

Abstract
Using a mouse predisposed to neoplasia by a germ line mutation in Apc (ApcMin), we tested whether induced hyperplasia is sufficient to increase intestinal tumor multiplicity or size in the intestine. We found that hyperplasia in the jejunum correlated with a significant increase in tumor multiplicity. However, tumor multiplicity was unchanged in the hyperplastic colon. This result indicates that even an intestine predisposed to neoplasia can, in certain regions including the colon, accommodate net increased cell growth without developing more neoplasms. Where hyperplasia correlated with increased tumor multiplicity, it did not increase the size or net growth of established tumors. This result suggests that the event linking hyperplasia and neoplasia in the jejunum is tumor establishment. Two novel observations arose in our study: the multiple intestinal neoplasia (Min) mutation partially suppressed both mitosis and transforming growth factor alpha-induced hyperplasia throughout the intestine; and zinc treatment alone increased tumor multiplicity in the duodenum of Min mice.
Introduction
‘Promotion’ is the second step in classic models of multistage carcinogenesis, following ‘initiation’ by DNA mutation or epigenetic change (1,2). Promotion, during which tumors undergo a net increase in cell number, is often associated with net proliferation of the tissue surrounding the initiated cells. This proliferation often causes hyperplasia, in which the tissue is enlarged but normal tissue architecture is preserved. Hyperplasia and tumor promotion are closely associated in many human tissues, including the thyroid gland, breast, prostate and colon (3–8).
In mice, tumor promotion has been analyzed most thoroughly in the skin, where it is tightly associated with hyperplasia (2,9,10). Invariably, chemical promoters of skin tumorigenesis cause hyperplasia, as do a variety of skin tumor-promoting signaling proteins expressed from transgenes in the epidermis (9,11,12). These hyperplasia-inducing signaling proteins include transforming growth factor alpha (TGFα). TGFα is also upregulated by various hyperplasia-inducing chemical tumor promoters (9).
TGFα overexpression from transgenes in mice causes uniform epithelial hyperplasia of several organs in addition to the skin, including the mammary gland, the liver and the intestine (13–15). In the mammary gland and the liver, TGFα overexpression leads to carcinoma development (13,14,16). In contrast, TGFα overexpression and the resulting hyperplasia do not, by themselves, lead to tumor development in the intestine (13).
Mice carrying the chain-terminating multiple intestinal neoplasia (Min) mutation in the mouse Apc gene on a susceptible genetic background develop scores of tumors in the intestine (17). Cell migration is reduced in non-neoplastic, heterozygous Apc-mutant tissue, as are both proliferation and apoptosis (18–21). These changes in the behavior of heterozygous tissue may either be autonomous to the heterozygous cells or else reflect a paracrine or systemic influence of the tumors arising in the intestines of heterozygous mice.
Tumors that form in Min mice, which are heterozygous for the ApcMin mutation, have silenced the wild-type allele or lost it through somatic recombination (17,22–24). Loss of normal Apc function, following loss of the wild-type Apc allele, might be sufficient for the development of adenomas in the small intestine (24–26). Apc loss leads to nuclear accumulation of β-catenin (27,28), which then forms a complex with lymphoid enhancer factor-1/T cell factor and transcriptionally activates a number of genes, including c-Myc (29–34). Loss of Apc leads to cellular changes, including failure to migrate and differentiate, that require c-Myc (35,36).
While Apc loss might be sufficient for tumorigenesis, genetic data suggest roles of several genes in enhancing or inhibiting the intestinal adenoma establishment (37–39). Deletion of Cox-1, Cox-2, Mmp-7 or PPARγ or mutation of Egfr dramatically reduces tumor multiplicity in Apc-mutant mice (40–44), whereas loss of Mlh1, Msh2, p53 or Atm increases tumor multiplicity (45–48).
To investigate the role of cell proliferation in intestinal adenoma formation, we have tested whether Min mice develop more larger or more advanced tumors when induced to show intestinal epithelial hyperplasia by transgenic TGFα. We have crossed mice carrying the Min mutation with mice carrying a germ line, zinc-inducible TGFα transgene (13). We show that TGFα from the transgene induces hyperplasia in mice carrying the Min mutation, in both the small and large intestines. In these hyperplastic regions, the establishment, but not the net growth, of intestinal tumors is enhanced in Apc-mutant mice. To our surprise, this tumor enhancement was limited to the small intestine. The comparably hyperplastic, transgenic colon developed no more tumors than its non-transgenic counterpart.
Materials and methods
Mice
Mice carrying a rat TGFα transgene controlled by the metallothionein promoter [Tg(Mt-1,rTgfα)Bril49; line 1745–8, abbreviated in the text as ‘Tg(MT-TGFα)’] (13), bred to C57BL/6J (B6) mice for 12 generations after their creation on a (B6 × SJL)F2 background, were a generous gift from Eric Sandgren. Mice carrying the ethylnitrosourea-induced ApcMin mutation had been bred to B6 mice for >45 generations after their detection on a (B6 × AKR/J)F1 background (17). All mice were maintained and bred on a Purina 5020 diet with 9% fat and 20% protein.
Zinc treatment
Progeny of Tg(Mt-TGFα) × ApcMin/+ crosses (Tables I, III and IV; Figures 1 and 2) were given 25 mM ZnSO4 (prepared using reverse osmosis-purified water and then autoclaved) in their drinking water from birth. Progeny of B6 × ApcMin/+ crosses (Table II) were given 40 μM, 1 mM and 25 mM ZnSO4 or 25 mM ZnCl2 (prepared using reverse osmosis-purified water and then autoclaved) starting at birth, from birth to 28 or 29 days, or starting at 28 or 29 days.
