

Abstract
Formation of a noncanonical base pair between dFTP, a dTTP analogue that cannot form H bonds, and the fluorescent base analogue 2-aminopurine (2AP) was studied in order to discover how the bacteriophage T4 DNA polymerase selects nucleotides with high accuracy. Changes in 2AP fluorescence intensity provided a spectroscopic reporter of the nucleotide binding reactions, which were combined with rapid-quench, pre-steady state reactions to measure product formation. These studies supported and extended previous findings that the T4 DNA polymerase binds nucleotides in multiple steps with increasing selectivity. With 2AP in the template position, initial dTTP binding was rapid, but selective: Kd(dTTP) 1st step = 31 μM, Kd(dCTP) 1st step ≈ 3 mM . In studies with dFTP, this step was revealed to have two components: formation of an initial pre-insertion complex in which H bonds between bases in the newly forming base pair were not essential, which was followed by formation of a final pre-insertion complex in which H bonds assisted. The second-nucleotide binding step was characterized by increased discrimination against dTTP binding opposite template 2AP, Kd 2nd step = 367 μM, and additional conformational changes were detected in ternary enzyme-DNA-dTTP complexes, as expected for forming closed complexes. We demonstrate here that the second binding step occurs before formation of the phosphodiester bond. Thus, the high fidelity of nucleotide insertion by T4 DNA polymerase is accomplished by the sequential application of selectivity in first forming accurate pre-insertion complexes and then additional conformational changes are applied that further increase discrimination against incorrect nucleotides.
Many DNA polymerases, including the bacteriophage T4 and T7 DNA polymerases, Escherichia coli DNA polymerases I and III, and eukaryotic DNA polymerases δ and ε rarely incorporate “wrong” nucleotides, the error frequency is only about 10−5 (reviewed in 1). Further accuracy of 100-fold or more is achieved by exonucleolytic proofreading, which is the ability of DNA polymerases to detect and remove misincorporated incorrect nucleotides at the primer terminus before further primer elongation (2, 3). Several questions remain, however, about how DNA polymerases avoid incorporating incorrect nucleotides in the first place. The ability of DNA polymerases to incorporate correct nucleotides is a fundamental aspect of molecular recognition that is shared by all enzymes. Linus Pauling pointed out the most critical feature of ligand recognition, which was acknowledged by A. Fersht (4): “a protein must keep ligands that are smaller than the correct one out of its binding site.” Molecules that are too large are excluded, but molecules with a similar shape or are smaller than the correct ligand may be bound mistakenly. While this deficiency in discrimination may not be a problem for many enzymes, especially if there are few competing substrate-like molecules present, DNA polymerases must achieve exceptionally high replication fidelity in order to maintain the genetic information. Furthermore, the polymerase active center must accommodate pairings with template C and incoming dGTP (and the reverse) as well as template A and incoming dTTP (and the reverse), while discriminating against C-A, G-T and other mispairs.
H bonds between Watson-Crick A-T and G-C base pairs were once thought to be the critical determinants for nucleotide selection by all DNA polymerases, but H bonds may serve this purpose for only certain polymerases such as the human DNA primase (5) and the translesion DNA polymerase, DNA pol η (6). For several other polymerases, including DNA polymerase α and the exonuclease-deficient forms of the bacteriophage T7 DNA polymerase and the Klenow fragment, the geometry of the newly forming base pair appears to be the principal determinant because these DNA polymerases can incorporate shape-correct nucleotide analogues that cannot form H bonds (7, 8). For example, dFTP, which resembles dTTP but 2,4-difluoro-5-toluene (F) replaces the thymine (T) base (Figure 1A), is incorporated opposite template A, but not G, C or T by these DNA polymerases (7, 8). Additional factors in nucleotide selection have also been proposed, which include hydrophobic interactions (9) and the ability of DNA polymerases to discriminate against “wrong” nucleotides, as opposed to selecting for the “correct” nucleotide (5, 10).
Figure 1.
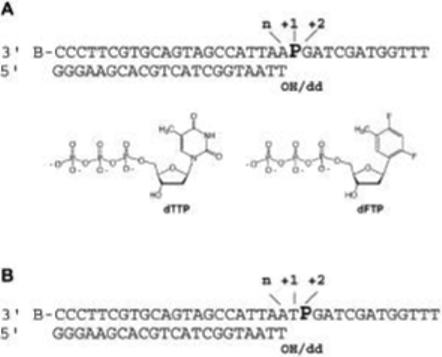
DNA substrates. 2-Aminopurine was placed in the templating, +1 position (A) or in the +2 position (B). The primers were either extendable and have a 3'-OH group or are terminated by a 2', 3'-dideoxynucleotide (dd). The structures of dTTP and dFTP are illustrated (A).
Increased fidelity can also be obtained if DNA polymerases check the accuracy of the bound nucleotide in more than one step (11, 12). Selection may be exerted in an initial binding step to form pre-insertion complexes, in a post-binding step that evaluates the accuracy of the newly forming base pair by an induced-fit mechanism (reviewed by Johnson, 13), and in the chemical step of phosphodiester bond formation (14). Evidence for the sequential application of at least two steps to achieve selectivity in nucleotide incorporation was obtained from kinetic studies of the bacteriophage T4 DNA polymerase in which the fluorescent base analogue 2-aminopurine (2AP) was the templating base (15).
Changes in 2AP fluorescence were used to report on both nucleotide binding and incorporation. A 20-fold increase in fluorescence intensity is detected when binary complexes are formed with T4 DNA polymerase and DNA substrates labeled with 2AP in the templating (+1) position (Figure 1A) (16). A large decrease in fluorescence intensity is detected when dTMP is incorporated opposite template 2AP; the rate of decrease corresponds to the reported nucleotide incorporation rate (17). 2AP fluorescence is also quenched when dTTP is bound opposite template 2AP in a Mg2+-dependent reaction (16). The DNA substrate is chain-terminated for the nucleotide-binding experiments to prevent incorporation (Figure 1A). The decrease in 2AP fluorescence with increasing dTTP concentration provides a direct assay to determine the Kd for nucleotide binding before incorporation. These studies revealed that the Kd for dTMP incorporation opposite template 2AP, 367 μM, is substantially greater than the Kd for dTTP binding, which is about 34 μM (15). Thus, not all of the DNA pol-DNA-dTTP ternary complexes formed initially go on to form product; additional discrimination is exerted in a subsequent step(s), either the post-binding step in which the geometric fit of the newly forming base pair is examined, the chemistry step or both steps.
