

Abstract
1H-imidazo[4,5-c]quinolin-4-amine derivatives have been synthesized as allosteric modulators of the human A3 adenosine receptor (AR). Structural modifications were made at the 4-amino and 2 positions. The compounds were tested in both binding and functional assays, and many were found to be allosteric enhancers of the action of A3AR agonists by several different criteria. First, a potentiation of the maximum efficacy of the agonist Cl-IB-MECA was observed for numerous derivatives. Also, a number of these compounds decreased the rate of dissociation of the agonist [125I]I-AB-MECA from the A3AR. Most prominently, compound 43 (LUF6000) was found to enhance agonist efficacy in a functional assay by 45% and decrease dissociation rate similarly without influencing agonist potency. The structural requirements for allosteric enhancement at the A3AR were distinct from the requirements to inhibit equilibrium binding. Thus, we have prepared allosteric enhancers of the human A3AR that have an improved allosteric effect in comparison to the inhibition of equilibrium binding at the orthosteric site.
Introduction
The adenosine receptors (ARs), a family of GPCRs consisting of the A1, A2A, A2B, and A3 subtypes, are activated by the ubiquitous modulator adenosine in our body. Selective agonists have been synthesized for three out of the four subtypes of adenosine receptors (ARs).1-3 Each of these groups of subtype-selective agonists has representative examples that have already entered clinical trials.1 A1AR selective agonists are under development for treating cardiac arrhythmia, pain, and pulmonary disorders.4-6 A2AAR agonists are in clinical trials for cardiac stress imaging and are also of interest for inflammation.7 A3AR agonists, such as IB-MECA (N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine, see Figure 1), are in clinical trials for colon carcinoma and rheumatoid arthritis and have potential for the treatment of myocardial infarction, stroke, and other disorders.1
Figure 1.

Structures of reference agonists (IB-MECA and 2-Cl-IB-MECA) and allosteric modulators (DU 124183, amiloride and VUF5455) of the A3AR.
Although newer and more selective AR agonists are being developed,1,2 there are inherent problems, in general, associated with the therapeutic use of GPCR agonists.8 The widespread occurrence of receptors such as the ARs makes selectivity for some organs or tissues nearly unachievable using directly-acting (orthosteric) agonists, and thus a number of agonists were discontinued after initial phases of clinical trials.1,2 In contrast to directly acting agonists, allosteric modulators act at a separate site on the receptor protein to modulate the effect of a native agonist.9 An advantage of a positive allosteric modulator of a membrane receptor over its native, orthosteric activator is that greater selectivity, in principle, may be achieved. The positive allosteric modulator would enhance the action of the native agonist, but may have no effect of its own on the unoccupied receptor. Thus, the effect of endogenous agonist, which may be insufficient in a particular disease state, may be magnified in a temporally specific manner through allosteric modulation. The higher subtype-selectivity commonly exerted by allosteric modulators and the fact that the allosteric action is ideally coupled to simultaneous presence of the endogenous ligand both help to prevent over-dosage compared to the administration of a conventional, and possibly nonselective, orthosteric agonist.
Allosteric modulation is considered an important mechanism of controlling receptor function.8,10-12 In the case of the ligand-gated ion channels, a series of benzodiazepines, including diazepam, enhance the CNS inhibitory function of the endogenous γ-aminobutyric acid, and have become the most widely prescribed sleep medications. In contrast, the directly acting agonists have not found clinical applications due to the inherent side effects. In the GPCR field, cinacalcet, a positive allosteric modulator of the calcium-sensing receptor (CaR), has recently been approved for the treatment of secondary hyperparathyroidism in dialysis patients suffering from chronic kidney disease.13,14 In the case of ARs, the A1AR has been the most studied, and one of its allosteric enhancers, T62 (2-amino-4,5,6,7-tetrahydrobenzo[b]thiophen-3-yl-(4-chlorophenyl)methanone), is now in Phase I clinical trials for the potential treatment of neuropathic pain.1,15
Several classes of compounds (see also Figure 1) have previously been reported to be allosteric modulators for the A3AR, including the 3-(2-pyridinyl)isoquinoline (VUF5455),16 the 1H-imidazo[4,5-c]quinolin-4-amine (DU124183, 28 in the current study),17 and analogues of amiloride such as N,N-hexamethylene-amiloride.18 One of the most interesting findings was that 28 and some of its analogues decreased agonist potency but enhanced maximum agonist efficacy.17,19
In the present study we have extended our earlier finding that 1H-imidazo[4,5-c]quinolin-4-amine derivatives act as allosteric modulators of the A3AR.17 We have synthesized and characterized the SAR (structure activity relationship) of a new set of analogues based on compound 28. A number of derivatives were found to be allosteric enhancers of the A3AR. One of the analogues of 28, compound 43, was found to enhance agonist efficacy in a functional assay and decrease dissociation rate in binding but without influencing agonist potency.
Results
Chemistry
The novel derivatives 28 - 45 were synthesized as shown in Scheme 1. Condensation of anthranilic acid hydrochloride (1) with 2-nitro-acetaldehyde-oxim (HON=CHCH2NO2), prepared in situ from CH3NO2 and NaOH20, resulted in 2-(2-nitro-ethylideneamino)-benzoic acid (2), which was dehydrated in acetic anhydride in the presence of potassium acetate to give 3-nitro-4-hydroxyquinoline (3).20 3-Nitro-4-hydroxyquinoline (3) was treated with phosphorus oxychloride to afford 3-nitro-4-chloroquinoline (4). This was converted to 3-nitro-4-aminoquinoline (5) with ammonia, which was subsequently reduced by catalytic hydrogenation to 3,4-diaminoquinoline (6) with 10 % palladium on charcoal as catalyst. The next step involved ring-closure, which was carried out on three different ways. Compounds 8-11 were prepared by ring closure of the appropriate carboxylic acids and 3,4-diaminoquinoline (6) in polyphosphoric acid.21 Compounds 12 and 13 were prepared by ring-closure of 2-furoyl chloride and hexanoyl chloride, respectively, with 3,4-diaminoquinoline (6).22 Compound 7 was prepared by ring-closure of 3,4-diaminoquinoline (6) with formic acid in trimethylorthoformate. Oxidation with 3-chloroperoxybenzoic acid afforded 5-oxides 14-20, which subsequently could be converted with phosphorous oxychloride into the respective 4-chloro compounds 21-27. Finally, treatment of compounds 21-27 with the appropriate amines afforded the desired compounds 28-45, respectively.23
Scheme 1.

