

1. Introduction
1.1 Overview of the hyaluronidases
The hyaluronidases (Hyals) are classes of enzymes that degrade predominantly hyaluronan (HA). The term “hyaluronidase” is somewhat of a misnomer since they have the limited ability to degrade chondroitin (Ch) and chondroitin sulfates (ChS), albeit at a slower rate. It is a common misconception that the bacterial Hyals have absolute specificity for HA. This is incorrect. Both bacterial 1 and vertebrate enzymes degrade Ch and ChS, albeit at a slower rate. The plausible reason for this broader specificity is that chondroitins preceded HA in evolution. For example, the nematode, Caenorhabditis elegans, contains only Ch and no HA, with only one Hyal-like sequence (unpublished observations). This is most likely a chondroitinase. It is plausible, therefore, that the vertebrate Hyals evolved originally from pre-existing chondroitinases 1. This may explain why Hyals, recognizing their ancestral substrate, retain limited ability to also degrade Ch and ChS.
The Hyals from bacteria have been well characterized, and much information is available (for representative publications see 2–5). The Hyals in vertebrate tissues, on the other-hand, have not been studied extensively, however, due to the lack of structural information. Such studies were more difficult and, therefore, more limited. In addition, vertebrate Hyals are present at exceedingly low concentrations. In human serum, e.g., Hyal1 is present at 60 ng/ml 6. They have high specific activities that are unstable during the course of purification, requiring the constant presence of detergents and protease inhibitors for their isolation. Many of such difficulties have been overcome, and a great deal of information is now available, facilitated in part by the Human Genome Project 7.
Six Hyal sequences occur in the human genome, constituting a newly recognized family of enzymes. They have similar catalytic mechanisms that contrast markedly with the bacterial Hyals. There is growing interest in these enzymes as their HA substrate is achieving much attention. An outstanding review of the hyaluronidases was published 50 years ago by Karl Meyer, who was also the first to describe the chemical structure of HA 8. Interestingly, a chapter on mucopolysaccharidases, the former name for the hyaluronidases, was included in Volume 1 of Methods in Enzymology 9. The most recent overview of all of the Hyals appeared in 1971 10. Since that time, no comprehensive review has appeared.
Karl Meyer classified the Hyals into three distinct classes of enzymes 8, based entirely on the biochemical analyses available at the time. With the advent of sequence and structural data, we can now appreciate how remarkably accurate Karl Meyer’s classification scheme was. No modification of his formulation is necessary. There are three major groups of Hyals, based on their mechanisms of action. Two of the groups are endo-β-N-acetyl-hexosaminidases. One group includes the vertebrate enzymes that utilize substrate hydrolysis 11,12. The second group, which is predominantly bacterial, includes the eliminases that function by β-elimination of the glycosidic linkage with introduction of an unsaturated bond 2–4,13–17. As these enzymes catalyze the breaking of chemical bond by means other than hydrolysis or oxidation, and with the forming a new double bond they are also termed lyases. Both terms, the eliminase (or β-eliminase) and the lyase, are used in the review interchangeably. The third group are the endo-β-glucuronidases. These are found in leeches, which are annelids 18, and in certain crustaceans 19. No sequence data are available, and little is known about this potentially interesting class of enzymes. However, their mechanism of action resembles that of the eukaryotic or vertebrate enzymes more closely than the bacterial enzymes.
Sequence data for vertebrate Hyals now provide opportunities to formulate structure-function relationships, to examine probable mechanisms of catalysis, to identify putative substrate binding sites, and to consider the additional non-enzymatic functions of this family of multifunctional enzymes for two of the three groups, for the hydrolase and lyase types of Hyals, respectively 2. Such a review is presented here, documenting some of the common and some of the unusual features that distinguish each of these families of enzymes.
The primary objective of this review is to clarify what is known about the structure and mode of action of all the Hyals. Since so little is known of the leech-type of Hyals, the β-endoglucuronidases, the emphasis will, by necessity, be upon two of the three classes of enzymes. Other aspects of these enzymes, such as their physiological activities, their dependence on reaction conditions, their role in cell biology and involvement in metabolism, and their use as reagents or as therapeutics, are not the concern presently. A review of these other aspects of the Hyals will appear separately (Stern and Jedrzejas, in preparation).
High levels of HA turnover occur in vertebrate tissues. Tight regulation of catabolism is crucial for modulating steady state levels, important for normal homeostasis, and for embryonic development, wound healing, regeneration, and repair. Under pathological conditions, as in severe stress, shock, septicemia, in burn patients, following major surgery, massive injury, circulating HA levels increases rapidly. HA also increases in association with aggressive malignancies. Determining the mechanism of action of the Hyals is critical for understanding their controls over such a wide range of functions (for reviews, see 20,21).
1.2. Assays for hyaluronidases activity
The Hyal enzymes have been assayed by classic turbidometric- and viscometry-based methods that require large amounts of enzymes, are relatively inaccurate, and are not suitable for isolation and purification procedures. This is affirmed by the observation that despite identification of the first vertebrate hyaluronidase in 1928 22 and their documentation in human serum in 1966 23 sufficient purification for sequence analysis was not achieved until 1997 6. A number of assays primarily for the hydrolase-type of Hyals developed in the last decade facilitated their characterization. These include microtiter-based enzyme linked immunosorbent essay (ELISA)-like assays, in which a highly specific HA-binding protein substitutes for the antibody component 24. Biotinylated HA bound to microtiter plates is subjected to Hyal activity, and the remaining HA quantified by an avidin-enzyme color reaction 25. HA substrate gel zymography procedures were also formulated that facilitated additional studies of these enzymes 26. The bacterial Hyals can, in addition, be assayed by spectrophotometric methods that detect the formation of an unsaturated bond during the catalysis of substrates of this enzyme 27.
1.3 The eukaryot hydrolase-type of hyaluronidases
The vertebrate hyaluronidases (EC 3.2.1.35) are a family of previously neglected enzymes 28. These are all endo-β-N-acetyl-hexosaminidases employing substrate hydrolysis for catalysis. The vertebrate Hyals also have transglycosidase activities, with the ability to cross -link chains of HA 29 and the potential ability to cross-link chains of HA with ChS or Ch. This reaction is not well understood, and the precise enzymatic mechanism is not known. No evidence has been obtained whether cross-linked chains of HA, or hybrid chains of HA and ChS or Ch exist, or whether they have biological significance. This reaction will not be discussed further. Whether it is merely a curious artifact of the isolated test tube reaction, or whether it occurs in nature and has major biological significance, remains to be established. The vertebrate Hyals degrade HA through a non-processive endolytic process, generating mostly tetrasaccharides.
1.4 The prokaryot eliminase-type of hyaluronidases
The second group, the bacterial enzymes, includes the eliminases, or lyases 2. The wealth of sequence, structural, and mechanistic information obtained during the last decade 13–16,30,31 allow for relatively precise formulation of the mechanisms for these glycoside eliminases, also known as HA lyases 2. They function by β-elimination with introduction of an unsaturated bond (EC 4.2.99.1) 2–4,13–17. The mechanism of action of HA lyases involving an acid/base type of proton acceptance and donation reaction is vastly different type from the hydrolysis mechanism of vertebrate Hyals 2. Many bacterial Hyals appear to degrade HA through a initial non-processive endolytic bites followed by exolytic processive degradation by generating unsaturated HA-disacchrides as products of exhaustive degradation 1,30,31. The degradation of Ch and ChS proceeds only through the non-processive endolytic method with generation of the same size product, unsaturated Ch/ChS-disacchrides.
1.5 The endo-β-glucuronidase type of hyaluronidases
The third group of Hyals are the endo-β-glucuronidases (EC 3.2.1.36) that cleave the β1,3 glycosidic bond. They are not considered here, as no sequence data are available. These enzymes, characteristic of annelids such as leeches (e.g., Herudo medicinalis) 32–35, and certain crustaceans 19,36 utilize the hydrolysis mechanism. They thus resemble the vertebrate enzymes more closely than the prokaryotic Hyals. The difference in specificity for the HA glycosidic bond remains unexplained, but may become evident once sequence and structural studies are performed.
1.6 Fungal hyaluronidases
There are hyaluronidase activities associated with several species of fungi 37–39. However, these enzyme activities should be approached with caution. In the absence of sequence data and without further characterization, no further discussion will be presented. Considering that opportunistic infections caused by Candida and other fungal organisms are increasing with the use of wide-spectrum antibiotics, antitumor drugs, immunosuppressive agents, and with the AIDS epidemic, these putative hyaluronidases may become important.
2. Hyaluronan as a substrate for catalysis
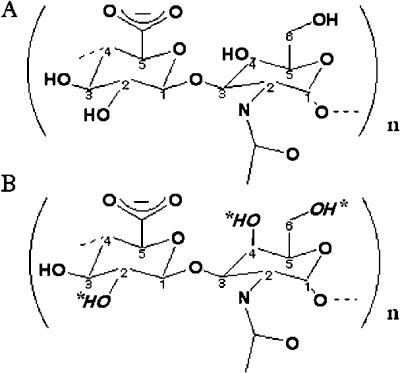
HA is a simple, unadorned high molecular weight glycosaminoglycan (GAG) linear polymer built of large numbers of repeating units consisting of [-D-glucuronic acid-β1,3-N-acetyl-D-glucosamine-β1,4-]n. The major families of all Hyals cleave the β1,4 glycosidic bond (Fig. 1). There is no clear explanation as to why the β1,3 glycosidic bond is resistant to their cleavage.
Fig. 1. Chemical structures of (A) hyaluronan, and (B) unsulfated chondroitin and chondroitin sulfates.

The substrate structures differ only in the anomericity at the C4 position of the N-acetyl-D-glycosamine, N-acetyl-D-glucosamine in hyaluronan and N-acetyl-D-galactosamine in chondroitin. Potential sulfation sites within the chondroitin molecule are indicated by italicized hydroxyl groups and asterisks. Both glycans are substrates for the human hyaluronidase enzymes. For Hyal-1 and -2, HA is the predominant substrate. However, binding and degradation of Ch/ChS also occurs, albeit at a slower rate, as observed experimentally in vitro. Hyal-4 appears to be a chondroitinase, with high specificity for Ch and ChS.
This propensity of all types Hyals to cleave specifically the β1,4 linkage might be related to structural properties of HA. Our data clearly correlates the presumed association of HA chains as a function of their molecular mass and the kinetics of HA degradation by bacterial Hyals. Such degradation appears to initiate as an endolytic, random bite process for high molecular mass HA (and presumably highly aggregated) and as HA chains become smaller (and presumably less aggregated) the processive, exolytic degradation takes over (see paragraph 8.3 for the complete discussion) (1 and M.J.J. unpublished results). All information to date on the structure of HA has been obtained from X-ray crystallography, nuclear magnetic resonance (NMR), or other biophysical and computational methods such as molecular dynamics (MD). However, while crystallography of HA reveals a variety of structures such as – 2-, 3-, and 4-fold helices for short chain HA 40, some NMR studies in D2O support a 2-fold helical conformation for HA in solution 41–44. The presence of such a 2-fold helical structure could explain the ability of these polymers to form higher order structures stabilized by hydrogen bonds and hydrophobic interactions, such as through antiparallel association of HA chains in β-sheet mesh-like structures 41–44. The protein-bound conformations of HA revealed by crystal structures of complexes of HA to HA lyases 30,31 are also 2-fold helices, although with small-scale bending and twisting. The binding of the HA substrate in conformations close to those found in solution presumably enhances binding. All these studies suggest that due to their extended helical conformation, the β1,4 linkage of HA is more exposed, and is therefore more accessible to HA-degrading enzymes, while their β1,3 linkage is hidden from such enzymes 1,45,46.
Other NMR studies do not support such a structure, at least for HA in aqueous solution. However, it may exist in D2O or DMSO solutions, or possibly in solid-phase at low pH 47–51. A dynamic/flexible character of HA emerges from such work. Furthermore, molecular dynamic simulations provide new insights into this question, suggesting that HA in solution is a highly dynamic molecule 52–54. Recent studies find no evidence for inter- or intra-molecular chain-chain association in concentrated HA solutions 55. Clearly, more studies are necessary to establish the structural properties of HA.
Finally, in addition to the dynamical studies of HA described above indicating high structural mobility of HA, studies by NMR and MD of HA octasaccharides in D2O suggest that the β1,4 and β1,3 linkages of HA have different dynamic properties: for β1,4 there are two readily exchangeable conformations. The β1,3 linkage, on the other hand, is more rigid, as it only has one conformation 49. This difference in dynamics can result in greater propensity and accessibility of the β1,4 linkage, as it has a greater degree of structural freedom to adjust to the active site of the HA degrading enzymes. In support of such possibilities, other studies confirm two conformations of dilute tetrasaccharides in water, with rapid movement occurring between them54, corresponding presumably to the two conformations of the β1,4 linkage. These conformations are stabilized by short-lived, highly dynamical, intramolecular hydrogen bonds (H-bonds) 53. Also, dilute decasaccharides in water studied by NMR and MD confirm its highly dynamic behavior 52. In conclusion, the selection of β1,4 linkage for cleavage might be due to: (1) the aforementioned HA helical structure with exposed β1,4 linkage and (2) greater flexibility compared to the β1,3 linkage, both of which make for easier accessibility to all the Hyal enzymes.
To complicate the matter even more, under physiological conditions, aggregation of high molecular weight HA can occur, though shorter HA chains may not aggregate. As a consequence, initial degradation of high molecular weight HA chains likely proceeds through a random endolytic cleavage at sites where such chains expose the β1,4 linkage in a proper conformation. Due to the presumed two-fold helical conformation of HA, the next β1,4 linkage would be rotated by ~180° and as such, is not likely to be accessible to the Hyal enzymes, due to the β-sheet mesh-like structures (see above). As the size of HA chains decreases, the ability of HA to aggregate also decreases. At a size of HA of molecular weight below 300 kDa (about 750 disaccharide units) the ability of HA to aggregate decreases, as shown by electron microscopy-rotary shadowing 56. At the same time, HA chains below ~50 disaccharides in length (~20 kDa), do not aggregate in salt solutions, as documented by light scattering 57.
These largely hypothetical properties of HA provide an explanation for the observed differences between bacterial and vertebrate Hyals. At some point below 300 kDa, the HA molecule can be degraded by bacterial hyaluronate lyases using a processive mechanism, due to decreased aggregation with chains of such size. This processive method predominates for HA chains at or below 20 kDa. In this model, as the average size of chains decrease, the processive mechanism takes over from the random cleavage mechanism. Such a model supports the observation of exponentially increasing rates of HA degradation with smaller polymer size by the bacterial Hyals (unpublished data and 1,3,4).
However, this model does not apply to the human Hyals, since these enzymes are non-processive, regardless of polymer size. To emphasize the importance of states of HA aggregation in relation to size, the human Hyal-2 enzyme rapidly degrades HA to approximately 20 kDa fragments, or about 50 disaccharide units, and then slows considerably 58. With continued incubation, Hyal-2 is able to generate even smaller reaction products, but does so at a much slower rate. Close inspection of the reaction reveals that a change in kinetics occurs at approximately 20 kDa for Hyal-1 also. Both enzymes have a decreased affinity for fragments smaller than 20 kDa, with Hyal-2 having a far lower affinity than Hyal-1 (A.D. Miller, personal communication). This appears to suggest that the HA and/or its structural properties as a substrate undergo a change at ~20 kDa in size.
