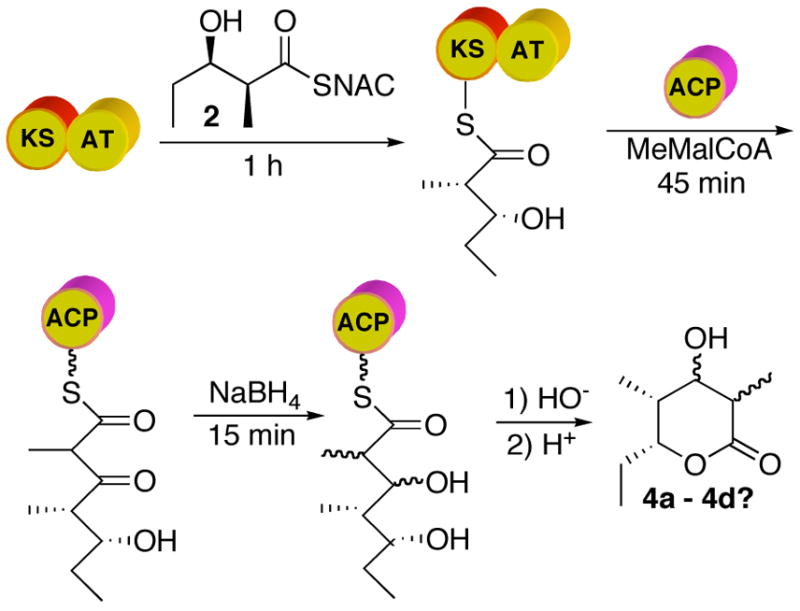
Trapping of ACP-bound 2,4-dimethyl-3-keto-5-hydroxytriketide by NaBH4 reduction.
Figure 8.
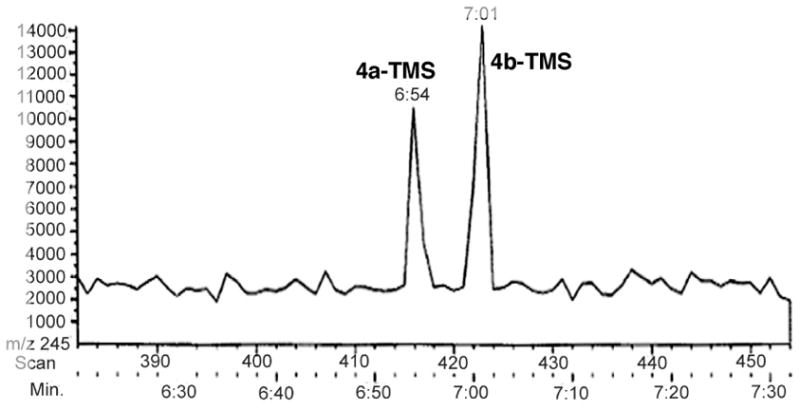
GC-CI-MS (XIC at m/z 245) of TMS-derivatized triketide lactones 4a–TMS and 4b–TMS isolated from incubation of [KS3][AT3] and [ACP3] with diketide-SNAC 2 and methylmalonyl-CoA, followed by quenching with NaBH4 and base hydrolysis.
To validate the reliability of the NaBH4 quenching protocol, a series of control experiments was carried out (Table 3). To rule out the possibility that the observed quenching products result from NaBH4 reduction of free triketide ketolactone 3 that may have been released from the ACP by buffer-catalyzed hydrolysis, a synthetic sample of 3 was treated with NaBH4 in either MeOH or in the normal pH 7.2 incubation buffer. In both cases, all 4 triketide lactones were formed, including significant proportions of 4c and 4d carrying an L-2-methyl group. In fact, under these conditions, the bulk of the unreduced ketolactone 3 was recovered, presumably due to the predominance of the enol-lactone tautomer. To rule out the alternative possibility that 4a is being formed by NaBH4 reduction of the free seco-acid form of prematurely released 2,4-dimethyl-3-keto-5-hydroxypentanoic acid, the enzymatic incubation mixture was subjected to ultrafiltration prior to NaBH4 reduction of protein-bound triketide in the retentate, resulting in exclusive formation of triketide lactones 4a and 4b. Interestingly, when the incubation mixture was pre-extracted with EtOAc, in an attempt to remove any protein-free organic side-products, reduction of the resulting cloudy solution gave all four triketide lactones, but with 4a and 4b still being the predominant components of the quenching mixture. Taken together, these experiments establish firmly that the triketide lactones 4a and 4b result exclusively from NaBH4 reduction of ACP-bound 2,4-dimethyl-3-keto-5-hydroxytriketide and that this intermediate carries exclusively a D-2-methyl group.
Table 3.
NaBH4 quenching of ACP6-bound 3-ketoacyl triketide.
| Treatment | Triketide lactones (ratio) |
|---|---|
| NaBH4 (15 min) | 4a, 4b (1.3:1) |
| 3 plus NaBH4 in MeOH | 4a, 4b, 4c, 4d (1:16:6.5:7) |
| 3 plus NaBH4 in 100 mM phosphate, pH 7.2 | 4a, 4b, 4c, 4d (1:5.5:29:5.8) |
| NaBH4 reduction of protein ultrafiltrate | 4a, 4b (2.6:1) |
| 1) EtOAc extraction; 2) NaBH4 treatment of aqueous phase | 4a, 4b, 4c, 4d (2:6:4:1) |
Incubations with DEBS [KR30]
The recently reported crystal structure of DEBS KR1 revealed a pseudodimeric domain organization, consisting of an N-terminal “structural” subdomain and a C-terminal “catalytic” subdomain.17a It was suggested that this catalytic subdomain might also harbor the requisite methyl epimerase activity that is associated with DEBS module 1.17a The epimerized L-methyl group at C-8 of 6-dEB is installed by DEBS module 3. When the 2-methyl-3-ketoacyl-ACP triketide intermediate of [KS3][AT3] incubations is reduced either enzymatically by [KR2] or [KR6] or by reductive trapping with NaBH4, however, it is clear that the initial product of KS3-catalyzed chain elongation has the unepimerized 2-D-methyl configuration. We therefore wished to examine the possibility that the reductively silent KR30 domain, which lacks a consensus NADPH binding site,2a,24 might harbor a cryptic methyl epimerase activity. Based on the previously defined consensus boundaries for KR domains,8,25a we used PCR to amplify the KR30 component of DEBS module 3 as an N-terminal His-tag protein, using DNA from plasmid pRSG34, encoding DEBS module 3+TE, as a template.5 Expression and purification as described for the DEBS KR2 and KR6 domains8 gave 30 mg/L culture of recombinant KR30, whose purity and molecular weight were verified by SDS-PAGE and MALDI-TOF MS.