Table I.
TGFα overexpression increases the number of neoplasms in the jejunum
| Genotype | Intestinal tumor multiplicity (95% confidence interval) |
||||
| n | Duodenum | Jejunum | Ileum | Colon | |
| ApcMin/+ | 28 | 55 ± 21 (47–62) | 13 ± 6.0 (11–15) | 7.8 ± 4.2 (6.3–9.4) | 2.0 ± 1.8 (1.4–2.7) |
| ApcMin/+;Tg(Mt-TGFα) | 28a | 61 ± 17 (55–67) | 32 ± 15 (27–37) | 7.4 ± 3.5 (6.2–8.7) | 1.4 ± 1.3 (0.9–1.9) |
| P = 0.068 | P < 10−6 | P = 0.95 | P = 0.20 | ||
Female mice carrying the TGFα transgene Tg(Mt-TGFα) were crossed with male ApcMin/+ mice to generate animals that segregated for both the mutation and the transgene. Pups were fostered to ICR mothers and given ZnSO4 in their drinking water at birth to induce expression of TGFα. Age-matched mice (60-90 days old) were killed and their intestines fixed. Tumors in 4 cm long sections representing the four major parts of the intestine were counted and measured using a dissecting microscope at ×20 magnification. P values were determined using the Wilcoxon rank-sum test.
n = 27 for proximal.
Table III.
Min animals carrying a TGFα transgene develop hyperplasia in the jejunum and colon, but not in the ileum
| Intestinal region | ApcMin/+ | ApcMin/+ Tg(Mt-TGFα) | Apc+/+ Tg(Mt-TGFα) | Apc+/+ | ||
| Cells per crypt, mean ± SD (n half-crypts scored; n mice) | ||||||
| Jejunum | 18 ± 2 (756; 10) | 20 ± 2 (602; 11) | P < 0.05 | 26 ± 3 (303; 5) | 19 ± 1 (496; 6) | P < 0.01 |
| Ileum | 17 ± 2 (553; 6) | 18 ± 4 (231; 4) | P = 0.26 | 20 ± 2 (308; 5) | 17 ± 1 (537; 7) | P < 0.05 |
| Colon | 25 ± 2 (298; 6) | 32 ± 8 (327; 7) | P < 0.05 | 40 ± 7 (247; 7) | 22 ± 5 (147; 3) | P = 0.01 |
| Mitoses per crypt, mean ± SD (n full crypts scored; n mice) | ||||||
| Jejunum | 1.1 ± 0.5 (378; 10) | 1.2 ± 0.4 (301; 11) | P = 0.40 | 1.9 ± 0.8 (127; 4) | 1.1 ± 0.2 (248; 6) | P < 0.05 |
| Ileum | 0.7 ± 0.3 (277; 6) | 0.6 ± 0.4 (168; 5) | P = 0.58 | 1.3 ± 0.2 (204; 6) | 1.0 ± 0.4 (269; 7) | P = 0.15 |
| Colon | 0.3 ± 0.2 (149; 6) | 0.4 ± 0.2 (155; 7) | P = 0.28 | 1.0 ± 0.4 (76; 4) | 0.2 ± 0.2 (74; 4) | P < 0.05 |
A 4 cm section of the jejunum and the entire colon of progeny of a Apc+/+;Tg(TGFα) × ApcMin/+ cross were sliced longitudinally into four or five sections. Paraffin blocks of these sections were cut to generate 5 μm sections that were stained with H&E. Crypt height (the number of cells from base to crypt edge in vertical cross sections of crypts) and mitotic index were assessed by examining these H&E-stained sections at ×600 magnification. P values determined using the Wilcoxon rank-sum test.
Table IV.
TGFα affects the rate of apoptosis in the jejunum
| Intestinal region | Apoptoses per crypt, mean ± SD (n full crypts scored; n mice) |
|||||
| ApcMin/+ | ApcMin/+ Tg(Mt-TGFα) | Apc+/+ Tg(Mt-TGFα) | Apc+/+ | |||
| Jejunum | 0.05 ± 0.05 (600; 12) | 0.01 ± 0.02 (550; 11) | P < 0.05 | 0.04 ± 0.08 (580; 12) | 0.02 ± 0.03 (500; 10) | P = 0.73 |
| Colon | 0.009 ± 0.02 (350; 7) | 0.03 ± 0.06 (472; 11) | P = 0.41 | 0.14 ± 0.07 (500; 10) | 0.03 ± 0.03 (300; 6) | P < 0.01 |
| P < 0.05 | P = 0.60 | P < 0.001 | P = 0.53 | |||
A 4 cm section of the jejunum and the entire colon of progeny of a Apc+/+;Tg(TGFα) × ApcMin/+ cross were sliced longitudinally into four or five sections. Paraffin blocks of these sections were cut to generate 5 μm sections that were stained with H&E. The number of apoptoses per crypt were assessed by examining these H&E-stained sections at ×600 magnification (A.B.). Reading of the transgenic and non-transgenic jejunum slides by an independent observer (R.S.) produced comparable results. P values were determined using the Wilcoxon rank-sum test.
Fig. 1.