Reagents: (i) HCl, HON=CHCH2NO2; (ii) (CH3CO)2O, CH3COOK; (iii) POCl3; (iv) NH3; (v) H2/Pd; (vi) a) polyphosphoric acid, R'COOH, b) trimethylorthoformate, HCOOH, c) 1. R'COCl, 2. NaOH; (vii) 3-chloroperoxybenzoic acid; (viii) POCl3; (ix) RNH2.
Biological Activity
Previously reported compound 28 (ref 17) and newly synthesized imidazoquinolines 29–45 affect AR binding to or function at ARs as shown in Table 1. We first tested the effect of these compounds on the equilibrium binding at A1, A2A, and A3ARs using standard agonist radioligands16,17 [3H]R-N6-[phenylisopropyl]adenosine, [3H]2-[p-(carboxyethyl)phenylethylamino]-5′-N-ethylcarboxamidoadenosine, and 125I-N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide, respectively. We then measured the effect of these compounds (at 10 μM) on the A2BAR with a cyclic AMP functional assay,24 as an agonist radioligand has not been readily available for this subtype. Most compounds only induced a rather weak inhibition of equilibrium binding at the concentration used. For compounds 28-32 we also determined Ki values, if possible. It is not known whether the observed inhibition is of allosteric or non-allosteric character.
Table 1.
Potency of 1H-imidazo [4,5-c]quinolin-4-amine derivatives in binding assays at human A1, A2A, A2B and A3ARs expressed in CHO cells and allosteric effects at the human A3AR. a
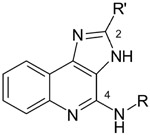 | ||||||||
|---|---|---|---|---|---|---|---|---|
| No. | R | R′ | Ki (hA1AR), nM a or % displ. at 10 μM | Ki (hA2AAR), nM a or % displ. at 10 μM | hA2BAR % inhib. at 10 μMb | Ki (hA3AR), nMa or % displ. at 10 μM | hA3AR ag. Dissociation (%) c at 10 μM | Relative efficacy (%) at hA3AR d at 10 μM |
| 28 | Ph | CP | 3420 ± 230 | 3150 ± 210 | -6.8% | 786 ± 67
(90%) |
174±5 | 138±8 |
| 29 | 4-CH3-Ph | CP | 3850 ± 500 | 5220 ± 320 | -4.1% | 1190 ± 107
(87%) |
153±4 | 128±4 |
| 30 | 4-CH3O-Ph | CP | 4170 ± 730 | >10,000
(16%) |
7.3% | 410 ± 64
(92%) |
166±11 | 132±4 |
| 31 | 3,4-Cl2-Ph | CP | >10,000
(15%) |
>10,000
(0%) |
-6.7% | 4690±970
(67%) |
144±9 | 141±5 |
| 32 | 4-Cl-Ph | CP | >10,000
(22%) |
>10,000
(17%) |
-10.2% | 1610 ± 550
(82%) |
159±5 | 136±3 |
| 33 | 3-HOCH2-Ph | CP | 51% | 49% | -4.1% | 56% | 129±15 | 118±4 |
| 34 |
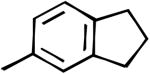
|
CP | 21% | 8% | -10.7% | 69% | 108±4 | 111±5 |
| 35 |
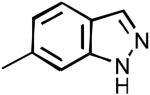
|
CP | 56% | 68% | 5.0% | 67% | 109±3 | 96±2 |
| 36 | 4-CH3O-PhCH2 | CP | 59% | 60% | 0.3% | 80% | 101±13 | 109±3 |
| 37 |
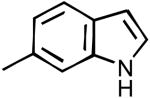
|
CP | 70% | 74% | 10.1% | 89% | 126±9 | 125±2 |
| 38 | PhCH2 | CP | 28% | 77% | -3.4% | 86% | 145±10 | 147±8 |
| 39 | Ph(CH2)2 | CP | 52% | 91% | -11.6% | 84% | 154±7 | 137±4 |
| 40 | 3,4-Cl2-Ph | cycloheptyl | -4% | -2% | -7.3% | 68% | 130±2 | 115±7 |
| 41 | 3,4-Cl2-Ph |
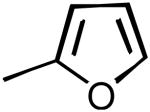
|
-4% | 70% | 9.0% | 78% | 98±3 | 95±4 |
| 42 | 3,4-Cl2-Ph | cyclobutyl | -5% | 0.4% | -5.5% | 52% | 116±3 | 126±3 |
| 43 | 3,4-Cl2-Ph (LUF6000) | cyclohexyl | -2% | -1% | -5.6% | 45% | 173±5 | 145±7 |
| 44 | 3,4-Cl2-Ph | H | 1.8% | -0.8% | 10.2% | 39% | 91±7 | 92±4 |
| 45 | 3,4-Cl2-Ph | n-pentyl | 43.9% | -1% | -9.3% | 84% | 116±5 | 102±5 |
All experiments were performed using adherent CHO cells stably transfected with cDNA encoding the human ARs. Binding at human A1, A2A and A3ARs in this study was carried out as described in Experimental Procedures using [3H]R-PIA, [3H]CGS 21680 or [125I]I-AB-MECA as a radioligand. Values from the present study are expressed as mean ± s.e.m., n = 3-5. Percentage inhibition at A1, A2A, or A3 receptors is expressed as the mean value from 2-4 separate experiments with similar results performed in duplicate.
