

Abstract
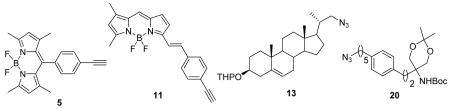
Two analogues (1, 2) of free cholesterol and one analogue (3) of the immunosuppressive sphingolipid FTY720 containing a boron dipyrromethene chromophore (BODIPY) were synthesized. The synthetic routes involved preparation of boron dipyrromethene moieties (5, 11) bearing a phenylethynyl group at different positions of the chromophore, and lipids (13, 20) bearing an azido group. The dye was tethered to the lipid via a 1,2,3-triazole in the linker by using the click reaction. Analogues derived from 11 (in which an (E)-styrylethynyl moiety is bonded to C-5 of BODIPY) exhibited a marked red-shift (~70–80 nm) compared with those derived from 5 (in which a phenylethynyl moiety is bonded to C-8 of BODIPY).
Introduction
The 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene fluorophore, better known as BODIPY, possesses many distinctive and desirable properties: it is relatively hydrophobic, and has a high molar absorption coefficient and fluorescence quantum yield.1 The photochemical and chemical stabilities of the boron dipyrromethene chromophore are higher than those of many other dyes, and the extent of π-electron conjugation can be modified by introduction of substituents, affording red-shifted BODIPY derivatives. BODIPY-linked reagents have been used in a wide range of applications, e.g., as fluorescent switches2 and chemosensors for protons,3 metal ions,4 nitric oxide,5 toxins,6 and peroxyl radicals.7 Biochemical applications of BODIPY include bioconjugates with proteins,8 DNA,9 and carbohydrates.10 Because BODIPY is relatively hydrophobic, its conjugates with lipids can partition efficiently into biomembranes.8,11 Indeed, BODIPY derivatives of many lipids, including fatty acids,12 triglycerides,13 sphingolipids,14 phospholipids,15 and glycolipids16 have been prepared and used for biological studies. When the local concentration of the probe increases, the characteristic green emission of the boron dipyrromethene chromophore is red-shifted via excimer formation, permitting visualization of probe accumulation within subcellular compartments of living cells by fluorescence microscopy.16c,d
We recently reported the synthesis of BODIPY-modified cholesterol analogues17 and found that one of the analogues can partition into liquid-ordered domains of model membranes.18 The spectroscopic and membrane properties of this BODIPY-cholesterol analogue and the commercially available 7-nitrobenz-2-oxa-1,3-diazolyl (NBD)-cholesterol have been summarized in a recent review.19 We now describe the preparation of two new red-shifted BODIPY conjugates of free cholesterol, compounds 1 and 2 (Figure 1).
Figure 1. Compounds 1 and 2.

We also report the introduction of the BODIPY chromophore into the synthetic sphingosine analogue known as FTY720 (Figure 2). FTY720, which was discovered during the optimization of the natural product myriocin (which inhibits the first enzyme in the sphingomyelin biosynthetic pathway),20 has attracted a great deal of attention in the past decade because it is a new immunosuppressant with a unique mechanism of action. FTY720-phosphate ((S)-FTY720-P), formed in vivo via sphingosine kinases 1 and 2,21 is an agonist for several G-protein coupled sphingosine 1-phosphate (S1P) receptors.22 FTY720-P exhibits immunosuppressive activity by redirecting the trafficking of circulating lymphocytes. FTY720 has been in phase III clinical trials as an immunosuppresant for organ transplants, and recently entered into a clinical trial for the treatment of multiple sclerosis.23 An FTY720 derivative bearing the relatively hydrophilic NBD fluorophore has been synthesized and used as to study the metabolism and mechanism of action of FTY720.24 However, since the NBD conjugates induce an “upside down orientation in the membrane”25 we directed our attention to the more lipophilic BODIPY conjugates. We report herein the synthesis of compound 3 (Figure 2), which contains a BODIPY fluorophore in the alkyl side chain and retains the intact hydrophilic head group of FTY720.
Figure 2. FTY720 and Compound 3.

The key step in these syntheses is the 1,3-dipolar cycloaddition between an alkyne and an azide, which was first studied and reviewed by Huisgen et al.26 The conventional procedure of 1,3-dipolar cycloaddition of an alkyne and an azide usually leads to a 1,2,3-triazole as a mixture of two regioisomers, which has limited the application of this reaction. The discovery that a catalytic amount of Cu(I) allows the 1,3-dipolar cycloaddition reaction to proceed under milder conditions with high regiospecificity27 spurred interest in using this reaction in various fields ranging from medicinal chemistry to material science.28 This reaction is often referred to as the “click” reaction.29 We have applied the click reaction to prepare BODIPY conjugates of lipids, compounds 1–3.
Results and Discussion
Synthesis of BODIPY-Substituted Alkynes
The synthesis of BODIPY lipid conjugates by click chemistry involved the preparation of BODIPY derivatives bearing a terminal acetylene group. We prepared two such derivatives with different emission wavelengths. Scheme 1 outlines the preparation of the first alkynyl-BODIPY derivative. Condensation of 4-ethynylbenzaldehyde (4) with 2,4-dimethylpyrrole in the presence of a catalytic amount of TFA, followed by oxidation with p-chloranil and chelation with BF3·OEt2 in the presence of NEt3, gave compound 5 in 28% overall yield.
Scheme 1. Synthesis of BODIPY-Acetylene Derivative 5a.

aReagents and conditions: (a) (i) TFA, CH2Cl2, rt, overnight; (ii) p-chloranil, rt, 30 min; (c) BF3·OEt2, NEt3, rt, 6 h.
We explored a variety of routes to the second alkynyl-linked BODIPY derivative. Generally, there are three routes for synthesizing BODIPY analogues with an extended conjugation: (1) de novo synthesis from a pyrrole derivative bearing a substituent with external conjugation;30 (2) condensation of the 3- and/or 5-methyl group of BODIPY with an aldehyde;31 and (3) a transition metal catalyzed coupling reaction of BODIPY via a halide substituent32 or direct activation of a C-H bond.33 Our initial attempt to prepare an alkynyl-linked BODIPY with extended conjugation through condensation of 4,4-difluoro-1,3,5,7-tetramethyl-2,6,8-triethyl-4-bora-3a,4a-diaza-s-indacene34 with 4 by following reported procedures31 failed, probably because of the instability of 4 under the relatively harsh reaction conditions (toluene/acetic acid/piperidine, reflux). We observed the total consumption of 4 by TLC (hexane/ethyl acetate 6:1) during the reaction and the complete recovery of the diaza-s-indacene starting material after work-up. We then turned to the synthesis of a TMS-substituted terminal alkyne as outlined in Scheme 2. A Wittig reaction of 635 with pyrrole-2-carboxaldehyde using potassium tert-butoxide as the base furnished 736 in 69% yield. After 7 underwent condensation37 with 3,5-dimethylpyrrole-2-carboxaldehyde in the presence of POCl3, chelation with BF3·OEt2 afforded BODIPY analog 8 in 47% yield. Unfortunately, the Sonogashira reaction for conversion of bromide 8 to alkyne 9 was unsuccessful, producing many unidentified products. After reversing the reaction sequences and performing the Sonogashira reaction on bromide 7, we were able to obtain 10 in 56% yield. The synthesis of 9 was accomplished by condensation of 10 with 3,5-dimethylpyrrole-2-carboxaldehyde, followed by chelation with BF3·OEt2 in 52% overall yield (Scheme 3). The TMS group in 9 was easily removed by treating 9 with potassium carbonate in a mixture of methanol and dichloromethane to give 11 in 64% yield.
Scheme 2. de novo Synthesis of BODIPY with Extended Conjugation.