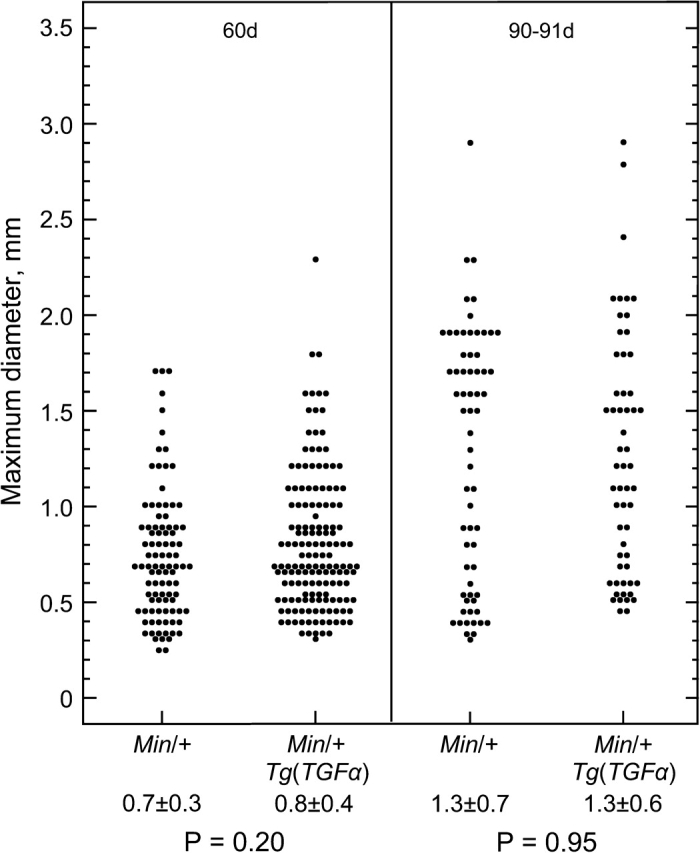
Tumor diameter at 60 and 90 days is almost identical between transgenic and non-transgenic animals. The average diameters of tumors in 4 cm sections of the jejunum obtained from ApcMin/+ or ApcMin/+;Tg(Mt-TGFα) mice at 60 days [ApcMin/+, seven mice and ApcMin/+;Tg(Mt-TGFα), six mice] and 90–91 days [ApcMin/+, five mice and ApcMin/+;Tg(Mt-TGFα), three mice] were measured under a dissecting microscope at ×20 power. P values were determined using the Wilcoxon rank-sum test.
Fig. 2.

Cross sections of crypts. Male progeny of an Apc+/+;Tg(Mt-TGFα)×ApcMin/+cross were given 25 mM ZnSO4 from birth until the age of 80–90 days. Intestines were fixed in formalin and 5 μm sections were stained with H&E. Note that the tunica muscularis (outer intestinal muscle) was cropped in photos of the colon. The average heights of crypts are given in units of cells per crypt in Table III. Scale bar, 100 μm.
Table II.
Zinc promotes tumorigenesis in the duodenum
| Treatment with 25 mM ZnSO4 in drinking water | No. of mice | Tumor multiplicity, mean ± SD |
|||
| Duodenum (P value relative to control) | Jejunum | Ileum | Colon | ||
| None (control) | 12 | 6.3 ± 2.5 | 10 ± 5.9 | 7.6 ± 4.4 | 3.2 ± 4.0 |
| Starting at 28/29 days of age | 6 | 67 ± 26 (P < 10−3) | ND | ND | ND |
| Starting at birth | 21 | 33 ± 15 (P < 10−5) | 9.8 ± 5.3 | 8.3 ± 4.5 | 2.3 ± 1.7 |
B6 mice carrying the ApcMin mutation were given ZnSO4 for the indicated duration (first column); otherwise they were given control drinking water. Mice were killed at ∼90 days of age. Tumors in 4 cm long sections representing the four major parts of the intestine were counted under a dissecting microscope at ×20 power. P values determined using the Wilcoxon rank-sum test. ND, not determined.
Tumor scoring and measurement
Four centimeter sections of small intestine from the duodenum, jejunum, ileum and the entire colon (17) were fixed in 10% formalin, generally for 24 h, and then rinsed and stored in 70% ethanol. Tumors in the fixed sections were counted and measured at ×20 power on an Olympus dissecting microscope with an ocular micrometer by an observer, blind with respect to genotype. To obtain the average tumor diameter, all tumors in the 4 cm jejunum were measured.
Genotyping
TGFα transgenic mice were identified by polymerase chain reaction analysis of genomic DNA obtained from 1 mm spleen pieces digested with Proteinase K (1 μg/μl in 45 mmol Tris–HCl, pH 8, 0.9 mM ethylenediaminetetraacetic acid and 0.45% Tween 20) and purified using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA) or from whole blood. Blood (50–100 μl) was mixed immediately with 200 μl of low-salt tris (LST) buffer (29 mM Tris, pH 7.4, 10 mM NaCl and 3 mM MgCl2) and then with 200 μl tris-nonidet P-40 lysis buffer (5% sucrose and 4% NP-40 in LST) until cells were lysed. Nuclei were precipitated by centrifugation, resuspended in water, frozen and then boiled. Polymerase chain reaction was performed using the primers TGF-pC3f (TGTCAGGCTCTGGAGAACAGC) and TGF-E4r (CACAGCGAACACCCACGTACC) for 1 min at 92°C, 2 min at 60°C and 3 min at 72°C for 40 cycles, followed by 10 min at 72°C. Apc allele status was assessed as described previously (49).
Histological assessment
Each of the formalin-fixed sections of gastrointestinal tract described above was sliced longitudinally into four or five sections. Paraffin blocks of these sections were cut to generate 5 μm sections that were stained with hematoxylin and eosin (H&E). The number of mitoses and apoptoses per crypt and crypt height (the number of cells from base to crypt edge in vertical cross sections of crypts) were assessed by examining these H&E-stained sections at ×600 magnification, blind with respect to genotype [A.B.; (50)].