We report here studies to identify which step or steps that follow formation of the initial DNA pol-DNA-dTTP complex increase discrimination against incorporation of dTMP opposite template 2AP. We also probed the role of H bonds in the nucleotide incorporation reaction catalyzed by the T4 DNA polymerase by using dFTP, which resembles dTTP but cannot form H bonds (7). Studies of binding and incorporation of dTTP opposite template 2AP have an important advantage compared to reactions in which only non-complimentary nucleotides are provided, such as for the incorporation of dTMP opposite template G. Although the T4 DNA polymerase discriminates in the incorporation of dTMP opposite template 2AP compared to template A (15, 18), binding and incorporation reactions can be done under pre-steady-state, single-turnover conditions (15-17). In contrast, multiple turnover conditions with high concentrations of DNA polymerase and the incorrect nucleotide are required for misincorporation experiments since polymerizing reactions with nucleotides in non-standard base pairing situations are not efficient. In these “forcing” conditions, DNA polymerases likely form numerous DNA pol-DNA-dNTP ternary complexes before an incorporation event occurs, which raises the possibility that only rare and perhaps aberrant complexes are capable of incorporating incorrect nucleotides. 2AP, as a pseudo natural template base, provides a way to challenge the nucleotide discrimination mechanisms used by the T4 DNA polymerase without overwhelming the process and, thus, provides an opportunity to determine how DNA polymerases normally select correct nucleotides for incorporation. Once the rules for the selection of correct nucleotides are learned, this information can be applied to determining how mismatches are formed.
The new findings reported here provide additional evidence that the T4 DNA polymerase achieves high fidelity in nucleotide selection by using multiple nucleotide binding steps, each with increased selectivity for the correct base pair. We reported previously that the initial binding step is rapid, but selective for the correct base and sugar (15), but studies with dFTP revealed that the initial binding step has at least two components. H bonds between bases in the newly forming base pair are not required in forming the initial pre-insertion complex, but assist in formation of the final pre-insertion complex. After formation of pre-insertion complexes, a conformational change in the template strand is detected by 2AP fluorescence. We report here that this conformational change occurs before the chemistry step of phosphodiester bond formation and is most likely connected with formation of closed complexes in which several residues in the polymerase active center make tight contacts with the newly forming base pair and a sharp bend is produced in the template strand (19, 20). The conformational change was linked to amino acids that surround the newly forming base pair in closed complexes, because the rate of conformational change was slowed for the mutant L412M-DNA polymerase, which has an amino acid substitution for one of the conserved residues that forms the base pair binding pocket (20).
These findings for the T4 DNA polymerase are discussed with respect to recent structural studies of the T7 RNA polymerase in which the incoming rNTP is observed to interact first with the template base in a pre-insertion site, which allows the “fit” of the newly forming base pair to be sampled prior to formation of the catalytically active closed conformation (21). We also discuss how mismatches may be formed when nucleotide binding accuracy is evaluated in multiple successive steps before phosphodiester bond formation.
Materials and Methods
DNA Polymerases
Expression and purification of the mutant T4-exo− (D112A/E114A) and L412M-exo− (D112A/E114A/L412M) T4 DNA polymerases were described previously (22-24).
DNA Substrates and Nucleotides
The 2AP DNA substrates have been described (15, 16) and are illustrated in Figure 1. The 2AP phosphoramidite was purchased from Glen Research. The 3'terminus of the template strand was protected from DNA polymerase binding (to direct enzyme binding to the primer terminus) by a biotin attachment (BiotinTEG-CPG, Glen Research). The primer and template DNAs were annealed in buffer containing HEPES (pH 7.6) and 50 mM NaCl with a 20% excess of the primer strand to ensure complete hybridization of the template strand. dFTP was synthesized as described (7).
Fluorescence Intensity Experiments
Emission data for 2AP-labeled DNAs and DNA polymerase complexes were obtained with a Photon Technology International scanning spectrofluorometer. Samples were excited at 315 nm and fluorescence emission was monitored at 368 nm. A 2 nm band-pass was used for both the excitation and emission monochromators. Solutions contained 200 nM 2AP-labeled DNA, 500 nM DNA polymerase, 25 mM HEPES (pH 7.6), 50 mM NaCl, 1 mM DTT, and 0.5 mM EDTA. Intrinsic protein fluorescence was subtracted. All experiments were preformed at 20 °C.
Equilibrium dissociation constants (Kds) for nucleotide binding opposite template 2AP were determined in reactions with the above buffer with the addition of 10 mM MgCl2, which is essential for nucleotide binding (16). The DNA substrates were chain-terminated by incorporation of a terminal dideoxynucleotide, which allowed nucleotide binding but prevented incorporation. Exonuclease-deficient DNA polymerases were used to prevent nuclease degradation of the DNA substrate. The decrease in 2AP fluorescence intensity produced as a function of dTTP concentration was plotted as an increase in quench. The data were fit to the hyperbolic equation, q = Q[dTTP]/(Kd + [dTTP]), where q is the observed quench and Q is the maximum quench observed at saturating dTTP concentration.
Ki values were determined in dTTP binding reactions as described above except that dFTP was added at the indicated concentrations at the beginning of the titration experiments. Note that dFTP at the highest concentration tested, 150 μM, did not quench the fluorescence of the binary complexes, but inhibited the ability of dTTP to form quenched, ternary complexes. Kis were determined from the equation Ki = [I] / [(Kdobs / Kd) – 1] where Kdobs is the Kd for dTTP binding detected in the presence of dFTP.
Determining Rates of Product Formation
dAMP incorporation opposite template template T with 2AP in the +2 position (Figure 1B) was determined by a rapid quench method with the same experimental conditions used to determine the rate of increase in 2AP fluorescence (15). A solution of binary complexes formed with 400 nM DNA (primer labeled at the 5'end with 32P), 1 μM T4-exo− or L412M-exo−, and buffer (0.5 mM DTT, 25 mM HEPES (pH 7.6), 50 mM NaCl, and 0. 5 mM EDTA) was mixed in the Kintek rapid quench instrument with a second solution containing 16 mM MgCl2, variable concentrations of dATP from 2 to 400 μM, and buffer. Reactions were quenched with 0.5 M EDTA. All experiments were performed at 20 °C. Reaction products were separated by PAGE and the bands were visualized using the Molecular Dynamics STORM PhosphorImager and quantitated using ImageQuant software. The nucleotide incorporation rate was determined by fitting the data to the hyperbolic equation, kobs = kproduct[dATP] / Kd + [dATP] where Kd is the equilibrium dissociation constant for dATP and kproduct is the maximum rate of primer extension. Note that the initial nucleotide binding is very rapid (15) and, thus, this step is not included in calculating the overall rate.