Overnight incubation of recombinant [KR30] with NADPH and trans-1-decalone, a useful surrogate substrate for DEBS KR domains,25 confirmed the complete absence of detectable KR30 reductase activity, as assayed by both UV and GC-MS. Attempted monitoring of any epimerase activity by incubation of [KR30] with trans-1-decalone in deuterated 100 mM sodium phosphate (pD 7.2) did not lead to any detectable deuterium exchange, as determined by GC-MS.
When recombinant [KR30] was coincubated with [KS6][AT6] and [ACP6] in the presence of diketide 2 and methylmalonyl-CoA, followed by quenching with NaBH4, a mixture of triketide lactones 4a and 4b was obtained, without any detectable formation of L-methyl products 4c and 4d. Alternatively, 45-min incubation of KR30 with the product of [KS6][ACP6]-catalyzed chain extension, followed by enzymatic reduction with [KR6] and NADPH resulted in exclusive formation of triketide lactone 4a. Identical results were obtained using the reconstituted [KS3][AT3] and [ACP3] mixture. The recombinant KR30 therefore has no detectable methyl epimerase activity, although we can not rigorously rule out the possibility that native KR30 in its natural DEBS module 3 context might still serve as an epimerase.
Discussion
Each module in a modular polyketide synthase is responsible for a discrete round of polyketide chain elongation and functional group modification. While the AT domain controls the substitution pattern through strict selection of the proper building block, the particular combination of KR, DH, and ER domains determines the ultimate oxidation level and stereochemistry of the growing polyketide chain. When only a KR domain is present in a given module, the reduction stereospecificity of the KR domain is preserved in the configuration of the corresponding hydroxyl group in the eventually formed macrolide or polyether product. For modules carrying additional tailoring domains, however, the stereochemistry of the β-hydroxyacyl-ACP intermediate is obscured by DH-catalyzed dehydration, while the double bond geometry of the resulting enoyl-ACP intermediate can be concealed as well by subsequent ER-catalyzed reduction.
Heterologous expression and in vitro characterization of individual PKS modules continues to be an extremely useful tool for biochemical investigation of PKS-catalyzed chain elongation, allowing detailed probing of the mechanism, stereochemistry, and substrate specificity of a variety of modules and their constituent domains. For example, by swapping heterologous KR domains into DEBS module 2, we first demonstrated that the stereochemistry of KR-catalyzed reductions is an intrinsic property of the individual KR domain, independent of substrate structure or modular context.14 More recently, using mutants of PICS module 2 in which the DH or KR domains had been selectively inactivated, we provided direct evidence for the sequence of biochemical reactions catalyzed by PICS module 2 and determined the previously cryptic stereochemistry of the D-hydroxy specific KR2 domain.16 Nonetheless, studies with intact modules still face a number of experimental obstacles, including the strict substrate specificity of the constituent AT domains, the labor-intensive effort required for the construction of mutant or, especially, hybrid modules, even with improved, structure-based insight into interdomain boundaries,6,26 and the challenges to analyzing single-turnover reactions. While recently developed FT-MS-MS methods allow the detection and structural identification of sub-nanomole levels of protein-bound polyketide chain elongation intermediates,27 such protein-MS methods cannot be used for the determination of the stereochemistry of protein-bound intermediates, nor are they yet suited for routine measurements of enzyme kinetics.
More recently, the ready availability of recombinant PKS domains and didomains has opened up a wealth of exciting opportunities that has already dramatically deepened our understanding of PKS structure and mechanism. By reconstituting chimeric PKS systems from combinations of heterologous domains, it has become possible to systematically probe the substrate specificity and stereochemistry of a variety of PKS domains.6–8,25a We have recently reported the results of individual incubations of a reconstituted PKS system consisting of the DEBS [KS3][AT3] didomain and its cognate acyl carrier partner [ACP3] with recombinant DEBS ketoreductase domains [KR1], [KR2], and [KR6] in the presence of diketide-SNAC 2 and methylmalonyl-CoA.8 Although KR1, which normally reduces a diketide substrate, failed to reduce the ACP-bound 2-methyl-3-keto triketide that was generated by [KS3][AT3], both [KR2] and [KR6] catalyzed the NADPH-dependent reduction to give, after base-catalyzed release from the ACP domain, triketide lactone 4. In experiments with [KR6], triketide lactone 4 was also accompanied by unreduced triketide ketolactone 3. Similar results were also obtained using the same set of KR domains in combination with DEBS [KS6][AT6] and [ACP6]. We have now established the stereochemistry of each of these reductions and shed light on the diastereoselectivity of each ketoreductase, using a sensitive method that can in principle be extended to any recombinant KR domain capable of reducing an ACP-bound 3-ketoacyl-ACP triketide.