In determining ‘normal’ crypt height, we avoided crypts adjacent to tumors. Our separate measurements of crypts adjacent to tumors confirmed previous reports that such crypts are unusually deep (20–200% deeper than in crypts further from the tumor). The depth of these adjacent crypts decreases exponentially with distance and stabilizes at about nine crypts from the tumor (data not shown).
Images of histological sections used in Figure 2 were obtained using a Zeiss Axiophot microscope at ×200 magnification.
Statistics
The significance of differences between data sets was determined by the Wilcoxon rank-sum test, using Mstat software (version 4.01; N.D., McArdle Laboratory for Cancer Research). All P values were calculated on the basis of a two-sided test, except in the case of crypt cell depth differences. Based on the known effect of the transgene on a wild-type background, the expected result in those comparisons was that the transgenic animals would have deeper crypts, so a one-sided test was performed. Power and correlation coefficient calculations were also performed using Mstat.
Results
TGFα overexpression increases the number of neoplasms in the jejunum, without increasing their size or progression
Mice that overexpress TGFα from a transgene driven by the metallothionein promoter develop hyperplasia throughout the intestine (13). To determine whether this hyperplastic stimulus would affect intestinal tumor development, we assessed the effect of TGFα overexpression on tumorigenesis in Min mice. These mice carry the ApcMin mutation and develop tumors throughout the intestine. We crossed female mice carrying the TGFα transgene with male Min mice (both on an inbred C57BL/6 genetic background) to generate animals that segregated for both the mutation and the transgene (Materials and Methods). These mice were fostered to ICR mothers and given ZnSO4 in their drinking water at birth to induce expression of TGFα.
ApcMin and ApcMin;Tg(Mt-TGFα) animals developed hallmarks of TGFα overexpression, including a fibrotic pancreas and a significantly more massive stomach. The additional burden of these abnormal organs might explain the earlier mortality of TGFα transgenic Min animals compared with the non-transgenic Min siblings and other Min mice in our colony. Transgenic Min mice became moribund at ∼80 days, whereas non-transgenic Min mice became moribund at ∼130 days.
At time points between 60 and 91 days, mice were killed, their intestines fixed and tumors were counted and measured in the entire colon and in 4 cm long sections representing the three major parts of the small intestine (in anterior–posterior order): the duodenum (or proximal small intestine), jejunum (or middle small intestine) and ileum (or distal small intestine).
The jejunum developed more than twice as many neoplasms in the presence of the TGFα transgene than in its absence (32 versus 13, P < 10−6; Table I). To determine whether this increase in multiplicity merely reflects an increase in tumor size, whereby tumors that would be undetectable in Min animals would grow to detectable size in TGFα transgenic Min animals, we measured the diameters of fixed tumors. Tumor diameters at 60 and 90 days were almost identical for transgenic and non-transgenic animals (Figure 1). This similarity in tumor size also indicates that TGFα overexpression does not dramatically affect the timing of tumor initiation. Histological analysis of the largest tumors indicates that, like their non-transgenic counterparts, TGFα transgenic Min tumors are rarely, if ever, invasive by 90 days of age (H.Pitot, personal communication).
The increase in tumor multiplicity caused by TGFα was limited to the jejunum. Relative to non-transgenic mice, the TGFα transgene had no detectable effect on tumor multiplicity either in the colon or in the ileum (see 95% confidence intervals; Table I). Tumor multiplicity in the duodenal region was only marginally elevated (P = 0.07).
Zinc promotes tumorigensis in the duodenum
Min mice lacking the TGFα transgene developed an average of 55 tumors in the duodenal region, ∼8-fold more tumors than we generally observe in Min mice [(17,51); A.Bilger and A.J.Prunuske, unpublished observations]. Further analyses revealed that most of the unusually high tumor multiplicity observed in the duodenal region of non-transgenic Min mice was completely independent of the transgene and was caused by the zinc administered in their drinking water.
When Min progeny of a B6 × Min mating were given 25 mM ZnSO4 (or 25 mM ZnCl2; data not shown), they developed 5- to 10-fold more tumors than those given control water (Table II). This effect required a high dose of zinc, as 1 mM caused only a 1.5-fold increase in tumor multiplicity (8.7 versus 6.3, P = 0.09) and 40 μM had no effect (6.2 versus 6.3, P = 0.8). Zinc affected tumor multiplicity whether given from birth or only after weaning, but the effect of zinc given lifelong was significantly weaker than when it was given only after weaning (P < 0.01; Table II). Mice wild-type for Apc developed no tumors when given ZnSO4. Importantly, the zinc effect in non-transgenic Min mice was strictly limited to the duodenal region: consumption of even 25 mM ZnSO4 had no effect on tumor multiplicity in the jejunum, ileum or colon in the absence of the TGFα transgene (Table II). To focus on effects caused by the transgene, we have omitted the duodenum from the analyses below.
The ApcMin mutation attenuates transgene-induced mitosis and hyperplasia
TGFα transgenic Min mice developed significant hyperplasia in both the jejunum and colon, with a crypt height in non-tumor crypts about two and seven cells longer, respectively, than in Min mice lacking the transgene (18 versus 20 cells per crypt in jejunum, P < 0.05 and 25 versus 32 in colon, P < 0.05; Table III). These results indicate that ApcMin intestinal cells in these regions responded to overexpressed TGFα by undergoing net excess growth. However, ileal ApcMin cells did not respond significantly to the transgene: ileal crypts were the same height in TGFα transgenic Min mice as in non-transgenc Min mice (17 versus 18 cells per crypt, P = 0.26; Table III).