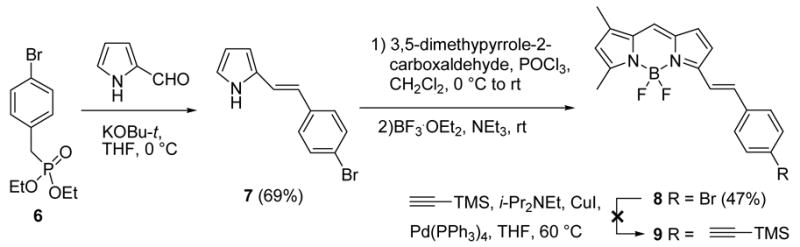
Scheme 3. Synthesis of BODIPY Acetylene with Extended Conjugation.

Synthesis of BODIPY-cholesterol Conjugates
We next explored the use of the two alkynyl-substituted BODIPY derivatives (5 and 11) in preparing fluorescent cholesterol conjugates. Known alcohol 1217 was converted by the conventional three-step sequence of mesylate to bromide to azide 13 in 85% overall yield (Scheme 4). BODIPY cholesterol conjugate 1 was obtained in 91% yield after deprotection of 14, which was prepared in 59% yield via a click reaction between 5 and 13 catalyzed by CuI in DMSO. A similar route was used to prepare BODIPY-cholesterol conjugate 2, which has a longer emission wavelength than 1, in 52% overall yield (Scheme 5).
Scheme 4. Synthesis of BODIPY-Cholesterol Conjugate 1.

Scheme 5. Synthesis of BODIPY-cholesterol Conjugate 2.

Synthesis of BODIPY-FTY720 Conjugate 3
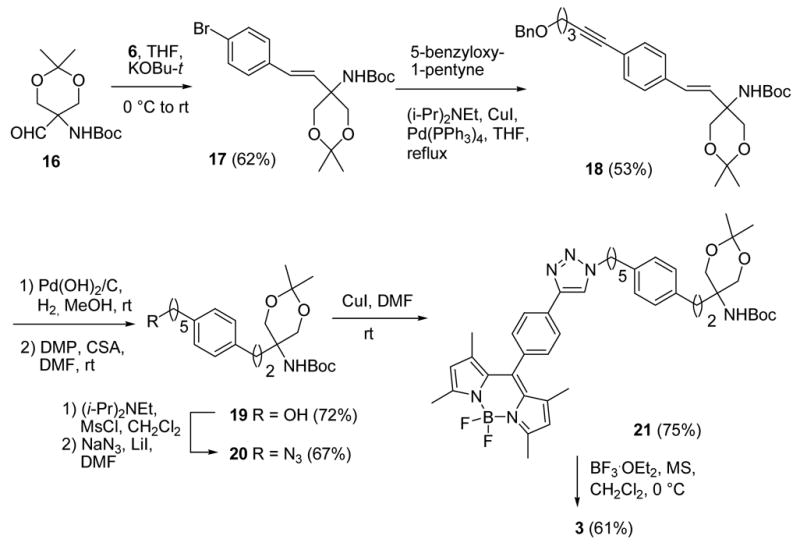
The synthesis of a BODIPY-FTY720 conjugate started with the Wittig reaction of 6 and aldehyde 1638 using potassium tert-butoxide as the base. The resulting bromide 17 was subjected to a Sonogashira reaction with 5-benzyloxy-1-pentyne39 to give 18 in 53% yield. The double and triple bonds were hydrogenated and the benzyl group was removed by hydrogenolysis with palladium hydroxide in methanol. The ketal protecting group was also partially removed under these conditions, but it was easily reinstalled by treating the resulting mixture with 2,2-dimethoxypropane in the presence of CSA to give 19 in 72% yield. After conversion of the hydroxy group in 19 to the mesylate and treatment with NaN3 in DMF, azide 20 was obtained in 67% yield. The click reaction of 20 with 5 catalyzed by CuI in DMF at room temperature gave protected BODIPY-FTY720 conjugate 21. Deprotection of 21 proved to be troublesome, although it has been reported that a NHBoc group can be removed with 4 M HCl in dioxane in the presence of a BODIPY moiety.14 When we treated 21 with proton acids, including HCl, trifluoroacetic acid, and p-toluenesulfonic acid, the ketal was removed but the Boc group remained intact under mild conditions. Harsher conditions destroyed the BF2 chelate of the BODIPY fluorophore. We then reasoned that since the BODIPY fluorophore is formed by chelation in the presence of BF3·OEt2, and that BF3·OEt2 has been reported to deprotect a NHBoc group under quite mild conditions,40 this Lewis acid might remove the NHBoc and ketal groups in one pot. Indeed, treatment of 21 with BF3·OEt2 in the presence of 4 Å molecular sieves in dichloromethane at 0 °C gave BODIPY-FTY720 conjugate 3 in 61% yield (Scheme 6).
Scheme 6. Synthesis of BODIPY-FTY720 Conjugate 3.
Photospectroscopic Properties
Spectroscopic data are presented in Table 1, and the emission spectra are shown in Figure 3. All compounds exhibited the characteristic absorption and emission patterns of the BODIPY fluorophore, with high extinction coefficients and small Stokes shifts. The absorption and emission maxima were red-shifted in chloroform (dielectric constant, 5.5) compared with ethanol (dielectric constant, 24.3), and the molar extinction coefficient (ε) was higher. Note that for compounds 1, 3, and 21, which have a phenyl substituent at the 8 position of the BODIPY moiety, the absorption and emission wavelengths are very similar to those of the parent 1,3,5,7-tetramethyl-BODIPY chromophore. The phenyl group at the 8 position is almost orthogonal to the BODIPY plane41 because of steric hindrance imposed by the methyl groups at the 1 and 7 positions, and thus does not contribute to the conjugation of the BODIPY fluorophore. The introduction of extended conjugation at the 5 position of the BODIPY moiety, however, did cause a significant red shift (~70 nm) in the absorption and emission wavelengths compared with the corresponding 5-methyl substituted chromophore, in good agreement with previous reports.31
TABLE 1. Spectroscopic Data in Ethanol and Chloroforma.
| Compd. | λmax (abs)
(nm) |
ε
(M−1 cm−1) |
λem (nm) |
|---|---|---|---|
| 1 | 499 | 110,000 | 508 |
| 2 | 569
(578) |
80,000
(89,000) |
583
(591) |
| 3 | 500
(502) |
72,000
(85,000) |
508
(512) |
| 8 | 562 | 118,000 | 572 |
| 9 | 568
(577) |
104,000
(117,000) |
579
(585) |
| 11 | 567
(572) |
114,000
(119,000) |
574
(580) |
| 15 | 568 | 76,000 | 577 |
| 21 | 495
(502) |
75,000
(85,000) |
500
(515) |
The parentheses indicate the data obtained with compounds dissolved in chloroform. The absorption spectra were recorded at 6.25 × 10−6 M at 25 °C. The emission spectra were recorded at 1.0 × 10−7 M.
Figure 3. Emission Spectra of Compounds 1–3, 8, 9, 11, 15, and 21 in EtOHa.