A2BAR: effect of compounds at 10 μM on NECA (150 nM)-induced cyclic AMP accumulation from one experiment performed in triplicate, CGS15943 (10 μM)=100%.
dissociation: % decrease of [125I]I-AB-MECA dissociation at 30 min (control = 100%)
increase of efficacy: compared to maximal effect by 2-Cl-IB-MECA alone (control)
We further measured the capability of these compounds to influence the dissociation of an agonist radioligand, e.g. [125I]I-AB-MECA at the A3AR (Figure 2), which is a commonly used approach in the study of allosteric modulators8,10 and perturbation of the dissociation rate reflects the potency of the allosteric modulators. It has been reported previously that compound 28 decreased the dissociation rate of [125I]I-AB-MECA from the human A3ARs.17 Here it is shown that the dissociation rates of [125I]I-AB-MECA from the A3AR in the presence of 10 μM 42 and 43 were 0.038 ± 0.004 and 0.036 ± 0.005 min-1, respectively, which were significantly reduced and different from control (no further compound present) and that in the presence of 10 μM 44 (0.061 ± 0.006 min-1). By comparing the ligand dissociation at 30 min in the absence or presence of 10 μM imidazoquinoline, a number of these newly synthesized compounds (compounds 28-33, 37-40, 43) were found to decrease the dissociation significantly (Table 1). The ability of these compounds to affect the dissociation was found not to correlate with their ability to displace the equilibrium ligand binding at the A3AR (Table 1, r = 0.34). For example, although 28 was more potent than 43 in displacing [125I]I-AB-MECA binding, their ability to affect the dissociation rate was similar (Table 1).
Figure 2.

Radioligand binding studies on the human A3AR. Study of the dissociation kinetics of the agonist radioligand [125I]I-AB-MECA under control conditions and in the presence of 10 μM of compound 42, 43 or 44.
We further examined the ability of these compounds to influence agonist function by using a cyclic AMP functional assay in intact CHO cells stably expressing the human A3AR. The selective A3 agonist Cl-IB-MECA concentration-dependently inhibited forskolin-stimulated cyclic AMP accumulation (Figure 3), corresponding to an EC50 of 3.8 ± 1.2 nM. Compound 28 concentration-dependently increased the maximum agonist efficacy but somewhat decreased agonist potency. Interestingly, one of the newly synthesized compounds (43) was found to increase agonist efficacy but did not influence agonist potency. The ability of compounds to influence agonist efficacy did not correlate with their ability to inhibit equilibrium ligand binding at the human A3AR (Table 1, r = 0.25), further evidence of a complex mechanism of interaction. However, the ability of each compound to decrease the dissociation rate was positively correlated with its efficacy-enhancing effect (Table 1, r = 0.88).
Figure 3.

Functional assay of the human A3AR. The % inhibition of forskolin-stimulated cAMP production by increasing concentrations of 2-Cl-IB-MECA under control conditions or in the presence of 10 μM of compound 28 or 43.
Discussion
Allosteric modulation is considered of growing importance in controlling biological systems.25 GPCRs are the single most important target in drug development. Thus, allosteric modulation of GPCRs is of particular importance.
The present study is actually an important extension of an earlier pharmacological study.17 Previously it was found that functional enhancement of agonist action at the A3AR by 28 (see Figure 1 and Table 1) and its analogues has potential for further development in the direction of therapeutic applications. A3AR mutagenesis studies18 have implicated amino acids F1825.43 and N2747.45 in the action of 28. The mutagenesis data were interpreted using a rhodopsin-based A3AR molecular model, suggesting multiple binding modes of the enhancers either at the orthosteric site or at a distinct putative allosteric site. Here we synthesized and systematically studied a new series of allosteric modulators for the A3AR. Allosteric enhancers of A3AR activation may be predicted to be useful against a number of disorders, including ischemic conditions and cancer.
It is interesting that compounds within this series showed a remarkable variety of allosteric modulatory effects, with positive, neutral, and maybe slightly negative effects on both agonist efficacy and ligand dissociation. It was shown that these compounds influenced agonist binding, potency and efficacy in a separate manner. The identification of the important correlation between the ability of this series of compounds to affect ligand dissociation and their capability to enhance agonist efficacy suggested the possibility of further structural refinement to design more potent and efficacious allosteric enhancers.
Allosteric modulators that separately influence agonist affinity and efficacy at several GPCRs have been reported. For example, T62, which is now under clinical trails for potential treatment of neuropathic pain, has recently been suggested to enhance agonist efficacy rather than agonist potency as demonstrated in a functional assay of GTPγS binding to rat brain membranes.15 CPCCOEt (7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester), a noncompetitive metabotropic glutamate receptor 1 antagonist, allosterically inhibits receptor signaling without affecting glutamate binding.28 PIT (2,2′-pyridylisatogen tosylate) antagonizes P2Y1 receptor signaling without inhibiting nucleotide binding.29 Compound 28 enhanced agonist efficacy but decreased agonist potency (ref. 17 and this study). In previous studies of modulation of various GPCRs by several series of compounds, allosteric potencies determined from dissociation kinetics have not been found to correlate with binding potency from equilibrium experiments.19,26,27 However, to our knowledge, the findings from the current study represent the first evaluation of the correlation among the effects on agonist efficacy, dissociation kinetics, agonist potency and affinity.
Agonist binding site(s) on a GPCR used to be conceived of as an orthosteric site in contrast to the site(s) at which allosteric modulators bind. In that framework, allosteric modulators are without effect in the absence of a ligand binding to the orthosteric site. However, recently, a number of compounds were found to be allosteric agonists. AC-42 (4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl]-piperidine) is an allosteric agonist for the M1 muscarinic receptor.30 Alcuronium has been shown to induce muscarinic receptor activation, which is not prevented by the classical muscarinic antagonist quinuclidinyl benzilate.31 Thus, the activation mechanisms and structural and pharmacological modeling of GPCRs should also be reconstructed to accommodate the complex nature of GPCR activation. Allosteric modulation of GPCRs has recently been modeled in pharmacological terms,32,33 to provide a theoretical basis for an allosteric modulator separately affecting affinity, potency and efficacy. Hall32 suggested that CPCCOEt is positively cooperative with the binding of glutamate but negatively cooperative with its functional activation of the receptor. There is also evidence that some allosteric enhancers of agonist binding to the A1AR have intrinsic activity themselves at that receptor.9,34 However, the allosteric enhancers in the present study were not demonstrated to be allosteric agonists, as they did not show any effects by themselves.