The ileum's failure to develop significant hyperplasia in transgenic Min animals seemed to be a regionally pronounced instance of a global suppression of hyperplasia in transgenic mice by the Min mutation (Table III). Less complete suppression of hyperplasia was also seen in the jejunum (20 versus 26 cells per crypt, P < 0.01) and in the colon (32 versus 40 cells per crypt, P < 0.05). This global suppression of hyperplasia correlated with a reduction in mitotic rates (and not with an increase in apoptotic rates; Table IV). In the jejunum, ileum and colon, crypts in transgenic animals carrying the Min mutation displayed only 40–64% of the mitotic figures seen in transgenic mice on a wild-type background (jejunum: 1.2 versus 1.9, P = 0.09; ileum: 0.6 versus 1.3, P < 0.05 and colon: 0.4 versus 1.0, P < 0.05). This suppression of mitosis in TGFα transgenic Min mice yielded mitotic rates that were indistinguishable from those for non-transgenic Min animals (jejunum: 1.2 versus 1.1, P = 0.40; ileum: 0.6 versus 0.7, P = 0.58 and colon, 0.4 versus 0.3, P = 0.28; Table III).
The near-complete resistance of ileal ApcMin cells to transgene-induced hyperplasia prevented evaluation of whether hyperplasia could contribute to tumor promotion in that region. However, the jejunum and the colon of TGFα transgenic Min mice both developed significant hyperplasia. Resistance to hyperplasia therefore cannot explain the difference in tumor promotion between the colon and the jejunum.
TGFα affects the rate of apoptosis in the medial small intestine
To begin to understand the jejunum's susceptibility relative to the colon, we viewed H&E-stained normal tissue sections of TGFα transgenic and non-transgenic Min crypts and scored apoptotic bodies in these two regions. While the transgenic and non-transgenic jejunums underwent similar numbers of mitoses (Table III), they differed in the incidence of apoptoses. In the normal jejunum, non-transgenic animals displayed 4-fold more apoptoses than transgenic animals (P < 0.05, A.B.; Table IV). (R.S., an independent observer using ×500 magnification, detected a 3.3-fold difference in apoptoses, P < 0.05; data not shown; correlation between observers = 0.45, P < 0.05.) The majority of apoptoses in the jejunum occurred in the proliferative region of the crypt. Apoptoses in colonic crypts assessed in H&E-stained sections were not suppressed by the transgene. (Indeed, in TGFα transgenic animals, wild-type for Apc, apoptotic rates were uniquely high; Table IV.) Thus, suppression of apoptosis occurred only in the TGFα transgenic jejunum and correlated with enhanced tumorigenesis in that region.
Discussion
We report three main results: (i) hyperplasia of a tissue ‘initiated’ by mutation does not necessarily promote tumorigenesis in that tissue; (ii) the Min mutation can suppress hyperplasia and (iii) zinc can promote tumorigenesis in the intestine.
Tumor promotion
Hyperplasia was associated with a significant increase in the number of tumors in the jejunum. Tumor size and progression were unaffected, indicating that enhancement of tumorigenesis in the jejunum occurred during tumor initiation or establishment, rather than during growth or progression. These effects on tumor establishment are consistent with the effects of mutations in the TGFα receptor, the epidermal growth factor receptor (EGFR), on Min tumorigenesis.
EGFR plays a critical role in the maintenance and/or expansion of microadenomas in the small intestine [not assessed in the colon; (43)]. Similarly, the survival or expansion of microadenomas might underlie the enhancement of tumorigenesis by transgenic TGFα. A requirement for a threshold level of EGFR ligand at this vulnerable stage of tumor development could explain the apparently high frequency of polyclonal tumors in Min mice (52,53). Specifically, microadenomas might fail to expand unless they are exposed to sufficient EGFR ligand, potentially supplied in a paracrine fashion by nearby microadenomas and transformed crypts.
The EGFR family is known to mediate migration, differentiation, cell polarity and survival (54,55). An effect of cell survival on tumorigenesis is suggested by our observation that apoptosis is suppressed in the normal TGFα transgenic Min jejunum. Thus, TGFα might enhance tumor establishment by preventing the apoptosis of aberrant cells in microadenomas, while independently promoting the hyperplastic growth of normal tissue.
We were surprised to find that hyperplasia induced by TGFα overexpression did not lead to enhanced tumorigenesis in the colon. Hyperplasia has long been associated with tumorigenesis, particularly in chemical carcinogenesis protocols that cause skin tumors (3–9). Our finding indicates that some tissues can accommodate a net increase of mutant cells without developing more tumors. Plausible explanations for the colon's resistance relative to the jejunum include regional variation in the timing or location of TGFα action or factors that modify its effect.
Work on skin carcinogenesis has shown that the timing of tumor promotion is often critical (10). Most tumors detected in Min are established within the first few weeks of life (56), and the time during which microadenomas can be effectively expanded or protected from loss might be limited. The colon might respond to TGFα later than the jejunum because of natural variation in the development of the intestine (57,58) or because slower zinc accumulation in the colon (59) delays activation of the TGFα transgene.
Alternatively, cellular or dietary factors or local flora that modify the effects of hyperplastic growth might differ between the jejunum and the colon. Cellular cofactors might include the products of genes that either require mutation or silencing in the colon but not the jejunum (39) or else enhance apoptosis in response to TGFα in the colon. We observed suggestive evidence for such an increase in apoptosis, but it was not statistically significant (Table IV).
Dietary cofactors might include zinc. Zinc accumulates to much higher levels in the proximal small intestine (duodenum and jejunum) than in the colon (59). Zinc can suppress apoptosis, its deficiency has been shown to affect tumorigenesis and excess zinc may increase mutagenicity by suppressing the levels of superoxide dismutase (60–62). However, zinc does not promote tumorigenesis in the jejunum of Min mice in the absence of the transgene (Table II) and its role in the jejunum would therefore be restricted to that of the cofactor.