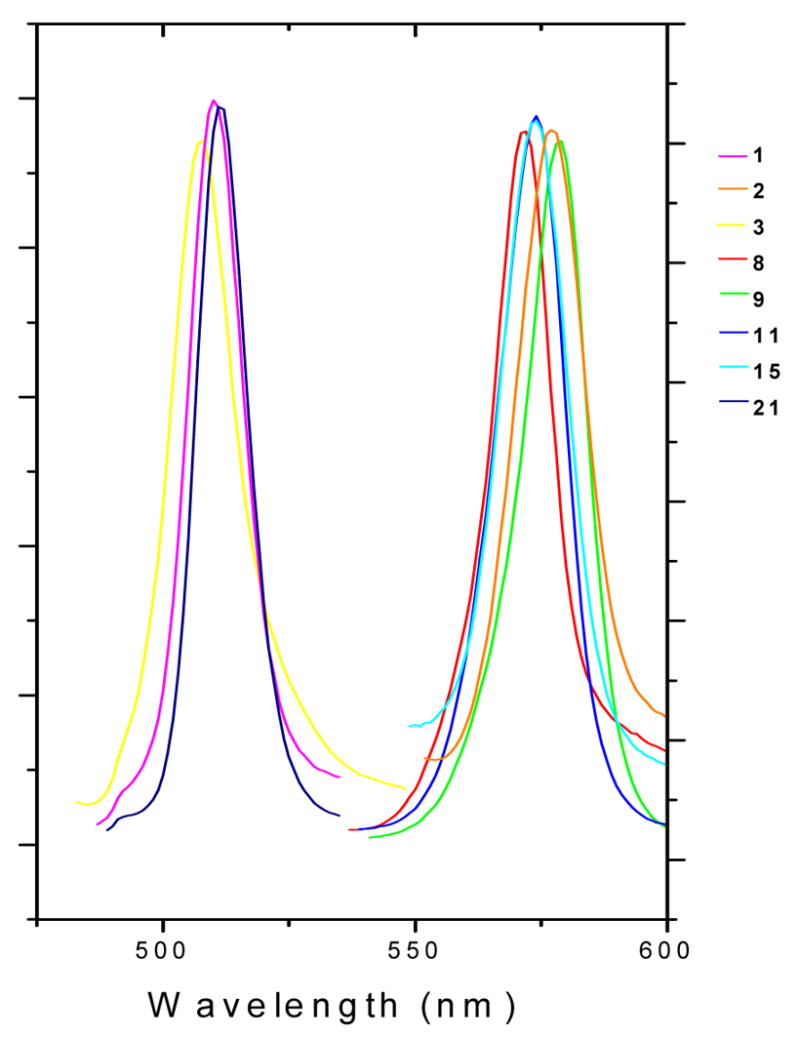
aThe spectra were recorded at 1.0 × 10−7 M at 25 °C; an excitation wavelength of 365 nm was used.
Conclusion
We have described the syntheses of two acetylenic-linked BODIPY fluorescent dyes (5 and 11) with different absorption and emission wavelengths. Lipids 13 and 20, which bear a terminal azido group, were covalently derivatized with one of these dyes to prepare BODIPY-conjugated cholesterol analogues 1 and 2 and FTY720 analogue 3 by utilizing the click reaction. As these new analogues exhibited intense absorption and strong fluorescence, they can serve as probes for monitoring the effects of cholesterol and FTY720 in model membranes and cells and for donor-acceptor resonance energy transfer studies. Their excellent luminescent properties make it possible to visualize the distribution and transport of these lipids in living cells by fluorescence microscopy.
Experimental Section
4,4-Difluoro-8-(4-ethynylphenyl)-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene (5)
To a solution of 4-ethynylbenzaldehyde (0.13 g, 1.0 mmol, compound 4) and 2,4-dimethylpyrrole (0.22 g, 2.3 mmol) in CH2Cl2 (50 mL) was added trifluoroacetic acid (7.6 μL, 0.1 mmol) under N2. After the mixture was stirred at room temperature overnight, a solution of p-chloranil (0.246 g, 1.0 mmol) in CH2Cl2 (10 mL) was added. The mixture was stirred at room temperature for an additional 30 min. BF3·OEt2 (2.60 g, 18.6 mmol) and NEt3 (1.7 g, 17.0 mmol) were added, followed by stirring at room temperature for 6 h. The reaction mixture was washed with water (4 × 60 mL) and dried (Na2SO4). The solvent was removed under vacuum and the residue was purified by chromatography (hexanes/EtOAc 20:1 to 5:1) to give compound 5 (98.8 mg, 28%). 1H NMR δ 7.63 (d, 2H, J = 7.8 Hz), 7.27 (d, 2H, J = 7.8 Hz), 5.99 (s, 2H), 3.18 (s, 1H), 2.55 (s, 6H), 1.40 (s, 6H); 13C NMR δ 155.8, 143.0, 140.6, 135.6, 132.9, 131.1, 128.2, 122.9, 121.4, 82.9, 78.6, 14.62, 14.58; 19F NMR δ -146.2 (m). HRMS m/z: calcd for C21H20BF2N2 (MH+), 349.1682; found, 349.1689.
4,4-Difluoro-5-[(E)-(4-bromophenyl)ethenyl]-1,3-dimethyl-4-bora-3a,4a-diaza-s-indacene (8)
A solution of 3,5-dimethylpyrrole-2-carboxaldehyde (0.12 g, 1.0 mmol) and 735 (0.248 g, 1.0 mmol) in CH2Cl2 (15 mL) was cooled to 0 °C. After a solution of POCl3 (0.15 g, 1.0 mmol) in CH2Cl2 (1 mL) was added with caution, the mixture was stirred at 0 °C for 1 h, and then at room temperature overnight. BF3·OEt2 (0.52 mL, 4.0 mmol) and N,N-diisopropylethylamine (0.70 mL, 4.0 mmol) were added sequentially at 0 °C, and the resulting mixture was stirred at room temperature for 6 h. The solution was washed with water (3 × 20 mL), dried (Na2SO4), and concentrated under vacuum. Compound 8 (0.19 g, 47%) was obtained by flash chromatography (hexanes/EtOAc 50:1 to 5:1). 1H NMR δ 7.60 (d, 1H, J = 16.0 Hz), 7.50-7.43 (m, 4H), 7.18 (d, 1H, J = 16.0 Hz), 7.06 (s, 1H), 6.94 (d, 1H, J = 4.4 Hz), 6.84 (d, 1H, J = 4.4 Hz), 6.13 (s, 1H), 2.60 (s, 3H), 2.26 (s, 3H); 13C NMR δ 160.0, 152.7, 143.3, 135.5, 134.8, 133.9, 131.9, 128.7, 128.1, 122.7, 122.2, 120.4, 119.9, 115.2, 15.1, 11.4; 19 F NMR δ -143.0 (m). HRMS m/z: calcd for C19H17BBrF2N2 (MH+), 401.0631; found, 401.0632.
4,4-Difluoro-1,3-dimethyl-5-[(E)-(4-trimethylethynylphenyl)ethenyl]-4-bora-3a,4a-diaza-s-indacene (9)
The synthesis of compound 9 (294 mg, 52%) was accomplished by using the procedure for compound 8, with 10 (362 mg, 1.35 mmol) as the starting material. 1H NMR δ 7.61 (d, 1H, J = 16.4 Hz), 7.50 (d, 2H, J = 8.4 Hz), 7.44 (d, 2H, J = 8.4 Hz), 7.22 (d, 1H, J = 16.4 Hz), 7.03 (s, 1H), 6.92 (d, 1H, J = 4.4 Hz), 6.83 (d, 1H, J = 4.4 Hz), 6.12 (s, 1H), 2.60 (s, 3H), 2.25 (s, 3H), 0.26 (s, 9H); 13C NMR δ 159.7, 152.8, 143.1, 136.6, 135.5, 134.9, 134.4, 132.3, 128.1, 127.1, 123.2, 122.1, 120.3, 120.0, 115.3, 105.1, 95.9, 15.0, 11.3, -0.06; 19F NMR δ -142.9 (m). HRMS m/z: calcd for C24H26BF2N2Si (MH+), 419.1920; found, 419.1928.