3. Properties of the hyaluronidases
3.1 Designation of hyaluronidases as a class within the glycosidase families of enzymes
Mammalian Hyals (EC 3.2.1.35) are members of the group of carbohydrate-active enzymes (CAZy), termed glycosidase family 56 59,60. The CAZy database (available at afmb.cnrs-mrs.fr/~cazy/CAZY/index.html) describes families of structurally related catalytic and carbohydrate-binding modules (or functional domains) of enzymes that degrade, modify, or create glycosidic bonds. The glycoside hydrolase family (EC 3.2.1.*) is classified in this way into 97 families. The authors of this regularly updated database imply that their system of classification reflects a common relationship in the sequence and in the folding for all enzymes that fall into the same group. Assignment of an enzyme into a CAZy family is expected to: (1) reflect the structural features of the enzymes better than relying solely on substrate specificity, (2) help to reveal evolutionary relationships between enzymes, and (3) provide a convenient tool to derive mechanistic information.
The vertebrate Hyal enzymes are of glycosidase family 56, defined as endo-β-acetyl-hexosaminidases that utilize hydrolysis in catalysis of HA at the β1,4 glycosidic linkages. On the other hand, bacterial Hyals are also endo-β-acetyl-hexosaminidases, but utilize the lyase mechanism. They belong to a different CAZy family, to polysaccharide lyase family 8. In general, these polysaccharide lyases (EC 4.2.2.*) cleave by β-elimination, resulting in a double bond at the new non-reducing end. The hyaluronate lyases (EC 4.2.2.1; bacterial Hyal) consists of only one subgroup within family 8 that also include: chondroitin ABC lyases (EC 4.2.2.4), chondroitin AC lyases (EC 4.2.2.5), and xanthan lyases (EC 4.2.2.12). All of these bacterial enzymes, Hyals, chondroitinases, and xanthanases, share significant sequence, structural, and mechanistic homology.
3.2 The term “hyaluronidase” is a misnomer
The Hyals of all origins do not have absolute substrate specificity, but digest additional substrates. Hyals of both vertebrate and bacterial origin can catabolize Ch and ChS, though degradation occurs more slowly 1. Chondroitins are polymeric GAGs differing from HA only in their anomeric configuration at the C4 atom of N-acetyl-D-glucosamine, making it N-acetyl-D-galactosamine. Degradation of ChS however is restricted to specific patterns of sulfation (Fig. 1). There is a common misconception that the bacterial Hyals have absolute substrate specificity for HA. But this is not correct. Digestion with a bacterial Hyal is often used to validate the occurrence of HA in histochemical and biochemical analyses. This practice should cease.
The ability of bacterial Hyals to digest Ch/ChS has structural limitations. The structural evidence for the Hyals of Streptococcus species, S. pneumoniae and S. agalactiae, correlates perfectly with biochemical data, and indicates that S. pneumoniae Hyal can only degrade Ch/ChS at the β1,4 linkage when the disaccharide on the non-reducing side of the bond to be cleaved is either unsulfated or 6-sulfated. The 4-sulfation configuration is not tolerated on the non-reducing side, although it is accepted on the reducing side of the bond to be cleaved. The 2-sulfated chondroitin is not cleaved under any circumstances 1,61. The reason for this specificity is directly related to steric clashes between enzyme and substrate. Therefore, the degradation of Ch/ChS proceeds, as a consequence, primarily by the endolytic ‘random bite’ mechanism. This is in contrast with the Hyal of S. pneumoniae, which is processive 3,5,15,16,30,31,62. The reaction is initiated by a random endolytic ‘initial bite,’ resulting in cleavage of the polymeric HA chain into two parts. This process is followed by processive, exolytic cleavage of one HA disaccharide at a time until the entire chain is degraded. However, the action on human Hyals seems to be entirely different. It is a non-processive event for both substrates, for HA as well as for Ch/ChS.
Structural studies of Ch/ChS suggest they also form higher order structures, similar to those formed by HA. Formation of such structures depends on the presence and position of the sulfate groups. Full sulfation at the 4 position, but not at 6, impedes association 63. This may explain why 4-sulfated ChS is not as good a substrate for the Hyals of all origins.
3.3 Evolution of hyaluronidases can be postulated from their substrate specificity
Considering this difference in mechanism of action of bacterial Hyals between HA and Ch/ChS, one might assume that the bacterial Hyals developed into processive enzymes from a non-processive chondroitinase predecessor. Evolutionary studies suggest that heparin-like polymers were the first GAG in metazoan life (reviewed in 64). The second GAG polymer to evolve was chondroitin with HA appearing significantly later. The first organism to produce chondroitin or the first to produce HA is still unknown. The recent availability of genomic sequences of many organisms allows for the inference, from their genetic content, of which GAGs they contain. These studies show, for example, that the worm Caenorhabditis elegans (Nematoda) and the fruit fly Drosophila melanogaster (Insecta) both have heparin and heparan sulfate. Only the unsulfated form of chondroitin is found in the worm. Neither of these organisms contain HA 65. Therefore, the single vertebrate Hyal-like sequence found in their genomes is likely to be exclusively a chondroitinase, rather than a Hyal, though this has not been demonstrated directly.
The deposition of HA is intimately linked to the development of the notochord in vertebrates 66,67. The first appearance of the notocord in evolution is in the free-swimming larval form of the tunicate. The complete sequence of the tunicate genome is now available 68, renewing interest in this most primitive of chordates. It may share a common ancestor with all vertebrates, or be itself a vertebrate predecessor. The notochord in tunicate larva is the critical structure that marks the evolutionary leap from the invertebrates. This notochord, a format for the later spinal column, becomes resorbed and is not present in the sessile adult tunicate.
It could be postulated that, in evolution, the first occurrence of HA is in tunicates, in association with notochord development. However, careful search of that genome demonstrates that there is only one Hyal-like sequence (unpublished observations), which probably codes for a chondroitinase. Thus, the duplication event that might have provided the first true Hyal does not occur in the tunicate. A search of the tunicate genome indicates that HA synthase (HAS) or HAS-like genes 69 are also not present. Amphioxus, a class of small flattened marine chordate organisms of the subphylum Cephalochordata, is a candidate for such an event. Unpublished evidence indicates that amphioxus does indeed contain HA (A. Spicer, personal communication), and this may be where the first true Hyal and HAS sequences occur.
The appearance of HA later in evolution than chondroitin suggests that hyaluronate lyase enzymes also developed from pre-existing chondroitin lyases, and that the original/ancestral hyaluronidase was actually a chondroitinase. Over time, with HA present in the higher host organisms, some of the bacterial chondroitinase acquired specificity for HA and lost the efficiency to break down Ch/ChS. The residual ability of bacterial Hyals to cleave Ch/ChS is an indication here also of their ancestral origin. Since HA does not have any sulfation, sulfation patterns cannot have an affect on catalysis. For this reason, the prokaryotic Hyal enzymes developed a processive mechanism of action 1,15,16,61, allowing for free sliding down the HA polymer chain, with exolytic cleavage of one HA disaccharide at a time 3,5,30,31,62. Evidence suggests that sulfation of Ch is responsible, in part, for the apparent inability of bacterial Hyals to cleave the Ch chain in a processive manner. Thus, processive cleavage of ChS is not possible by either bacterial Hyals or chondroitinases. Both enzymes cleave ChS by the same random endolytic process.
The appearance of a HA in evolution may correspond to the necessity for stem cell-like pluripotential cells to travel some distance in organisms during the course of embryonic development. Such motility may have been and continues to be dependent on a HA-rich matrix that, through the hydrated solution volume, creates spaces through which such cells can migrate. The areas in which such cells maintain their embryonic pluripotential state may also be a HA-rich environment, such as observed in bone marrow. Within such protected areas, stem cells continue to proliferate, without the danger of signals that would initiate differentiation. The HA in such areas would function to suppress cell-cell and cell-matrix communication, impeding ligand access to cell surface receptors. Such a situation parallels the semi-autonomous state that describes malignancies. The increased incidence of malignancies in organisms might be predicted to occur in parallel with the appearance of HA, a high price to pay in evolution.
Another facet of the same scenario stems from the requirement for HA to be removed before differentiation commences. Hyaluronidase activity is involved in eradicating the HA 70 (a free radical bust in order to degrade HA cannot be, however, excluded). How is the decision made to initiate activation of hyaluronidase or HA degradation in general? This of course reflects on the problem that virtually nothing is known about the control mechanisms that modulate hyaluronidase activity and HA degradation.
3. 4 Digestion products of human and bacterial hyaluronidase catalysis
There are profound differences in Hyal digestion products between the vertebrate and prokaryotic Hyals. The bacterial enzymes generate unsaturated disaccharides as the predominant products 15,16,71. These disaccharides appear to have limited biological function and primarily serve as an energy supply for the organism, and are not known to participate in the pathogenesis of infection 1,3. Other enzymes, of either bacterial or host origin, may then cleave the disaccharides to monosaccharides.
Bacterial pathogens such as S. pneumoniae, can utilize Hyals to overcome human host defense mechanisms, to facilitate invasion, and to reach cryptic sites essential for successful infection. These exogenous pressures force the host to diversify its glycan structures as part of their defense and survival strategies. As described earlier, all bacterial Hyals studied to date are lyases, and may have evolved such catalytic mechanisms precisely for overcoming host defenses as a counter-strategy 2,5,17. Bacterial Hyals may have evolved two separate functions in their survival counter-strategy. First, bacterial spread is facilitated by degradation of the one of the major component of host tissue extracellular matrix (ECM), which is HA. The second is to provide a source for their carbon and energy requirements. The identity of such a disaccharide transport system essential in this case, presumably of the ABC type, has not been revealed by bioinformatics/sequence methods of analysis of bacterial genomic sequences (unpublished data). Therefore, further degradation of HA disaccharides to individual sugars, such as glucose, glucose-P, or similar sugars, would allow for utilization of standard glucose transport mechanisms. For example, the two β-exoglycosidases can generate N-acetyl-D-glucosamine and D-glucuronic acid as the monosaccharide end products. The N-acetyl-D-glucosamine can be phosphorylated by a kinase, followed by a deacetylase to generate glucosamine-6-P.
Of recent interest are the riboswitches in bacteria that bind glucosamine-6-P, one of the two terminal products of HA catabolism. The generation of such products by ribozymes and riboswitches can control gene expression by using a cassette mechanism in response to variations in levels of available metabolites 72,73. As mentioned earlier, HA has a high rate of turnover in vertebrate tissues. 15 g of HA occur in the 70 Kg individual, 5 g of which turn over daily. Therefore, 2.5 g of glucosamine-6-P are generated daily, a not inconsiderable amount. Variations on such levels can have profound effects on homeostatic mechanisms.
This source of carbon and energy might be the reason why bacteria degrade HA to small disaccharide molecules, as they are (1) easier to transport or to degrade further, and (2) they introduce an unsaturated bond in the glucuronic part of HA between carbon atoms C4 and C5 (standard scheme for nomenclature/numbering of atoms). Such unsaturation allows for additional chemistry to take place, facilitating degradation of these disaccharides, compared to the presence of the saturated bonds that occur in vertebrate Hyal digestion products. The HA disaccharides with C4-C5 unsaturation are distinct in their structural properties compared to conventional HA disaccharides. They have different three-dimensional (3D) structures, compared to regular HA disaccharides, and this difference is exhibited by a different puckering of the ring of the glucuronate moiety that assumes a distorted half-chair conformation 14,74. For free HA, this sugar is clearly in chair conformation 30,31. The puckered ring structure of the unsaturated HA glucuronate might be more amenable to further metabolic reactions due to its putative resemblance of transition state(s) of the pyranose-like ring.
The vertebrate Hyal’s however, in marked contrast, can generate a range of HA oligomers with a wide spectrum of size-specific biological activities 21. The high molecular weight substrate HA is anti-angiogenic 75 and immunosuppressive 76,77. The products of human Hyal-2 digestion, the 20 kDa fragments, are highly angiogenic 78, inflammatory 79, induce transcription of matrix metalloproteinases 80, and stimulate vascular endothelial recognition of injury 81. The human Hyal-1 digests HA oligomers to low molecular weight fragments that are immuno-stimulatory, participate in oncogenesis, and are degraded further down to the tetrasaccharide level. Even these small digestion products, i.e., tetrasaccharides, have biological activity, being anti-apoptotic and inducers of heat shock proteins 82. The tetrasaccharides are probably cleaved to yet smaller fragments by the β-exoglycosidases. The exoglycosidases, β-glucuronidase and β-N-acetyl-D-glucosaminidase participate in degradation to a greater degree than previously appreciated. It is the monosaccharides that have the ability in all likelihood to exit the lysosome for participation in other metabolic pathways.
4. Identification of mechanism of hyaluronan-degradation by vertebrate-like hyaluronidases
4.1 Catalytic mechanism and its importance
By degrading HA, the human Hyals participate in the tight regulation of HA catabolism, which has been shown to be crucial not only for preserving its biophysical properties, so important in mammalian tissues. The detailed understanding of the enzymatic mechanisms involved in HA degradation is crucial for a better understanding of the entire process of HA catabolism and the steady-state deposition of HA. The precise mechanism for modulation of hydrolysis of HA by vertebrate (including human), and vertebrate-like Hyals is still under investigation. However, based on structural investigations of human-like bee venom hyaluronidase (BVHyal) by Markovic-Housley et al., and the inclusion in CAZy glycosidase family 56 59,60 it involves a double-displacement mechanism at the C1 anomeric carbon atom of the N-acetyl-D-glucosamine of HA substrate at the β1,4 glycosidic linkage with D-glucuronic acid 11,83. Such a mechanism results in a net retention of the anomeric configuration of the substrate at the carbon C1 position 84,85. In many other glycan degrading enzymes a covalent glycosyl-enzyme intermediate is created as a part of this reaction.
4.2 Three-dimensional structures of the bee venom and bovine PH-20 hyaluronidases suggest a possible mechanism of action
In order for vertebrate Hyals to degrade HA, the enzymes must bind their substrates. The binding site in the 3D structure, an elongated cleft traversing the enzymes, is large enough to accommodate the polymer. There is only one 3D structure of a vertebrate-like Hyal enzyme available, which is ironically the bee venom enzyme. The structure was elucidated by X-ray crystallography in both its native form, and in a complex with an HA tetrasaccharide (protein data bank structure depository (pdb) code: 1FCQ, 1FCU, 1FCV) (Fig. 2A, B) 11. A mechanistically insightful structure of the enzyme-tetrasaccharide HA complex is available (pdb code: 1FCV). In addition, it should be noted that of the entire family 56 glycosidases to which vertebrate-like Hyal hydrolases belong, BVHyl is the only one with an established 3D structure 59,60. A 3D model of bovine PH-20 has been derived recently that can also serve as a model for human PH-20 as well as the other vertebrate-like Hyals 86.
Fig. 2. X-ray structure of the native bee venom Hyal and of its complex with a tetrasaccharide fragment of the HA substrate.
The only experimental structure of Hyal available is that of the bee venom enzyme. The historic secondary structure of Hyal is that of a homology model for BPH-20 and its complex with HA 86, which are similar to that of the native X-ray crystal structure of bee venom hyaluronidase and its complex with HA (pdb code: 1FCQ and 1FCV, respectively) 11.
(A) The overall distorted (β/α)8 TIM barrel fold and the cleft are shown.