The timing of a cofactor's accumulation might also determine its ability to act as a promoter. In their study of tumors induced by the food mutagen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in Min mice, Steffensen et al. (63) found that sensitivity to 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine, which causes inactivation of the wild-type Apc allele (64), was greatest between postpartum days 3–12 in the small intestine. In the colon, sensitivity was greatest from day 3 before birth to day 3 after birth (63). Thus, in our experiments, the colon might have responded to zinc and TGFα overexpression by developing more tumors if zinc had been provided starting in late gestation. Initial attempts to induce TGFα expression in pregnant females throughout gestation have resulted in overgrown, stillborn fetuses (A.Bilger and A.J.Prunuske, unpublished observations).
Min suppresses TGFα-induced hyperplasia
A second unexpected result from our study was the general suppression by the Min mutation of TGFα-induced hyperplasia throughout the intestine. Although this suppression was incomplete in the jejunum and colon, it was significant and correlated with suppression of mitosis. The suppression of mitosis and hyperplasia seen here might reflect an effect of Apc mutation on TGFα expression or signaling. However, suppression of cell division in normal, non-transgenic tissue by Apc mutations has been noted previously (51,65). It is not clear whether the suppression is a cell-autonomous result of heterozygosity for Apc or, instead, whether it is a paracrine or systemic effect of tumors.
Zinc as tumor promoter
We have found that chronic consumption of 25 mM zinc in drinking water causes a 5- to 10-fold increase in tumor multiplicity in Min mice on an otherwise entirely B6 background (Table II). These novel results demonstrate that zinc can act as a tumor promoter in vivo. The consumption of 25 mM zinc in drinking water by mice is equivalent to the consumption of ∼1500 mg/day of zinc by humans. Recently, a study of men taking zinc supplements suggested that consumption of >100 mg of zinc per day may play a role in prostate carcinogenesis (66). Our observation of zinc's effect on Min mice strengthens the study’s conclusion that high doses of zinc supplements might promote carcinogenesis.
Summary
We have shown that the jejunum of Min mice responded to a hyperplastic environment by developing more tumors, but the colon, significantly, did not. In addition, our novel observations that the Min mutation can suppress hyperplasia and that zinc can promote tumorigenesis in the Min intestine each warrant further study.
Funding
National Research Service Award and a National Cancer Institute Postdoctoral Fellowship to A.B.; National Cancer Institute (P30-CA014520) to UW Comprehensive Cancer Center and (R37-CA63677) to W.F.D.
Acknowledgments
We thank Eric Sandgren for providing transgenic mice; Michael Newton for assistance with statistics; Henry Pitot for assessing pathology; Rich Halberg for comments on the manuscript and Natalie Borenstein, Xiaodi Chen, Dawn Albrecht, Brianna Laube, Kathy Krentz and Laura Schorrak for assistance with experiments; and all for their advice.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- EGFR
epidermal growth factor receptor
- H&E
hematoxylin and eosin
- Min
multiple intestinal neoplasia
- TGFα
transforming growth factor alpha
- Tg(MT-TGFa)
Tg(Mt-1,rTgfa)Bril49
References
- 1.Miller EC, et al. Mechanisms of chemical carcinogenesis. Cancer. 1981;47:1055–1064. doi: 10.1002/1097-0142(19810301)47:5+<1055::aid-cncr2820471302>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Slaga TJ, et al. The mouse skin carcinogenesis model. J. Investig. Dermatol. Symp. Proc. 1996;1:151–156. [PubMed] [Google Scholar]
- 3.Derwahl M, et al. Hyperplasia versus adenoma in endocrine tissues: are they different? Trends Endocrinol. Metab. 2002;13:23–28. doi: 10.1016/s1043-2760(01)00519-7. [DOI] [PubMed] [Google Scholar]
- 4.Li Z, et al. Increased risk of prostate cancer and benign prostatic hyperplasia associated with transforming growth factor-beta 1 gene polymorphism at codon10. Carcinogenesis. 2004;25:237–240. doi: 10.1093/carcin/bgg197. [DOI] [PubMed] [Google Scholar]
- 5.Salam MT, et al. Associations between polymorphisms in the steroid 5-alpha reductase type II (SRD5A2) gene and benign prostatic hyperplasia and prostate cancer. Urol. Oncol. 2005;23:246–253. doi: 10.1016/j.urolonc.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 6.Shaaban AM, et al. Prognostic significance of estrogen receptor Beta in epithelial hyperplasia of usual type with known outcome. Am. J. Surg. Pathol. 2005;29:1593–1599. doi: 10.1097/01.pas.0000184807.38037.75. [DOI] [PubMed] [Google Scholar]
- 7.Collins LC, et al. The influence of family history on breast cancer risk in women with biopsy-confirmed benign breast disease: results from the Nurses’ Health Study. Cancer. 2006;107:1240–1247. doi: 10.1002/cncr.22136. [DOI] [PubMed] [Google Scholar]
- 8.Rubio CA, et al. Hyperplastic polyposis coli syndrome and colorectal carcinoma. Endoscopy. 2006;38:266–270. doi: 10.1055/s-2006-925026. [DOI] [PubMed] [Google Scholar]
- 9.DiGiovanni J, et al. Role of the epidermal growth factor receptor and transforming growth factor alpha in mouse skin carcinogenesis. Prog. Clin. Biol. Res. 1994;387:113–138. [PubMed] [Google Scholar]