2-[(E)-(4-Trimethylethynylphenyl)ethenyl]pyrrole (10)
To a solution of trimethylsilylacetylene (0.98 g, 10 mmol), 7 (0.50 g, 2.0 mmol), and N,N-diisopropylethylamine (4 mL, 22.9 mmol) in THF (12 mL) were added CuI (58 mg, 0.30 mmol) and tetrakis(triphenylphosphine)palladium (0.36 g, 0.3 mmol) under N2 at room temperature. After the mixture was stirred at 60 °C overnight under N2 and then cooled to room temperature, it was passed through a short pad of silica gel to remove insoluble salts or by-products. The filtrate was concentrated under vacuum, and the crude product was purified by chromatography (hexanes/EtOAc 20:1 to 5:1) to give 10 (0.30 g, 56%). 1H NMR δ 8.28 (br s, 1H), 7.40 (d, 2H, J = 8.4 Hz), 7.30 (d, 2H, J = 8.4 Hz), 6.92 (d, 1H, J = 16.4 Hz), 6.76 (m, 1H), 6.55 (d, 1H, J = 16.4 Hz), 6.36 (m, 1H), 6.24 (m, 1H), 0.25 (s, 9H); 13C NMR δ 137.3, 132.2, 130.5, 125.5, 122.4, 121.2, 119.8, 119.6, 110.1, 109.7, 105.3, 94.8, −0.02. HRMS m/z: calcd for C17H20NSi (MH+), 266.1359; found, 266.1356.
4,4-Difluoro-1,3-dimethyl-5-[(E)-(4-ethynylphenyl)ethenyl]-4-bora-3a,4a-diaza-s-indacene (11)
Compound 9 (42 mg, 0.10 mmol) was dissolved in a mixture of CH2Cl2 (5 mL) and MeOH (5 mL). After addition of K2CO3 (41 mg, 0.30 mmol), the mixture was stirred at room temperature under N2 until the disappearance of 9 (~ 4 h, monitored by TLC: hexanes/EtOAc, 5:1). The solvent was removed under vacuum. The residue was distributed in CH2Cl2 (20 mL) and water (10 mL), and then neutralized with acetic acid. The organic layer was separated, washed with water (3 × 10 mL), dried (Na2SO4), and concentrated to give a blue crude product. Compound 11 (22.1 mg, 64%) was obtained by chromatographic purification with gradient elution (hexanes/EtOAc 20:1 to 5:1). 1H NMR δ 7.63 (d, 1H, J = 16.4 Hz), 7.54 (d, 1H, J = 8.4 Hz), 7.48 (d, 1H, J = 8.4 Hz), 7.22 (d, 1H, J = 16.4 Hz), 7.06 (s, 1H), 6.94 (d, 1H, J = 4.0 Hz), 6.85 (d, 1H, J = 4.0 Hz), 6.13 (s, 1H), 3.17 (s, 1H), 2.61 (s, 3H), 2.26 (s, 3H); 13C NMR δ 160.0, 152.6, 143.2, 137.0, 135.6, 134.9, 134.2, 132.5, 128.1, 127.1, 122.2, 122.1, 120.4, 120.3, 115.3, 83.7, 78.6, 15.1, 11.4; 19F NMR −143.0 (m). HRMS m/z: calcd for C21H18BF2N2 (MH+), 347.1526; found, 347.1528.
22-Azido-5β-tetrahydropyranyloxy-23,24-bisnorchol-5-ene (13)
A solution of alcohol 12 (2.2 g, 5.3 mmol) and NEt3 (5.3 g, 53 mmol) in CH2Cl2 (150 mL) was cooled to 0 °C. Methanesulfonyl chloride (3.0 g, 26.4 mmol) was added dropwise. After the addition was complete, the mixture was stirred at 0 °C for 3 h and then at room temperature overnight. The solution was washed with water (3 × 100 mL), dried, and concentrated to give a viscous oil, which solidified after storing at low temperature. To the solution of the mesylate in THF (100 mL) was added lithium bromide (0.92 g, 10.6 mmol). The mixture was heated at reflux for one day, and then concentrated to give a white solid, which was distributed in CH2Cl2 (100 mL) and water (100 mL). The organic layer was washed with water (2 × 100 mL), dried over Na2SO4, and concentrated to give the corresponding bromide (2.48 g, 98% from 12). 1H NMR δ 5.34 (m, 1H), 4.71 (m, 1H), 3.91 (m, 1H), 3.60-3.40 (m, 2H), 3.37-3.32 (m, 2H), 2.44-0.88 (m, 33H), 0.70 (s, 3H); 13C NMR δ 140.9, 140.7, 121.3, 121.2, 96.8, 96.7, 75.79, 75.77, 62.7, 62.6, 56.2, 53.6, 49.91, 49.88, 43.4, 42.2, 40.1, 39.3, 38.6, 37.6, 37.3, 37.1, 36.61, 36.57, 31.8, 31.7, 31.2, 29.5, 27.8, 27.4, 25.4, 24.1, 20.9, 19.93, 19.88, 19.2, 18.6, 12.1.
The above bromide (0.30 g, 0.63 mmol), sodium azide (0.122 g, 1.88 mmol), and a catalytic amount of lithium iodide (10 mg, 75 μmol) were mixed in dry DMF (10 mL). The mixture was heated at 80 °C for 18 h, and then cooled to room temperature. Water (30 mL) was added, and the precipitate was collected by filtration. The solid was washed with water (4 × 5 mL), and then dried to give 13 (0.235 g, 85%). 1H NMR δ 5.34 (m, 1H), 4.72 (m, 1H), 3.92 (m, 1H), 3.57-3.45 (m, 2H), 3.37 (dd, 1H, J = 12.0 Hz, 3.2 Hz), 3.04 (dd, 1H, J = 12.0 Hz, 7.2 Hz), 2.38-0.88 (m, 33H), 0.69 (s, 3H); 13C NMR δ 141.1, 140.9, 121.4, 97.0, 96.8, 76.0, 62.9, 58.0, 56.5, 53.2, 50.1, 42.5, 39.5, 38.7, 37.4, 37.2, 36.9, 36.8, 31.9, 31.3, 27.9, 25.5, 24.3, 21.0, 20.1, 19.4, 17.8, 11.9. HRMS m/z: calcd for C27H44NO2 ([M − N2 + H]+), 414.3366; found, 414.3354.
3β-Tetrahydropyranyloxy-22-{4-[4-(4,4-difluoro-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacen-8-yl)phenyl]-1,2,3-triazol-1-yl}-23,24-bisnorchol-5-ene (14)
A mixture of 5 (16.8 mg, 48 μmol), 13 (21.3 mg, 48 μmol), and CuI (0.9 mg, 4.8 μmol) in DMSO (3 mL) was stirred at 80 °C until the disappearance of alkyne 5 (~ 5 h). Water (10 mL) was added, and the mixture was extracted with Et2O (4 × 20 mL). The ether layers were combined, washed with water (2 × 20 mL), and dried (Na2SO4). The solvent was removed under vacuum, and the residue was purified by chromatography (hexanes/EtOAc 10:1 to 2:1) to give 14 (22.4 mg, 59%). 1H NMR δ 7.99 (d, 2H, J = 8.0 Hz), 7.81 (s, 1H), 7.35 (d, 2H, J = 8.0 Hz), 5.99 (s, 2H), 5.35 (m, 1H), 4.72 (m, 1H), 4.45 (dd, 1H, J = 13.6 Hz, 3.2 Hz), 4.14 (dd, 1H, J = 13.6 Hz, 9.2 Hz), 3.95-3.88 (m, 1H), 3.58-3.43 (m, 2H), 2.56 (s, 6H), 2.40-0.82 (m, 39H), 0.76 (s, 3H); 13C NMR δ 155.6, 146.8, 143.1, 141.3, 141.1, 141.0, 134.7, 131.5, 131.4, 128.6, 126.3, 121.3, 120.5, 97.0, 96.9, 76.0, 62.9, 56.5, 56.3, 53.7, 50.1, 42.7, 39.6, 38.8, 38.1, 36.78, 36.75, 31.9, 31.3, 29.69, 29.65, 29.4, 28.2, 28.0, 25.5, 24.4, 22.7, 21.0, 20.1, 19.4, 17.2, 14.64, 14.59, 14.1, 12.0; 19F NMR δ 146.2 (m). HRMS m/z: calcd for C48H63BF2N5O2 (MH+), 790.5037; found, 790.5042.