GC-MS analysis of triketide lactone diastereomers
The synthetic reference samples of each of the 4 triketide lactone diastereomers 4a–4d can be readily separated and analyzed by capillary GC-MS, with sensitivity down to 1 nmol or better using chemical ionization (CI) and extracted ion monitoring (XIM) of the parent [M+H]+ ion at m/z 245 for the corresponding TMS derivatives.28 At this scale, each enzyme incubation can be carried out in a convenient volume of 500 μL, using 10–20 nmol of [KS][AT] didomain in combination with 100–150 nmol of each of the other PKS domains. Even [KS][AT] didomains with exceptionally low turnover numbers of 0.01 min−1 readily generate the requisite amounts of triketide lactone in a 1–2-h incubation. With sufficient quantities of product from a given incubation, both the free lactones and the corresponding TMS-derivatives can be subjected to the calibrated GC-MS analysis to reinforce to the reliability of the analysis.
Analysis of KR stereospecificity
Recombinant DEBS KR1 has been shown to reduce racemic 2-methyl-3-ketopentanoyl-SNAC (13) exclusively to the syn-(2S,3R)-diketide-SNAC 2, thereby demonstrating that this reductase is completely specific not only for reduction on the natural diastereotopic face of the ketone carbonyl but also for the natural L-configuration of the adjacent 2-methyl group, as expected based on the overall stereospecificity of DEBS module 1 (Scheme 7).25a These results were also completely consistent with earlier results obtained using monomodular and bimodular DEBS proteins harboring KR1.29 Recombinant KR1 from the tylactone synthase reduces 13 exclusively to the anti-(2R,3R)-diketide-SNAC 14, again completely consistent with the expected diastereoselectivity for the D-2-methyl substrate.25a Unfortunately, reduction of 3-ketodiketide-SNAC 13 with recombinant DEBS KR2, KR5, and KR6 resulted in each case in both an apparent erosion in the intrinsic stereospecifity of ketoreduction, as evidenced by the formation of 2–20% of the unnatural D-(3R)-hydroxydiketides-SNAC products along with the expected L-(3S)-hydroxy-diastereomers, and a significant reversal in diastereoselectivity, unnatural L-(2S)-methyl diketides being generated in 3-fold excess over the predicted D-(2R)-methyl diketide-SNAC diastereomers.25a A similar degradation in stereoselectivity was observed when 13 was incubated with intact DEBS module 2 or with DEBS3, harboring both modules 5 and 6.30 Since each of these modules is known to act completely stereospecifically when processing both natural and unnatural enzyme-bound substrates,5,22b it is likely that the loss of stereochemical control in reduction of the ketodiketide-SNAC analog 13 may reflect the requirement that the substrate be covalently anchored to the ACP domain for proper positioning and orientation in the KR active site. Indeed, the results reported here confirm that recombinant DEBS KR2 and KR6 both catalyze completely stereospecific reductions of ACP-bound 2-methyl-3-ketotriketides that have been generated in situ by the action of a [KS][AT] didomain. Whether the observed ACP requirement reflects the existence of an ACP-specific binding site on the surface of the KR domain, as has been reported for the interaction of the Type II ketoreductase FabG with ACP,31 or whether the size of the ACP protein simply prevents the misbinding observed with the SNAC analogs, is not yet clear, although recombinant KR domains exhibit no apparent preference for ACP domains from different DEBS modules.8
Scheme 7.
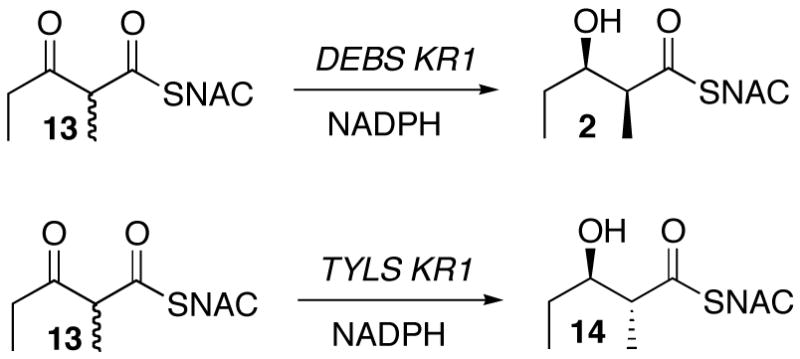
Reduction of 2-methyl-3-ketodiketide-SNAC 13 by recombinant KR domains.
The exclusive formation of a single triketide lactone 4a in reductions catalyzed by DEBS KR2 or KR6 in combination with either the [KS3][AT3] or [KS6][AT6] didomains does not establish whether either KR2 or KR6 is capable of discriminating against ACP-bound substrates with the unnatural L-2-methyl configuration, since only the D-2-methyl substrate generated by either KS3 or KS6 is presented to the KR domain, as established conclusively by the reductive quenching with NaBH4. The same is also true of the diastereoselectivity of KR5, as deduced from the formation of triketide lactone 4a by the chimeric [KS5][AT5][KR5][ACP5][TE] module. Similarly, the reported generation of the syn-(2R,3S)-2-methyl-3-hydroxypentanoic acid by the chimeric module DKS1-2, corresponding to DEBS [ATL][ACPL][KS1][AT2][KR2][ACP2][TE], established that epimerization occurs after KS-catalyzed condensation (Pathway B of Scheme 1) and that KR2 can stereospecifically reduce the initially-generated intermediate D-2-methyl-3-ketopentanoyl-ACP.32 No conclusions could be drawn as to whether KR2 exercises any selection against the diastereomeric L-2-methyl-3-ketopentanoyl-ACP, which may or may not have been transiently formed by the hybrid DKS1-2 enzyme.