- 10.Miller EC. Some current perspectives on chemical carcinogenesis in humans and experimental animals: Presidential Address. Cancer Res. 1978;38:1479–1496. [PubMed] [Google Scholar]
- 11.Greenhalgh DA, et al. Multistage epidermal carcinogenesis in transgenic mice: cooperativity and paradox. J. Investig. Dermatol. Symp. Proc. 1996;1:162–176. [PubMed] [Google Scholar]
- 12.Muller-Decker K, et al. Differential protein expression in the epidermis of wild-type and COX-2 transgenic mice. Skin Pharmacol. Physiol. 2006;19:89–94. doi: 10.1159/000091975. [DOI] [PubMed] [Google Scholar]
- 13.Sandgren EP, et al. Overexpression of TGF alpha in transgenic mice: induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell. 1990;61:1121–1135. doi: 10.1016/0092-8674(90)90075-p. [DOI] [PubMed] [Google Scholar]
- 14.Jhappan C, et al. TGF alpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell. 1990;61:1137–1146. doi: 10.1016/0092-8674(90)90076-q. [DOI] [PubMed] [Google Scholar]
- 15.Matsui Y, et al. Development of mammary hyperplasia and neoplasia in MMTV-TGF alpha transgenic mice. Cell. 1990;61:1147–1155. doi: 10.1016/0092-8674(90)90077-r. [DOI] [PubMed] [Google Scholar]
- 16.Sandgren EP, et al. Inhibition of mammary gland involution is associated with transforming growth factor alpha but not c-myc-induced tumorigenesis in transgenic mice. Cancer Res. 1995;55:3915–3927. [PubMed] [Google Scholar]
- 17.Moser AR, et al. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 18.Mahmoud NN, et al. Apc gene mutation is associated with a dominant-negative effect upon intestinal cell migration. Cancer Res. 1997;57:5045–5050. [PubMed] [Google Scholar]
- 19.Nathke I. Relationship between the role of the adenomatous polyposis coli protein in colon cancer and its contribution to cytoskeletal regulation. Biochem. Soc. Trans. 2005;33:694–697. doi: 10.1042/BST0330694. [DOI] [PubMed] [Google Scholar]
- 20.Hanson CA, et al. Non-traditional roles for the adenomatous polyposis coli (APC) tumor suppressor protein. Gene. 2005;361:1–12. doi: 10.1016/j.gene.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 21.Akiyama T, et al. Wnt signalling and the actin cytoskeleton. Oncogene. 2006;25:7538–7544. doi: 10.1038/sj.onc.1210063. [DOI] [PubMed] [Google Scholar]
- 22.Su LK, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 23.Shoemaker AR, et al. A resistant genetic background leading to incomplete penetrance of intestinal neoplasia and reduced loss of heterozygosity in ApcMin/+ mice. Proc. Natl Acad. Sci. USA. 1998;95:10826–10831. doi: 10.1073/pnas.95.18.10826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haigis KM, et al. A Robertsonian translocation suppresses a somatic recombination pathway to loss of heterozygosity. Nat. Genet. 2003;33:33–39. doi: 10.1038/ng1055. [DOI] [PubMed] [Google Scholar]
- 25.Yamada Y, et al. Microadenomatous lesions involving loss of Apc heterozygosity in the colon of adult Apc(Min/+) mice. Cancer Res. 2002;62:6367–6370. [PubMed] [Google Scholar]
- 26.Preston SL, et al. Bottom-up histogenesis of colorectal adenomas: origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. 2003;63:3819–3825. [PubMed] [Google Scholar]
- 27.Korinek V, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 28.Rubinfeld B, et al. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- 29.He TC, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 30.Ignatenko NA, et al. Role of c-Myc in intestinal tumorigenesis of the ApcMin/+ mouse. Cancer Biol.Ther. 2006;5:1658–1664. doi: 10.4161/cbt.5.12.3376. [DOI] [PubMed] [Google Scholar]
- 31.Behrens J. The role of the Wnt signalling pathway in colorectal tumorigenesis. Biochem. Soc. Trans. 2005;33:672–675. doi: 10.1042/BST0330672. [DOI] [PubMed] [Google Scholar]
- 32.Karim R, et al. The significance of the Wnt pathway in the pathology of human cancers. Pathology. 2004;36:120–128. doi: 10.1080/00313020410001671957. [DOI] [PubMed] [Google Scholar]
- 33.Clements WM, et al. Adenomatous polyposis coli/beta-catenin interaction and downstream targets: altered gene expression in gastrointestinal tumors. Clin. Colorectal Cancer. 2003;3:113–120. doi: 10.3816/ccc.2003.n.018. [DOI] [PubMed] [Google Scholar]
- 34.Kaiser S, et al. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol. 2007;8:R131. doi: 10.1186/gb-2007-8-7-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sansom OJ, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sansom OJ, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–679. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- 37.Bilger A, et al. Manipulation of the mouse germline in the study of Min-induced neoplasia. Semin. Cancer Biol. 1996;7:249–260. doi: 10.1006/scbi.1996.0033. [DOI] [PubMed] [Google Scholar]
- 38.Dove WF, et al. The intestinal epithelium and its neoplasms: genetic, cellular and tissue interactions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1998;353:915–923. doi: 10.1098/rstb.1998.0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamada Y, et al. Multistep carcinogenesis of the colon in Apc(Min/+) mouse. Cancer Sci. 2007;98:6–10. doi: 10.1111/j.1349-7006.2006.00348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oshima M, et al. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 41.Wilson CL, et al. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc. Natl Acad. Sci. USA. 1997;94:1402–1407. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chulada PC, et al. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000;60:4705–4708. [PubMed] [Google Scholar]