22-{4-[4-(4,4-Difluoro-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacen-8-yl)phenyl]-1,2,3-triazol-1-yl}-23,24-bisnorchol-5-en-3β-ol (1)
To a solution of 14 (25.0 mg, 31.7 μmol) in a mixture of CH2Cl2 (2 mL) and MeOH (6 mL) was added PPTS (2.0 mg, 8.0 μmol). After being stirred at room temperature overnight, the mixture was concentrated and redistributed between CH2Cl2 (20 mL) and water (10 mL). The organic layer was washed with water (2 × 10 mL) and dried (Na2SO4). The solvent was removed under vacuum, and the residue was purified by chromatography (hexanes/EtOAc 4:1 to 2:1) to give 14 (8.8 mg) and 1 (13.2 mg, 91% based on reacted 14). 1H NMR δ 7.99 (d, 2H, J = 8.4 Hz), 7.81 (s, 1H), 7.35 (d, 2H, J = 8.4 Hz), 5.99 (s, 2H), 5.36 (m, 1H), 4.45 (dd, 1H, J = 13.6 Hz, 4.0 Hz), 4.14 (dd, 1H, J = 13.6 Hz, 8.8 Hz), 3.58-3.48 (m, 1H), 2.56 (s, 6H), 2.48-0.82 (m, 33H), 0.76 (s, 3H); 13C NMR δ 155.6, 146.8, 143.1, 141.3, 140.8, 134.7, 131.5, 131.4, 128.6, 126.3, 121.5, 121.3, 120.5, 71.7, 56.5, 56.2, 53.7, 50.0, 42.7, 42.2, 39.5, 38.0, 37.2, 36.5, 31.9, 31.8, 31.6, 29.7, 29.6, 29.3, 28.2, 24.4, 22.7, 21.0, 20.3, 19.4, 17.2, 14.62, 14.57, 14.1, 12.0; 19F NMR δ 146.1 (m). HRMS m/z: calcd for C43H55BF2N5O (MH+), 706.4462; found, 706.4464.
3β-Tetrahydropyranyloxy-22-{4-[4-((E)-(4,4-difluoro-1,3-dimethyl-4-bora-3a,4a-diaza-s-indacen-5-yl)ethenyl)phenyl]-1,2,3-triazol-1-yl}-23,24-bisnorchol-5-ene (15)
Compound 15 (10.2 mg, 54%) was prepared from 11 (8.8 mg, 25.5 μmol), 13 (10.0 mg, 22.3 μmol), CuI (1.0 mg, 5.3 μmol), and DMF (3 mL) by a procedure similar to that used to prepare compound 14. 1H NMR δ 7.85 (d, 2H, J = 8.8 Hz), 7.76 (s, 1H), 7.68 (s, 1H), 7.65 (d, 2H, J = 8.8 Hz), 7.31 (s, 1H), 7.06 (s, 1H), 6.97 (d, 1H, J = 4.4 Hz), 6.88 (d, 1H, J = 4.4 Hz), 6.13 (s, 1H), 5.36 (t, 1H, J = 6.0 Hz), 4.74-4.70 (m, 1H), 4.43 (dd, 1H, J = 13.6 Hz, 4.0 Hz), 4.12 (dd, 1H, J = 13.6 Hz, 9.6 Hz), 3.95-3.88 (m, 1H), 3.57-3.44 (m, 2H), 2.62 (s, 3H), 2.27 (s, 3H), 2.39-0.80 (m, 33H), 0.75 (s, 3H); 13C NMR δ 159.3, 153.3, 147.1, 142.7, 140.9, 136.2, 135.3, 135.1, 134.9, 131.0, 128.2, 127.9, 125.9, 122.0, 121.4, 120.3, 120.2, 119.2, 115.3, 97.0, 96.9, 75.9, 62.9, 56.4, 56.2, 53.7, 50.0, 42.7, 40.2, 39.5, 38.7, 38.0, 37.4, 36.75, 36.71, 31.8, 31.3, 29.68, 29.64, 28.2, 25.5, 24.4, 22.7, 21.0, 20.1, 20.0, 19.4, 17.1, 15.0, 14.1, 11.9, 11.4, 11.3; 19F NMR δ -143.1 (m). HRMS m/z: calcd for C48H61BF2N5O2 (MH+), 788.4881; found, 788.4888.
22-{4-[4-((E)-(4,4-Difluoro-1,3-dimethyl-4-bora-3a,4a-diaza-s-indacen-5-yl)ethenyl)-phenyl]-1,2,3-triazol-1-yl}-23,24-bisnorchol-5-en-3β-ol (2)
Compound 2 was synthesized in 97% yield by using the procedure to prepare 1. 1H NMR δ 7.85 (d, 2H, J = 8.0 Hz), 7.75 (s, 1H), 7.68 (s, 1H), 7.65 (d, 2H, J = 8.0 Hz), 7.31 (s, 1H), 7.06 (s, 1H), 6.96 (d, 1H, J = 4.0 Hz), 6.88 (d, 1H, J = 4.0 Hz), 6.13 (s, 1H), 5.38-5.33 (m, 1H), 4.43 (dd, 1H, J = 13.6 Hz, 3.6 Hz), 4.12 (dd, 1H, J = 13.6 Hz, 8.8 Hz), 3.58-3.48 (m, 1H), 2.62 (s, 3H), 2.27 (s, 3H), 2.37-0.81 (m, 27H), 0.76 (s, 3H); 13C NMR δ 159.3, 153.3, 147.1, 142.8, 140.7, 136.2, 135.3, 135.1, 134.9, 131.0, 128.3, 127.9, 125.9, 122.0, 121.5, 120.3, 120.2, 119.2, 115.3, 71.7, 56.4, 56.2, 53.7, 49.9, 42.7, 39.5, 38.0, 37.2, 36.4, 31.9, 31.6, 29.7, 24.4, 22.7, 21.0, 19.4, 17.1, 15.0, 14.1, 11.9, 11.3; 19F NMR δ -143.1 (m). HRMS m/z: calcd for C43H53BF2N5O (MH+), 704.4306; found, 704.4289.
5-tert-Butoxycarbonylamino-5-[(E)-(4-bromophenyl)ethenyl]-2,2-dimethyl-1,3-dioxane (17)
To a solution of aldehyde 16 (0.82 g, 3.14 mmol) and phosphonate 6 (1.45 g, 4.7 mmol) in dry THF (10 mL) was added potassium tert-butoxide (1.58 g, 14.1 mmol) slowly at 0 °C. After the mixture was stirred at 0 °C for 3 h and at room temperature overnight, ice-water (20 mL) was added and the suspension was extracted with CH2Cl2 (3 × 30 mL). The combined organic layer was washed with water (2 × 20 mL) and brine (2 × 20 mL), dried (Na2SO4), and concentrated under vacuum. The residue was purified by chromatography with gradient elution (hexanes/EtOAc 20:1 to 8:1) to give 17 (0.80 g, 62%). 1H NMR δ 7.37 (d, 2H, J = 8.4 Hz), 7.19 (d, 2H, J = 8.4 Hz), 6.46 (d, 1H, J = 16.4 Hz), 6.21 (d, 1H, J = 16.4 Hz), 5.42 (s, 1H), 3.97 (d, 2H, J = 11.2 Hz), 3.88 (d, 2H, J = 11.2 Hz), 1.50-1.39 (m, 15H); 13C NMR δ 154.5, 135.3, 131.2, 129.0, 128.7, 127.6, 121.1, 97.9, 79.1, 65.6, 52.7, 28.1, 27.0, 19.5. HRMS m/z: calcd for C19H27BrNO4 (MH+), 412.1118; found, 412.1120.