Timing of the epimerization of the 2-methyl group
The exclusive formation of triketide lactone 4a, whether resulting from mixed incubations of [KS3][AT3] with either [KR2] or [KR6] or when [KS3] is paired with KR5 in the chimeric module, establishes firmly that the methyl group epimerization normally associated with DEBS module 3, which ultimately leads to generation of the L-8-methyl of 6-dEB (1), must occur subsequent to and not prior to the KS3-catalyzed condensation reaction. The absence of epimerization by the [KS3][AT3] didomain, either alone or combination with a KR domain, suggests that the epimerization associated with DEBS module 3 is not mediated by the KS3 domain. Based on analysis of the KR1 crystal structure, it was recently suggested that epimerization might in fact be catalyzed by the KR1 domain itself.17a A mechanism involving enolization of the β-ketoacyl-ACP ester and conjugate reduction of the resulting β-hydroxyenoyl-ACP was advanced to account for diastereoselective formation of the observed diketide. Subsequent bioinformatics analysis has claimed to support of this proposal, but direct experimental evidence is so far lacking.33 It was also suggested that reductively silent domains such as DEBS KR30, which lacks a functional NADPH binding site, might in fact play a role in methyl group epimerization. Since addition of recombinant KR30 to incubation mixtures containing either the [KS3][AT3] or [KS6][AT6] didomains did not result in any detectable epimerization, however, the proposed role for KR30 in mediating epimerization is so far unsupported.
Role of the ACP domain
For many years it has been assumed that the acyl carrier protein in fatty acid and polyketide biosynthesis merely serves to solubilize the growing hydrophobic fatty acid or polyketide chain while acting as an essentially passive platform for the flexible, 18-Å phosphopantetheinate prosthetic group whose role is to deliver attached chain elongation intermediates to the successive FAS or PKS reaction centers. Recent structural studies of both the DEBS [KS3][AT3] and [KS5][AT5] didomains,2a,26,34 which show that the active sites of the constituent KS and AT domains are separated by more than 80 Å. The simplistic “swinging-arm” model for the ACP must therefore be discarded. A structure-based, dynamic model of ACP biochemistry is now required, in which the ACP protein itself undergoes significant segmental motion in order to deliver the phosphopantetheinyl arm and its tethered substrates to the various active sites of each module. Considerable support for the notion of a dynamic ACP has also come from the recently reported crystal structure of the topologically distinct yeast FAS and a closely related fungal FAS.35 Furthermore, although all DEBS ACP domains appear to share a modest level of sequence similarity and are predicted to have a common 4-helix topology,2a,36 there is a growing body of evidence that ACP domains are not simply interchangeable and that individual ACP domains preferentially interact with the KS domains from the same module.7,34
The unexpected finding that KS-generated 2,4-dimethyl-3-keto-5-hydroxyheptanoyl-ACP triketides are both chemically and configurationally stable has a number of important implications, both practical and theoretical: 1) The ACP-bound 5-hydroxyacylthioester is considerably more resistant to buffer-catalyzed lactonization than the corresponding free thioester. Earlier attempts in our laboratory to prepare 2,4-dimethyl-3,5-dihydroxyheptanoyl-SNAC were repeatedly thwarted by spontaneous formation of the corresponding δ-lactone 4a. Although precise kinetic measurements were not carried out, we estimated the t1/2 for lactonization as <1–2 min. By comparison, although buffer-catalyzed release of triketide lactones 3 or 4 from the ACP is measurable, it generally does not exceed 5–10% of the recovered product even after 1 h incubation.6 Indeed DEBS and related macrolide synthases ordinarily generate little if any detectable triketide lactones such as 4 as coproducts of complete polyketide chain elongation.37 2) The remarkable configurational stability of the ACP-bound 2-methyl-3-ketoacylthioester intermediates, which retain their configurational integrity even after 1 h at pH 7.2, conditions under which free β-ketoesters undergo complete exchange of α-protons, makes the enzymatic and chemical reductive trapping of these otherwise configurationally labile intermediates possible. The enhanced stability of these species might also facilitate eventual NMR determination of the structure and conformation of the ACP-bound polyketide intermediates.36
The simplest explanation of the remarkable chemical and configurational stability of the ACP-bound polyketides is that the phosphopantetheinate-tethered substrate does not swing freely in solution but is bound by the acyl-carrier protein itself. An extended binding conformation of the substrate would suppress spontaneous lactonization or other intramolecular reactions. Similarly, an appropriately placed kink in the polyketide chain can prevent the β-ketone and thioester carbonyl groups from becoming coplanar, thereby raising the apparent pKa by 10–15 orders of magnitude, more than sufficient to retard or completely suppress undesirable loss of stereochemical integrity during the normal assembly-line synthesis of the polyketide.38 In fact, there have been several recent reports of ACP binding of fatty acid chain elongation intermediates. E. coli ACP contains a cavity in which acyl chains 6, 7 or 10 carbon atoms in length can be observed interacting with hydrophobic amino acid residues, well shielded from the buffer.39 The crystal structure of a heptanoyl-ACP clearly shows the conformation of the acyl chain in which the β-methylene and the thioester carbonyl are out of plane. The recently reported crystal structure of the Saccharomyces cerevisiae FAS exhibits a stalled ACP at the active site of KS, with ACP-bound substrate well buried in a groove on the ACP with only the reactive portion of the substrate exposed.35a This observation has led to a proposed “switch-blade” model, in which the phosphopantetheinyl residue folds back on the protein in order to allow the attached substrate to be bound in a groove in the ACP, with the tethered substrate being flicked out upon binding to the appropriate FAS domain.
Conclusions
In summary, we have described a convenient, sensitive, and robust system for the analysis of PKS ketoreductase stereospecificity. In principle, the method can be extended to any combination of [KS][AT] and [KR] domains, provided that the KS domain be able to utilize the diketide-SNAC 2 or related substrate analog and that the paired recombinant KR domain be capable of processing the KS-generated, ACP-bound intermediate. The experimental system can be further expanded by using the Sfp phosphopantetheinyl transferase to generate unnatural malonyl-ACP derivatives. In each case, the relevant triketide lactone standards can be readily prepared using variants of the Evans oxazolidinone chemistry or related methods for acyclic stereoselection. The use of reconstituted PKS systems made up of dissected domains has already established the stereochemical course of KS and KR-catalyzed reactions, shed light on the relative timing of methyl group epimerization, and revealed unexpected configurational stability of ACP-bound polyketide intermediates. Further studies will contribute to the solution of critical questions in PKS enzymology and should provide valuable guidelines for the rational design of combinatorial biosynthetic strategies.