Structure-activity relationships for the allosteric enhancing effects, based on slower dissociation of an A3AR agonist radioligand, for the present series were evident. At the 4-amino position, a phenyl ring or substituted monocyclic phenyl ring was more enhancing than bicyclic aryl rings. Also, by the same criterion a phenyl group was more highly enhancing than the corresponding benzyl and phenylethyl groups. As noted previously17 and consistent with compound 44, substitution at the 2-position was necessary for allosteric enhancement. At this position, medium sized cycloalkyl substituents (cyclopentyl and cyclohexyl) were most favorable for enhancement. Analogues bearing smaller or larger rings or an acyclic alkyl group were considerably less enhancing, and similarly an aromatic 5-membered ring was not conducive to enhancement. With the criterion of increased maximal efficacy of a potent A3AR agonist, benzyl and 3,4-dichlorophenyl groups at the 4-amino position were most enhancing.
In summary, by chemical structural modulation we have surpassed the allosteric enhancement observed in the previous series of 1H-imidazo[4,5-c]quinolin-4-amine derivatives. Two compounds, 2-cyclopentyl-4-benzylamino 38 and 2-cyclohexyl-4-(3,4-dichlorophenyl)amino 43 analogues, potentiated the maximum efficacy of the agonist Cl-IB-MECA by 45-50%. Moreover, 43 enhanced agonist efficacy in a functional assay and decreased agonist dissociation rate without influencing agonist potency, probably because of its experimentally observed decreased interaction with the orthosteric binding site on the A3AR (compare DU124128 (28) and 43). In fact, most allosteric enhancers in the present study were generally weak in inhibition of ligand binding, an otherwise complicating factor in pharmacological evaluation. Thus, members of this series are useful as pharmacological probes for further studies of the A3AR. Since the structural requirements for allosteric enhancement at the A3AR are distinct from the requirements to inhibit equilibrium binding, there is hope of achieving even greater selectivity upon future structural manipulation.
Experimental Procedures
Instrumentation and Analysis
Microwave reactions were performed in an Emrys™ Optimizer (Biotage AB). Wattage was automatically adjusted so as to maintain the desired temperature.
1H-NMR spectra were measured at 200 MHz with a Bruker AC 200 or Bruker DMX 600 spectrometer. Chemical shifts for 1H are given in ppm (d) relative to tetramethylsilane (TMS) as internal standard, coupling constants are given in Hz. Melting points were determined with a Büchi capillary melting point apparatus and are uncorrected. Combustion analyses of new target compounds were performed by the analytical department of the Gorlaeus Laboratories, Leiden University (The Netherlands) and are within 0.4 % of theoretical values unless otherwise specified.
Synthesis
2-(2-Nitro-ethylideneamino)-benzoic acid (2)
Prepared as described in literature.20 Yield: 18.40 g (89%): mp 196-197 °C.
3-Nitro-4-hydroxyquinoline (3)
Prepared as described in literature.20 Yield: 6.93 g (49%): mp >300 °C.
3-Nitro-4-chloroquinoline (4)
Prepared as described in literature.23 Yield: 5.05 g (81%): mp 118-119 °C.
3-Nitro-4-aminoquinoline (5)
Prepared as described in literature.23 Yield: 6.1 g (95%): mp 255-257 °C.
3,4-Diaminoquinoline (6)
Prepared as described in literature.23 Yield: 2.66 g (98%): mp 183-185 °C.
1H-Imidazo[4,5-c]quinoline (7)
Prepared as described in literature.23 Yield: 0.44 g (81%): mp 263-265 °C.
General procedure for 2-substituted 1H-imidazo[4,5-c]quinolines (8-11)
Polyphosphoric acid (1.3 mL/mmol) was added to 3,4-diaminoquinoline (6) and the appropriate carboxylic acid (1.2 equiv). The mixture was stirred at 100 °C for 5 hours. Then it was cooled to 0 °C and to it was added slowly NH4OH till pH = 8-9. The mixture was extracted with ethyl acetate (3 × 15 mL), washed with water, brine and again water and dried over MgSO4. The solution was filtered, the solvent was evaporated and the residue was dried.
2-Cyclobutyl-1H-imidazo[4,5-c]quinoline (8)
Scale: 6.2 mmol. Eluent for column chromatography was ethyl acetate:petroleum ether = 1:1. Yield: 0.88 g (63%): mp 191-192 °C.
2-Cyclopentyl-1H-imidazo[4,5-c]quinoline (9)
Scale: 4.0 mmol. Eluent for column chromatography was 5% methanol in dichloromethane. Yield: 0.77 g (81%): mp 191-192 °C.
2-Cyclohexyl-1H-imidazo[4,5-c]quinoline (10)
Scale: 6.3 mmol. Eluent for column chromatography was 1-5% methanol in dichloromethane. Yield: 0.60 g (38%): mp 205-206°C.
2-Cycloheptyl-1H-imidazo[4,5-c]quinoline (11)
Scale: 6.3 mmol. Eluent for column chromatography was 1-5% methanol in dichloromethane. Yield: 0.45 g (27%): mp 225-226 °C.
2-(2-Furyl)-1H-imidazo[4,5-c]quinoline (12)
2-Furoyl chloride (1.1 g, 0.8 mL, 8.1 mmol) in dry dichloromethane (15 mL) was added drop wise to a solution of to 3,4-diaminoquinoline (1.0 g, 6.0 mmol) in dry pyridine (6.2 mL) under an atmosphere of nitrogen. The solution was stirred for 2 hours at room temperature. Water (15 mL) was added to quench the reaction and the solvent was evaporated under reduced pressure to afford an orange solid. This crude solid was refluxed for 2 hours in 2 M NaOH (15 mL). After cooling on ice the pH was adjusted to 7 using concentrated HCl. The solid was filtered off and washed with water and ether. Then extracted with ethyl acetate (3 × 15 mL) and washed with water (3 × 15 mL) and dried over MgSO4. After evaporation the residue was dried. Eluent for column chromatography was 1-5% methanol in dichloromethane. Yield: 0.62 g (44%): mp 236-238 °C.