Results
T4 DNA Polymerase Incorporates dFMP Opposite Template 2AP
Incorporation of dTMP opposite template 2AP by the T4 DNA polymerase is detected by a decrease in 2AP fluorescence intensity; the decrease in fluorescence intensity corresponds to the rate of nucleotide incorporation (17). Thus, incorporation of dFMP opposite template 2AP is also expected to decrease 2AP fluorescence intensity and the rate of decrease will correspond to the incorporation rate. Binary complexes were formed with the exonuclease-deficient D112A/E114A-DNA polymerase (T4-exo−) and the DNA substrate illustrated in Figure 1A. When fluorescent, binary complexes (100 nM) were mixed with 250 nM dTTP and 10 mM Mg2+, 2AP fluorescence intensity decreased at the rate of about 0.09 s−1 (Figure 2). Under the same experimental conditions, except that dFTP replaced dTTP, fluorescence intensity decreased at a 30-fold slower rate (0.003 s−1) (Figure 2). The slower rate of dFMP incorporation parallels previous studies in which several DNA polymerases were observed to incorporate dFMP with accuracy opposite template A, but with reduced efficiency compared to dTMP (7). Thus, we have extended these studies to the incorporation of dFMP opposite template 2AP.
Figure 2.
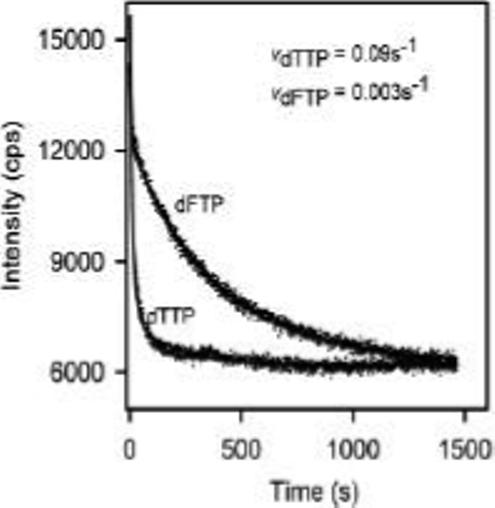
Incorporation of dTMP or dFMP opposite template 2AP by T4-exo−. Preformed enzyme-DNA complexes were made by combining 100 nM of the DNA substrate (2AP at the fluorescence reporting +1 position) with 250 nM DNA polymerase in buffer containing 25 mM HEPES (pH 7.6), 50 mM NaCl, 1 mM DTT, and 0.5 mM EDTA. The addition of 250 nM dTTP (or 250 nM dFTP) and 10 mM Mg2+ resulted in nucleotide incorporation, which quenched 2AP fluorescence. The experiments were performed at 20 °C. The data were fit to a single-exponential equation (solid line) to give the incorporation rate.
Formation of DNA pol-DNA-dNTP Ternary Complexes with dTTP and dFTP
One possible explanation for the slower incorporation rate for dFMP compared to dTMP is that T4 DNA polymerase discriminates against dFTP during one or more nucleotide binding steps. To test this proposal, nucleotide binding assays were performed as for incorporation, but the primer end was chain-terminated to prevent extension (Figure 1A). Since dTTP binding in the presence of Mg2+ quenches 2AP fluorescence as observed for incorporation (15, 16), the Kd for dTTP binding opposite template 2AP by T4-exo− can be determined from the quench curve (Figure 3A) and is 31 ± 4 μM. A similar Kd for dTTP binding, 34 ± 3 μM, was obtained with a different DNA substrate that was terminated by a ddA (15). We have also observed similar values for a variety of A+T- and G+C-rich DNA substrates (unpublished data). Thus, these experiments further support the conclusion that the Kd for dTTP binding opposite template 2AP is an order of magnitude less than the Kd for incorporation of dTMP (15).
Figure 3.
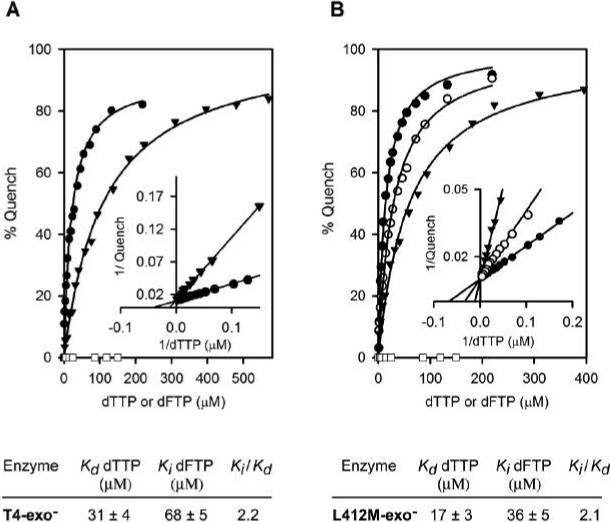
Nucleotide binding assays. Formation of ternary Enzyme-DNA-dNTP complexes with T4-exo− (A) and L412M-exo− (B) were measured as a function of dTTP concentration using the chain-terminated DNA substrate illustrated in Figure 1A. 10 mM MgCl2 was present. dTTP binding quenches 2AP fluorescence in a concentration-dependent manner (•), but dFTP does not at the concentrations tested (□); however, dFTP at 50 (○) and 150 μM (▼) reduced the ability of dTTP to quench 2AP fluorescence. Equilibrium dissociation constants (Kds) for dTTP binding were determined by fitting the data (solid line) to the hyperbolic equation, q = [Q] [dTTP]/Kd + [dTTP], where q is the observed fluorescence quench at various dTTP concentrations and Q is the maximum fluorescence quench observed at saturating dTTP. Kis for dFTP were determined in dTTP binding reactions with either 50 or 150 μM dFTP; dFTP was a competitive inhibitor of dTTP binding as demonstrated by the Lineweaver-Burk plots (insets). Kd values for dTTP binding and Ki values for dFTP for T4-exo-− and L412M-exo− are given below the quench curves.
In experiments similar to the dTTP binding experiments except with dFTP, no decrease in fluorescence intensity was detected with dFTP at concentrations up to 150 μM, a concentration that produced > 80% quench with dTTP (Figure 3A). The inability of dFTP to drive formation of quenched ternary complexes does not mean that dFTP is not bound, because dFMP is incorporated and, thus, must be bound at some point in the nucleotide incorporation pathway (Figure 2). We tested the possibility that dFTP is bound but that this binding does not produce quenched complexes, by assaying dFTP as an inhibitor of dTTP binding. The addition of 150 μM dFTP to dTTP titration experiments significantly decreased the ability of dTTP to quench 2AP fluorescence (Figure 3A) by a competitive mechanism, Ki = 68 ± 5 μM (Figure 3A, inset). The ability of dFTP to strongly inhibit dTTP binding is noteworthy because dCTP and rUTP are not strong competitive inhibitors of dTTP binding (data not shown) or for dTMP incorporation (15).