5-tert-Butoxycarbonylamino-5-{(E)-[4-(5-benzyloxy-pent-1-ynyl)phenyl]ethenyl}-2,2-dimethyl-1,3-dioxane (18)
To a solution of 17 (0.21 g, 0.51 mmol) and 5-benzyloxy-1-pentyne (133 mg, 0.77 mmol) in N,N-diisopropylethylamine (1 mL, 5.7 mmol) and dry THF (5 mL) were added CuI (9.7 mg, 51 μmol) and tetrakis(triphenylphosphine)palladium(0) (29.5 mg, 25.5 μmol) under nitrogen. The mixture was heated at reflux under nitrogen for one day, and then was cooled to room temperature and filtered through a short pad of silica gel to remove insoluble and very polar components in the mixture. The filtrate was concentrated and dissolved in CH2Cl2 (50 mL). The solution was washed with water (2 × 20 mL) and brine (2 × 20 mL), and dried over Na2SO4. The solvent was removed under vacuum and the residue was purified by column chromatography (hexanes/EtOAc 20:1 to 6:1) to give 18 (0.14 g, 53%). 1H NMR δ 7.43-7.19 (m, 4H), 6.49 (d, 1H, J = 16.4 Hz), 6.21 (d, 1H, J = 16.4 Hz), 5.43 (s, 1H), 4.50 (s, 2H), 3.98 (d, 2H, J = 11.2 Hz), 3.87 (d, 2H, J = 11.2 Hz), 3.59 (t, 2H, J = 6.0 Hz), 2.53 (t, 2H, J = 6.8 Hz), 1.89 (m, 2H), 1.45 (s, 15H); 13C NMR δ 154.4, 138.1, 135.5, 131.3, 129.3, 127.9, 127.2, 127.1, 125.9, 122.8, 97.8, 90.1, 80.6, 79.0, 72.5, 68.3, 65.6, 52.7, 28.5, 28.0, 27.1, 19.3, 15.9. HRMS m/z: calcd for C31H39NNaO5 (MNa+), 528.2720; found, 528.2727.
5-tert-Butoxycarbonylamino-5-{2-[4-(5-hydroxyl-1-pentyl)phenyl]ethyl}-2,2-dimethyl-1,3-dioxane (19)
Palladium hydroxide on carbon (20%) (36 mg, 0.05 mmol) was added to a solution of 18 (0.361 g, 0.71 mmol) in MeOH (8 mL). Hydrogen was bubbled through the suspension with stirring until 18 was completely consumed and converted to a polar substance (monitored by TLC: hexanes/EtOAc 2:1). The mixture was filtered through a short pad of silica gel and concentrated under vacuum. To a solution of the resulting yellow oil in dry DMF (5 mL) were added 2,2-dimethoxypropane (90.5 mg, 0.84 mmol) and camphorsulfonic acid (10 mg, 0.043 mmol). After the mixture was stirred at room temperature for 1 day, it was diluted with saturated aqueous NaHCO3 solution (15 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organic layer was washed sequentially with saturated aqueous NaHCO3 solution (2 × 20 mL), water (2 × 20 mL), and brine (20 mL), and was then dried (Na2SO4). The solvent was removed under vacuum, and the residue was purified by chromatography (hexanes/EtOAc 8:1 to 2:1) to give 19 (217 mg, 72%). 1H NMR δ 7.15-7.05 (m, 4H), 5.09 (s, 1H), 3.89 (d, 2H, J = 11.6 Hz), 3.67 (d, 2H, J = 11.6 Hz), 3.60 (t, 2H, J = 6.8 Hz), 2.60-2.50 (m, 4H), 2.40 (br s, 1H), 1.96 (t, 2H, J = 8.0 Hz), 1.65-1.36 (m, 21H); 13C NMR δ 154.7, 139.9, 139.0, 128.2, 128.0, 98.2, 79.1, 66.1, 62.4, 51.5, 35.5, 33.5, 32.4, 31.2, 28.4, 28.2, 27.2, 25.3, 19.7. HRMS m/z: calcd for C24H40NO5 (MH+), 422.2901; found, 422.2904.
5-tert-Butoxycarbonylamino-5-{2-[4-(5-azido-1-pentyl)phenyl]ethyl}-2,2-dimethyl-1,3-dioxane (20)
A solution of 19 (168 mg, 0.40 mmol) and N,N-diisopropylethylamine (155 mg, 1.2 mmol) in CH2Cl2 (10 mL) was cooled to 0 °C. Methanesulfonyl chloride (66 mg, 0.60 mmol) was added, and the mixture was stirred at 0 °C for 2 h and then at room temperature overnight. The mixture was washed with saturated aqueous NaHCO3 solution (2 × 10 mL), water (2 × 10 mL), and brine (2 × 10 mL), and then dried (Na2SO4). After the solvent was removed, the residue was dissolved in dry DMF (5 mL) and lithium iodide (7 mg, 48 μmol) and sodium azide (78 mg, 1.2 mmol) were added. The reaction mixture was stirred at 80 °C for 18 h and then was cooled to room temperature. Water (15 mL) was added, and the suspension was extracted with CH2Cl2 (3× 20 mL). The combined organic layer was washed with water (3× 30 mL) and brine (2× 20 mL), dried (Na2SO4), and concentrated under vacuum. The residue was purified by chromatography (hexanes/EtOAc 4:1) to give 20 (119 mg, 67%). 1H NMR δ 7.14-7.04 (m, 4H), 5.02 (s, 1H), 3.89 (d, 2H, J = 11.6 Hz), 3.67 (d, 2H, J = 11.6 Hz), 3.24 (t, 2H, J = 7.2 Hz), 2.60-2.50 (m, 4H), 1.97 (t, 2H, J = 8.0 Hz), 1.66-1.56 (m, 4H), 1.47 (s, 9H), 1.43 (s, 3H), 1.41 (s, 3H), 1.40-1.36 (m, 2H); 13C NMR δ 154.7, 139.7, 139.2, 128.3, 128.2, 98.2, 79.1, 66.2, 51.6, 51.2, 35.2, 33.5, 30.9, 28.6, 28.5, 28.3, 27.3, 26.2, 19.6. HRMS m/z: calcd for C24H38N4NaO4 (MNa+), 469.2785; found, 469.2786.
5-tert-Butoxycarbonylamino-5-{2-[4-(5-(4-(4-(4,4-difluoro-1,3,5,7-tetramethyl-3a,4a-diaza-s-indacen-8-yl)phenyl)-1,2,3-triazol-1-yl)pentyl)phenyl]ethyl}-2,2-dimethyl-1,3-dioxane (21)
To a solution of 20 (22.3 mg, 50 μmol) and 5 (17.4 mg, 50 μmol) in dry DMF (1 mL) was added CuI (1 mg, 5 μmol). The mixture was stirred at room temperature until the starting materials are completely consumed (monitored by TLC: hexanes/EtOAc 2:1). Water (5 mL) was added, and the mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic layer was washed with water (2 × 20 mL) and brine (2 × 20 mL), and dried over Na2SO4. After the solvent was removed under vacuum, the residue was purified by chromatography (hexanes/EtOAc 4:1 to 2:1) to afford 21 (29.8 mg, 75%). 1H NMR δ 7.99 (d, 2H, J = 8.0 Hz), 7.84 (s, 1H), 7.35 (d, 2H, J = 8.0 Hz), 7.13-7.04 (m, 4H), 5.99 (s, 2H), 5.00 (br s, 1H), 4.42 (t, 2H, J = 6.8 Hz), 3.90 (d, 2H, J = 11.6 Hz), 3.68 (d, 2H, J = 11.6 Hz), 2.56 (s, 6H), 2.63-2.49 (m, 4H), 2.03-1.93 (m, 4H), 1.71-1.63 (m, 2H), 1.47 (s, 9H), 1.44 (s, 6H), 1.43 (s, 3H), 1.42 (s, 3H); 13C NMR δ 155.5, 154.8, 146.9, 143.0, 141.2, 139.5, 139.4, 134.6, 131.4, 131.3, 128.5, 128.4, 128.3, 126.2, 121.2, 119.8, 98.3, 66.3, 51.6, 50.4, 35.1, 33.6, 30.8, 30.3, 28.6, 28.4, 27.4, 26.1, 19.6, 14.6; 19F NMR δ -146.1 (m). HRMS m/z: calcd for C45H57BF2N6NaO4 (MNa+), 817.4395; found, 817.4401.