2-Pentyl-1H-imidazo[4,5-c]quinoline (13)
Prepared as described for the furyl compound (12) using hexanoyl chloride (1.75 g, 13 mmol). Eluent for column chromatography was ethyl acetate:petroleum ether 1:4 to 4:1. Yield: 0.85 g (41%): mp 142-143 °C.
General procedure for 2-substituted 1H-imidazo[4,5-c]quinolin-5-oxide (14-20)
Starting material was almost completely dissolved (with heating) in chloroform (2.5 mL/mmol), dichloromethane (2.5 mL/mmol) and methanol (0.25 mL/mmol). 3-Chloroperoxybenzoic acid (2.5 eq.) was added and the solution was refluxed. After 30 minutes Na2CO3 (0.04 g/mmol) was added and the mixture was refluxed for one more hour. The reaction mixture was cooled and the solvent was evaporated. Column chromatography was needed for purification and removal of 3-chloroperoxybenzoic acid.
1H-Imidazo[4,5-c]quinolin-5-oxide (14)
Scale: 8.3 mmol. Eluent for column chromatography was 2% methanol in dichloromethane. Yield: 0.55 g (36%): mp 290-295 °C.
2-Cyclobutyl-1H-imidazo[4,5-c]quinolin-5-oxide (15)
Scale: 1.8 mmol. Eluent for column chromatography was 2-6% methanol in dichloromethane. Yield: 0.18 g (42%): mp 123-130 °C.
2-Cyclopentyl-1H-imidazo[4,5-c]quinolin-5-oxide (16)
Scale: 0.8 mmol. Eluent for column chromatography was 5% methanol in dichloromethane. Yield: 0.19 g (95%): mp 155-157 °C.
2-Cyclohexyl-1H-imidazo[4,5-c]quinolin-5-oxide (17)
Scale: 2.4 mmol. Eluent for column chromatography was 5-10% methanol in dichloromethane. Yield: 0.59 g (92%): mp 160-165 °C.
2-Cycloheptyl-1H-imidazo[4,5-c]quinolin-5-oxide (18)
Scale: 2.0 mmol. Eluent for column chromatography was 3-8% methanol in dichloromethane. Yield: 0.22 g (39%): mp 115-120 °C.
2-(2-Furyl)-1H-imidazo[4,5-c]quinolin-5-oxide (19)
Scale: 2.2 mmol. Eluent for column chromatography was 1-10% methanol in dichloromethane. Yield: 0.36 g (66%): mp >280 °C.
2-Pentyl-1H-imidazo[4,5-c]quinolin-5-oxide (20)
Scale: 3.34 mmol. Eluent for column chromatography was 1-5% methanol in dichloromethane. Yield: 0.12 g (14%).
General procedure for substituted 4-chloro-1H-imidazo[4,5-c]quinolines (21-27)
A mixture of dry toluene (0.45 mL/mmol) and dry dimethylformamide (0.90 mL/mmol) was cooled in an ice bath and phosphorus oxychloride (2.6 eq.) was added. After 10 minutes the appropriate 1H-imidazo[4,5-c]quinolin-5-oxide was added and the solution was stirred at room temperature for 10 minutes. Subsequently the solution was heated to 100 °C for 30 minutes. Upon cooling, the solvent was evaporated and the resulting syrup was poured on chipped ice while stirring. The mixture was then warmed to room temperature and carefully adjusted to pH 6-7 with solid NaHCO3. After 2 hours, the formed solid was filtered off, washed with water and diisopropylether and subsequently dried.
4-Chloro-1H-imidazo[4,5-c]quinoline (21)
Scale: 3.5 mmol. Eluent for column chromatography was 2% methanol in dichloromethane. Yield: 0.31 g (44%): mp 257-258 °C.
4-Chloro-2-cyclobutyl-1H-imidazo[4,5-c]quinoline (22)
Scale: 0.6 mmol. Yield: 0.16 g (99%): mp 142-145 °C.
4-Chloro-2-cyclopentyl-1H-imidazo[4,5-c]quinoline (23)
Scale: 12.7 mmol. Yield: 2.65 g (75%): mp > 265 °C.
4-Chloro-2-cyclohexyl-1H-imidazo[4,5-c]quinoline (24)
Scale: 2.2 mmol. Yield: 0.65 g (97%): mp 245-250 °C.
4-Chloro-2-cycloheptyl-1H-imidazo[4,5-c]quinoline (25)
Scale: 0.8 mmol. Yield: 0.38 g (86%): mp 195-200 °C.
4-Chloro-2-(2-furyl)-1H-imidazo[4,5-c]quinoline (26)
Scale: 1.4 mmol. Eluent for column chromatography was ethyl acetate: petroleum ether = 25:75. Yield: 0.1 g (26%): mp 235-238 °C.
4-Chloro-2-pentyl-1H-imidazo[4,5-c]quinoline (27)
Scale: 0.45 mmol. Yield: 0.052 g (41%): mp 236-237 °C.
General procedure for N-substituted 1H-imidazo[4,5-c]quinolin-4-amines (28-45)
These compounds were prepared by means of microwave-assisted chemistry. Absolute ethanol (2.5-3.0 mL) was added to the appropriate 4-chloro-1H-imidazo[4,5-c]quinoline and the appropriate aniline (2-3 eq.) under nitrogen. Conditions: prestirring 60 seconds, temperature 120 °C, time 2400 seconds, normal sample absorption, fixed hold time. After the reaction was completed the solvent was evaporated and the remaining product was purified by column chromatography and recrystallized (MeOH/H2O).
N-Phenyl-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (28)
Scale: 0.8 mmol. Eluent for column chromatography was 2.5-10% methanol in dichloromethane. Yield: 40 mg (15%): mp 155-157 °C.
N-(4-Methyl-phenyl)-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (29)
Scale: 0.7 mmol. Eluent for column chromatography was 1% methanol in dichloromethane. Yield: 80 mg (35%): mp 125-126 °C.
N-(4-Methoxy-phenyl)-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (30)
Scale: 0.7 mmol. Eluent for column chromatography was 1% methanol in dichloromethane. Yield: 90 mg (38%): mp 106-107 °C.