Material and methods
Material
Reagents, solvents and substrates purchased from Sigma-Aldrich were of the highest quality available and were used without further purification. Restriction enzymes were from Promega. N,O-(bistrimethylsilyl)trifluoroacetamide (BSTFA) was purchased from Supelco and trimethylsilyl (TMS)-imidazole, was from Pierce. Isopropylthio-D-galactopyranoside (IPTG) was purchased from Invitrogen. Preparations of plasmids pAYC02 ([KS3][AT3]),7 pAYC11 ([KS6][AT6]),7 pVYA05 ([ACP3]),7 pPK223 ([ACP6]),7 pAYC60 ([KR2]),8 pAYC62 ([KR6]),8 pRSG34 (DEBS module 3 + TE),5 and pAYC46 ([KS3][AT3][KR5][ACP5][TE])7 have been previous described. Ni-NTA affinity resin was from Qiagen. PD-10 columns were from GE Healthcare and ultrafiltration devices (UltraFree-MC 5,000 MWCO and Amicon Ultra-15 10,000 and 50,000 MWCO) were purchased from Millipore. (2S,3R)-2-Methyl-3-hydroxypentanoic acid N-acetylcysteamine thioester (2) was synthesized as described.40
Methods
All DNA manipulations were performed following standard procedures.41 The sequence of plasmid pRCD1 was verified directly (U. C. Davis Sequencing Facility, Davis, CA). GC-CI-MS (CH4) and GC-EI-MS spectra were recorded on a Jeol JMS-600H mass spectrometer using an HP-5MS capillary column (30 m × 0.25 mm, 0.25 μm film thickness) with the injector port at 250 °C and a temperature program of 60 °C for 1 min, then 25 °C/min up to 280 °C, and 280 °C for 5 min. Previously published procedures were employed for the purification of [KS3][AT3], [KS6][AT6], [ACP3], [ACP6], [KR2], and [KR6] proteins, and for DEBS TE.7,8,9a Protein concentrations were determined by Bradford assay using bovine serum albumin as standard.42
Derivatization and mass spectrometric analysis of triketide lactones 4a–4d
Triketide lactones 4a–4d (0.5 mg) were dissolved directly in 100 μL BSTFA containing 1μL of TMS-imidazole and agitated under nitrogen for 10 min. The sample was then used directly for GC-MS analysis.
Incubation of DEBS domains and extraction of enzymatic products
[KS][AT] didomain (40 μM) was incubated with diketide-SNAC (2) (5 mM) and TCEP (5 mM) in 100 mM sodium phosphate, pH 7.2 at 30 °C for 1 h in a typical volume of 500 μL. [ACP] (200 μM), [KR2] (200 μM) or [KR6] (300 μM), methylmalonyl-CoA (300 μM) and NADPH (2 mM) were added and the mixture incubated at 30 °C for 1.5 h. Alternatively, all enzymes and substrates were combined for 1.5 h or there was a delay of 20–45 min between addition of the [ACP] and [KR] proteins along with their respective substrates. The triketide was hydrolytically released from the ACP by addition of 200 μL of aq. 0.5 M NaOH and incubation at 65 °C for 20 min, followed by addition of 100 μL of 1.5 M HCl. Alternatively, the mixture was treated with DEBS TE (100 μM) for 50 min at 30 °C. The reaction products were isolated by extraction with ethyl acetate (4 × 700 μL), the organic phase was concentrated in vacuo, and the residue was derivatized prior to GC-MS analysis.
NMR analysis of ethyl 2-methylacetoacetate racemization
Ethyl 2-methylacetoacetate was dissolved in 100 mM sodium phosphate in deuterated water, pD 7.2. 1H NMR spectra were recorded at 1, 3, 6, 10, 14 and 20 min. The intensity of the α proton signal was measured as a function of time and the data were fit to the first-order decay expression to calculate the t1/2 for proton exchange. 1H, 13C, COSY and HSQC spectra NMR in CDCl3 on a 35 mM sample of ethyl 2-methylacetoacetate were used to fully assign the 1H and 13C NMR spectra.
Incubation with hybrid module [KS3][AT3][KR5][ACP5][TE]
The plasmid pAYC46 harboring the hybrid module [KS3][AT3][KR5][ACP5][TE] was transformed into E. coli BAP1 cells (incorporating the sfp gene for in vivo phosphopantetheinylation of ACP)43 and used for expression and purification of the module as previously described.7 The hybrid protein (40 μM), in a volume of 15 mL, was incubated with diketide-SNAC 2 (4 mM), methylmalonyl-CoA (1.5 mM, added in three portions separated by 1 h) and NADPH (2 mM) in 100 mM sodium phosphate, pH 7.2 overnight at 30 °C. The reaction was quenched by addition of 2 mL of 1 M HCl. The protein was removed by centrifugation, the supernatant was extracted with ethyl acetate (5 × 20 mL), and the organic extract was concentrated in vacuo. The residue was dissolved in BSTFA (100 μL) and TMS-imidazole (1 μL) and analyzed by GC-MS.