Nucleotide binding experiments were repeated with the exonuclease-deficient D112A/E114A/L412M-DNA polymerase (L412M-exo−), which has a Leu to Met substitution in the Motif A sequence in the polymerase active center. The L412M-DNA polymerase was identified by genetic selection as a mutant DNA polymerase with reduced ability to initiate the proofreading pathway (25). As observed for T4-exo−, the addition of dFTP did not decrease the fluorescence intensity of L412M-exo− binary complexes (Figure 3B), but dFTP was a strong competitive inhibitor of dTTP binding, Ki = 36 ± 5 μM (Figure 3B). Although the Kd for dTTP and the Ki for dFTP were lower for L412M-exo− than for T4-exo−, the Ki to Kd ratios for both DNA polymerases were nearly the same, 2.1 − 2.2 (Figure 3), which indicates similar dTTP/dFTP discrimination by both DNA polymerases.
Conformational Changes in the Template Strand During the Nucleotide Incorporation Reaction
We have detected conformational changes in the template strand during the nucleotide incorporation reaction catalyzed by the T4 DNA polymerase that are reported by changes in 2AP fluorescence (16). In reactions in which 2AP is placed initially in the +2 position in the template strand (Figure 1B), an increase in fluorescence intensity is detected when the primer is extended to place 2AP in the +1 (fluorescence reporting) position. In reactions with T4-exo−, fluorescence intensity increases with rates of about 314 s−1 for incorporation of dAMP opposite template T (16) and about 318 s−1 for incorporation of dTMP opposite template A (15). There are at least three possible steps in the nucleotide incorporation pathway in which this conformational change reported by 2AP fluorescence could occur: (1) nucleotide binding to form pre-insertion complexes, (2) a conformational change to form closed complexes that occurs after initial nucleotide binding but before formation of the phosphodiester bond, or (3) a conformational change that occurs after the chemistry step.
The first possibility – formation of pre-insertion complexes is ruled out by previous experiments (16). When T4 DNA polymerase forms complexes with DNA labeled at the +2 position with 2AP (Figure 1B), only a small increase in fluorescence intensity is detected in contrast to the large increase observed for complexes formed with DNA labeled at the +1 position with 2AP (Figure 1A). A similar small increase is detected for complexes formed with chain-terminated DNA substrates with 2AP in the +2 position and no further increase is observed when the correct nucleotide is bound (16). Thus, the conformational change that increases fluorescence intensity for 2AP in the +2 position must occur after the initial nucleotide-binding step.
In order to determine if the conformational change happens before or after formation of the phosphodiester bond, product formation was measured in rapid-quench, stopped-flow experiments with the same DNA substrate that was used in the fluorescence stopped-flow studies (Figure 1B) except that the 5'end of the primer was labeled with 32P so that primer elongation could be detected by gel electrophoresis as described in Materials and Methods. For T4-exo−, product was formed at the rate of about 164 s−1 (Figure 4A), which is clearly slower than the 314 s−1 rate measured for the increase in 2AP fluorescence. Thus, the conformational change that produces 2AP fluorescence occurs before formation of the phosphodiester bond.
Figure 4.
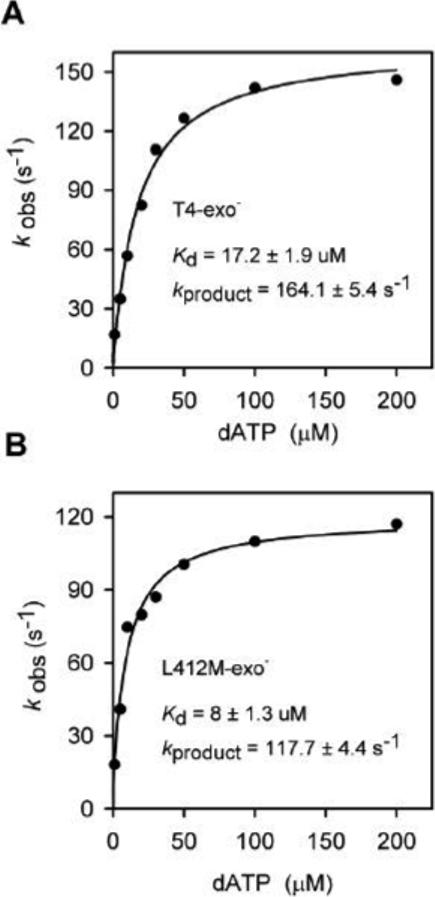
Determination of the rate of product formation by T4-exo− (A) and L412M-exo− (B). Rates for product formation for incorporation of dAMP opposite template T were determined in rapid-quench, stopped flow studies as described in Experimental Procedures.
The pre-steady state rate for the conformational change detected by the increase in 2AP fluorescence is called kunstack, to indicate that the increase in fluorescence intensity is due to a conformational change that reduces 2AP base-base interactions in the template strand (Table 1). The product formation rate is a composite of all of the microscopic rate constants (forward and reverse) for each step in the nucleotide incorporation pathway beginning with the binary complex (Scheme 1). Since binding the correct nucleotide to form the quenched ternary complex is rapid (15), the chemistry rate can be calculated from the equation, 1/kproduct = 1/kunstack + 1/kchem, which yields a kchem rate of about 345 s−1 (Table 1).
Table 1.
Kinetic Parameters for Nucleotide Incorporation Catalyzed by T4-exo− and L412M-exo− DNA polymerases
| Incorporation of dAMP opposite template T |
dTTP binding and incorporation of dTMP opposite template 2AP |
|||
|---|---|---|---|---|
| DNA polymerase | 2AP Fluorescence assaya | Rapid quench assayb | dTTP bindingc | dTMP incorporationd |
| T4-exo− | kunstack = 314 ± 18.5 s−1 | kproduct = 164.1 ± 5.4 s−1 | kbind = very rapid | kproduct = 165 ± 5 s−1 |
| Kd = 16 ± 3 μM | Kd = 17.2 ± 1.9 μM | Kd = 31 ± 4 μM | Kd = 367 ± 36 μM | |
| kchem = 345 s−1 | ||||
| L412M-exo− | kunstack = 177 ± 5 s−1 | kproduct = 117 ± 4.4 s−1 | Kd = 17 ± 3 μM | Kd = 252 ± 15 μM |
| Kd = 11 ± 1 μM | Kd = 8 ± 1.3 μM | |||
| kchem = 345 s−1 | ||||
The rates for base unstacking in the template strand and Kds, determined by the 2AP fluorescence assay, are from previous studies (15, 16).
The rates for product formation were determined under the same conditions as the 2AP fluorescence assays and are derived from the data presented in Figure 4.