The fold of this globular protein is characteristic of all glycoside hydrolases of carbohydrate active enzyme (CAZy) family 56. The molecule (based on coordinates of BVHyal having the pdb code: 1FCV) is color-coded by the secondary structure elements (helices in red, β-sheets in yellow, others in green). The HA molecule bound to the enzyme is located in the HA-binding cleft and is depicted in ball and stick fashion, and is colored in purple. This figure and other structural figures in this work were made with PyMol 156.
(B) Surface of the molecule and a tetrasaccharide HA substrate bound within the cleft.
The orientation of BVHyal molecule is similar to that in panel A. The protein surface is colored by the atomic element (C – green, N - blue, O - red, S - yellow). A large and positive charge and the hydrophobic character of the surface of the cleft allows for binding of negatively charged and also hydrophobic substrates. The cleft is where the catalytic function of hyaluronidases is performed. The HA tetrasaccharide is shown bound to the enzyme, also color coded by the atomic element.
The structural properties of BVHyal, as well as those of bovine PH-20 (BPH-20) encompass a large cleft (see above) that allows for substrate binding, followed by catalysis. As these two structures demonstrate, vertebrate-like Hyals are globular proteins that encompass a large groove or cleft that traverses the entire enzyme (Fig. 2). These two structures display an enzyme with a classical distorted (β/α)8 triose phosphate isomerase (TIM) barrel fold 87. The central β-sheets deviate the most from the standard TIM barrel fold, as three of the β-sheets appear to be missing from the BVHyal enzyme complex (pdb code: 1FCV). But only one is missing in the high resolution native BVHyal structure (pdb code: 1FCQ), suggesting that the substrate induces an allosteric modification of the secondary structure of the enzyme. The cleft that transverses the wider end of the TIM barrel is built primary of residues originating from the loops between the major secondary structure elements, the sheets and helices. As expected, the cleft is where HA (or Ch/ChS) is bound and catalyzed, and is large enough to accommodate an entire hexasaccharide unit of HA, which is the smallest HA fragment that can be degraded by these enzymes. The surface of the cleft is lined with conserved aa residues that are mostly positively charged or hydrophobic. This allows for binding of elongated HA substrate chains that are negatively charged and have hydrophobic sugar ring surfaces (Fig. 2B). On one side of such a cleft, a catalytic site is located that encompasses the catalytic Glu113 aa residue (BVHyal numbering)/Glu149 (BPH-20 numbering) (Table 1) 11,86
Table 1. Numbering scheme for conserved residues among the vertebrate Hyal hydrolases involved in the catalytic process and in essential positioning of the substrate’s carbonyl of the acetamido group.
The residues were divided into the catalytic Glu and supporting residues that position the HA’s carbonyl of the acetamido group for catalysis, as reported by Jedrzejas and Stern 12. The Cys264 residue of Hyal-4 interrupts the conserved scheme and likely reflects this Hyal’s specificity for chondroitin and its chondroitinase function. All other residues are strictly conserved (Fig. 3).
| BVHyal | BPH-20 | Hyal-1 | Hyal-2 | Hyal-3 | Hyal-4 | HPH-20 |
|---|---|---|---|---|---|---|
| Catalytic residue: Glu113 | 149 | 131 | 135 | 129 | 147 | 148 |
| Positioning residues: | ||||||
| Asp111 | 147 | 129 | 133 | 127 | 145 | 146 |
| Tyr184 | 220 | 202 | 206 | 202 | 218 | 219 |
| Tyr227 | 265 | 247 | 253 | 246 | Cys263 | 264 |
| Trp301 | 341 | 321 | 327 | 319 | 339 | 339 |
4.3 Identity of the nucleophile and the catalytic acid in the active site
A covalent glycosyl-enzyme intermediate seems not to be involved in the vertebrate-like Hyal-catalyzed reaction, due to the involvement of the HA substrate(s)’s own N-acetyl group. This is one of the unusual features of this class of enzymes. The reaction involves an intermediate, an oxocarbonium ion-like transition state and retention of configuration at the C1 carbon atom 88 (standard HA nomenclature scheme). Again, as in many other glycan degrading enzymes, two carboxylic acids residues (glutamic acid/aspartic acid) are intrinsic to the process. One of these aa residues functions as an acid/base catalyst, involved directly in the cleavage of glycosidic linkages. The other residue functions as a nucleophile, and is involved in the creation of a covalently-linked glycosyl-enzyme intermediate. For steric reasons, such catalytic residues must be separated by ~6 Å 2. Glu212 and Glu217 are examples of such aa residues for cellbiohydrolase I from Trichoderma reesei 85, or Glu139 and Glu228 from the Bacillus agaradherans 5 endoglucanase family of enzymes 84. Based on the analysis of known sequences of vertebrate-like Hyal hydrolases of mammalian origin and in their comparison to the bee venom Hyal structure 11 and to the three-dimensional model structure of bovine PH-20 hyaluronidase 86, there are only two conserved carboxylic aa residues in the active site cleft of these enzymes that could be directly involved in the mechanism described above, Asp147 and Glu149 (bovine PH-20 numbering; equivalent to Asp111 and Glu113 of the bee venom Hyal) (Table 1). However, due to their physical proximity, it is not possible for both to participate in such a mechanism. It is more likely that only one of the two residues is involved. Structural 11 and biochemical 89 data are consistent with Glu149 of BPH-20 being the identity of the catalytically involved acid/base residue. The site-directed mutagenesis in human sperm PH-20 enzyme of Glu148 and Asp146 (equivalent of Glu131 and Asp129 in human Hyal-1) to Gln and Asn, respectively, resulted in total loss of activity for E148Q mutant and only 3% activity for D146N construct 89. These results are entirely consistent with the proposed mechanism, and support the catalytic importance of this single Glu aa residue. The residual low level of activity of the D146N mutant in HPH-20 is also consistent with it having only a supportive role, being only one positioning residue out of a total of four.
The functional nucleophilic residue is assigned to the HA substrate itself, to its carbonyl oxygen (acetamido oxygen) of the N-acetyl-D-glucosamine residue. In order for this carbonyl to be able to participate in catalysis, it must be positioned precisely in order to interact with C1 carbon of the HA substrate. Such positioning is accomplished by a set of highly conserved aa residues in the cleft of the enzyme: Asp147, Tyr220, Tyr265, and Trp 341 (Asp111, Tyr184, Tyr227, and Trp301 for BPH-20) (Table 1). Based on the structure of BVHyal, such interactions between enzyme and substrate induce a change in the conformation of the pyranose ring of N-acetyl-D-glucosamine, from the normal chair conformation into a distorted boat conformation. This process brings the glycosidic bond into nearly an equatorial position and closer to the catalytic Glu149 for the presumed catalytic H-donation 11. Similar mechanistic behavior with respect to the nucleophile identity is observed in chitinolytic enzymes 90,91. In the structure of BVHyal complex with tetrasaccharide units of HA, this carbonyl oxygen is in an interaction distance with the anomeric carbon C1 of HA, a distance of ~2.9 Å.
5. The human hyaluronidases
5.1 Properties of human hyaluronidases
In the human genome there are six known genes coding for hyaluronidase-like sequences, all of which have high degree of homology (Table 2). They include human Hyal-1, Hyal-2, Hyal-3, Hyal-4, and PH-20/Spam1, as well as a pseudogene Phyal1 that is transcribed in the human but not translated. They each have a unique tissue distribution, and except for PH-20, are widely expressed. The first three human Hyals, Hyal-1, -2, and –3, are tightly clustered on chromosome 3p21.3 and the latter are similarly clustered on chromosome 7q31.3 21. Human Hyal-1 and Hyal-2 are the two major hyaluronidases for the degradation of HA in somatic tissues. Hyal-2 of human origin degrades high molecular mass HA to an approximately 20 kDa product (~50 disaccharide units), whereas Hyal-1 can degrade high molecular weight HA to small oligomers, primarily to tetrasaccharides. Human Hyal-3 is an acid-active enzyme (B. Triggs-Raine and R. Hammond, personal communication) as is Hyal-1. Hyal -2 has a broader pH optimum. A product of the human hyal-4 gene, Hyal-4, based on preliminary studies is also a chondroitinase with a predominant activity towards Ch and ChS. The detailed properties of neither Hyal-3 nor Hyal-4 have been documented to date. Details of Hyal-3 catalysis are currently being investigated, and will be forthcoming (B. Triggs-Raine and R. Hammond, personal communication).
Table 2. Sequence identity among human hyaluronidases Hyal-1–4, and PH-20.
The data is based on Clastal W 1.82 101 as reported by Jedrzejas and Stern 12. The alignments are as shown in Fig. 3.
| Enzyme | Hyal-1 | Hyal-2 | Hyal-3 | Hyal-4 | HPH-20 |
|---|---|---|---|---|---|
| Hyal-1 | 100% | 38.3% | 38.3% | 38.5% | 35.1% |
| Hyal-2 | 100% | 38.0% | 36.4% | 34.5% | |
| Hyal-3 | 100% | 33.1% | 33.7% | ||
| Hyal-4 | 100% | 41.7% | |||
| HPH-20 | 100% |
There are also six Hyal-like sequences in the mouse. The homology between similar sequences in mouse and human genomes are far greater than that between individual members of the paralog (see figure in the supplementary data). Separation of mice and humans from a common ancestor occurred 75 to 80 million years ago, suggesting that the original divergence of members of the Hyal-like sequences occurred before the emergence of modern mammals. There may have been one original sequence; followed by two duplication events to form three tightly linked sequences. This was followed by en masse duplication to form the current pattern of three sequences on each of two chromosomal locations, 3p21.3 and 7q31.3 in the human, and 9F1-F2 and 6A2 in the mouse. Evidence from expression profiles suggests that the en masse duplication event, from three to six genes, corresponded to the divergence of placental from non-placental mammals. Due to the high homology between human Hyals and those of other vertebrates such as the mouse, properties of the human Hyals are presented here as exemplars for all vertebrates.
Human PH-20 or SPAM1 (SPerm Adhesion Molecule 1) (HPH-20), necessary for fertilization, is associated with testes, and facilitates penetration of sperm through the cumulus mass to the ovum. It was first detected in testicular extracts, and is the identity of the “spreading factor” demonstrated by Duran-Reynals 22. The localization of PH-20 has now been refined to the acrosome of spermatids, a structure that is related to lysosomes. PH-20 is a bifunctional protein, and is also an adhesion protein, with binding properties to the cumulus mass surrounding the ovum 92.
Testicular hyaluronidase or PH-20 was originally assumed to be tissue-specific, with expression limited to the testes. By the more sensitive technique of polymerase chain reaction (PCR) analysis, PH-20 can be detected in the epididymis 93, breast 94 in the female reproductive tract 95, as well as in association with a number of malignancies 94,96,97 The presence of PH-20 in the female reproductive tract and breast speaks to the multi-functionality of the enzyme, and possibly to all of the enzymes of the Hyal family.
Bovine PH-20 is the most commonly used Hyal for experimental purposes, and is available commercially in a reasonably pure form (Sigma catalog no. H3631, Type VI-S, from bovine testes, with an activity of 3,000 to 15,000 national formulary units (NFU) units/mg). This protein is approximately 20–25% pure. The preparation has a bimodal pH activity curve, with activity at both neutral and acid pH, attributed to isoforms, as described 98.
A seventh human gene has been proposed to have hyaluronidase activity 99 but its identity as a hyaluronidase remains questionable, and will not be discussed here in further detail. The primary, secondary, and tertiary structure analysis of this protein yielded markedly different results than that for human Hyal-1–4 and HPH-20, suggesting its different function that is not consistent with hyaluronidase. There is no resemblance of this protein to BVHyal at all as well as to any protein/domain with known 3D structure (unpublished results). A splice-variant of this gene mRNA produces a nuclear protein with β-N-acetylglucosaminidase activity 100.
The human Hyals have significant degrees of sequence conservation, indicative of their presumed common structural and catalytic properties (Table 2, Fig. 3). The sequences of known human Hyals described above (Hyal-1–4 and HPH-20) are relatively uniform in their amino acid (aa) length and range from the shortest, Hyal-1 with 435 aa, to the longest, HPH-20, with 510 aa. Interestingly, PH-20 and Hyal-4, together with Hyal-2, are all glycosylphosphatidylinositol- (GPI-) anchored proteins. The GPI-membrane attachment sites all occur in the C-terminus, close to the extreme C-terminal aa residues. The pair wise and multiple sequence alignments of known human Hyals described above (Hyal-1–4 and HPH-20) 12,101 show their sequence identity to range from 33.1% (between Hyal-4 and Hyal-3) to 41.2% (between Hyal-4 and HPH-20) (Table 2). There are a number of absolutely conserved regions of amino acid sequence, suggesting the functional and mechanistic importance of these residues. The major differences are located to the extreme C-termini of all these proteins, the possible locations of the additional functions attributed to these multifunctional enzymes.
Fig. 3. Sequence analysis of human hyaluronidases.

(A) Sequence alignment of the human hyaluronidases Hyal-1-Hyal-4, PH-20/SPAM1 based on the known X-ray structure of bee venom Hyal.
The identity between all sequences varies from 33.1% between Hyal-3 and -4 to 41.2% between Hyal-4 and HPH-20 12,101. The identity of the sequence of the BVHyal enzyme in the aligned region ranges from 22.9% for PH-20 to 25.2% for Hyal-1. The catalytic Glu H-donor residue and the residues positioning the nucleophile/base of the substrate are strictly conserved (Table 1), with the exception of Cys264 residue of Hyal-4, which may reflect specificity of this enzyme for Ch/ChS. The types of amino acid residues are color coded as follows: red - AVFPMILW small residue, green - STYHCNGQ hydroxyl, amine, or basic, blue - DE acidic, magenta - RK basic, and gray = others. The conserved residues are marked with ‘*’ – identical in entire column, ‘:’ – conserved according to color scheme above, and ‘.’ – semi-conserved substitutions are observed. The proposed catalytic Glu H-donating residue is in addition marked with ‘σ‘ whereas residues positioning the carbonyl nucleophile/base is marked with ‘λ‘. The sequence of the portion of the BVHyal enzyme that was crystallized is underlined. The sequences were aligned and the figure was made using Clastal W 1.82 101.
(B) Schematic diagram of domain composition of human Hyals.
Human Hyals are composed of two domains, a major catalytic domain followed by a C-terminal one of unknown function. These domains are connected by a probably flexible peptide linker. The short segment at the extreme N-terminus is independent of the catalytic domain and assumes an α-helical conformation 12. The secondary structure elements for each domain are also indicated.
5.2 Considerations at the genomic level
The smallest gene among the six human sequences is hyal-1 at approximately 3.7 kb, and the largest gene is hyal-4 at over 32 kb, a nearly ten-fold variation in gene size. There is no explanation at present for such differences. Human Hyal-1 has also been shown to retain intron 1, occurring within exon 1, in its mRNA. This generates two mRNA species, only one of which is translated into protein. This does not occur with any of the other hyal sequences. The mRNA with the retained intron cannot be translated, as it has a number of stop codons 102–104.