2-Amino-2-{2-[4-(5-(4-(4-(4,4-difluoro-1,3,5,7-tetramethyl-3a,4a-diaza-s-indacen-8-yl)phenyl)-1,2,3-triazol-1-yl)pentyl)phenyl]ethyl}propane-1,3-diol (3)
To a cold solution (0 °C) of 21 (40 mg, 50.3 μmol) in dry CH2Cl2 (10 mL) were added 4 Å molecular sieves (0.40 g) and BF3·OEt2 (0.18 g, 1.27 mmol) with vigorous stirring. The mixture was stirred at 0 °C, and the reaction was monitored by TLC (hexanes/EtOAc 2:1). On the disappearance of 21 (~ 5 h), the reaction was quenched by adding saturated aqueous NaHCO3 solution (10 mL). After the mixture was stirred for 30 min, CH2Cl2 (30 mL) was added. The organic layer was separated and washed with saturated aqueous NaHCO3 solution (20 mL), water (2 × 20 mL), and brine (2 × 20 mL). The solution was dried (Na2SO4) and concentrated under vacuum. The residue was purified by chromatography (CH2Cl2/MeOH 9:1) to give 3 (20.1 mg, 61%). 1H NMR δ 7.96 (d, 2H, J = 8.4 Hz), 7.84 (s, 1H), 7.34 (d, 2H, J = 8.4 Hz), 7.11-7.01 (m, 4H), 5.98 (s, 2H), 4.39 (t, 2H, J = 7.2 Hz), 3.85-3.57 (m, 4H), 3.56-3.01 (br s, 4H), 2.61-2.51 (m, 4H), 2.55 (s, 6H), 2.00-1.92 (m, 2H), 1.70-1.56 (m, 2H), 1.43 (s, 6H), 1.46-1.05 (m, 4H); 13C NMR δ 155.6, 147.0, 143.0, 141.2, 139.8, 138.6, 134.7, 131.4, 128.6, 128.5, 128.3, 126.3, 122.5, 121.3, 119.9, 50.4, 35.1, 30.8, 30.3, 29.7, 29.4, 26.0, 14.6, 14.1; 19F NMR δ -146.1 (m). HRMS m/z: calcd for C37H46BF2N6O2 (MH+), 655.3737; found, 655.3746.
Supplementary Material
Copies of 1H, 13C, and 19F NMR spectra for all new compounds described herein. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by NIH Grant HL-083187.
References
- 1.Karolin J, Johansson LBA, Strandberg L, Ny T. J Am Chem Soc. 1994;116:7801. [Google Scholar]
- 2.(a) Golovkova TA, Kozlov DV, Neckers DC. J Org Chem. 2005;70:5545. doi: 10.1021/jo050540k. [DOI] [PubMed] [Google Scholar]; (b) Trieflinger C, Rurack K, Daub J. Angew Chem, Intl Ed Engl. 2005;44:2288. doi: 10.1002/anie.200462377. [DOI] [PubMed] [Google Scholar]
- 3.(a) Baki CN, Akkaya EU. J Org Chem. 2001;66:1512. doi: 10.1021/jo005706q. [DOI] [PubMed] [Google Scholar]; (b) Baruah M, Qin W, Basariæ N, De Borggraeve WM, Boens N. J Org Chem. 2005;70:4152. doi: 10.1021/jo0503714. [DOI] [PubMed] [Google Scholar]
- 4.(a) Baruah M, Qin W, Vallée RAL, Beljonne D, Rohand T, Dehaen W, Boens N. Org Lett. 2005;7:4377. doi: 10.1021/ol051603o. [DOI] [PubMed] [Google Scholar]; (b) Wu Y, Peng X, Guo B, Fan J, Zhang Z, Wang J, Cui A, Gao Y. Org Biomol Chem. 2005;3:1387. doi: 10.1039/b501795e. [DOI] [PubMed] [Google Scholar]; (c) Peng X, Du J, Fan J, Wang J, Wu Y, Zhao J, Sun S, Xu T. J Am Chem Soc. 2007;129:1500. doi: 10.1021/ja0643319. [DOI] [PubMed] [Google Scholar]; (d) Zeng L, Miller EW, Pralle A, Isacoff EY, Chang CJ. J Am Chem Soc. 2006;128:10. doi: 10.1021/ja055064u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Qi X, Jun EJ, Xu L, Kim SJ, Hong JSJ, Yoon YJ, Yoon J. J Org Chem. 2006;71:2881. doi: 10.1021/jo052542a. [DOI] [PubMed] [Google Scholar]; (f) Coskun A, Yilmaz MD, Akkaya EU. Org Lett. 2007;9:607. doi: 10.1021/ol062867t. [DOI] [PubMed] [Google Scholar]; (g) Yuan M, Li Y, Li J, Li C, Liu X, Lü J, Xu J, Liu H, Wang S, Zhu D. Org Lett. 2007;9:2313. doi: 10.1021/ol0706399. [DOI] [PubMed] [Google Scholar]; (h) Wang J, Qian X. Org Lett. 2006;8:3721. doi: 10.1021/ol061297u. [DOI] [PubMed] [Google Scholar]; (i) Coskun A, Akkaya EU. J Am Chem Soc. 2005;127:10464. doi: 10.1021/ja052574f. [DOI] [PubMed] [Google Scholar]
- 5.Gabe Y, Urano Y, Kikuchi K, Kojima H, Nagano T. J Am Chem Soc. 2004;126:3357. doi: 10.1021/ja037944j. [DOI] [PubMed] [Google Scholar]
- 6.Gawley RE, Mao H, Haque MM, Thorne JB, Pharr JS. J Org Chem. 2007;72:2187. doi: 10.1021/jo062506r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oleynik P, Ishihara Y, Cosa G. J Am Chem Soc. 2007;129:1842. doi: 10.1021/ja066789g. [DOI] [PubMed] [Google Scholar]
- 8.Johnson ID, Kang HC, Haugland RP. Anal Biochem. 1991;198:228. doi: 10.1016/0003-2697(91)90418-s. [DOI] [PubMed] [Google Scholar]
- 9.Kurata S, Kanagawa T, Yamada K, Torimura M, Yokomaku T, Kamagata Y, Kurane R. Nucleic Acids Res. 2001;29:e34. doi: 10.1093/nar/29.6.e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y, Prestwich GD. Bioconj Chem. 1999;10:755. doi: 10.1021/bc9900338. [DOI] [PubMed] [Google Scholar]
- 11.Kaiser RD, London E. Biochim Biophys Acta. 1998;1375:13. doi: 10.1016/s0005-2736(98)00127-8. [DOI] [PubMed] [Google Scholar]
- 12.Kasurinen J. Biochem Biophys Res Commun. 1992;187:1594. doi: 10.1016/0006-291x(92)90485-4. [DOI] [PubMed] [Google Scholar]
- 13.Ellena JF, Le M, Cafiso DS, Solis RM, Langston M, Sankaram MB. Drug Delivery. 1999;6:97. [Google Scholar]
- 14.Peters C, Billich A, Ghobrial M, Högenauer K, Ullrich T, Nussbaumer P. J Org Chem. 2007;72:1842. doi: 10.1021/jo062347b. [DOI] [PubMed] [Google Scholar]
- 15.Hendrickson HS, Hendrickson EK, Johnson ID, Farber SA. Anal Biochem. 1999;276:27. doi: 10.1006/abio.1999.4280. [DOI] [PubMed] [Google Scholar]