The data for binding reactions with template 2AP and dTTP are from Figure 3. The rate of dTTP binding was determined in stopped-flow studies (15).
The kinetic parameters for dTMP incorporation opposite template 2AP are from 15.
Scheme 1.
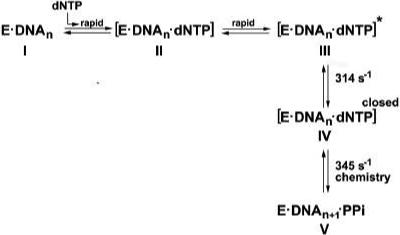
The nucleotide incorporation pathway catalyzed by the T4 DNA polymerase. Complexes II and III are pre-insertion complexes that are formed very rapidly in the presence of the correct nucleotide. If 2AP is the templating base, complex II is fluorescent, but 2AP fluorescence is quenched in complex III, which indicates conformational changes between complexes II and III. The pre-insertion complexes are likely open complexes that facilitate rapid nucleotide binding. Further conformational changes are required to produce complex IV, a closed complex, which is formed before the chemistry step of phosphodiester bond formation.
Additional evidence in support of this pathway is provided by primer-extension reactions with L412M-exo−. Product formation is slower for this mutant enzyme than detected for T4-exo−, about 117−118 s−1 (Figure 4B), but the conformational change that reduces 2AP base-base interactions is also slower, about 177 s−1 (15). The calculated kchem rate, however, is about 345 s−1 as observed for T4-exo− (Table 1). Thus, the conformational change detected by the increase in 2AP fluorescence is compromised by the L412M substitution in the polymerase active center, but the chemistry rate is apparently not affected.
Discussion
Three steps in the nucleotide incorporation pathway have been proposed to be important for ensuring high fidelity nucleotide incorporation (11, 12): (1) formation of a pre-insertion complex (for example formation of complexes II and III, Scheme 1), (2) a post-binding conformational change that evaluates the accuracy of the base pair (formation of closed complexes, complex IV Scheme 1), and (3) the chemical step of phosphodiester bond formation (chemistry step, Scheme 1). DNA polymerases may also apply two or more steps sequentially to filter out incorrect nucleotides and in this case each of the three steps may be considered a checkpoint (12). Most of the experimental evidence to date, however, suggests that accuracy in nucleotide binding is achieved primarily by evaluation of the “geometric fit” of newly forming base pair in closed complexes. This view is exemplified by structural studies of ternary complexes of the T7 DNA polymerase in which several protein contacts are made to the newly forming base pair that seem to allow formation only of correct base pairs (19). There is also evidence that the chemistry step can exert fidelity (14, and note review by Joyce and Benkovic, 26). In contrast, there are only a few experiments that demonstrate that DNA polymerases bind nucleotides in apparent open complexes with selectivity (15, 27) and that DNA polymerases can achieve increased discrimination by employing multiple steps (15). Studies reported here for the T4 DNA polymerase and provide additional evidence for multiple checkpoints in nucleotide selection.
Formation of Nucleotide Pre-insertion Complexes; Selective Nucleotide Binding to an Apparent Open Complex
In previous experiments, dTTP binding to fluorescent DNA pol-DNA complexes with 2AP in the template position produced a rapid drop in fluorescence intensity within the dead time of the stopped flow instrument (15). Rapid dTTP binding was also demonstrated for the Klenow fragment in similar experiments (27). Because the fluorescence changes were rapid, dTTP binding appears to bind to a complex in an open conformation in which nucleotides have ready access. Although binding was rapid, dCTP was not bound, which indicates selectivity in forming the ternary complex (15).
In studies reported here with dFTP and template 2AP, the initial nucleotide-binding step was found to have two components. While dTTP binding in the presence of Mg2+ quenches 2AP fluorescence in a concentration dependent manner, dFTP does not at the highest concentration tested, 150 μM (Figure 3). Yet, dFTP is bound since dFTP is a strong, competitive inhibitor of dTTP binding that produces quenched, ternary complexes (Figure 3). Thus, there must be ternary complexes formed with dFTP that do not quench 2AP fluorescence, but formation of these dFTP complexes interferes with formation of quenched complexes with dTTP. We propose the following model for nucleotide binding by the T4 DNA polymerase (Figure 5), which is adapted from structural studies of the T7 RNA polymerase (21).
Figure 5.
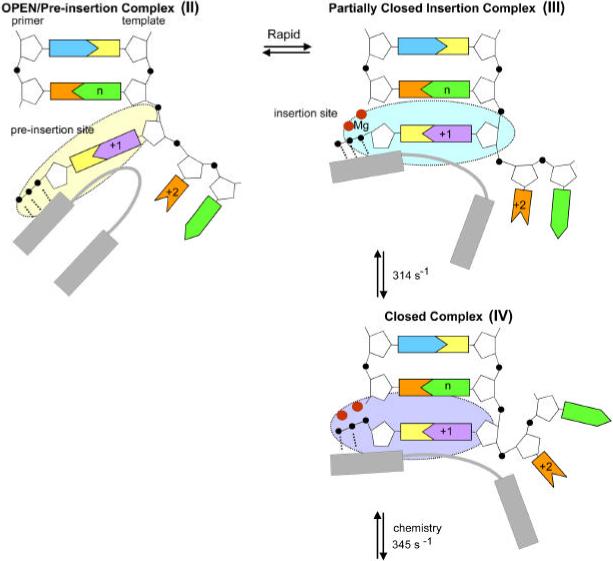
Illustrations of potential T4 DNA polymerase pre-insertion and closed complexes. Complexes II, III and IV correspond to the same complexes in Scheme 1. Structural data for pre-insertion complexes for T4 DNA polymerase or other DNA polymerases are not available; however the structures indicated for pre-insertion complexes II and III are consistent with the 2AP fluorescence data and complex II resembles the T7 RNA polymerase pre-insertion complex (21). Complex IV resembles closed complexes observed for several DNA polymerases.
Initial nucleotide binding by the T7 RNA polymerase is observed in an open complex that resembles complex II (Figure 5). The incoming nucleotide is in position to allow the accuracy of the base, sugar and phosphates to be examined. Note that the templating base in the +1 position in the template strand is not stacked with neighboring bases (21). If the +1 base were 2AP, this complex would be predicted to be fluorescent because there are no nearby bases in position to quench 2AP fluorescence by base-base interactions. We propose that a similar complex is formed with dFTP bound opposite template 2AP. dFTP binding does not change 2AP fluorescence, but this complex is not detected with dTTP because dTTP binding drives formation of quenched complexes that may resemble complex III (Figure 5). The quench in 2AP fluorescence is due to base stacking interactions with the base in the “n” position; Mg2+ ions are illustrated for complex III since Mg2+ is required to form quenched complexes (16). Note that Mg2+ ions are proposed to act in regulating the transition between closed and open states as well as the chemical reaction (28), but metal ions may also be necessary for formation of the partially closed complex III.