N-(3,4-Dichloro-phenyl)-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (31)
Scale: 0.7 mmol. Eluent for column chromatography was 0-2% methanol in dichloromethane. Yield: 150 mg (54%): mp 114-115 °C.
N-(4-Chloro-phenyl)-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (32)
Scale: 1.0 mmol. Eluent for column chromatography was 0.5-2% methanol in dichloromethane. Yield: 120 mg (33%): mp 189-190 °C.
N-(3-Hydroxymethyl-phenyl)-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (33)
Scale: 0.4 mmol. Eluent for column chromatography was ethyl acetate:petroleum ether = 1:1, later increased to 7:3. Yield: 33 mg (25%): mp 228-230 °C.
N-([3,4-c]Indan-5-yl)-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (34)
Scale: 0.4 mmol. Eluent for column chromatography was 1% methanol in dichloromethane. Yield: 120 mg (81%): mp 142-145 °C.
N-(1H-indazol-6-yl)-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (35)
Scale: 0.4 mmol. Eluent for column chromatography was 4% methanol in dichloromethane. Yield: 110 mg (74%): mp 235-236 °C.
N-(4-Methoxy-benzyl)-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (36)
Scale: 0.4 mmol. Eluent for column chromatography was diisopropylether. Yield: 33 mg (22%): mp 245-247 °C.
N-(1H-Indol-6-yl)-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (37)
Scale: 0.4 mmol. Eluent for column chromatography was ethyl acetate:petroleum ether = 1:4, increased to 3:7. Yield: 30 mg (20%): mp 260-261 °C.
N-(Benzyl)-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (38)
Scale: 0.8 mmol. Eluent for column chromatography was 2% methanol in dichloromethane. Yield: 70 mg (25%), product was oily.
N-(Phenethyl)-2-cyclopentyl-1H-imidazo[4,5-c]quinolin-4-amine (39)
Scale: 0.8 mmol. Eluent for column chromatography was 1-4% methanol in dichloromethane. Yield: 160 mg (56%): mp 95-97 °C.
N-(3,4-Dichloro-phenyl)-2-cycloheptyl-1H-imidazo[4,5-c]quinolin-4-amine (40)
Scale: 0.8 mmol. Eluent for column chromatography was ethyl acetate:petroleum ether = 1:9, increased to 100% ethyl acetate. Yield: 164 mg (50%): mp 236-238 °C.
N-(3,4-Dichloro-phenyl)-2-(2-furyl)-1H-imidazo[4,5-c]quinolin-4-amine (41)
Scale: 0.4 mmol. Eluent for column chromatography was ethyl acetate:petroleum ether = 1:4. Yield: 96 mg (64%): mp 135-138 °C.
N-(3,4-Dichloro-phenyl)-2-cyclobutyl-1H-imidazo[4,5-c]quinolin-4-amine (42)
Scale: 0.7 mmol. Eluent for column chromatography was ethyl acetate:petroleum ether = 1:4. Yield: 121 mg (48%): mp 131-134 °C.
N-(3,4-Dichloro-phenyl)-2-cyclohexyl-1H-imidazo[4,5-c]quinolin-4-amine (43)
Scale: 0.9 mmol. Eluent for column chromatography was ethyl acetate:petroleum ether = 15:85, increased to 30:70. Yield: 160 mg (44%): mp 237-240 °C.
N-(3,4-Dichloro-phenyl)-1H-imidazo[4,5-c]quinolin-4-amine (44)
Scale: 1.0 mmol. Eluent for column chromatography was ethyl acetate:petroleum ether = 1:4. Yield: 160 mg (50%): mp 167-172 °C.
N-(3,4-Dichloro-phenyl)-2-pentyl-1H-imidazo[4,5-c]quinolin-4-amine (45)
Scale: 0.2 mmol. Eluent for column chromatography was dichloromethane. Yield: 44 mg (73%): mp 195-200 °C.
Pharmacological Methods
[125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide (I-AB-MECA; 2000 Ci/mmol), [3H]R-PIA (R-N6-[phenylisopropyl]adenosine, 34 Ci/mmol), [3H]CGS21680 (2-[p-(2-carboxyethyl)phenylethylamino]-5′-N-ethylcarboxamido-adenosine, 47 Ci/mmol) and [3H]cyclic AMP (40 Ci/mmol) were from Amersham Pharmacia Biotech (Buckinghamshire, UK).
Cell culture and membrane preparation
CHO (Chinese hamster ovary) cells expressing the recombinant human ARs (HEK-293 cells were used for the human A2AAR) were cultured in DMEM and F12 (1:1) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin and 2 μmol/ml glutamine. Cells were harvested by trypsinization. After homogenization and suspension, cells were centrifuged at 500 g for 10 min, and the pellet was re-suspended in 50 mM Tris·HCl buffer (pH 7.4) containing 10 mM MgCl2. The suspension was homogenized with an electric homogenizer for 10 sec, and was then re-centrifuged at 20,000 g for 20 min at 4°C. The resultant pellets were resuspended in buffer in the presence of 3 Units/mL adenosine deaminase, and the suspension was stored at -80°C until the binding experiments. The protein concentration was measured using the Bradford assay.35
Binding Assays to the Human A1 and A2A ARs
For binding to the human A1 AR, [3H]R-PIA (2 nM) was incubated with membranes (40 μg/tube) from CHO cells stably expressing the human A1 AR at 25 °C for 60 min in 50 mM Tris.HCl buffer (pH 7.4; MgCl2, 10 mM) in a total assay volume of 200 μL. Nonspecific binding was determined using 10 μM of N6-cyclopentyladenosine. For human A2A AR binding, membranes (20 μg/tube) from HEK-293 cells stably expressing the human A2A AR were incubated with 15 nM [3H]CGS21680 at 25 °C for 60 min in 200 μL of 50 mM Tris.HCl, pH 7.4, containing 10 mM MgCl2. N-5′-ethyluronamidoadenosine (10 μM) was used to define nonspecific binding. Reaction was terminated by filtration with GF/B filters.