- 16.(a) Dahim M, Mizuno NK, Li XM, Momsen WE, Momsen MM, Brockman HL. Biophys J. 2002;83:1511. doi: 10.1016/S0006-3495(02)73921-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu Y, Bittman R. Chem Phys Lipids. 2006;142:58–69. doi: 10.1016/j.chemphyslip.2006.03.001. [DOI] [PubMed] [Google Scholar]; (c) Singh RD, Liu Y, Wheatley CL, Holicky EL, Makino A, Marks DL, Kobayashi T, Subramaniam G, Bittman R, Pagano RE. J Biol Chem. 2006;281:30660. doi: 10.1074/jbc.M606194200. [DOI] [PubMed] [Google Scholar]; (d) Coban O, Burger M, Laliberte M, Ianoul A, Johnston LJ. Langmuir. 2007;23:6704. doi: 10.1021/la0635348. [DOI] [PubMed] [Google Scholar]
- 17.Li Z, Mintzer E, Bittman R. J Org Chem. 2006;71:1718. doi: 10.1021/jo052029x. [DOI] [PubMed] [Google Scholar]
- 18.Shaw JE, Epand RF, Epand RM, Li Z, Bittman R, Yip CM. Biophys J. 2006;90:2170. doi: 10.1529/biophysj.105.073510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wüstner D. Chem Phys Lipids. 2007;146:1. doi: 10.1016/j.chemphyslip.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Adachi K, Kohara T, Nakao N, Arita M, Chiba K, Mishina T, Sasaki S, Fujita T. Bioorg Med Chem Lett. 1995;5:853. [Google Scholar]
- 21.(a) Billich A, Bornancin F, Dévay F, Mechtcheriakova D, Urtz N, Baumruker T. J Biol Chem. 2003;278:47408. doi: 10.1074/jbc.M307687200. [DOI] [PubMed] [Google Scholar]; (b) Paugh SW, Payne SG, Barbour SE, Milstien S, Spiegel S. FEBS Lett. 2003;554:189. doi: 10.1016/s0014-5793(03)01168-2. [DOI] [PubMed] [Google Scholar]
- 22.Chiba K. Pharmacol Ther. 2005;108:308. doi: 10.1016/j.pharmthera.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Brinkmann V. Pharmacol Ther. 2007;115:84. doi: 10.1016/j.pharmthera.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 24.Ettmayer P, Baumruker T, Guerini D, Mechtcheriakova D, Nussbaumer P, Streiff MB, Billich A. Bioorg Med Chem Lett. 2006;16:84. doi: 10.1016/j.bmcl.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 25.(a) Scheidt HA, Müller P, Herrmann A, Huster D. J Biol Chem. 2003;278:45563. doi: 10.1074/jbc.M303567200. [DOI] [PubMed] [Google Scholar]; (b) Loura LMS, Fedorov A, Prieto M. Biochim Biophys Acta. 2001;1511:236. doi: 10.1016/s0005-2736(01)00269-3. [DOI] [PubMed] [Google Scholar]
- 26.Huisgen R, Szeimies G, Moebius L. Chem Ber. 1967;100:2494. [Google Scholar]
- 27.(a) Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]; (b) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem, Int Ed Engl. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 28.For a few excellent reviews, see: Lutz J-F. Angew Chem, Int Ed Engl. 2007;46:1018. doi: 10.1002/anie.200604050.Binder WH, Sachsenhofer R. Macromol Rapid Commun. 2007;28:15.Wang Q, Chittaboina S, Barnhill HN. Lett Org Chem. 2005;2:293.Kolb HC, Sharpless KB. Drug Disc Today. 2003;8:1128. doi: 10.1016/s1359-6446(03)02933-7.
- 29.Kolb HC, Finn MG, Sharpless KB. Angew Chem, Int Ed Engl. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 30.(a) Rurack K, Kollmannsberger M, Daub J. New J Chem. 2001;25:289. [Google Scholar]; (b) Chen J, Burghart A, Derecskei-Kovacs A, Burgess K. J Org Chem. 2000;65:2900. doi: 10.1021/jo991927o. [DOI] [PubMed] [Google Scholar]; (c) Burghart A, Kim H, Welch MB, Thoresen LH, Reibenspies J, Burgess K, Bergstroem F, Johansson LBA. J Org Chem. 1999;46:7813. [Google Scholar]
- 31.(a) Rurack K, Kollmannsberger M, Daub J. Angew Chem, Int Ed Engl. 2001;40:385. [PubMed] [Google Scholar]; (b) Coskun A, Akkaya EU. J Am Chem Soc. 2005;127:10464. doi: 10.1021/ja052574f. [DOI] [PubMed] [Google Scholar]; (c) Saki N, Dinc T, Akkaya EU. Tetrahedron. 2006;62:2721. [Google Scholar]; (d) Dost Z, Atilgan S, Akkaya EU. Tetrahedron. 2006;62:8484. [Google Scholar]
- 32.Rohand T, Qin W, Boens N, Dehaen W. Eur J Org Chem. 2006:4658. [Google Scholar]
- 33.Thivierge C, Bandichhor R, Burgess K. Org Lett. 2007;9:2135. doi: 10.1021/ol0706197. [DOI] [PubMed] [Google Scholar]
- 34.Boyer JH, Haag AM, Sathyamoorthi G, Soong ML, Thangaraj K. Heteroatom Chem. 1993;4:39. [Google Scholar]
- 35.(a) Lei Y, Wrobleski AD, Golden JE, Powell DR, Aubé JJ. Am Chem Soc. 2005;127:4552. doi: 10.1021/ja050214m. [DOI] [PubMed] [Google Scholar]; (b) Viau L, Maury O, Bozec HL. Tetrahedron Lett. 2004;45:125. [Google Scholar]
- 36.Jones RA, Pojarlieva T, Head RJ. Tetrahedron. 1968;24:2013. [Google Scholar]
- 37.Malan SF, Van Marle A, Menge WM, Zuliana V, Hoffman M, Timmerman H, Leurs R. Bioorg Med Chem. 2004;12:6495. doi: 10.1016/j.bmc.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 38.(a) Lane JW, Halcomb RL. Tetrahedron. 2001;57:6531. [Google Scholar]; (b) Kang JH, Chung HE, Kim SY, Kim Y, Lee J, Lewin NE, Pearce LV, Blumberg PM, Marquez VE. Bioorg Med Chem. 2003;11:2529. doi: 10.1016/s0968-0896(03)00156-1. [DOI] [PubMed] [Google Scholar]
- 39.Marshall JA, Chobanian HR, Yanik MM. Org Lett. 2001;3:4107. doi: 10.1021/ol016899m. [DOI] [PubMed] [Google Scholar]
- 40.(a) Evans EF, Lewis NJ, Kapfer I, Macdonald G, Taylor RJK. Synth Commun. 1997;27:1819. [Google Scholar]; (b) Looper RE, Williams RM. Tetrahedron Lett. 2001;42:769. [Google Scholar]
- 41.Ziessel R, Bonardi L, Retailleau P, Ulrich G. J Org Chem. 2006;71:3093. doi: 10.1021/jo0600151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Copies of 1H, 13C, and 19F NMR spectra for all new compounds described herein. This material is available free of charge via the Internet at http://pubs.acs.org.