Human hyal-1 is a tumor suppressor gene, particularly for tobacco-related cancers. It is inactivated in most lung cancers in a conventional manner, by loss of heterozygosity or by homozygous deletion, at the DNA level. Interestingly, this hyal-1 is also inactivated in many head and neck carcinomas that are also tobacco-related by aberrant splicing of the mRNA, so that only the non-translatable form is transcribed 102. Thus it appears that tumor suppressor genes can be inactivated not only at the level of DNA, but occasionally at the level of RNA. It is not known why it is only the gene for human Hyal-1 among the six sequences that has the retained intron permitting alternative splicing, with transcription of multiple mRNA species. Recent analyses indicate that the Hyals can also function as oncogenes. Hyal-1 of humans is an oncogene in many cancers of the prostate and urinary tract. Hyal-2 can also function as an oncogene. Evidence from the laboratory of the Lokeshwars (University of Miami) clarifies some of these apparent discrepancies. These are dose-dependent effects 98. The bimodal curve indicates that tumor suppressor function and oncogenic effects occur at different concentrations or with different gene doses.
The hyal genes on human chromosome 3 show considerable complexity, and are located in a region densely packed with genes, in an occasionally overlapping manner 105. Co-transcription of several genes occurs, in a polycistronic manner, with coordinated tissue expression. This may have physiological significance, particularly in the mouse, but is lost to some degree in the human 105. The adage that only bacteria utilize polycistronic mRNA’s must be reconsidered.
Another curious feature of the vertebrate-like hyals is the presence of such a gene in bee venom. As discussed previously, HA occurs late in evolution, at some point early within the development of the chordates. The PH-20-like gene in the bee and hornet may be an example of lateral gene transfer. The enzyme may have been commandeered as a virulence factor, the hyaluronidase activity promoting penetration of other venom components. Such gene transfers appears to have occurred more frequently than realized previously, and may explain the presence of Has genes and HA in chlorella 106, or cellulose in the body wall of the tunicate 107.
Another example of putative lateral gene transfer in the presence of a collagenous sequence might be represented by a bacteriophage-coded hyaluronan lyase in Streptococcus pyogenes 108. This is a virulence factor, obviously of vertebrate origin, facilitating adhesion of the enzyme to the collagenous tissues of the host. The collagenous sequence codes for the minimum size needed for forming a stable collagen triple helix. Very high levels of hyaluronidase activities have been associated with only certain lysogenic strains of bacteriophage that infect S. pyogenes (group A streptococci), but not with virulent strains. They are termed HylP and HylP2 109,110. Virulent strains are unable to infect S. pyogenes, unless the HA capsule has been removed. This suggests that the function of the hyaluronidases of lysogenic phage is to penetrate the HA capsule 111. These hyaluronidases, referred to as the HylP-type have the lowest molecular mass of any hyaluronidases, ranging from 36 to 40 kDa 110 (see also section “8.4. Streptomyces hyalurolyticus hyaluronan lyase“). Both classes are lyase enzymes with absolute specificity for HA. One major difference is that while the HylP type contain a collagen-like repeat sequence 112, the HylP2 do not.
Yet another example of possible lateral gene transfer of Hyal hydrolase, with specific activity for HA, appear to be the enzyme of Mycoplasma alligatoris, a flesh-eating mycoplasma 113. It is a spreading factor of this organism113,114. It appears to be a very large enzyme composed of 1442 aa residues and an apparent multi domain composition. This Hyal belongs to CAZy glycoside hydrolase family 84, unlike the vertebrate, and specifically human, enzymes that belong to a different glycoside hydrolase group, family 56. The mechanism of this enzyme also appears to involve substrate as it is the case form vertebrate Hyals 115. In addition, the enzyme has an apparent carbohydrate-binding module that belongs to CAZy carbohydrate-binding module family 32
6. Sequence analysis, and homology and ab initio three-dimensional modeling for human hyaluronidases
The protein sequences of all full length human Hyal genes hyal-1–4, and PH-20 identified either experimentally or from the human genome (available at www.ncbi.nlm.nih.gov) were used in sequence homology and structural studies 12. These studies provide significant new information regarding primary, secondary structure and 3D structural properties of these proteins. Such sequence analyses lead to establishing additional functional properties and mechanisms for the human Hyals. The primary sequence alignments (Table 2, Fig. 3) indicate that all human Hyals analyzed are homologous to one another, as well as to the BVHyal and BPH-20 enzymes 11,86 (Fig. 3). Such analyses, and comparison with sequences of mouse Hyals, suggest that human Hyals are representative of all vertebrate Hyal enzymes (data not shown, and figure in supplementary materials).
6.1 Fold recognition studies
The state of the art fold recognition analyses 12 indicate that the catalytic portions, the major component of this group of enzymes, of all human Hyals, are very similar to one another at the primary, secondary, and tertiary levels, and are similar to those of the BVHyal enzyme (pdb code: 1FCQ). This similarity is consistently recognized by all fold recognition methods, even by the BLAST and PDB-BLAST approaches 116. The BVHyal structure is the top-scoring for all the individual consensus fold recognition methods utilized 12,117,118. This supports the hypothesis that all human Hyal enzymes conform to the single CAZY glycoside hydrolase family 56 59,60 and classification by SCOP database 119,120 as members of the single (trans)glycosidase superfamily of enzymes C.1.8. Other members of such SCOP superfamily include the catalytic domain of amylase 121, and type II chitinase enzymes 122 that are partially related to vertebrate-like Hyal enzymes. It should be recalled that their substrates have parallels. Amylase is a glycogen-degrading enzyme, glycogen being a polymer of glucose containing exclusively α bonds. Chitinase degrades chitin, which is a polymer of N-acetyl-D-glucosamine, comprised exclusively of β-links.
Additional support for the structural and functional correspondence for all these proteins comes from the excellent match between all predicted human Hyal secondary structures and to the actual secondary structure of the BVHyal obtained from X-ray crystallography (data not shown). Therefore, the fold recognition studies suggest that all human Hyals have 3D structures that are similar to one another, to that of BVHyal, and presumably to all other vertebrate Hyals.
6.2 Homology and ab initio modeling of the human hyaluronidases’ structures
Although the human Hyal sequences share only moderate (22–25%) sequence identity with that of BVHyal, the fold recognition studies described above indicate that the 3D structure of the BVHyal enzyme is the best, and the only structural template available for modeling these proteins. The relatively low sequence identity is not reflective of the structural similarity between these Hyals. Protein structures with such low identities are often found appropriate for homology modeling 123,124. The fold recognition methods clearly and unambiguously suggest significantly higher structural conservation than that suggested by sequence identities. Because of the homology with BVHyal structures, the structures of human Hyals can be modeled. All models must be viewed with caution, compared to experimental structures obtained using either X-ray crystallography or NMR. However, the significant structural homology with BVHyal structure allows for reliable 3D structure model determination for human Hyal-1–4 and HPH-20 enzymes; and may be considered as structurally representative of all vertebrate Hyals. Obtaining such models facilitated exploration of the mechanisms by which the vertebrate-like Hyals degrade HA, and to ascertain how differences between the Hyals occur. In particular, such models facilitate (1) establishing how human Hyal-1 degrades HA to tetrasaccharides, whereas (2) Hyal-2 degrades the same substrate to a limit digestion product of approximately 50 disaccharides, and (3) for HPH-20, the ability to facilitate sperm entry through the decidua and into the human ovum. And (4) insight is obtained potentially into more functional and mechanistic details of catalysis for human Hyal-3 and Hyal-4, since they are conspicuously lacking, compared to the other Hyals.
All models of human Hyal-1 through -4 as well as for HPH-20 are of high quality and are essentially identical to one another in the structure of their main domain (Figs. 4, 5). These five models, therefore, represent reliable structural models for all five human Hyal enzymes. For all human Hyals, a two domain structure is predicted for each enzyme (Table 3). The major, catalytic domain contains aa residues starting from the very N-terminus of each protein (Fig. 3), and appears to be always accompanied by a significantly smaller C-terminal domain of unknown function. The extreme N-terminal segments extending from the catalytic domain, ranging in length from 28 aa for human Hyal-1 to 41 aa for HPH-20, are not included in the BVHyal structure (Fig. 3). This segment’s model is predicted, therefore, by the ab initio approach. Due to the short length of this N-terminal segment, the modeling is likely to be reliable. The sequence similarity among this portion of all human Hyals is minimal, compared to the remaining portion. For the C-terminal domain, searches in structural databases showed no significant similarity to any known structures. As a consequence, this portion of each of the enzymes was also constructed by the ab initio approach 125,126. Even though the prediction methods have recently improved, such models still need to be looked upon with caution.
Fig. 4. Ribbon diagrams of three-dimensional structural model of human hyaluronidase Hyal-1.

The structure of the model is based primarily on the comparative/homology modeling (for the main, catalytic domain) 125 utilizing the structure of bee venom hyaluronidase (pdb code: 1FCQ) 11 as described by Jedrzejas and Stern 12. The modeled C-terminal domain was obtained using the ab initio method and its primary, secondary, or tertiary structure is not similar to any other molecule’s sequence of 3D domain structure currently available, which implies a novel fold and possibly novel function. A characteristic (β/α)8 TIM barrel fold of the main domain, which is the catalytic domain that supports the binding to the substrate, and its hydrolytic degradation by means of double-displacement, retaining mechanism (described in the text). Reprinted with permission from Reference 12. Copyright 2005 John Wiley & Sons, Inc.
(A) Three-dimensional model of Hyal-1 with bound hyaluronan.
The molecule is color-coded by the secondary structure elements (same as in Fig. 2A). The bound HA molecule is located in the HA-binding cleft and is depicted in ball and stick fashion colored in purple. The catalytic residue, Glu131 is also shown in ball and stick fashion colored in red (labeled); other residues that position the carbonyl of the acetylamido group of HA are also shown and are colored in blue (not labeled). The catalytic and C-terminal domains, as well as the N- and C-termini are shown and labeled.
(B) Comparison of 3D structure of Hyal-1 and BVHyl enzymes.
The Hyal-1 molecule is shown in the same color and orientation as in panel A (including HA). The BVHyal enzyme structure is overlaid on the structure of the Hyal-1 model and colored in blue. The positions of the catalytic Glu and carbonyl positioning residues are essentially identical in the two structures (data not shown). The BVHyal does not have a C-terminal domain.
(C) Surface of the Hyal-1 molecule and a tetrasaccharide HA substrate bound in the cleft.
The orientation of the molecule is similar to that in panels A-B. The protein surface is colored as in Fig. 2B. The surface of the HA tetrasaccharide bound to the enzyme is shown and color in purple.
Fig. 5. Ribbon diagrams of structural models of human hyaluronidase Hyal-2, Hyal-3, Hyal-4, and HPH-20.

The molecule is sown in similar orientation and using the same color scheme and labeling as that of Hyal-1 in Fig. 4 A-B. The structures of the catalytic domains are similar to the same domain of Hyal-1 and to BVHyal enzyme. The structure and the size of the C-terminal domain are different between every Hyal and different from the same domain of Hyal-1. Reprinted with permission from Reference 12. Copyright 2005 John Wiley & Sons, Inc.
(A) Model of Hyal-2, which degrades hyaluronan in human tissues to the approximate size of 50 disaccharides (~20 kDa).
(B) Structural model of human PH-20/SPAM1 hyaluronidase found in sperm, involved in fertilization, as well as facilitating the penetration of sperm through the cumulus mass to reach the ovum.
(C) Structural model of a relatively little investigated Hyal-3.
(D) Model of Hyal-4 with the putative chondroitinase activity.
Table 3. Domain arrangement of human Hyals.
The domains were predicted by the Ginzu domain parsing and fold detection method 126 and unanimously confirmed with very high confidence implemented in the META server/3D Jury method 118,155 as reported by Jedrzejas and Stern 12. All Hyals have a two-domain architecture with the major domain being the catalytic one with a high primary, secondary, structural and functional homology to the bee venom hyaluronidase of known 3D structure (pdb code: 1FCQ). The structure of BVHyal consists of aa residues 33–382 of the full length mature protein. The second and minor domain is located at the extreme C-terminus, and does not have homology with any known proteins. Sequence conservation of the C-termini among all hyaluronidases is significantly lower than that for their catalytic domains. The function of these C-termini is unknown.
| Hyal | Hyal AA span | Domain | Domain AA span | Parent | Parent AA Span | Functional annotations |
|---|---|---|---|---|---|---|
| Hyal-1 | 1–435 | 1 catalytic | 1–371 | 1FCQb | 33–360 | hydrolase/hyaluronidase |
| 2 C-terminal | NAa | NAa | Unknownd | |||
|
| ||||||
| Hyal-2 | 1–471 | 1 catalytic | 1–376 | 1FCQb | 33–366 | hydrolase/hyaluronidase |
| 2 C-terminal | 377–471 | NAa | NAa | Unknownd | ||
|
| ||||||
| Hyal-3 | 1–463 | 1 catalytic | 1–373 | 1FCQb | 41–366 | hydrolase/hyaluronidase |
| 2 C-terminal | 374–463 | NAa | NAa | Unknownd | ||
|
| ||||||
| Hyal-4 | 1–481 | 1 catalytic | 1–384 | 1FCQb | 41–366 | hydrolase/hyaluronidase |
| 2 C-terminal | 385–481 | NAa | NAa | Unknownd | ||
|
| ||||||
| HPH-20 | 1–510 | 1 catalytic | 1–389 | 1FCQb | 41–366 | hydrolase/hyaluronidase |
| 2 C-terminal | 390–510 | NAa | NAa | Unknownd | ||
|
| ||||||
| BVHyal | 1–382 | 1 catalytic | 1–382 | 1FCQb | 33–382c | hydrolase/hyaluronidase |
NA - Not Available or unknown.
pdb code is reported (3D structure coordinates available at www.rcsb.org/pdb under this code).
The crystallized BVHyal enzyme consists of aa residues 33–382 of the full length protein. Residues 33–43, 97–103, and 363–382 were not identified in the 1FCQ X-ray crystal structure11.
The state of art sequence comparisons and structural analyses of the ab initio 3D model has not revealed any protein with similar sequence or 3D structure to the C-terminal domain.12
7. Structures and mechanism of action of human hyaluronidases
7.1 Description of the model structures
For all five human Hyal structure models, the major catalytic domain (Fig. 4, 5) closely resembles the (β/α)8 TIM barrel of the parent BVHyal structure. The number of sheets constituting the central part of the TIM barrel varies, however, but is similar to that of various forms of the BVHyal structures. The wider end of the TIM barrel assumes a conformation that creates a large and elongated cleft. Its sides are built primarily from aa residues of loops formed between sheets and helices of the TIM barrel. Based on similarity to BVHyal 11 and mutation analysis of HPH-20 89, this region is responsible for binding the HA substrate and for catalysis, and is large enough to bind at least an octasaccharide segment of HA. The extreme N-terminal segment of the catalytic domain, ranging in length from 28 aa for human Hyal-1 to 41 aa for HPH-20, always assumes a helical conformation. It takes on a structure of a single helix for Hyal-1, and Hyal-2 (Hyals with the shortest segment at their N-terminus) or a set of two helices connected by a short loop for Hyal-3, -4 and HPH-20. Hyal-4 and HPH-20 have the longest aa segments at their N-termini (Fig. 3). In all five cases, however, the helices are located on the outside of the TIM barrel core structure, are not incorporated into the distorted TIM barrel fold, and are primarily associated with the catalytic domain only; the interactions with the C-terminal domain are minimal, if any (Fig. 4, 5). The sequence similarity in this region of all human Hyals is very low, compared to the remaining portion of these Hyals, and therefore allows for some variations in structure.