Binding Assay to the Human A3 AR
Each tube in the competitive equilibrium binding assay contained 100 μl membrane suspension (20 μg protein), 50 μl [125I]I-AB-MECA (0.5 nM), and 50 μl of increasing concentrations of the test ligands in Tris·HCl buffer (50 mM, pH 8.0) containing 10 mM MgCl2, 1 mM EDTA. Nonspecific binding was determined using 10 μM of 5′-N-ethylcarboxamidoadenosine in the buffer. The mixtures were incubated at 25°C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using a MT-24 cell harvester (Brandell, Gaithersburgh, MD, USA). Filters were washed three times with 9 mL ice-cold buffer. Radioactivity was determined in a Beckman 5500B γ-counter.
Dissociation kinetics of [125I]I-AB-MECA from human A3ARs
The dissociation of [125I]I-AB-MECA was measured as follows. Membranes (20 μg) were preincubated at 25°C with 0.5 nM [125I]I-AB-MECA, in a total volume of 100 μl of Tris-HCl buffer (50 mM, pH 8.0) containing 10 mM MgCl2, and 1 mM EDTA for 60 min. The dissociation was then initiated by the addition of 3 μM Cl-IB-MECA with or without allosteric modulators. The time course of dissociation of total binding was measured by rapid filtration at appropriate time intervals. Nonspecific binding was measured after 60-min incubation in the presence of 3 μM Cl-IB-MECA. Binding reactions were terminated as described above.
Cyclic AMP accumulation assay
Intracellular cyclic AMP levels were measured with a competitive protein binding method.24 CHO cells that expressed recombinant human A3ARs were harvested by trypsinization. After centrifugation and resuspension in medium, cells were plated in 24-well plates in 0.5 mL medium. After 24 hr, the medium was removed and cells were washed three times with 1 mL DMEM, containing 50 mM HEPES, pH 7.4. Cells were then treated with agonists and/or test compounds in the presence of rolipram (10 μM) and adenosine deaminase (3 units/mL). After 45 min forskolin (10 μM) was added to the medium, and incubation was continued an additional 15 min. The reaction was terminated by removing the supernatant, and cells were lysed upon the addition of 200 μL of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at -20°C. For determination of cyclic AMP production, protein kinase A (PKA) was incubated with [3H]cyclic AMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 μL of the cell lysate, and 30 μL 0.1 M HCl or 50 μL of cyclic AMP solution (0-16 pmol/200 μL for standard curve). Bound radioactivity was separated by rapid filtration through Whatman GF/C filters and washed once with cold buffer. Bound radioactivity was measured by liquid scintillation spectrometry.
Statistical analysis
Binding and functional parameters were calculated using Prism 5.0 software (GraphPAD, San Diego, CA, USA). IC50 values obtained from competition curves were converted to Ki values using the Cheng-Prusoff equation.36 Data were expressed as mean ± standard error. The Pearson correlation coefficients (r) between efficacy, binding and dissociation were calculated using SYSTAT 11 (SYSTAT Software Inc.). The Pearson correlation coefficient is between -1 and 1, with 1 meaning that two series are positively correlated, 0 meaning that they are completely uncorrelated, and -1 meaning they are perfectly negatively correlated.
Supplementary Material
Tables with 1H- and 13C-NMR spectroscopic data and elemental analyses are available for selected compounds including all target compounds (28-45). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases and by the European Union (Marie Curie fellowship to A.G., QLK3-CT-2001-51963).
References
- 1.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Reviews Drug Discovery. 2006 doi: 10.1038/nrd1983. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan L, Burbiel JC, Maass A, Müller CE. Adenosine receptor agonists: from basic medicinal chemistry to clinical development. Expert Opin Emerging Drugs. 2003;8:537–576. doi: 10.1517/14728214.8.2.537. [DOI] [PubMed] [Google Scholar]
- 3.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 4.Zablocki JA, Wu L, Shryock J, Belardinelli L. Partial A1 adenosine receptor agonists from a molecular perspective and their potential use as chronic ventricular rate control agents during atrial fibrillation (AF) Curr Top Med Chem. 2004;4:839–854. doi: 10.2174/1568026043450998. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Conklin D, Pan HL, Eisenach JC. Allosteric adenosine receptor modulation reduces hypersensitivity following peripheral inflammation by a central mechanism. J Pharmacol Exp Ther. 2003;305:950–955. doi: 10.1124/jpet.102.047951. [DOI] [PubMed] [Google Scholar]
- 6.Sun CX, Young HW, Molina JG, Volmer JB, Schnermann J, Blackburn MR. A protective role for the A1 adenosine receptor in adenosine-dependent pulmonary injury. J Clin Invest. 2005;115:35–43. doi: 10.1172/JCI22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, Ohta A, Thiel M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. 2004;22:657–682. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 8.Birdsall NJ, Lazareno S. Allosterism at muscarinic receptors: ligands and mechanisms. Mini Rev Med Chem. 2005;5:523–543. doi: 10.2174/1389557054023251. [DOI] [PubMed] [Google Scholar]
- 9.Bruns RF, Fergus JH. Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol Pharmacol. 1990;38:939–949. [PubMed] [Google Scholar]
- 10.Christopoulos A. Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nature Reviews Drug Discov. 2002;1:198–210. doi: 10.1038/nrd746. [DOI] [PubMed] [Google Scholar]
- 11.Soudijn W, van Wijngaarden I, IJzerman AP. Allosteric modulation of G protein-coupled receptors: perspectives and recent developments. Drug Discov Today. 2004;9:752–758. doi: 10.1016/S1359-6446(04)03220-9. [DOI] [PubMed] [Google Scholar]