- 43.Roberts RB, et al. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc. Natl Acad. Sci. USA. 2002;99:1521–1526. doi: 10.1073/pnas.032678499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McAlpine CA, et al. Intestinal-specific PPARgamma deficiency enhances tumorigenesis in ApcMin/+ mice. Int. J. Cancer. 2006;119:2339–2346. doi: 10.1002/ijc.22115. [DOI] [PubMed] [Google Scholar]
- 45.Reitmair AH, et al. MSH2 deficiency contributes to accelerated APC-mediated intestinal tumorigenesis. Cancer Res. 1996;56:2922–2926. [PubMed] [Google Scholar]
- 46.Shoemaker AR, et al. Mlh1 deficiency enhances several phenotypes of ApcMin/+ mice. Oncogene. 2000;19:2774–2779. doi: 10.1038/sj.onc.1203574. [DOI] [PubMed] [Google Scholar]
- 47.Halberg RB, et al. Tumorigenesis in the multiple intestinal neoplasia mouse: redundancy of negative regulators and specificity of modifiers. Proc. Natl Acad. Sci. USA. 2000;97:3461–3466. doi: 10.1073/pnas.050585597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwong LN, et al. Atm is a negative regulator of intestinal neoplasia. Oncogene. 2008;27:1013–1018. doi: 10.1038/sj.onc.1210708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dietrich WF, et al. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- 50.Potten CS, et al. Regulation and significance of apoptosis in the stem cells of the gastrointestinal epithelium. Stem Cells. 1997;15:82–93. doi: 10.1002/stem.150082. [DOI] [PubMed] [Google Scholar]
- 51.Cormier RT, et al. Dnmt1N/+ reduces the net growth rate and multiplicity of intestinal adenomas in C57BL/6-multiple intestinal neoplasia (Min)/+ mice independently of p53 but demonstrates strong synergy with the modifier of Min 1(AKR) resistance allele. Cancer Res. 2000;60:3965–3970. [PubMed] [Google Scholar]
- 52.Thliveris AT, et al. Polyclonality of familial murine adenomas: analyses of mouse chimeras with low tumor multiplicity suggest short-range interactions. Proc. Natl Acad. Sci. USA. 2005;102:6960–6965. doi: 10.1073/pnas.0502662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Halberg RB, et al. Polyclonal tumors in the mammalian intestine: are interactions among multiple initiated clones necessary for tumor initiation, growth, and progression? Cell Cycle. 2007;6:44–51. doi: 10.4161/cc.6.1.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh AB, et al. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal. 2005;17:1183–1193. doi: 10.1016/j.cellsig.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 55.Aranda V, et al. Par6-aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat. Cell Biol. 2006;8:1235–1245. doi: 10.1038/ncb1485. [DOI] [PubMed] [Google Scholar]
- 56.Shoemaker AR, et al. N-ethyl-N-nitrosourea treatment of multiple intestinal neoplasia (Min) mice: age-related effects on the formation of intestinal adenomas, cystic crypts, and epidermoid cysts. Cancer Res. 1995;55:4479–4485. [PubMed] [Google Scholar]
- 57.Sidhu NK, et al. Regional variation in ontogeny of class II antigens in enterocytes of mouse small intestine. Histol. Histopathol. 1992;7:137–142. [PubMed] [Google Scholar]
- 58.Penney L, et al. Regional variation in the proliferative rate and lifespan of alpha beta TCR+ and gamma delta TCR+ intraepithelial lymphocytes in the murine small intestine. Immunology. 1995;86:212–218. [PMC free article] [PubMed] [Google Scholar]
- 59.Tran CD, et al. Regional distribution of metallothionein and zinc in the mouse gut: comparison with metallothionien-null mice. Biol. Trace Elem. Res. 1998;63:239–251. doi: 10.1007/BF02778942. [DOI] [PubMed] [Google Scholar]
- 60.Walsh CT, et al. Zinc: health effects and research priorities for the 1990s. Environ. Health Perspect. 1994;102(suppl. 2):5–46. doi: 10.1289/ehp.941025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Truong-Tran AQ, et al. Cellular zinc fluxes and the regulation of apoptosis/gene-directed cell death. J. Nutr. 2000;130:1459S–1466S. doi: 10.1093/jn/130.5.1459S. [DOI] [PubMed] [Google Scholar]
- 62.Carter JW, et al. Zinc deprivation promotes progression of 1,2-dimethylhydrazine-induced colon tumors but reduces malignant invasion in mice. Nutr. Cancer. 1997;27:217–221. doi: 10.1080/01635589709514529. [DOI] [PubMed] [Google Scholar]
- 63.Steffensen IL, et al. Intestinal tumorigenesis in multiple intestinal neoplasia mice induced by the food mutagen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine: perinatal susceptibility, regional variation, and correlation with DNA adducts. Cancer Res. 2001;61:8689–8696. [PubMed] [Google Scholar]
- 64.Mollersen L, et al. Adenomatous polyposis coli truncation mutations in 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced intestinal tumours of multiple intestinal neoplasia mice. Mutat. Res. 2004;557:29–40. doi: 10.1016/j.mrgentox.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 65.Mahmoud NN, et al. Genotype-phenotype correlation in murine Apc mutation: differences in enterocyte migration and response to sulindac. Cancer Res. 1999;59:353–359. [PubMed] [Google Scholar]
- 66.Leitzmann MF, et al. Zinc supplement use and risk of prostate cancer. J. Natl Cancer Inst. 2003;95:1004–1007. doi: 10.1093/jnci/95.13.1004. [DOI] [PubMed] [Google Scholar]