For all human Hyals at their C-termini, the catalytic domain is followed by the second domain, which is not present in the BVHyal homolog structure. This part of human Hyals is significantly smaller than that of the catalytic portion, but larger than the N-terminal segment of the catalytic domain, ranging in size from 68 aa for Hyal-1, to 122 aa for HPH-20 (Table 3, Fig. 3). The sequence similarity along this portion is minimal, compared to the remaining part of the enzymes, including the extreme N-terminus. The structure of this domain varies significantly among different human Hyals. It assumes a three-stranded antiparallel β-sheet flanked on one side by a helix, and low complexity structures on the other extreme C-terminus side for Hyal-1 (Fig. 4A, B). In Hyal-2, it is composed of a 2-helix group, whereas in HPH-20 it is a group of 8 helices, 3 of the helices being very short. In Hyal-3 it is composed of two helices with a significant portion of a coiled region. Hyal-4’s C-terminal domain assumes a structure of three helices flanked on one side by an antiparallel, two stranded β-sheet (Fig. 5). There is one commonality however. The C-terminal domain is always separated from the catalytic part by a linker peptide. This presumably flexible peptide is of significant length, making this domain relatively independent of the rest of the protein, and certainly with its own separate motion characteristics (Fig. 4, 5) 12.
7.2 Structure of the active site
The active site of the human Hyals is located within the substrate binding cleft that traverses the wider end of the TIM barrel structure of the catalytic domain. The cleft does not extend to the C-terminal domain. It is lined by a number of positive and hydrophobic aa residues. As such, it is ideally suited to bind the negative and hydrophobic HA/Ch/ChS substrates. The active site is composed of one catalytic residue, Glu131, and a group of several residues that position the carbonyl nucleophile for catalysis. Structural alignment of all the available Hyals, including all the modeled human Hyal structures, the BVHyal X-ray structure, its X-ray structure complexed with HA substrate, plus BPH-20 demonstrate consistent structural conservation. The same positioning of the catalytic Glu131 (human Hyal-1 numbering) residue recurs, and is accompanied by a group of substrate positioning residues: Asp129, Tyr202, Tyr247, and Trp321 (human Hyal-1 numbering) residues. All these residues are also strictly conserved in sequence (Table 1, Fig. 3, 6) (and data not shown). The Tyr247 aa residue is replaced by Cys263 in human Hyal-4, an enzyme that may have different substrate preferences, as it degrades predominantly Ch and ChS.
Fig. 6. Catalytic part of the Hyal-1 HA binding cleft.

The scheme is based on the structure of Hyal-1 enzyme (based on structure of Hyal-1 as reported by Jedrzejas and Stern 12) with HA tetrasaccharide positions as in the structure of BVHyal homolog (pad coordinate code: 1FCV). The reducing end of bound HA molecule is located in the HA-binding cleft and is depicted in ball and stick fashion colored in by atom type as in Fig. 2B. The catalytic residue, Glu131 is also shown in ball and stick fashion colored in red (labeled); other residues that position the carbonyl of the acetylamido group (labeled) of HA are also shown and are colored in black (labeled). The C1 atom and carbonyl group (marked as C=O) of HA’s N-acetyl-D-glucosamine are labeled.
(A) View from above HA and HA-binding cleft down towards the enzyme’s catalytic domain.
The catalytic H-donor (acid) Glu131, in red, is shown in position to interact with the C1 carbon. The carbonyl positioning residues, in blue, are deeper in the enzyme’s cleft in which the acetamido group is located.
(B) Alternate view from down the HA chain along the HA binding cleft.
The carbonyl positioning residues are clearly in position to modify the position of the acetamido group to allow interaction of the carbonyl group with C1 carbon.
(C) Schematic double displacement, retaining mechanism common for human hyaluronidases.
This mechanism is characteristic of polysaccharide hydrolases and it involves the five depicted steps, 1–5, and one aa residue, Glu, of the enzyme (as described in the text). It involves a double displacement mechanism with the retention of C1 carbon (marked with an asterisks) configuration (retaining enzyme) 2. The catalytic process proceeds through an intermediate reaction step involving oxocarbonium ion at the C1 position of N-acetyl-D-glucosamine of the substrate. The catalytic acid function is performed by an absolutely conserved mechanism among Hyal enzymes Glu residue. The nucleophile/base function is attributes to the carbonyl oxygen of C-2 acetamido group of the substrate: HA, Ch, or ChS.
As a part of catalysis, Glu113 acts as an acid, H-donor. The substrate positioning residues are involved in precise orientation of the carbonyl of the acetamido group of HA, which serves as a nucleophile for performing this function, instead of the usual second acidic residue. In such a situation, no enzyme-substrate covalent intermediate is formed.
7.3 Catalytic mechanism of hyaluronidase activity
Based on high primary, secondary, and tertiary homology to BVHyal and site directed mutagenesis of HPH-20 enzyme, it is proposed that degradation by human Hyal hydrolases may proceed via a double-displacement mechanism, with retention of HA substrate conformation. Such a mechanism involves one glutamic acid aa residue as an H donor (Glu149 in BPH-20, equivalent to Glu131 of human Hyal-1, (Table 1)) and a carbonyl oxygen of N-acetyl group of the HA performing the function usually assigned to another carboxylic aa. The usual two carboxylic acid mechanism can be modified to reflect the different nucleophilic residues. The steps involved in this mechanism are (Fig. 6C): (1) binding of Hyal to the HA substrate; (2) residues around the catalytic site positioning the carbonyl oxygen nucleophile of the HA’s N-acetyl group next to the to-be-cleaved β1,4 glycosidic bond, attacking the C1 carbon of the same sugar to form a covalent intermediate between them. This leads to cleavage of the glycosidic bond on the non-reducing side of the glycosidic oxygen. The process also results in the inversion of the anomeric C1 atom configuration; (3) at the same time the protonated Glu149 donates its H (deprotonation, acid function) to the glycosidic oxygen, leaving part of HA (glycan on the reducing side of the cleaved glycosidic bond); (4) hydrolytic cleavage of the carbonyl oxygen-C1 intermediate bond by a water molecule in the active site, leads to re-protonation of Glu149, readying it for the next catalytic step, and to the second inversion of the configuration of C1; and (5) release of the HA product from the Hyal’s active site (glycan on the non-reducing side of the cleaved glycosidic bond).
The anomeric configuration of the C1 carbon atom of the substrate is retained because it is inverted twice during catalysis (in steps 2 and 4 above). The formation of an oxocarbonium-ion transition state has been implicated in this process at step 2 described above. Structural evidence from the BVHyal-HA tetrasaccharide complex suggests that (1) as the carbonyl nucleophile moves into place to interact with the C1 carbon of HA, it results in (2) change in puckering of the pyranose ring of N-acetyl-D-glucosamine on the non-reducing side of the bond to be cleaved, from regular chair to distorted boat and (3) consequently, in moving the glycosidic bond into nearly an equatorial position, which results in (4) its positioning closely to the Glu149 (BVHyal numbering) to allow for the donation of its H to this glycosidic O as the bond is being cleaved 11.
7.4 An endolytic, random cut pattern of hyaluronidase activity
This process of HA degradation occurs through a random bite type of catalysis. This is an endolytic, non-processive mechanism, with the products of each degradation reaction becoming substrates for further cleavage. At each step, following cleavage of the glycosidic bond, the catabolic product leaves the active site of the enzyme.
The shortest HA fragment that can still be cleaved by the mammalian enzymes is a hexasaccharide 21,127. As has been established, the final end-products of HA digestion are tetrasaccharides. These however can be further degraded by the lysosomal β-exoglycosidases, β-glucuronidase and β-N-acetyl-glucosaminidase. As mammalian Hyals are also able to degrade Ch and ChS, albeit at slower rates, the mechanism described above should hold for these substrates as well. The sulfation pattern of ChS can presumably be accommodated by these human Hyal enzymes, allowing for binding and catalysis. The prediction would be that these enzymes cleave ChS in regions of low or of no sulfation.
7.5 The putative functions of the C-terminal domains
The lack of experimental data and of sequence/structural homolog(s) for the C-terminal 128 make functional studies of this human Hyal domain purely speculative. However, the peptide linker between the catalytic and the C-terminal is indicative of a dynamic flexibility. This also indicates an independent role for the C-terminal domain. The different structures of this domain for each of the five human Hyals imply a different function for each Hyal. Analysis of their structural and surface properties suggest HA-binding ability as a component of their function. In general, all models of this domain have large grooves/clefts that could easily accommodate the HA substrate. In the absence of additional evidence, no clear account can be established for possible differences between them. This is in marked contrast with the additional domain of the bacterial enzymes (which in this case are all located at the N-termini). However, sequence and structural homologies do exist for these additional N-termini of the bacterial Hyals. In these enzymes, N-terminal domains have homologies to other glycan-binding modules found in other catabolic carbohydrate-active enzymes, such as cellulases and xylanases 128,129.
The possibilities are that these domains are involved in interactions with HA and as such: (1) might increase the general affinity of Hyals for the HA matrix and, therefore (2), participate in simple co-localization of enzyme and substrate. A further possible consequence of binding to HA might be (3) disruption of non-covalent interactions between the substrate chains or between substrate and other polysaccharides 130. Also, these domains may play a role in (4) the appropriate orientation of the substrate to the catalytic domain, to further facilitate HA degradation e.g., by directional positioning of the HA’s reducing end. This is clearly the case for the bacterial Hyals, which have been studied in much greater detail than the human enzymes 2,31,128. Each of these functions improves the efficiency of HA cleavage. Flexibility and solvent exposure of the linker peptide suggests it would be most vulnerable to protease degradation, as has been observed for the bacterial Hyals 71,131,132 and the BPH-20 enzyme 98. Such degradation indeed occurs, but does not change the specific activity of Hyals in vitro (both bacterial-and vertebrate-like). Similar arguments can be made for the extreme N-terminus of the catalytic domain. That domain is only loosely associated with the TIM barrel. Its proteolytic removal should not affect in vitro specific activity. However, in vivo activity of such N- or C-terminal truncated enzymes in tissues would likely be affected, to their individual physiological requirements.
The bioinformatics/structural results demonstrate the need for further research, in order to determine C-terminal domain specific function(s), and to delineate avenues for such studies. It is, however, not coincidental that the bacterial Hyals are also multidomain enzymes. In their case, however, it is the extreme N-terminal domain that is implicated in HA-binding in the variable processes described above 128. This is in contrast with the vertebrate Hyals, in which it is the functions of the C-terminal domains that await exploration.
8. Bacterial hyaluronidases, their structure and mechanism of action
8.1 Polymeric glycan-binding cleft is a common feature of the hyaluronidases and other glycan-degrading enzymes
A common structural feature of all HA-degrading enzymes is their elongated cleft for accommodating substrate. Such a cleft is found in the Hyals from Streptococci, the enzyme assuming a distorted (α/α)5–6 barrel composed of α-helices inside and out 13,14. HA binding is facilitated by the cleft located at the top of the wider end of the barrel nested among helices and inter-helix loops. On the other hand, Flavobacterium heparinum chondroitinase B enzyme that degrades dermatan sulfate adopts a right-handed parallel β-helix fold 133. This fold is similar to that found in pectin lyases 134,135 and pectate lyases 136–139. In these structures, the cleft consists of loops extending from the surface of the strands of the β-helix. Such a β-helix fold has also been identified in several polysaccharide hydrolases, such as in rhamnogalacturonase A 140, polygalacturonase A 141, and the tail spike protein of phage P22 (polysaccharide hydrolase) 142,143, implying that the cleft is specific for binding of elongated polysaccharides, and is not necessarily for their catalysis. In some enzymes, the cleft is covered to form a tunnel, a structure specific for cellbiohydrolases 144,145.
This important feature of such clefts are their predominantly positive and hydrophobic character, and as such, facilitate binding of negatively charged hydrophobic HA/Ch/ChS substrates 30,31. This process is therefore driven by interactions of two molecules, enzyme and polymer, and the charge potential, which they generate. The energy for the reaction is likely generated by the exothermic reaction that occurs with degradation of the glycosidic bond.
8.2 Domain structure bacterial hyaluronidases
Structural and bioinformatics studies of streptococci (namely S. pneumoniae and S. agalactiae) demonstrate a four-domain bacterial Hyal enzyme anchored to the surface of these bacteria through a covalent linkage to the cross bridges of peptidoglycan structures 13,128. The four Hyal domains are an N-terminal carbohydrate binding domain, a spacer, followed by the catalytic, and finally the C-terminal domain (Fig. 7A) 13,128. The X-ray crystal structure of the three major streptococcal domains have been elucidated (Fig. 7B) 13,14.
Fig. 7. Three-dimensional structure of S. pneumoniae hyaluronate lyase.

(A) Schematic diagram of domain composition of bacterial Hyals.
Bacterial Hyals are composed of four domains, a HA-binding module, a spacer domain, a major catalytic domain (α-domain), followed by a C-terminal one (β-domain) that regulates HA access to the cleft 128. These N-terminal HA-binding module and the most C-terminal domains are connected to the rest of the protein by presumably flexible peptide linkers. The module at the extreme N-terminus is independent of the rest of the protein and assumes an all β-sheet structure. The secondary structure elements for each domain are indicated.
(B) Overall structure.
The crystallized enzyme is built from an α-helical catalytic domain and a supportive β-sheet domain (based on structure coordinates, pdb code: 1LOH) 31. Both domains are connected by one flexible peptide linker. The most N-terminal two domains, a spacer domain (composed of β-sheets) and the additional HA-binding one (α/β fold) are included in this structure 128. Two perpendicular views of the enzyme (rotated along the axis parallel to the labeled N- - C-termini) are shown. The hexasaccharide hyaluronan substrate is depicted in the enzyme’s cleft in a ball and stick fashion color coded by atomic element as in Fig. 2B. The molecule is colored by the secondary structure elements (α-helices- blue, 3/10-helices- purple, β-sheets- green, coil regions- brown). N-and C-termini are labeled.
(C) Catalytic residues of the enzyme.
The residues directly involved in catalysis are Asn349, His399, and Tyr408 (catalytic group); positioning of the substrate are Trp291, Trp292, and Phe343 (hydrophobic patch); release of the product are Glu388, Asp398, and Thr400 (negative patch) are shown together with the hyaluronan hexasaccharide substrate (based on structure coordinates, pdb code: 1LOH) 31. The most essential enzyme-substrate interactions for the catalytic process are shown as black lines. Consecutive HA disacchrides from the reducing to non-reducing end are labeled HA1-HA3. All residues and HA hexasaccharide are shown in ball and stick fashion and color coded by atomic element (as in Fig. 2B).
(D) Proton acceptance and donation mechanism of bacterial hyaluronan lyases.
This mechanism consists of a five steps process and involved three residues of the enzyme, Asn, His, and Tyr, as described in the text 4,13. The glycosidic oxygen as well as C4 and C5 carbon atoms directly involved in catalysis are also marked by asterisks. There is no direct water molecule involvement in this catalytic process except during H exchange to ready enzyme for the next round of catalysis. Smaller HA chains are primarily degraded in a processive manner, whereas large aggregated HA molecules, Ch and ChS with selected sulfation patterns (as described in the text) of any length are degraded non-processively 1.