Thus, we propose that bound nucleotides are first “sampled” in an open complex that resembles complex II that is observed for the T7 RNA polymerase (21) and then scrutinized again by a subsequent conformational change that produces a partially closed complex III (Figure 5), which is still relevantly open compared to the closed complexes discussed below. Alternatively, complexes II and III may be in rapid equilibrium and complex III is trapped by dTTP binding, but not dFTP binding. We favor the first model because of the T7 RNA polymerase structural studies and because of experiments in which we determined fluorescence lifetimes for T4 DNA polymerase complexes formed as a function of dTTP concentration (29). When T4-exo− forms complexes with DNA labeled in the templating position with 2AP (Figure 1A), four types of complexes with different fluorescence lifetimes are detected. Interestingly, the amplitude of one of the more fluorescent species, fluorescence lifetime of 8.3 ns, initially increased as the concentration of dTTP increased and then declined at higher dTTP concentrations. We proposed that complexes identified by the 8.3 ns lifetime are pre-insertion complexes. While low concentrations of dTTP favor formation of complexes with a lifetime of 8.3 ns that may resemble complex II (Figure 5), higher concentrations of dTTP drive formation of much less fluorescent complexes, characterized by a fluorescence lifetime of 0.8 ns, that may resemble complex III (Figure 5).
The Role of H bonds in Formation of Pre-insertion Complexes
T4 DNA polymerase displays little discrimination against dFTP in the initial binding step as demonstrated by the ability of dFTP to strongly inhibit dTTP binding (Figure 3). Thus, H bonds between bases in the newly forming base pair are not essential in forming pre-insertion complexes. The lack of H bonding capability, however, is not the only difference between dFTP and dTTP. The F base lacks a hydration shell and is effectively smaller than the standard bases. Thus, dFTP may be accepted as a pairing partner with 2AP, and is able to exclude dTTP binding, if the absence of a hydration shell means that its smaller size can be accommodated within the constraints of the pre-insertion site. This reasoning can explain why dFMP is incorporated opposite template F as the F-F base pair is smaller than Watson-Crick base pairs with standard bases (7). Thus, experiments with new base analogs are required to determine if dFTP is a strong inhibitor of dTTP binding because of the similar shapes of the T and F bases or if any nucleotide analog with a small base is accepted.
H bonds are also not essential for subsequent steps because dFMP is incorporated opposite template 2AP, but the incorporation rate for dFMP is 30-fold slower than the rate for dTMP incorporation (Figure 2). This discrimination may stem from the reduced ability of dFTP to form quenched, ternary complexes as observed for ternary complexes formed with dTTP (complex III, Figure 5) or any subsequent step. Note, the inability to detect quenched, ternary complexes with dFTP does not mean that these complexes are not formed, but only if they are formed, the level is below detection. dFTP concentrations > 150 μM could not be tested because of limited availability of the nucleotide analogue.
Formation of Closed Complexes; Increased Nucleotide Discrimination
While formation of complex III (Figure 5) is rapid, a slower conformational change is detected before the chemistry step of phosphodiester bond formation. In reactions in which 2AP is initially in the +2 position (Figure 1B), primer extension produces an increase in fluorescence intensity at the rate of about 314 s−1 (16). In parallel reactions except that product formation was measured (Figure 4), product was formed at the rate of about 164 s−1 (Table 1), which indicates that the chemistry step follows the conformational change. We propose that the rate of increase in 2AP fluorescence at about 314 s−1 (Table 1) is measuring conformational changes that are associated with formation of closed complexes that may resemble complex IV in Figure 5. This proposal is consistent with structural studies of DNA polymerase closed complexes that show base unstacking in the template strand similar to complex IV (19, 20).
It is important to note that besides the large differences in rate of formation of complex III compared to complex IV, there are also significant differences in structure. While incorporation of a nucleotide produces an increase in 2AP fluorescence for 2AP at the +2 position, which occurs before formation of the phosphodiester bond, nucleotide binding does not (16). Thus, the conformational changes that produce base unstacking at the +2 position must occur after formation of the pre-insertion complex (complex III in Figure 5) and the chemistry step. This point is illustrated in Figure 5 by indicating base stacking interactions between the base at the +2 position and the neighboring +3 base for complex III, but not for complex IV.
Additional information about conformational changes involved in forming closed complexes was revealed by studies of L412M-exo−. Based on structural studies of the T4-like RB69 DNA polymerase closed complex (20), residue L412 is one of the conserved amino acids that form the nucleotide-binding pocket that surrounds the newly forming base pair. Because the base unstacking rate for 2AP at the +2 position is slower in reactions with L412M-exo− than in reactions with T4-exo− (Table 1), a methionine substitution for L412 alters interactions between amino acids within the polymerase active center that affect DNA polymerase-induced conformational changes in the template strand. The L412M substitution also affects nucleotide binding. The Kd for dTMP incorporation opposite template 2AP is less for L412M-exo− (252 μM) compared to T4-exo− (367 μM) and the Kd for dTTP binding opposite template 2AP is also less for L412M-exo− (17 μM) compared to T4-exo− (31 μM) (Figure 3; Table 1). Thus, residue L412 plays a role in determining nucleotide selectivity in forming pre-insertion and closed complexes as well as the transition from the open to closed conformations.
Does the Chemistry Step Exert Further Discrimination?
We have no evidence that further nucleotide discrimination is applied in the chemistry step. The Kd for dAMP incorporation opposite template T is the same measured by the fluorescence and rapid quench assays, 16 and 17 μM, respectively (Table 1), which indicates that no additional discrimination was applied at the chemistry step. Differences were also not detected for the overall rates of product formation by T4-exo− for incorporation of dAMP opposite template T, about 164 s−1 (Table 1, Figure 4), and for incorporation of dTMP opposite template 2AP, about 165 s−1 (15). These studies, however, do not rule out the possibility that the chemistry step may play role for aberrant mismatches.
How Are Mismatches Formed?
As demonstrated here, in order for T4 DNA polymerase to misincorporate a nucleotide, the “wrong” nucleotide must escape detection first during formation of pre-insertion complexes, which may have two components, and then formation of closed complexes. dCTP is efficiently excluded from binding opposite template 2AP in the formation of pre-insertion complexes. dCTP, unlike dFTP, is not an effective competitor of dTTP binding. Thus, under normal physiological conditions in the presence of all four dNTPs, dCTP is not expected to be readily bound or, if bound, to be incorporated opposite template 2AP.