- 12.Gao ZG, Jacobson KA. Allosterism in membrane receptors. Drug Discov Today. doi: 10.1016/S1359-6446(05)03689-5. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Block GA, Martin KJ, de Francisco AL, Turner SA, Avram MM, Suranyi MG, Hercz G, Cunningham J, Abu-Alfa AK, Messa P, Coyne DW, Locatelli F, Cohen RM, Evenepoel P, Moe SM, Fournier A, Braun J, McCary LC, Zani VJ, Olson KA, Drueke TB, Goodman WG. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med. 2004;350:1516–25. doi: 10.1056/NEJMoa031633. [DOI] [PubMed] [Google Scholar]
- 14.Nagano N. Pharmacological and clinical properties of calcimimetics: Calcium receptor activators that afford an innovative approach to controlling hyperparathyroidism. Pharmacol Ther. 2006;109:339–65. doi: 10.1016/j.pharmthera.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 15.Childers SR, Li X, Xiao R, Eisenach JC. Allosteric modulation of adenosine A1 receptor coupling to G-proteins in brain. J Neurochem. 2005;93:715–723. doi: 10.1111/j.1471-4159.2005.03044.x. [DOI] [PubMed] [Google Scholar]
- 16.Gao ZG, van Muijlwijk-Koezen JE, Chen A, Müller CE, IJzerman AP, Jacobson KA. Allosteric modulation of A3 adenosine receptors by a series of 3-(2-pyridinyl)isoquinoline derivatives. Mol Pharmacol. 2001;60:1057–1063. [PMC free article] [PubMed] [Google Scholar]
- 17.Gao ZG, Kim SG, Soltysiak KA, Melman N, IJzerman AP, Jacobson KA. Selective allosteric enhancement of agonist binding and function at human A3 adenosine receptors by a series of imidazoquinoline derivatives. Mol Pharmacol. 2002;62:81–89. doi: 10.1124/mol.62.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao ZG, Kim SK, Gross AS, Chen A, Blaustein J, Jacobson KA. Identification of essential residues involved in the allosteric modulation of the human A3 adenosine receptor. Mol Pharmacol. 2003;63:1021–1031. doi: 10.1124/mol.63.5.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao ZG, Kim SK, IJzerman AP, Jacobson KA. Allosteric modulation of the adenosine family of receptors. Mini Rev Med Chem. 2005;5:545–553. doi: 10.2174/1389557054023242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bachman GB, Welton DE, Jenkins GL, Christian JE. Quinoline derivatives from 3-nitro-4-hydroxyquinoline. J Am Chem Soc. 1947;69:365–371. doi: 10.1021/ja01194a060. [DOI] [PubMed] [Google Scholar]
- 21.Young RC, Jones M, Milliner KJ, Rana KK, Ward JG. Purine derivatives as competitive inhibitors of human erythrocyte membrane phosphatidylinositol 4-kinase. J Med Chem. 1990;33:2073–2080. doi: 10.1021/jm00170a005. [DOI] [PubMed] [Google Scholar]
- 22.Scammells PJ, Baker SP, Belardinelli L, Olsson RA. Substituted 1,3-dipropylxanthines as irreversible antagonists of A1 adenosine receptors. J Med Chem. 1994;37:2704–2712. doi: 10.1021/jm00043a010. [DOI] [PubMed] [Google Scholar]
- 23.van Galen PJM, Nissen P, van Wijngaarden I, IJzerman AP, Soudijn W. 1-H-imidazo[4,5c]quinolin-4-amines: novel non-xantine adenosine antagonists. J Med Chem. 1991;34:1202–1206. doi: 10.1021/jm00107a046. [DOI] [PubMed] [Google Scholar]
- 24.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 25.Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 26.Gao ZG, Liu CG. Competitive and allosteric binding of 2α-DHET and its optical isomers to rat cardiac muscarinic receptors. Eur J Pharmacol. 1995;289:369–373. doi: 10.1016/0922-4106(95)90115-9. [DOI] [PubMed] [Google Scholar]
- 27.van der Klein PA, Kourounakis AP, IJzerman AP. Allosteric modulation of the adenosine A1 receptor. Synthesis and biological evaluation of novel 2-amino-3-benzoylthiophenes as allosteric enhancers of agonist binding. J Med Chem. 1999;42:3629–3635. doi: 10.1021/jm991051d. [DOI] [PubMed] [Google Scholar]
- 28.Litschig S, Gasparini F, Rueegg D, Stoehr N, Flor PJ, Vranesic I, Prezeau L, Pin JP, Thomsen C, Kuhn R. (1999) CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol. 1999;55:453–461. [PubMed] [Google Scholar]
- 29.Gao ZG, Mamedova L, Tchilibon S, Gross AS, Jacobson KA. 2,2′-Pyridylisatogen tosylate antagonizes P2Y1 receptor signaling without affecting nucleotide binding. Biochem Pharmacol. 2004;68:231–237. doi: 10.1016/j.bcp.2004.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langmead CJ, Fry VA, Forbes IT, Branch CL, Christopoulos A, Wood MD, Herdon HJ. Probing the Molecular Mechanism of Interaction between 4-n-Butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl]-piperidine (AC-42) and the Muscarinic M1 Receptor: Direct Pharmacological Evidence That AC-42 Is an Allosteric Agonist. Mol Pharmacol. 2006;69:236–246. doi: 10.1124/mol.105.017814. [DOI] [PubMed] [Google Scholar]
- 31.Jakubik J, Bacakova L, Lisa V, el-Fakahany EE, Tucek S. Activation of muscarinic acetylcholine receptors via their allosteric binding sites. Proc Natl Acad Sci USA. 1996;93:8705–8709. doi: 10.1073/pnas.93.16.8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hall DA. Modeling the functional effects of allosteric modulators at pharmacological receptors: an extension of the two-state model of receptor activation. Mol Pharmacol. 2000;58:1412–1423. doi: 10.1124/mol.58.6.1412. [DOI] [PubMed] [Google Scholar]
- 33.Ehlert FJ. Analysis of allosterism in functional assays. J Pharmacol Exp Ther. 2005;315:740–754. doi: 10.1124/jpet.105.090886. [DOI] [PubMed] [Google Scholar]
- 34.Figler H, Olsson RA, Linden J. Allosteric enhancers of A1 adenosine receptors increase receptor-G protein coupling and counteract guanine nucleotide effects on agonist binding. Mol Pharmacol. 2003;64:1557–1564. doi: 10.1124/mol.64.6.1557. [DOI] [PubMed] [Google Scholar]
- 35.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 36.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables with 1H- and 13C-NMR spectroscopic data and elemental analyses are available for selected compounds including all target compounds (28-45). This material is available free of charge via the Internet at http://pubs.acs.org.