Sodium borohydride reduction of ACP-bound 3-keto-2-methyltriketide
Incubations using mixtures of DEBS [KS][AT] and [ACP] were carried out as described above without addition of KR domain. Following addition of [ACP] and methylmalonyl-CoA, the mixture was incubated for 45 min before quenching with sodium borohydride (0.3 mg) which was added directly to the buffered solution followed by slow rotation for 15 min at room temperature. The resultant mixture of triketide lactones was released from the ACP by the usual basic hydrolysis – acidification protocol. In a control incubation, 45 min after the addition of [ACP] and methylmalonyl-CoA protein-free side products were removed by ultrafiltration using an UltraFree-MC unit (5,000 MWCO). The reaction mixture was concentrated to a final vol. of ~40 μL and the retentate was diluted to a volume of 500 μL with 100 mM sodium phosphate buffer, pH 7.2 before reductive quenching with NaBH4 for 15 min. Protein-free side products were also removed by extraction with ethyl acetate. After 45 min of incubation with [ACP] and methylmalonyl-CoA, the aqueous phase was extracted with 2 × 700 μL ethyl acetate, followed by reduction of the aqueous phase with NaBH4 and workup in the usual manner. In another control reaction, synthetic triketide ketolactone 3 (0.25 mg) in 100 μL of methanol or phosphate buffer (100 mM, pH 7.2), and reacted with NaBH4 at room temperature under agitation for 30 min. The reaction was then quenched with 1 M HCl (50 μL), the reaction mixture was extracted with ethyl acetate and the organic residue derivatized with BSTFA containing 1% (v/v) TMS-imidazole.
Cloning, expression, and purification of DEBS [KR30] from DEBS
Plasmid pRSG34 harboring DEBS module 3+TE was used as a template for amplification of [KR30] by PCR using the primers 5′-CACACACATATGGCCTCCGACGAGCTGGCCTACC-3′ (NdeI restriction site underlined) and 5′-CACACAGAATTCTTAAAGCCCCGCGAGCCGTTGTGCC-3′ (EcoRI underlined and stop codon in italics). The resulting PCR amplicon was digested with NdeI and EcoRI and cloned into the corresponding restriction sites in the expression vector pET28a. The resulting plasmid, pRCD1, encodes the [KR30] with an N-terminal His-tag and the amino acids from the YRVSW to the RLAGL consensus sequences.5 The plasmid pRCD1 was transformed into E. coli BL21(DE3) for protein expression. Cells were grown with 50 μg/mL kanamycin in superbroth (SB) medium at 37 °C up to an OD600 of 0.6, then were cooled to 18 °C and induced with 0.2 mM IPTG for 20 h. The cells were harvested by centrifugation (2,700g, 10 min), then washed and resuspended in lysis buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 10 mM β-mercaptoethanol, 10 % glycerol, 10 mM imidazole) supplemented with 1 μg/mL each of pepstatin and leupeptin. The cells were lysed by incubation with lysozyme (1 mg/mL) for 30 min on ice, then sonicated (3 × 30 s cycles), and cellular debris was removed by centrifugation (20,000g, 45 min). The supernatant was mixed with Ni-NTA resin (2 mL/L culture) for 1.5 h at 4 °C, and the resin was poured into a column fitted with a fritted filter. The resin was washed with lysis buffer (10 column volumes), then eluted with elution buffer (lysis buffer with 250 mM imidazole, 10 column volumes). The eluted protein was concentrated by ultrafiltration, the buffer was exchanged for storage buffer (100 mM sodium phosphate, pH 7.2, 10 % glycerol) on a PD-10 column and the protein was concentrated by ultrafiltration and flash frozen at −78 °C. MALDI-TOF of the purified protein (bovine serum albumin as internal standard): predicted (without N-terminal Met): 51,111 Da, observed: doublet of peaks at 51,040 and 51,191 Da.
Incubations with added DEBS [KR30] protein
[KS][AT] didomain was incubated with diketide-SNAC 2 for 1 h as described above. [ACP] and [KR30] were then added along with methylmalonyl-CoA and NADPH. After 45 min, [KR6] was added and the incubation continued for 1.5 h. Alternatively, the incubation was quenched with NaBH4 for 15 min, and the resulting products isolated and derivatized as described above.
Supplementary Material
Synthesis and spectroscopic data for triketide lactones 3 and 4a–4d and SDS-PAGE of recombinant DEBS [KR30]. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by NIH grants GM22172 (D.E.C.) and CA66736 (C.K.). R.C. is a recipient of a NSERC postdoctoral fellowship and A.Y.C. is a recipient of a Stanford Graduate Fellowship. We thank Dr. Tun-Li Shen for assistance with the mass spectrometric analysis.
References and Notes
- 1.Walsh CT. Chembiochem. 2002;3:125–134. doi: 10.1002/1439-7633(20020301)3:2/3<124::AID-CBIC124>3.0.CO;2-J.Cane DE, Walsh CT, Khosla C. Science. 1998;282:63–68. doi: 10.1126/science.282.5386.63.Walsh CT. Science. 2004;303:1805–1810. doi: 10.1126/science.1094318.For a comprehensive review of PKS and NRPS biochemistry and molecular biology as of 1997, see the thematic issue of Chem Rev. 1997;97:2463–2706. doi: 10.1021/cr970097g.Khosla C, Gokhale RS, Jacobsen JR, Cane DE. Ann Rev Biochem. 1999;68:219–253. doi: 10.1146/annurev.biochem.68.1.219.