The crystallized native S. pneumoniae enzyme molecule 71,131 is composed of the major, catalytic domain having an (α/α)5–6 α-helical barrel structure, and the most C-terminal domain, comprised mainly of β-sheets, arranged in an antiparallel, large three layer β-sandwich. The structure of the S. agalactiae Hyal is similar to that of S. pneumoniae Hyal but, in addition, encompasses an additional domain at the N-terminus, functioning as a spacer domain, which is absent in the pneumococcal enzyme 14,132. This spacer domain is also composed of β-sheets. The function of the C-terminal β-sheet domain is to modulate the polymeric HA/Ch/ChS substrate’s access to the catalytic cleft, present in the α-helical catalytic domain. The catalytic cleft traverses the α-helical domain. Only three substrate disaccharide building blocks (i.e., a hexasaccharide) can be fitted inside the cleft 30,31. The active site of the enzyme is also located within the barrel domain’s cleft and is composed of three residues; Asn349, His399, and Tyr408 (S. pneumoniae Hyal numbering) (Fig. 7C) 5,13,31,62. However, bioinformatic studies reveal that the full length mature enzyme contains additional residues at the N-terminal, arranged in an additional domain 128. This domain is clearly a carbohydrate binding module that presumably acts to enhance overall affinity of all known bacterial Hyals for their substrates. As a result, the second, small spacer domain likely acts as a spacer, to distance the C-terminal domains from the carbohydrate-binding domain located at the extreme N-terminus.
8.3 The mechanism of hyaluronan-degradation by bacterial hyaluronan lyases
The mechanism of streptococcal, and presumably of all bacterial Hyal lyase enzymes is the β-elimination, acid/base, processive type, and is termed proton acceptance and donation (PAD), as described by Li et al. 13. The catalytic mechanism elucidated for S. pneumoniae and S. agalactiae Hyals involves several discreet steps: (1) binding of the HA substrate contained in the cleft; followed by (2) the acidification of C5 carbon atom of HA’s glucuronate residue by an Hyal’s Asn aa acting as an electron sink (Fig. 1, 7C, D); (3) extraction of this C5 carbon proton by the enzyme’s His residue, followed by the formation of an unsaturated bond between C4 and C5 of the glucuronate on the reducing side of the glycosidic bond; and (4) cleavage of the glycosidic bond after a proton is donated from the enzyme’s Tyr residue, and finally, (5) departure of the HA disaccharide product from the active site and balancing of the hydrogen ions by an enzyme exchange with the water environment (Fig. 7C, D) 13,14,74. The enzyme is then ready for the next round of catalysis. For processive degradation of substrate(s), HA is translocated by one disaccharide unit toward the reducing end of the chain and endolytically degraded using the PAD mechanism. During the process, the C4 and C5 carbon atoms change their hybridization from sp3 to sp2 with respective changes in the product conformation of the sugar ring, involving a puckering of the sugar ring, leading then to a distorted half chair conformation (Fig. 7D).
In addition to the three catalytic residues, additional residues are involved in important aspects of the enzyme’s action: a patch of hydrophobic residues, Trp291, Trp292, and Phe343, as well as a patch of residues generating negative potential at the end of the cleft, Glu388, Asp398, and Thr400 (Fig. 7C). The hydrophobic patch interacting with substrate positions it for catalysis, whereas the negative patch facilitates release of product. The structure of the enzyme complex with substrates and products of degradation reveal the exact substrate positioning with respect to the residues identified in the cleft, and show that the enzyme degrades the substrates starting from the reducing end. In this model, the degradation commences by initial endolytic binding of enzyme to substrate, followed by an initial cut of the polymer; this ‘initial bite’ is followed for HA, but not for Ch/ChS, by progressive exolytic degradation from the reduced end towards the non-reducing end, one disaccharide unit at a time, until the polymeric chain is fully degraded. The degradation of Ch and ChS, on the other hand, seems to proceed only by the endolytic, non-processive method.
As a consequence of HA aggregation, initial degradation of high molecular weight HA is likely to proceed through a random endolytic cleavage, but only at sites where the β1,4 linkage in the chain is exposed. As the size of HA decreases, its ability to aggregate also decreases. At molecular masses below 300 kDa, the ability of HA to aggregate, as shown by electron microscopy-rotary shadowing, decreases 56. At the same time, HA chains below ~50 disaccharides, as shown by light scattering evidence, do not aggregate in salt solutions 57. These properties indicate that at some size below 300 kDa, the molecule can be degraded by streptococcal, and possibly other bacterial hyaluronate lyases using a purely processive mechanism, due to decreased aggregation. As the average size decreases, the processive mechanism takes over from random cleavage, leading to faster exponential degradation 1,3.
8.4. Streptomyces hyalurolyticus hyaluronan lyase
Streptomyces hyalurolyticus Hyal is yet another bacterial with apparently different properties that those of streptococcal Hyals 27,37. This enzyme appears to act on HA in a random endolytic action pattern and produces unsaturated products of varied size 146,147. It also utilizes a β-elimination cleavage mechanism as the streptococcal enzymes. However, the most abundant products are primarily the HA disaccharides, then tetra-, and then hexasaccharide units of HA 15,27,31,146. The sequence of this enzyme is, however, not known preventing its additional analyses. The peculiarity of this Hyal is that unlike majority of other Hyals it is specific for HA only. It does not appear to degrade chondroitins 37,146. The reasons of this unexpected and surprising specificity are at present not known. Certainly, more studies are needed to confirm this specific degradation under broad range of conditions and utilizing unsulfated chondroitins as well as those with varying sulfation patterns. In addition, certain size of HA digestion products are under-represented, namely those comprised of 5 to 6 disaccharide units. Park et al., 1997 suggested that this resistance might be the result of secondary or higher order structures of HA 146. The enzyme is readily available from Sigma (catalog no. H1136, with an activity of ~0.1 NFU/vial) and as such is often utilized in studies of HA degradation and as means of producing of short HA fragments, namely di-, tetra-, and hexasaccharides.
More recently anther hyaluronan lyase of microbial/viral origin with specificity only for HA from Streptococcus pyogenes bacteriophage H4489A (termed HylP) 111,148 was reported (see also section “5.2 Considerations at the genomic level”). As its sequence is know it allowed for an additional insight in the properties of this Hyal. The enzyme is very short in sequence, only 371 aa residues, suggesting different molecular structure and architecture than that of streptococcal Hyals described above 149. The phage Hyal belongs to CAZy polysaccharide lyase family 16 unlike the streptococcal enzyme which belong to a different polysaccharide lyase group, family 8. More studies are clearly needed to elucidate the properties of this apparently new enzyme.
9. Summary
9.1 Conclusions
This study provides the first descriptions of 3D models s for the human Hyal enzymes: Hyal-1 through –4, and PH-20. The models indicate that they are very similar, and that they differ primarily in their C-terminal domains, whose functions are unknown. The catalytic clefts and the active sites are highly conserved. The models provide insight into their catalytic mechanisms. This involves an acidic residue, Glu131 (human Hyal-1 numbering) that functions as an H-donor. No other acidic residue, such as another Glu or an Asp in the active site could support such a function. Therefore, as is the case for some chitinases, these enzymes utilize the substrate carbonyl group of the acetamido moiety as a nucleophile. No enzyme-substrate covalent intermediate is created in this process, in marked contrast with all other glycan-degrading hydrolases.
The sequence analysis, fold recognition, and modeling studies make possible the identification of the residues involved in catalysis and their mechanism of action. The structures of the C-terminal domain in particular must be viewed with caution. Further experimental evidence is needed for delineating the function of this part of the human Hyals. Additional data must be obtained and a larger database generated of 3D structures with annotated functions for the various domains, to further enhance our understanding of this class of enzymes.
Information regarding the Hyals, both the hydrolases and the lyases, has accumulated rapidly. The human and microbial genome projects, the structural studies, and the number of computer-assisted bioinformatic approaches have provided a fund of information that facilitates our understanding of how these enzymes operate. However there are many areas that require continued investigation. The vertebrate and vertebrate-like Hyals are multi-functional enzymes. The functions of the C-termini, separated from the catalytic domains by linker peptides, have the greatest variability and the least homology. It is these C-termini that may help identify these additional functions.
9.2 Future directions
Other questions abound. What is the identity of the organism in evolution in which the first Hyal sequence duplication event occurred, or the second, to generate the three closely associated genes? And when did the en masse block duplication occur to form the resulting total of six sequences that occur presently? Evidence suggests this may have occurred when placental mammals separated from non-placental mammals.
Silencing of the sixth sequence, the pseudogene, Phyal1, with premature stop codons, occurs only in the human, among those that have been sequenced to date. This suggests that the Hyals are continuing to evolve. It would be intriguing to determine if silencing of this gene has occurred in the chimpanzee, whose genomic sequencing is now underway.
Why are the introns for the sequences on chromosome 7 up to ten times the size of those on chromosome 3 (e.g., human hyal-4 compared to hyal-1)? Do these introns have functions of which we are not aware? Do these functions include regulatory activities for transcription? Do such large introns reflect older sequences or do they represent more recent events? This question can be restated. Is it the Hyals on chromosome 3 or on 7 that are more closely reflect the original cluster of three sequences?
It has been established that hyal-1 has two major transcripts, only one of which can be translated 104, the other having a retained intron that prevents translation. It appears that most tissues contain both pre-mRNA’s. What is the intelligence behind the splice mechanisms that determines the ratio for these pre-mRNAs? Increases in the non-translatable transcripts is one of the mechanisms for silencing this tumor suppressor gene, particularly in tobacco-related cancers. How is this accomplished? The answer to how alternative splices are selected by the splice mechanism is a major unsolved problem. Are there other sequences in the genome that code for hyaluronidase-like enzymes, in addition to and unrelated to the six (or seven) that have been identified?
We are still unable to ascertain why human Hyal-2 catalysis ceases, or slows, when the HA substrate reached 50 disaccharide units. Is it a function of the enzyme, or a change in conformation of the HA substrate at this critical size? HA exists in vertebrate tissues in a number of states: as a freely circulating molecule, associated with a number of HA-binding proteins or hyaladherins, tightly intercalated within proteoglycan aggregates, loosely associated with tissues, or tethered to the cell surface by a number of membrane receptors. HA is now also recognized as being an intracellular molecule, within cytoplasm, nucleus, and also in the nucleolus 150,151. In some cases of severe ER (endoplasmic reticulum) stress, HA can form cables that appear to originate from the perinuclear Golgi, and project out into the ECM. The question becomes, how do the vertebrate Hyals recognize their HA substrate in such multiple formats? How do the structures of the Hyals participate in this recognition process?
The vertebrate Hyals function at either acid or neutral pH, yet they retain the same basic structure. How is this accomplished? Both human Hyal-1 and Hyal-2 can function as either tumor suppressors, or as oncogenes. This is in part a gene-dose-dependent phenomenon 152 but also depends on the type of malignancy, as well as other, as yet, unknown factors. How is this accomplished, and how does substrate-enzyme interactions participate in this process? The bee venom Hyal is a major antigen of bee venom, involved in allergic reactions. Some individuals are extraordinarily sensitive to these antigens. What epitopes within this Hyal is involved in the hyper allergenic process?
Human monocytes contain a chitinase activity. Chitin is a polymer of β-N-acetylglucosamine, bearing some resemblance to HA. This monocytes/macrophage chitinase occurs in a precursor higher molecular weight form requiring two endoproteolytic steps to generate the final product 153. This is not a zymogen-active enzyme relationship, since they have the same specific activity. Of interest is that Hyal-1, and possibly other Hyals undergo similar processing 154. Does this processing also occur with other β-chain sugar polymer-degrading enzymes?
Clearly, there is still much to be done within the Hyal field. Only a coordinated approach that includes biochemistry, cell biology, structural chemistry, crystallography, computer-assisted bioinformatics, immunology, and molecular genetics can provide the answers to the many questions that remain.
Supplementary Material
Acknowledgments
The authors would like to acknowledge assistance and helpful discussions with present and past members of their laboratories. This work in the authors’ laboratories has been supported by a grant from the National Institutes of Health AI 44078 (to MJJ) and the Department of Pathology, UCSF (RS).
Abbreviations
- 3D
three-dimensional
- aa
amino acid
- Ch
unsulfated chondroitin
- ChS
chondroitin sulfate
- BVHyal
bee venom Hyal
- BPH-20
bovine PH-20
- ECM
extracellular matrix
- ER
endoplasmic reticulum
- GAG
glycosaminoglycan
- GPI
glycosylphosphatidylinositol
- HA
hyaluronan, hyaluronic acid
- HPH-20
human PH-20
- Hyal
hyaluronidase
- MD
molecular dynamics
- NMR
nuclear magnetic resonance
- PCR
polymerase chain reaction
- pdb
protein data bank (3D structure depository)
- TIM
triose phosphate isomerase
Biographies
Mark J. Jedrzejas: Dr. Jedrzejas received his B.A./M.S. in Physics from Jagellonian University, Krakow, Poland in 1988, a M.S. in Chemistry in 1992 from Cleveland State University, and a Ph.D. in Structural Chemistry from Cleveland State University/Cleveland Clinic Foundation in 1993. He was appointed an Assistant Professor of Microbiology at the University of Alabama at Birmingham in 1995. In 2001, Dr. Jedrzejas moved to the Children’s Hospital Oakland Research Institute as an Associate Scientist where he directs a structural biology group and continues his studies of the structure and mechanisms of macromolecules using predominantly tools of structural biology with emphasis on X-ray crystallography. His research interests include structural aspects of Gram-positive bacterial pathogens, mainly bacteria-host interactions, with focus on Streptococcus species, and mechanisms of essential processes leading to the formation of bacterial spores, their germination and outgrowth, with an emphasis on Bacillus and Clostridium species. Recent years his laboratory determined crystal structures of hyaluronan lyases from Streptococcus species and modeled structures of human hyaluronidases. These studies lead to the determination of their mechanisms of degradation of hyaluronan as a part of a broad effort to understand the role and function of exo- and endogenous pressures exerted on this polymeric molecule of vertebrate animals.
Robert Stern left Germany in 1938 for Seattle, Washington. He graduated from Harvard College in 1957, and obtained the M.D. degree from the University of Washington (Seattle) in 1962. While a medical student, he worked in the laboratories of Drs. Krebs and Fisher, who became Nobel laureates. He received his resident training in Pathology at the NCI, and was a research scientist at the NIH t for 10 years. Since 1977, he has been a member of the Pathology Department at the University of California, San Francisco. He is a board-certified Anatomic Pathologist, participating in the research, teaching, administrative, and diagnostic activities of the Department. He directed the Ph.D. program in Experimental Pathology for ten years. For the past decade, his research has focused on hyaluronan and the hyaluronidases, an outgrowth of an interest in malignancies of connective tissue, stromal-epithelial interactions in cancer, and biology of the tumor extracellular matrix. His laboratory was the first to identify the family of six hyaluronidase sequences in the human genome. These enzymes were then sequenced, expressed, and characterized. Subsequent work has identified a catabolic pathway for hyaluronan. Dr. Stern was a Fulbright scholar in Germany (1984–5, He is currently a member of the editorial boards of Matrix Biology, and the University of California Press.