Yet, 2AP is mutagenic in phage T4 infections and in Escherichia coli. AT to GC and GC to AT transitions are increased, which suggests that 2AP-C base pairs are formed in vivo and are formed more readily than A-C base pairs. Furthermore, because 2AP pairs with C more frequently as the template base rather than as the incoming deoxynucleoside triphosphate (30), two incorporation steps may be required for 2AP mutagenesis: (1) incorporation of the 2AP nucleotide opposite template T, but the 2AP-T base pair is subject to proofreading (31) and (2) misincorporation of dCMP opposite template 2AP, in which severe discrimination is expected as reported here and the 2AP-C base pair is also subject to proofreading (31). Thus, it is difficult to imagine how any 2AP-C mismatches are formed and persist unless there are additional factors that facilitate formation and/or stabilization of mismatches. One likely factor is the surrounding DNA sequence, which may create “hotspots” for formation of mismatches (30). Another possibility is that base modifications may promote formation of 2AP mismatches. Bacteriophage T4 uses the hydroxymethylcytosine (HMC) nucleotide instead of dCTP, which may promote increased formation of HMC-2AP mismatches if the HMC nucleotide is accepted more readily than dCTP as a base-pairing partner of 2AP. The probability of incorporating the HMC nucleotide opposite template 2AP in vivo is estimated to be large, about 2% per round of replication (32). The HMC nucleotide, however, is not essential for 2AP mutagenesis since 2AP is mutagenic in bacteria, which lack the HMC nucleotide, but 5-methylcytosines are hotspots for 2AP mutagenesis (33). Another consideration is that local differences in pH may affect formation of 2AP-C mismatches by increasing H bonds; two H bonds have been detected for protonated 2AP-C mismatches (33). Thus, an important future line of experimentation is to design new 2AP DNA substrates that recapitulate 2AP mutation hotspots observed in vivo, in order to determine what factor(s) facilitate formation of 2AP mismatches.
Summary
The accuracy of nucleotide selection by T4 DNA polymerase is achieved by at least two nucleotide binding steps: (1) formation of pre-insertion complexes, which may be composed of two components, where the accuracy of the incoming nucleotide is first evaluated (complexes II and III, Scheme 1, Figure 5) and (2) formation of closed complexes in which the geometry of the newly forming base pair is further examined (complex IV, Scheme 1, Figure 5). Only bound nucleotides that pass both steps are substrates for phosphodiester bond formation. Not all DNA polymerases may use the sieve process to filter out incorrect nucleotides, but highly accurate DNA polymerases, such as the T4 DNA polymerase, achieve high fidelity in nucleotide selection by using multiple binding steps with each successive step having increased stringency.
Joyce and Benkovic stated in a recent review (26) that “Further understanding of the mechanism underlying polymerase fidelity would be helped immensely by a more detailed structural characterization of the species on the misincorporation reaction pathway. However, the probable instability of many of these complexes may handicap crystallographic approaches.” The spectroscopic studies reported here with 2AP as the template base provide a method to study the dynamics of nucleotide incorporation in solution with both pseudo-correct pairings with dTTP and mismatches with other nucleotides.
ACKNOWLEDGMENT
We thank Robert Campbell for his helpful comments on the manuscript.
Abbreviations
- 2AP
2-aminopurine
- dFTP
difluorotoluene deoxynucleoside triphosphate
- F
the difluorotoluene base
- dTTP
2'-deoxythymidine 5'-triphosphate
- T
the thymine base
- dATP
2'-deoxyadenosine 5'-triphosphate
- A
the adenine base
- dCTP
2'-deoxycytidine 5'-triphosphate
- C
the cytosine base
- dNTP
2'-deoxynucleoside 5'-triphosphate
- T4-exo−
exonuclease-deficient D112A/E114A-DNA polymerase
- L412M-exo−
exonuclease and proofreading-deficient D112A/E114A/L412M-DNA polymerase
- O6meG
O6-methylguanine
- HMC
hydroxymethylcytosine
Footnotes
This research was supported by grants from the Canadian Institutes of Health Research (14300), the Natural Sciences and Engineering Research Council of Canada and the Canadian Foundation for Innovation to L.R-K., and NIH grants GM072705 and GM55596 to E.T.K. and L.B.B., respectively. L.R-K. is a Scientist of the Alberta Heritage Foundation for Medical Research.
REFERENCES
- 1.Kunkel TA. DNA replication fidelity. J. Biol. Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 2.Muzyczka N, Poland R, Bessman MJ. Studies on the biochemical basis of spontaneous mutation. I. A comparison of the deoxyribonucleic acid polymerases of mutator, antimutator, and wild type strains of bacteriophage T4. J. Biol. Chem. 1972;247:7116–7122. [PubMed] [Google Scholar]
- 3.Brutlag D, Kornberg A. Enzymatic synthesis of deoxyribonucleic acid. XXXVI. A proofreading function for the 3'→5' exonuclease activity in deoxyribonucleic acid polymerases. J. Biol. Chem. 1972;247:2410248. [PubMed] [Google Scholar]
- 4.Fersht AR. Sieves in sequence. Science. 1998;280:541. doi: 10.1126/science.280.5363.541. [DOI] [PubMed] [Google Scholar]
- 5.Moore CL, Zivkovic A, Engels JW, Kuchta RD. Human DNA primase uses Watson-Crick Hydrogen Bonds to distinguish between correct and incorrect nucleoside triphosphates. Biochemistry. 2004;43:12367–12374. doi: 10.1021/bi0490791. [DOI] [PubMed] [Google Scholar]
- 6.Washington MT, Helquist SA, Kool ET, Prakash L, Prakash S. Requirment of Watson-Crick hydrogen bonding for DNA synthesis by yeast DNA polymerase eta. Mol. Cell Biol. 2003;23:5107–5112. doi: 10.1128/MCB.23.14.5107-5112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moran S, Ren RX-F, Kool ET. A thymidine triphosphate shape analog lacking Watson-Crick pairing ability is replicated with high sequence selectivity. Proc. Natl. Acad. Sci. U.S.A. 1997;94:10506–10511. doi: 10.1073/pnas.94.20.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morales JC, Kool ET. Varied molecular interactions at the active site of several DNA polymerases: nonpolar nucleoside isosteres as probes. J. Am. Chem. Soc. 2000;122:1001–1007. doi: 10.1021/ja993464+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu Y, Ogawa AK, Berger M, McMinn DL, Schultz PG, Romesberg FE. Efforts toward expansion of the genetic alphabet: optimization of interbase hydrophobic interactions. J. Am. Chem. Soc. 2000;122:7621–7632. [Google Scholar]