- 2.a) Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Annu Rev Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]; b) Staunton J, Wilkinson B. In: Comprehensive Natural Products Chemistry, Polyketides and Other Secondary metabolites Including Fatty Acids and Their Derivatives. Sankawa U, editor. Vol. 1. Elsevier; Oxford: 1999. pp. 495–532. [Google Scholar]
- 3.Kumar P, Khosla C, Tang Y. Methods Enzymol. 2004;388:269–293. doi: 10.1016/S0076-6879(04)88023-6. [DOI] [PubMed] [Google Scholar]
- 4.Wu N, Kudo F, Cane DE, Khosla C. J Am Chem Soc. 2000;122:4847–4852. [Google Scholar]
- 5.Gokhale RS, Tsuji SY, Cane DE, Khosla C. Science. 1999;284:482–485. doi: 10.1126/science.284.5413.482. [DOI] [PubMed] [Google Scholar]
- 6.Kim CY, Alekseyev VY, Chen AY, Tang Y, Cane DE, Khosla C. Biochemistry. 2004;43:13892–13898. doi: 10.1021/bi048418n. [DOI] [PubMed] [Google Scholar]
- 7.Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. J Am Chem Soc. 2006;128:3067–3074. doi: 10.1021/ja058093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen AY, Cane DE, Khosla C. Chem Biol. 2007;14:784–792. doi: 10.1016/j.chembiol.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Gokhale RS, Hunziker D, Cane DE, Khosla C. Chem Biol. 1999;6:117–125. doi: 10.1016/S1074-5521(99)80008-8. [DOI] [PubMed] [Google Scholar]; b) Lu H, Tsai SC, Khosla C, Cane DE. Biochemistry. 2002;41:12590–12597. doi: 10.1021/bi026006d. [DOI] [PubMed] [Google Scholar]; c) Schnarr N, Cronin T, Khosla C, Cane DE. unpublished observations. [Google Scholar]
- 10.Hans M, Hornung A, Dziarnowski A, Cane DE, Khosla C. J Am Chem Soc. 2003;125:5366–5374. doi: 10.1021/ja029539i. [DOI] [PubMed] [Google Scholar]
- 11.Walsh CT, Gehring AM, Weinreb PH, Quadri LE, Flugel RS. Curr Opin Chem Biol. 1997;1:309–315. doi: 10.1016/s1367-5931(97)80067-1. [DOI] [PubMed] [Google Scholar]
- 12.Celmer WD. J Am Chem Soc. 1965;87:1801–1802. doi: 10.1021/ja01086a038. [DOI] [PubMed] [Google Scholar]
- 13.a) McPherson M, Khosla C, Cane DE. J Am Chem Soc. 1998;120:3267–3268. [Google Scholar]; b) Yin Y, Gokhale R, Khosla C, Cane DE. Bioorg Med Chem Lett. 2001;11:1477–1479. doi: 10.1016/s0960-894x(00)00529-1. [DOI] [PubMed] [Google Scholar]
- 14.Kao CM, McPherson M, McDaniel RN, Fu H, Cane DE, Khosla C. J Am Chem Soc. 1998;120:2478–2479. [Google Scholar]
- 15.a) Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. Biochemistry. 2003;42:72–79. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]; b) Caffrey P. ChemBioChem. 2003;4:654–657. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 16.Wu J, Zaleski TJ, Valenzano C, Khosla C, Cane DE. J Am Chem Soc. 2005;127:17393–17404. doi: 10.1021/ja055672+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keatinge-Clay AT, Stroud RM. Structure. 2006;14:737–748. doi: 10.1016/j.str.2006.01.009.Baerga-Ortiz A, Popovic B, Siskos AP, O’Hare HM, Spiteller D, Williams MG, Campillo N, Spencer JB, Leadlay PF. Chem Biol. 2006;13:277–285. doi: 10.1016/j.chembiol.2006.01.004.Based on the determination of the crystal structure of the tylosin KR1 domain and comparison with that of the DEBS KR1 domain, Keatinge-Clay has proposed a binding model to account for the ketone facial stereospecificity of ketoreductases, as well as their preference for unepimerized or epimerized 2-methyl-3-ketoacyl-ACP substrates. Cf. Keatinge-Clay A. Nat Chem Biol. 2007;14:898–908. doi: 10.1016/j.chembiol.2007.07.009.
- 18.Marsden AF, Caffrey P, Aparicio JF, Loughran MS, Staunton J, Leadlay PF. Science. 1994;263:378–380. doi: 10.1126/science.8278811. [DOI] [PubMed] [Google Scholar]; Wiesmann KE, Cortes J, Brown MJ, Cutter AL, Staunton J, Leadlay PF. Chem Biol. 1995;2:583–589. doi: 10.1016/1074-5521(95)90122-1. [DOI] [PubMed] [Google Scholar]
- 19.a) Weissman KJ, Timoney M, Bycroft M, Grice P, Hanefeld U, Staunton J, Leadlay PF. Biochemistry. 1997;36:13849–13855. doi: 10.1021/bi971566b. [DOI] [PubMed] [Google Scholar]; b) Cane DE, Liang TC, Taylor PB, Chang C, Yang CC. J Am Chem Soc. 1986;108:4957–4964. [Google Scholar]
- 20.Note on stereochemical conventions: Although strictly speaking it is incorrect to mix Fischer D/L designations with the more rigorous Cahn, Ingold, Prelog R/S nomenclature, it is often convenient to retain the D- and L- labels when discussing a homologous series of polyketides, for which the specific R or S designation for the same absolute configuration at a given site might vary in response to the substitution pattern on neighboring carbons. We have there mixed the two stereochemical conventions in the interests of greater clarity.