References
- 1.Rigden DJ, Jedrzejas MJ. J Biol Chem. 2003;278:50596–606. doi: 10.1074/jbc.M307596200. [DOI] [PubMed] [Google Scholar]
- 2.Jedrzejas MJ. Crit Rev Biochem Mol Biol. 2000;35:221–51. doi: 10.1080/10409230091169195. [DOI] [PubMed] [Google Scholar]
- 3.Jedrzejas MJ. Front Biosci. 2004;9:891–914. doi: 10.2741/1299. [DOI] [PubMed] [Google Scholar]
- 4.Jedrzejas MJ. In: Hyaluronan 2003. Hascall VC, Balazs EA, editors. Matrix Bilogy Institute; Edgewater, NJ: 2004. www.matrixbiologyinstitute.org/ha03. [Google Scholar]
- 5.Jedrzejas MJ. In: Recent Research Developments in Biophysics and Biochemistry. Pandali SG, editor. Vol. 2 Research Signpost; Trivandrum, India: 2002. [Google Scholar]
- 6.Frost GI, Csoka AB, Wong T, Stern R, Csoka TB. Biochem Biophys Res Commun. 1997;236:10–5. doi: 10.1006/bbrc.1997.6773. [DOI] [PubMed] [Google Scholar]
- 7.Hascall VC, Yanagishita M. Science of Hayluronan Today. 2004 Glycoforum www.glycoforum.gr.jp/science/hyaluronan.
- 8.Meyer K, Rapport MM. Adv Enzymol. 1952;13:199–236. doi: 10.1002/9780470122587.ch6. [DOI] [PubMed] [Google Scholar]
- 9.Dorfman A. Methods Enzymol. 1955;1:166–73. [Google Scholar]
- 10.Meyer K. In: The Enzymes. Boyer PL, editor. V Academic Press; New York: 1971. [Google Scholar]
- 11.Markovic-Housley Z, Miglierini G, Soldatova L, Rizkallah PJ, Muller U, Schirmer T. Structure Fold Des. 2000;8:1025–35. doi: 10.1016/s0969-2126(00)00511-6. [DOI] [PubMed] [Google Scholar]
- 12.Jedrzejas MJ, Stern R. Proteins Struct Funct Genet. 2005;61:227–38. doi: 10.1002/prot.20592. [DOI] [PubMed] [Google Scholar]
- 13.Li S, Kelly SJ, Lamani E, Ferraroni M, Jedrzejas MJ. EMBO J. 2000;19:1228–40. doi: 10.1093/emboj/19.6.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S, Jedrzejas MJ. J Biol Chem. 2001;276:41407–16. doi: 10.1074/jbc.M106634200. [DOI] [PubMed] [Google Scholar]
- 15.Kelly SJ, Taylor KB, Li S, Jedrzejas MJ. Glycobiology. 2001;11:297–304. doi: 10.1093/glycob/11.4.297. [DOI] [PubMed] [Google Scholar]
- 16.Nukui M, Taylor KB, McPherson DT, Shigenaga MK, Jedrzejas MJ. J Biol Chem. 2003;278:3079–88. doi: 10.1074/jbc.M204999200. [DOI] [PubMed] [Google Scholar]
- 17.Jedrzejas MJ. Microbiol Mol Biol Rev. 2001;65:187–207. doi: 10.1128/MMBR.65.2.187-207.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuki H, Fishman WH. J Biol Chem. 1963;238:1877–9. [PubMed] [Google Scholar]
- 19.Karlstam B, Ljungloef A. Polar Biol. 1991;11:501–7. [Google Scholar]
- 20.Csoka AB, Frost GI, Stern R. Matrix Biol. 2001;20:499–508. doi: 10.1016/s0945-053x(01)00172-x. [DOI] [PubMed] [Google Scholar]
- 21.Stern R. Glycobiology. 2003;13:105R–15R. doi: 10.1093/glycob/cwg112. [DOI] [PubMed] [Google Scholar]
- 22.Duran-Reynals F. CR Soc Biol. 1928;99:6–7. [Google Scholar]
- 23.Bonner WM, Jr, Cantey EY. Clin Chim Acta. 1966;13:746–52. doi: 10.1016/0009-8981(66)90142-2. [DOI] [PubMed] [Google Scholar]
- 24.Tengblad A. Biochim Biophys Acta. 1979;578:281–9. doi: 10.1016/0005-2795(79)90158-2. [DOI] [PubMed] [Google Scholar]
- 25.Frost GI, Stern R. Anal Biochem. 1997;251:263–9. doi: 10.1006/abio.1997.2262. [DOI] [PubMed] [Google Scholar]
- 26.Guntenhoner MW, Pogrel MA, Stern R. Matrix. 1992;12:388–96. doi: 10.1016/s0934-8832(11)80035-1. [DOI] [PubMed] [Google Scholar]
- 27.Shimada E, Matsumura G. J Biochem (Tokyo) 1980;88:1015–23. doi: 10.1093/oxfordjournals.jbchem.a133052. [DOI] [PubMed] [Google Scholar]
- 28.Kreil G. Protein Sci. 1995;4:1666–9. doi: 10.1002/pro.5560040902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffman P, Meyer K, Linker A. J Biol Chem. 1956;219:653–63. [PubMed] [Google Scholar]
- 30.Mello LV, de Groot BL, Li S, Jedrzejas MJ. J Biol Chem. 2002;277:36678–88. doi: 10.1074/jbc.M205140200. [DOI] [PubMed] [Google Scholar]
- 31.Jedrzejas MJ, Mello LV, de Groot BL, Li S. J Biol Chem. 2002;277:28287–97. doi: 10.1074/jbc.M112009200. [DOI] [PubMed] [Google Scholar]
- 32.Hovingh P, Linker A. Comp Biochem Physiol B Biochem Mol Biol. 1999;124:319–26. doi: 10.1016/s0305-0491(99)00128-5. [DOI] [PubMed] [Google Scholar]
- 33.Linker A, Hoffman P, Meyer K. Nature. 1957;180:810–1. doi: 10.1038/180810b0. [DOI] [PubMed] [Google Scholar]
- 34.Linker A, Meyer K, Hoffman P. J Biol Chem. 1960;235:924–7. [PubMed] [Google Scholar]
- 35.Rigbi M, Levy H, Iraqi F, Teitelbaum M, Orevi M, Alajoutsijarvi A, Horovitz A, Galun R. Comp Biochem Physiol B. 1987;87:567–73. doi: 10.1016/0305-0491(87)90053-8. [DOI] [PubMed] [Google Scholar]
- 36.Karlstam B, Vincent J, Johansson B, Bryno C. Prep Biochem. 1991;21:237–56. doi: 10.1080/10826069108018576. [DOI] [PubMed] [Google Scholar]
- 37.Ohya T, Kaneko Y. Biochim Biophys Acta. 1970:198. doi: 10.1016/0005-2744(70)90139-7. [DOI] [PubMed] [Google Scholar]
- 38.Shimizu MT, Jorge AO, Unterkircher CS, Fantinato V, Paula CR. J Med Vet Mycol. 1995;33:27–31. [PubMed] [Google Scholar]
- 39.Coutinho SD, Paula CR. Med Mycol. 2000;38:73–6. doi: 10.1080/mmy.38.1.73.76. [DOI] [PubMed] [Google Scholar]
- 40.Winter WT, Smith PJ, Arnott S. J Mol Biol. 1975;99:219–35. doi: 10.1016/s0022-2836(75)80142-2. [DOI] [PubMed] [Google Scholar]
- 41.Scott JE, Heatley F, Wood B. Biochemistry. 1995;34:15467–74. doi: 10.1021/bi00047a011. [DOI] [PubMed] [Google Scholar]
- 42.Scott JE. Pathol Biol (Paris) 2001;49:284–9. doi: 10.1016/s0369-8114(01)00152-3. [DOI] [PubMed] [Google Scholar]
- 43.Scott JE, Heatley F. Proc Natl Acad Sci USA. 1999;96:4850–5. doi: 10.1073/pnas.96.9.4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scott JE, Heatley F. Biomacromolecules. 2002;3:547–53. doi: 10.1021/bm010170j. [DOI] [PubMed] [Google Scholar]
- 45.Haxaire K, Braccini I, Milas M, Rinaudo M, Perez S. Glycobiology. 2000;10:587–94. doi: 10.1093/glycob/10.6.587. [DOI] [PubMed] [Google Scholar]
- 46.Tratar Pirc E, Zidar J, Bukovec P, Hodoscek M. Carbohydr Res. 2005;340:2064–9. doi: 10.1016/j.carres.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 47.Toffanin R, Kvam JB, Flaibani A, Atzori M, Biviano F, Paoletti S. Carboh Res. 1993;245:113–28. doi: 10.1016/0008-6215(93)80064-l. [DOI] [PubMed] [Google Scholar]
- 48.Sicinska W, Adams B, Lerner LE. Carboh Res. 1993;242:29–51. doi: 10.1016/0008-6215(93)80020-f. [DOI] [PubMed] [Google Scholar]
- 49.Holmbeck SM, Petillo PA, Lerner LE. Biochemistry. 1994;33:14246–55. doi: 10.1021/bi00251a037. [DOI] [PubMed] [Google Scholar]
- 50.Cowman MK, Feder-Davis J, Hittner DM. Macromolecules. 2001;34:110–5. [Google Scholar]
- 51.Cowman MK, Hittner DM, Feder-Davis J. Macromolecules. 1996;29:2894–902. [Google Scholar]
- 52.Almond A, Brass A, Sheehan JK. J Phys Chem B. 2000;104:5634–40. [Google Scholar]
- 53.Almond A, Brass A, Sheehan JK. Glycobiology. 1998;8:973–80. doi: 10.1093/glycob/8.10.973. [DOI] [PubMed] [Google Scholar]
- 54.Almond A, Brass A, Sheehan JK. J Mol Biol. 1998;284:1425–37. doi: 10.1006/jmbi.1998.2245. [DOI] [PubMed] [Google Scholar]
- 55.Gribbon P, Heng BC, Hardingham TE. Biochem J. 2000;350:329–35. [PMC free article] [PubMed] [Google Scholar]
- 56.Scott JE, Thomlinson AM. J Anat. 1998;192:391–405. doi: 10.1046/j.1469-7580.1998.19230391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turner RE, Lin PY, Cowman MK. Arch Biochem Biophys. 1988;265:484–95. doi: 10.1016/0003-9861(88)90153-1. [DOI] [PubMed] [Google Scholar]
- 58.Lepperdinger G, Mullegger J, Kreil G, Winter WT, Smith PJ, Arnott S. Matrix Biol. 2001;20:509–14. doi: 10.1016/s0945-053x(01)00170-6. [DOI] [PubMed] [Google Scholar]
- 59.Coutinho PM, Henrissat B. 1999 [Google Scholar]
- 60.Coutinho PM, Henrissat B. In: Recent Advances in Carbohydrate Bioengineering. Gilbert HJ, Davies G, Henrissat B, Svensson B, editors. The Royal Society of Chemistry; Cambridge, UK: 1999. [Google Scholar]
- 61.Baker JR, Pritchard DG. Biochem J. 2000;348:465–71. [PMC free article] [PubMed] [Google Scholar]
- 62.Jedrzejas MJ. Hascall VC, Yanagishita M, editors. Science of Hayluronan Today. 2002 Glycoforum www.glycoforum.gr.jp/science/hyaluronan.
- 63.Scott JE, Chen Y, Brass A. Eur J Biochem. 1992;209:675–80. doi: 10.1111/j.1432-1033.1992.tb17335.x. [DOI] [PubMed] [Google Scholar]
- 64.DeAngelis PL. Anat Rec. 2002;268:317–26. doi: 10.1002/ar.10163. [DOI] [PubMed] [Google Scholar]
- 65.Toyoda H, Kinoshita-Toyoda A, Selleck SB. J Biol Chem. 2000;275:2269–75. doi: 10.1074/jbc.275.4.2269. [DOI] [PubMed] [Google Scholar]
- 66.Hay ED, Meier S. J Cell Biol. 1974;62:889–98. doi: 10.1083/jcb.62.3.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Solursh M, Fisher M, Meier S, Singley CT. J Embryol Exp Morphol. 1979;54:75–98. [PubMed] [Google Scholar]
- 68.Dehal P, Satou Y, Campbell RK, Chapman J, Degnan B, De Tomaso A, Davidson B, Di Gregorio A, Gelpke M, Goodstein DM, Harafuji N, Hastings KE, Ho I, Hotta K, Huang W, Kawashima T, Lemaire P, Martinez D, Meinertzhagen IA, Necula S, Nonaka M, Putnam N, Rash S, Saiga H, Satake M, Terry A, Yamada L, Wang HG, Awazu S, Azumi K, Boore J, Branno M, Chin-Bow S, DeSantis R, Doyle S, Francino P, Keys DN, Haga S, Hayashi H, Hino K, Imai KS, Inaba K, Kano S, Kobayashi K, Kobayashi M, Lee BI, Makabe KW, Manohar C, Matassi G, Medina M, Mochizuki Y, Mount S, Morishita T, Miura S, Nakayama A, Nishizaka S, Nomoto H, Ohta F, Oishi K, Rigoutsos I, Sano M, Sasaki A, Sasakura Y, Shoguchi E, Shin-i T, Spagnuolo A, Stainier D, Suzuki MM, Tassy O, Takatori N, Tokuoka M, Yagi K, Yoshizaki F, Wada S, Zhang C, Hyatt PD, Larimer F, Detter C, Doggett N, Glavina T, Hawkins T, Richardson P, Lucas S, Kohara Y, Levine M, Satoh N, Rokhsar DS. Science. 2002;298:2157–67. doi: 10.1126/science.1080049. [DOI] [PubMed] [Google Scholar]
- 69.Itano N, Sawai T, Yoshida M, Lenas P, Yamada Y, Imagawa M, Shinomura T, Hamaguchi M, Yoshida Y, Ohnuki Y, Miyauchi S, Spicer AP, McDonald JA, Kimata K. J Biol Chem. 1999;274:25085–92. doi: 10.1074/jbc.274.35.25085. [DOI] [PubMed] [Google Scholar]
- 70.Toole BP. In: Cell Biology of Extracellular Matrix. Hay ED, editor. Plenum Press; New York: 1991. [Google Scholar]
- 71.Jedrzejas MJ, Mewbourne RB, Chantalat L, McPherson DT. Protein Expr Purif. 1998;13:83–9. doi: 10.1006/prep.1997.0864. [DOI] [PubMed] [Google Scholar]
- 72.Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, Collins J, Lee M, Roth A, Sudarsan N, Jona I, Wickiser JK, Breaker RR. Proc Natl Acad Sci USA. 2004;101:6421–6. doi: 10.1073/pnas.0308014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Nature. 2004;428:281–6. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- 74.Ponnuraj K, Jedrzejas MJ. J Mol Biol. 2000;299:885–95. doi: 10.1006/jmbi.2000.3817. [DOI] [PubMed] [Google Scholar]
- 75.Feinberg RN, Beebe DC. Science. 1983;220:1177–9. doi: 10.1126/science.6857242. [DOI] [PubMed] [Google Scholar]
- 76.Delmage JM, Powars DR, Jaynes PK, Allerton SE. Ann Clin Lab Sci. 1986;16:303–10. [PubMed] [Google Scholar]
- 77.McBride WH, Bard JB. J Exp Med. 1979;149:507–15. doi: 10.1084/jem.149.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.West DC, Hampson IN, Arnold F, Kumar S. Science. 1985;228:1324–6. doi: 10.1126/science.2408340. [DOI] [PubMed] [Google Scholar]
- 79.Noble PW. Matrix Biol. 2002;21:25–9. doi: 10.1016/s0945-053x(01)00184-6. [DOI] [PubMed] [Google Scholar]
- 80.Fieber C, Baumann P, Vallon R, Termeer C, Simon JC, Hofmann M, Angel P, Herrlich P, Sleeman JP. J Cell Sci. 2004;117:359–67. doi: 10.1242/jcs.00831. [DOI] [PubMed] [Google Scholar]