- 10.Chiaramonte M, Moore CL, Kincaid K, Kuchta RD. Facile polymerization of dNTPs bearing unnatural base analogues by DNA polymerase α and Klenow fragment (DNA polymerase I) Biochemistry. 2003;42:10472–10481. doi: 10.1021/bi034763l. [DOI] [PubMed] [Google Scholar]
- 11.Goodman MF. Hydrogen bonding revisited: geometric selection as a principal determinant of DNA replication fidelity. Proc. Natl. Acad. Sci. U.S.A. 1997;94:10493–10495. doi: 10.1073/pnas.94.20.10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Echols H, Goodman MF. Fidelity mechanisms in DNA replication. Ann. Rev. Biochem. 1991;60:477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- 13.Johnson KA. Conformational coupling in DNA polymerase fidelity. Ann. Rev. Biochem. 1993;62:685–713. doi: 10.1146/annurev.bi.62.070193.003345. [DOI] [PubMed] [Google Scholar]
- 14.Florián J, Goodman MF, Warshel A. Computer simulation of the chemical catalysis of DNA polymerases: discriminating between alternative nucleotide insertion mechanisms for T7 DNA polymerase. J. Am. Chem. Soc. 2003;125:8163–8177. doi: 10.1021/ja028997o. [DOI] [PubMed] [Google Scholar]
- 15.Fidalgo da Silva E, Mandal SS, Reha-Krantz LJ. Using 2-aminopurine fluorescence to measure incorporation of incorrect nucleotides by wild type and mutant bacteriophage T4 DNA polymerases. J. Biol. Chem. 2002;277:40640–40649. doi: 10.1074/jbc.M203315200. [DOI] [PubMed] [Google Scholar]
- 16.Mandal SS, Fidalgo da Silva E, Reha-Krantz LJ. Using 2-aminopurine fluorescence to detect base unstacking in the template strand during nucleotide incorporation by the bacteriophage T4 DNA polymerase. Biochemistry. 2002;41:4399–4406. doi: 10.1021/bi015723p. [DOI] [PubMed] [Google Scholar]
- 17.Frey MW, Sower,s LC, Millar DP, Benkovic SJ. The nucleotide analog 2-aminopurine as a spectroscopic probe of nucleotide incorporation by the Klenow fragment of Escherichia coli polymerase I and bacteriophage T4 DNA polymerase. Biochemistry. 1995;34:9185–9192. doi: 10.1021/bi00028a031. [DOI] [PubMed] [Google Scholar]
- 18.Reha-Krantz LJ, Bessman MJ. Studies on the biochemical basis of mutation. VI. Selection and characterization of a new bacteriophage T4 mutator DNA polymerase. J. Mol. Biol. 1981;145:677–695. doi: 10.1016/0022-2836(81)90309-0. [DOI] [PubMed] [Google Scholar]
- 19.Doublié S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of the bacteriophage T7 DNA replication complex at 2.2Å resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 20.Franklin MC, Wang J, Steitz T. Structure of the replicating complex of a pol α family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 21.Temiakov D, Patlan P, Aniken M, McAllister WT, Yokoyama S, Vassylyev DG. Structural basis for substrate selection by T7 RNA polymerase. Cell. 2004;116:381–391. doi: 10.1016/s0092-8674(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 22.Reha-Krantz LJ, Nonay RL, Stocki SA. Bacteriophage T4 DNA polymerase mutations that confer sensitivity to the PPi analog phosphonoacetic acid. J. Virol. 1993;67:60–66. doi: 10.1128/jvi.67.1.60-66.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reha-Krantz LJ, Nonay RL. Genetic and biochemical studies of bacteriophage T4 DNA polymerase 3'→5' exonuclease activity. J. Biol. Chem. 1993;268:27100–27108. [PubMed] [Google Scholar]
- 24.Reha-Krantz LJ, Nonay RL. Motif A of bacteriophage T4 DNA polymerase: role in primer extension and DNA replication fidelity. J. Biol. Chem. 1994;269:5635–5643. [PubMed] [Google Scholar]
- 25.Stocki SA, Nonay RL, Reha-Krantz LJ. Dynamics of bacteriophage T4 DNA polymerase function: identification of amino acid residues that affect switching between polymerase and 3'→5' exonuclease activities. J. Mol. Biol. 1995;254:15–28. doi: 10.1006/jmbi.1995.0595. [DOI] [PubMed] [Google Scholar]
- 26.Joyce CM, Benkovic SJ. DNA polymerase fidelity: kinetics, structure and checkpoints. Biochemistry. 2004;43:14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 27.Purohit V, Grindley NDF, Joyce CM. Use of 2-aminopurine fluorescence to examine conformational changes during nucleotide incorporation by DNA polymerase I Klenow fragment) Biochemistry. 2003;42:10200–10211. doi: 10.1021/bi0341206. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Arora K, Beard WA, Wilson SH, Schlick T. Critical role of magnesium ions in DNA polymerase β's closing and active site assembly. J. Am. Chem. Soc. 2004;126:8441–8453. doi: 10.1021/ja049412o. [DOI] [PubMed] [Google Scholar]
- 29.Hariharan C, Reha-Krantz LJ. Using 2-aminopurine fluorescence to detect bacteriophage T4 DNA Polymerase-DNA complexes that are important for primer extension and proofreading reactions. Biochemistry. 2005;44:15674–15684. doi: 10.1021/bi051462y. [DOI] [PubMed] [Google Scholar]
- 30.Hopkins RL, Goodman MF. Deoxyribonucleotide pools, base pairing, and sequence configuration affecting bromodeoxyuridine- and 2-aminopurine-induced mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 1980;77:1801–1805. doi: 10.1073/pnas.77.4.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bloom LB, Otto M,R, Eritja R, Reha-Krantz LJ, Goodman MF, Beechem JM. Pre-steady-state kinetic analysis of sequence-dependent nucleotide excision by the 3'-exonuclease activity of bacteriophage T4 DNA polymerase. Biochemistry. 1994;33:7576–7586. doi: 10.1021/bi00190a010. [DOI] [PubMed] [Google Scholar]
- 32.Hopkins R, Goodman MF. Asymmetry in forming 2-aminopurine·hydroxymethylcytosine heteroduplexes; a model giving misincorporation frequencies and rounds of DNA replication from base-pair populations in vivo. J. Mol. Biol. 1979;135:1–22. doi: 10.1016/0022-2836(79)90337-1. [DOI] [PubMed] [Google Scholar]
- 33.Sowers LC, Fazakerley V, Eritja R, Kaplan BE, Goodman MF. Base pairing and mutagenesis: observation of a protonated base pair between 2-aminopurine and cytosine in an oligonucleotides by proton NMR. Proc. Natl. Acad. Sci. U.S.A. 1986;83:5434–5438. doi: 10.1073/pnas.83.15.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]