- 21.a) Evans DA, Ennis MD, Le T, Mandel N, Mandel G. J Am Chem Soc. 1984;106:1154–1156. [Google Scholar]; b) Evans DA, Kim AS, Metternich R, Novack VJ. J Am Chem Soc. 1998;120:5921–5942. [Google Scholar]; c) Evans DA, Britton TC, Ellman JA. Tetrahedron Lett. 1987;28:6141–6144. [Google Scholar]; d) Beck BJ, Aldrich CC, Fecik RA, Reynolds KA, Sherman DH. J Am Chem Soc. 2003;125:12551–12557. doi: 10.1021/ja034841s. [DOI] [PubMed] [Google Scholar]
- 22.a) Kao CM, Luo G, Katz L, Cane DE, Khosla C. J Am Chem Soc. 1994;116:11612–11613. [Google Scholar]; b) Jacobsen JR, Cane DE, Khosla C. Biochemistry. 1998;37:4928–4934. doi: 10.1021/bi9729920. [DOI] [PubMed] [Google Scholar]
- 23.Luo G, Pieper R, Rosa A, Khosla C, Cane DE. Bioorg Med Chem. 1996;4:995–999. doi: 10.1016/0968-0896(96)00096-x. [DOI] [PubMed] [Google Scholar]
- 24.Donadio S, Staver MJ, Mcalpine JB, Swanson SJ, Katz L. Science. 1991;252:675–679. doi: 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]
- 25.a) Siskos AP, Baerga-Ortiz A, Bali S, Stein V, Mamdani H, Spiteller D, Popovic B, Spencer JB, Staunton J, Weissman KJ, Leadlay PF. Chem Biol. 2005;12:1145–1153. doi: 10.1016/j.chembiol.2005.08.017. [DOI] [PubMed] [Google Scholar]; b) Bali S, O’Hare HM, Weissman KJ. Chembiochem. 2006;7:478–484. doi: 10.1002/cbic.200500430. [DOI] [PubMed] [Google Scholar]
- 26.Tang Y, Kim CY, Mathews II, Cane DE, Khosla C. Proc Natl Acad Sci U S A. 2006 [Google Scholar]
- 27.a) Hicks LM, Mazur MT, Miller LM, Dorrestein PC, Schnarr NA, Khosla C, Kelleher NL. Chembiochem. 2006;7:904–907. doi: 10.1002/cbic.200500416. [DOI] [PubMed] [Google Scholar]; b) Chan YA, Boyne MT, II, Podevels AM, Klimowicz AK, Handelsman J, Kelleher NL, Thomas MG. Proc Natl Acad Sci U S A. 2006;103:14349–14354. doi: 10.1073/pnas.0603748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Although GC-MS has previously been used to characterize tetrasubstituted polyketide δ-lactones, the diastereomers cannot be differentiated on the basis of their MS fragmentation patterns. Cf. Weissman KJ, Kearney GC, Leadlay PF, Staunton J. Rapid Commun Mass Spectrom. 1999;13:2103–2108. doi: 10.1002/(SICI)1097-0231(19991115)13:21<2103::AID-RCM760>3.0.CO;2-Y.
- 29.Ostergaard LH, Kellenberger L, Cortes J, Roddis MP, Deacon M, Staunton J, Leadlay PF. Biochemistry. 2002;41:2719–2726. doi: 10.1021/bi0117605. [DOI] [PubMed] [Google Scholar]; Holzbaur IE, Harris RC, Bycroft M, Cortes J, Bisang C, Staunton J, Rudd BA, Leadlay PF. Chem Biol. 1999;6:189–195. doi: 10.1016/S1074-5521(99)80035-0. [DOI] [PubMed] [Google Scholar]
- 30.A hybrid triketide synthase based on DEBS3, in which the native KS5 domain was replaced by fusion with the DEBS ATL-ACPL loading didomain and KS1, has been reported to generate a mixture of the expected triketide lactone and a diastereomer of unknown absolute configuration whose mechanism of formation has not yet been satisfactorily explained. Cf. Holzbaur IE, Ranganathan A, Thomas IP, Kearney DJ, Reather JA, Rudd BA, Staunton J, Leadlay PF. Chem Biol. 2001;8:329–340. doi: 10.1016/s1074-5521(01)00014-x.
- 31.Zhang YM, Wu B, Zheng J, Rock CO. J Biol Chem. 2003;278:52935–52943. doi: 10.1074/jbc.M309874200. [DOI] [PubMed] [Google Scholar]
- 32.Bohm I, Holzbaur IE, Hanefeld U, Cortes J, Staunton J, Leadlay PF. Chem Biol. 1998;5:407–412. doi: 10.1016/s1074-5521(98)90157-0. [DOI] [PubMed] [Google Scholar]
- 33.Starcevic A, Jaspars M, Cullum J, Hranueli D, Long PF. Chembiochem. 2007;8:28–31. doi: 10.1002/cbic.200600399.The specific mechanism suggested for enoyl-ACP reduction is unlikely, since it posits nucleophilic attack of NADPH on a carbon bearing a negatively-charged enolate oxygen. It is also implausible that different KR domains would reduce 2-methyl-3-ketoacyl-ACP substrates by two distinct mechanisms, depending on the reaction stereospecificity.
- 34.Tang Y, Kim CY, Chen AY, Cane DE, Khosla C. Chem Biol. 2007;14:931–943. doi: 10.1016/j.chembiol.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) Leibundgut M, Jenni S, Frick C, Ban N. Science. 2007;316:288–290. doi: 10.1126/science.1138249. [DOI] [PubMed] [Google Scholar]; b) Jenni S, Leibundgut M, Boehringer D, Frick C, Mikolasek B, Ban N. Science. 2007;316:254–261. doi: 10.1126/science.1138248. [DOI] [PubMed] [Google Scholar]
- 36.Alekseyev VY, Liu CW, Cane DE, Puglisi JD, Khosla C. Protein Sci. 2007;16:0000. doi: 10.1110/ps.073011407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aromatic polyketide synthases generate long-chain, poly-β-ketoacyl-ACP intermediates that do not undergo premature aldol cylizations, in contrast to the highly reactive protein-free substrates which rapidly cyclize at the triketide or tetraketide stage unless prevented by masking of the ketone groups. Cf. Harris TM, Harris CM, Hindley KB. Fortschr Chem Org Naturst. 1974;31:217–282. doi: 10.1007/978-3-7091-7094-6_5.
- 38.The Evans β-ketoimides 5 and 9 are widely used for alkylation due to their high enantiomeric purity and configurational stability which due to conformational constraints that suppress epimerization of the C-2 methyl group. cf ref. 21.
- 39.Roujeinikova A, Simon WJ, Gilroy J, Rice DW, Rafferty JB, Slabas AR. J Mol Biol. 2007;365:135–145. doi: 10.1016/j.jmb.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 40.Cane DE, Kudo F, Kinoshita K, Khosla C. Chem Biol. 2002;9:131–142. doi: 10.1016/s1074-5521(02)00089-3. [DOI] [PubMed] [Google Scholar]
- 41.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning, A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 42.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 43.Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthesis and spectroscopic data for triketide lactones 3 and 4a–4d and SDS-PAGE of recombinant DEBS [KR30]. This material is available free of charge via the Internet at http://pubs.acs.org.