- 81.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. J Biol Chem. 2004;279:17079–84. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 82.Xu H, Ito T, Tawada A, Maeda H, Yamanokuchi H, Isahara K, Yoshida K, Uchiyama Y, Asari A. J Biol Chem. 2002;277:17308–14. doi: 10.1074/jbc.M112371200. [DOI] [PubMed] [Google Scholar]
- 83.Koshland DE. Biol Rev Camb Philos. 1953;28:416–36. [Google Scholar]
- 84.Davies GJ, Dauter M, Brzozowski AM, Bjornvad ME, Andersen KV, Schulein M. Biochemistry. 1998;37:1926–32. doi: 10.1021/bi972162m. [DOI] [PubMed] [Google Scholar]
- 85.Divne C, Stahlberg J, Teeri TT, Jones TA. J Mol Biol. 1998;275:309–25. doi: 10.1006/jmbi.1997.1437. [DOI] [PubMed] [Google Scholar]
- 86.Botzki A, Rigden DJ, Braun S, Nukui M, Salmen S, Hoechstetter J, Bernhardt B, Dove S, Jedrzejas MJ, Buschauer A. J Biol Chem. 2004;279:45990–7. doi: 10.1074/jbc.M406146200. [DOI] [PubMed] [Google Scholar]
- 87.Rigden DJ, Jedrzejas MJ, de Mello LV. FEBS Lett. 2003;544:103–11. doi: 10.1016/s0014-5793(03)00481-2. [DOI] [PubMed] [Google Scholar]
- 88.Sinnott ML. Biochem J. 1984;224:817–21. doi: 10.1042/bj2240817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arming S, Strobl B, Wechselberger C, Kreil G. Eur J Biochem. 1997;247:810–4. doi: 10.1111/j.1432-1033.1997.t01-1-00810.x. [DOI] [PubMed] [Google Scholar]
- 90.Tews I, Perrakis A, Oppenheim A, Dauter Z, Wilson KS, Vorgias CE. Nat Struct Biol. 1996;3:638–48. doi: 10.1038/nsb0796-638. [DOI] [PubMed] [Google Scholar]
- 91.Tews I, Terwisscha von Scheltinga AC, Perrakis A, Wilson KS, Dijkstra BW. J Am Chem Soc. 1997;119:7954–9. [Google Scholar]
- 92.Hunnicutt GR, Primakoff P, Myles DG. Biol Reprod. 1996;55:80–6. doi: 10.1095/biolreprod55.1.80. [DOI] [PubMed] [Google Scholar]
- 93.Deng X, He Y, Martin-Deleon PA. J Androl. 2000;21:822–32. [PubMed] [Google Scholar]
- 94.Beech DJ, Madan AK, Deng N. J Surg Res. 2002;103:203–7. doi: 10.1006/jsre.2002.6351. [DOI] [PubMed] [Google Scholar]
- 95.Zhang H, Martin-DeLeon PA. Biol Reprod. 2003;69:446–54. doi: 10.1095/biolreprod.102.013854. [DOI] [PubMed] [Google Scholar]
- 96.Godin DA, Fitzpatrick PC, Scandurro AB, Belafsky PC, Woodworth BA, Amedee RG, Beech DJ, Beckman BS. Arch Otolaryngol Head Neck Surg. 2000;126:402–4. doi: 10.1001/archotol.126.3.402. [DOI] [PubMed] [Google Scholar]
- 97.Madan AK, Yu K, Dhurandhar N, Cullinane C, Pang Y, Beech DJ. Oncol Rep. 1999;6:607–9. doi: 10.3892/or.6.3.607. [DOI] [PubMed] [Google Scholar]
- 98.Oettl M, Hoechstetter J, Asen I, Bernhardt G, Buschauer A. Eur J Pharm Sci. 2003;18:267–77. doi: 10.1016/s0928-0987(03)00022-8. [DOI] [PubMed] [Google Scholar]
- 99.Heckel D, Comtesse N, Brass N, Blin N, Zang KD, Meese E. Hum Mol Genet. 1998;7:1859–72. doi: 10.1093/hmg/7.12.1859. [DOI] [PubMed] [Google Scholar]
- 100.Comtesse N, Maldener E, Meese E. Biochem Biophys Res Commun. 2001;283:634–40. doi: 10.1006/bbrc.2001.4815. [DOI] [PubMed] [Google Scholar]
- 101.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Frost GI, Mohapatra G, Wong TM, Csoka AB, Gray JW, Stern R. Oncogene. 2000;19:870–7. doi: 10.1038/sj.onc.1203317. [DOI] [PubMed] [Google Scholar]
- 103.Junker N, Latini S, Petersen LN, Kristjansen PE. Oncol Rep. 2003;10:609–6. [PubMed] [Google Scholar]
- 104.Triggs-Raine B, Salo TJ, Zhang H, Wicklow BA, Natowicz MR. Proc Natl Acad Sci USA. 1999;96:6296–300. doi: 10.1073/pnas.96.11.6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shuttleworth TL, Wilson MD, Wicklow BA, Wilkins JA, Triggs-Raine BL. J Biol Chem. 2002;277:23008–18. doi: 10.1074/jbc.M108991200. [DOI] [PubMed] [Google Scholar]
- 106.DeAngelis PL, Jing W, Graves MV, Burbank DE, Van Etten JL. Science. 1997;278:1800–3. doi: 10.1126/science.278.5344.1800. [DOI] [PubMed] [Google Scholar]
- 107.Nakashima K, Yamada L, Satou Y, Azuma J, Satoh N. Dev Genes Evol. 2004;214:81–8. doi: 10.1007/s00427-003-0379-8. [DOI] [PubMed] [Google Scholar]
- 108.Stern M, Stern R. Mol Biol Evol. 1992;9:1179–80. doi: 10.1093/oxfordjournals.molbev.a040786. [DOI] [PubMed] [Google Scholar]
- 109.Mylvaganam H, Bjorvatn B, Hofstad T, Osland A. Microb Pathog. 2000;29:145–53. doi: 10.1006/mpat.2000.0378. [DOI] [PubMed] [Google Scholar]
- 110.Hynes WL, Hancock L, Ferretti JJ. Infect Immun. 1995;63:3015–20. doi: 10.1128/iai.63.8.3015-3020.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hynes WL, Ferretti JJ. Infect Immun. 1989;57:533–9. doi: 10.1128/iai.57.2.533-539.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stern M, Stern R. Matrix. 1992;12:397–403. doi: 10.1016/s0934-8832(11)80036-3. [DOI] [PubMed] [Google Scholar]
- 113.Brown DR, Zacher LA, Farmerie WG. J Bacteriol. 2004;186:3922–7. doi: 10.1128/JB.186.12.3922-3927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hunt ME, Brown DR. Clin Diagn Lab Immunol. 2005;12:1370–7. doi: 10.1128/CDLI.12.12.1370-1377.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Macauley MS, Whitworth GE, Debowski AW, Chin D, Vocadlo DJ. J Biol Chem. 2005;280:25313–22. doi: 10.1074/jbc.M413819200. [DOI] [PubMed] [Google Scholar]
- 116.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ginalski K, Rychlewski L. Nucleic Acids Res. 2003;31:3291–2. doi: 10.1093/nar/gkg503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ginalski K, Elofsson A, Fischer D, Rychlewski L. Bioinformatics. 2003;19:1015–8. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 119.Andreeva A, Howorth D, Brenner SE, Hubbard TJ, Chothia C, Murzin AG. Nucleic Acids Res. 2004;32(Database issue):D226–9. doi: 10.1093/nar/gkh039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Murzin AG, Brenner SE, Hubbard T, Chothia C. J Mol Biol. 1995;247:536–40. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 121.Aleshin A, Golubev A, Firsov LM, Honzatko RB. J Biol Chem. 1992;267:19291–8. doi: 10.2210/pdb1gly/pdb. [DOI] [PubMed] [Google Scholar]
- 122.Perrakis A, Tews I, Dauter Z, Oppenheim AB, Chet I, Wilson KS, Vorgias CE. Structure. 1994;2:1169–80. doi: 10.1016/s0969-2126(94)00119-7. [DOI] [PubMed] [Google Scholar]
- 123.Rigden DJ, Bagyan I, Lamani E, Setlow P, Jedrzejas MJ. Protein Sci. 2001;10:1835–46. doi: 10.1110/ps.15701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rigden DJ, Setlow P, Setlow B, Bagyan I, Stein RA, Jedrzejas MJ. Protein Sci. 2002;11:2370–81. doi: 10.1110/ps.0216802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chivian D, Kim DE, Malmstrom L, Bradley P, Robertson T, Murphy P, Strauss CE, Bonneau R, Rohl CA, Baker D. Proteins. 2003;53(Suppl 6):524–33. doi: 10.1002/prot.10529. [DOI] [PubMed] [Google Scholar]
- 126.Kim DE, Chivian D, Baker D. Nucleic Acids Res. 2004;32:W526–31. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stern R. Hascall VC, Yanagishita M, editors. Science of Hayluronan Today. 2004 Glycoforum www.glycoforum.gr.jp/science/hyaluronan.
- 128.Rigden DJ, Jedrzejas MJ. Proteins Struct Funct Genet. 2003;52:203–11. doi: 10.1002/prot.10405. [DOI] [PubMed] [Google Scholar]
- 129.Rigden DJ, Galperin MY, Jedrzejas MJ. Crit Rev Biochem Mol Biol. 2003;38:143–68. doi: 10.1080/713609215. [DOI] [PubMed] [Google Scholar]
- 130.Tomme P, Warren RA, Miller RC, Jr, Kilburn DG, Gilkes NR. In: Enzymatic Degradation of Insoluble Polysaccharides. Sadler JN, Penner M, editors. American Chemical Society; Washington, D.C: 1995. [Google Scholar]
- 131.Jedrzejas MJ, Chantalat L, Mewbourne RB. J Struct Biol. 1998;121:73–5. doi: 10.1006/jsbi.1998.3963. [DOI] [PubMed] [Google Scholar]
- 132.Jedrzejas MJ, Chantalat L. Acta Crystallogr D Biol Crystallogr. 2000;56:460–3. doi: 10.1107/s0907444900000706. [DOI] [PubMed] [Google Scholar]
- 133.Huang W, Matte A, Li Y, Kim YS, Linhardt RJ, Su H, Cygler M. J Mol Biol. 1999;294:1257–69. doi: 10.1006/jmbi.1999.3292. [DOI] [PubMed] [Google Scholar]
- 134.Mayans O, Scott M, Connerton I, Gravesen T, Benen J, Visser J, Pickersgill R, Jenkins J. Structure. 1997;5:677–89. doi: 10.1016/s0969-2126(97)00222-0. [DOI] [PubMed] [Google Scholar]
- 135.Vitali J, Schick B, Kester HC, Visser J, Jurnak F. Plant Physiol. 1998;116:69–80. doi: 10.1104/pp.116.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pickersgill R, Jenkins J, Harris G, Nasser W, Robert-Baudouy J. Nat Struct Biol. 1994;1:717–23. doi: 10.1038/nsb1094-717. [DOI] [PubMed] [Google Scholar]
- 137.Scavetta RD, Herron SR, Hotchkiss AT, Kita N, Keen NT, Benen JA, Kester HC, Visser J, Jurnak F. Plant Cell. 1999;11:1081–92. doi: 10.1105/tpc.11.6.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yoder MD, Lietzke SE, Jurnak F. Structure. 1993;1:241–51. doi: 10.1016/0969-2126(93)90013-7. [DOI] [PubMed] [Google Scholar]
- 139.Yoder MD, Keen NT, Jurnak F. Science. 1993;260:1503–7. doi: 10.1126/science.8502994. [DOI] [PubMed] [Google Scholar]
- 140.Petersen TN, Kauppinen S, Larsen S. Structure. 1997;5:533–44. doi: 10.1016/s0969-2126(97)00209-8. [DOI] [PubMed] [Google Scholar]
- 141.Pickersgill R, Smith D, Worboys K, Jenkins J. J Biol Chem. 1998;273:24660–4. doi: 10.1074/jbc.273.38.24660. [DOI] [PubMed] [Google Scholar]
- 142.Steinbacher S, Baxa U, Miller S, Weintraub A, Seckler R, Huber R. Proc Natl Acad Sci USA. 1996;93:10584–8. doi: 10.1073/pnas.93.20.10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Steinbacher S, Seckler R, Miller S, Steipe B, Huber R, Reinemer P. Science. 1994;265:383–6. doi: 10.1126/science.8023158. [DOI] [PubMed] [Google Scholar]
- 144.Kleywegt GJ, Zou JY, Divne C, Davies GJ, Sinning I, Stahlberg J, Reinikainen T, Srisodsuk M, Teeri TT, Jones TA. J Mol Biol. 1997;272:383–97. doi: 10.1006/jmbi.1997.1243. [DOI] [PubMed] [Google Scholar]
- 145.Rouvinen J, Bergfors T, Teeri T, Knowles JK, Jones TA. Science. 1990;249:380–6. doi: 10.1126/science.2377893. [DOI] [PubMed] [Google Scholar]
- 146.Park Y, Cho S, Linhardt RJ. Biochim Biophys Acta. 1997;1337:217–26. doi: 10.1016/s0167-4838(96)00167-7. [DOI] [PubMed] [Google Scholar]
- 147.Price KN, Al T, Baker DC, Chisena C, Cysyk RL. Carbohydr Res. 1997;303:303–11. doi: 10.1016/s0008-6215(97)00171-7. [DOI] [PubMed] [Google Scholar]
- 148.Baker JR, Dong S, Pritchard DG. Biochem J. 2002;365:317–22. doi: 10.1042/BJ20020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Smith NL, Taylor EJ, Lindsay AM, Charnock SJ, Turkenburg JP, Dodson EJ, Davies GJ, Black GW. Proc Natl Acad Sci U S A. 2005;102:17652–7. doi: 10.1073/pnas.0504782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Evanko SP, Parks WT, Wight TN. J Histochem Cytochem. 2004;52:1525–35. doi: 10.1369/jhc.4A6356.2004. [DOI] [PubMed] [Google Scholar]
- 151.Hascall VC, Majors AK, De La Motte CA, Evanko SP, Wang A, Drazba JA, Strong SA, Wight TN. Biochim Biophys Acta. 2004;1673:3–12. doi: 10.1016/j.bbagen.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 152.Lokeshwar VB, Cerwinka WH, Isoyama T, Lokeshwar BL. Cancer Res. 2005;65:7782–9. doi: 10.1158/0008-5472.CAN-05-1022. [DOI] [PubMed] [Google Scholar]
- 153.Renkema GH, Boot RG, Strijland A, Donker-Koopman WE, van den Berg M, Muijsers AO, Aerts JM. Eur J Biochem. 1997;244:279–85. doi: 10.1111/j.1432-1033.1997.00279.x. [DOI] [PubMed] [Google Scholar]
- 154.Csoka AB, Frost GI, Wong T, Stern R, Csoka TB. FEBS Lett. 1997;417:307–10. doi: 10.1016/s0014-5793(97)01309-4. [DOI] [PubMed] [Google Scholar]
- 155.Bujnicki JM, Elofsson A, Fischer D, Rychlewski L. Bioinformatics. 2001;17:750–1. doi: 10.1093/bioinformatics/17.8.750. [DOI] [PubMed] [Google Scholar]
- 156.DeLano WL. The PyMOL Molecular Graphics System. 2003 www.pymol.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.