

Abstract
We present a new convenient method for quantitative three-dimensionally resolved diffusion measurements based on the photobleaching (FRAP) or photoactivation (FRAPa) of a disk-shaped area by the scanning laser beam of a multiphoton microscope. Contrary to previously reported spot-photobleaching protocols, this method has the advantage of full scalability of the size of the photobleached area and thus the range of diffusion coefficients, which can be measured conveniently. The method is compatible with low as well as high numerical aperture objective lenses, allowing us to perform quantitative diffusion measurements in three-dimensional extended samples as well as in very small volumes, such as cell nuclei. Furthermore, by photobleaching/photoactivating a large area, diffusion along the optical axis can be measured separately, which is convenient when studying anisotropic diffusion. First, we show the rigorous mathematical derivation of the model, leading to a closed-form formula describing the fluorescence recovery/redistribution phase. Next, the ability of the multiphoton FRAP method to correctly measure absolute diffusion coefficients is tested thoroughly on many test solutions of FITC-dextrans covering a wide range of diffusion coefficients. The same is done for the FRAPa method on a series of photoactivatable green fluorescent protein solutions with different viscosities. Finally, we apply the method to photoactivatable green fluorescent protein diffusing freely in the nucleus of living NIH-3T3 mouse embryo fibroblasts.
INTRODUCTION
Obtaining quantitative information on the mobility of molecules and particles in biological matrices is an important aspect in many research areas. In the biomedical and pharmaceutical field, for example, successful delivery of (macromolecular) therapeutics, such as peptides, proteins, and polynucleotides, to their target site in the body requires overcoming several biological barriers (1). Substantial efforts are being made to develop smart carrier materials capable of protecting the therapeutic molecules against degradation and facilitating their transport during the various phases of the delivery process (2). A detailed understanding of the dynamics of such carrier materials in tissues and inside cells is a prerequisite for an efficient and rational optimization of their design.
Nowadays, several complementary advanced fluorescence microscopy methods are available for studying the dynamic behavior of molecules and particles on the micro- and nanoscale, such as fluorescence correlation spectroscopy (FCS), single particle tracking (SPT), and fluorescence recovery after photobleaching (FRAP) (2–4). FCS is based on the temporal measurement of fluorescence intensities in a very small volume (<1 femtoliter). The movement of fluorescently labeled molecules in and out of this detection volume gives rise to fluorescence fluctuations whose duration is directly related to the velocity of the molecules. By autocorrelation analysis it is possible to calculate the (ensemble average) diffusion coefficient from the fluorescence fluctuation trace (5). In SPT, the transport of individual molecules or particles is directly imaged at a high tempospatial resolution (6,7). Complementary to FCS and SPT, which both require very dilute samples (typically in the nanomolar range), FRAP has proven to be a very useful and convenient tool for measuring diffusion of fluorescently labeled molecules at typical imaging concentrations (usually >100 nM) in a micron-sized area (8–10). A typical FRAP experiment involves three distinct steps, namely registration of the fluorescence before photobleaching; fast photobleaching within a defined area using a high power laser beam; and subsequent imaging of the fluorescence recovery arising from the diffusional exchange of photobleached molecules by intact ones from the immediate surroundings. It is then possible to extract the diffusion coefficient and a local (im)mobile fraction from the recovery curve by fitting of a suitable mathematical FRAP model. FRAP has been used, for example, to study the mobility of molecules in cells (11–14), as well as in extracellular matrices, such as mucus, (tumor) cell interstitium, and vitreous (2).
During the first period since its introduction by Peters et al. in 1974 (15), FRAP experiments were mainly performed with a stationary laser beam focused to a small spot by the microscope objective lens (16–18). As the confocal laser-scanning microscope became a popular and widespread tool, spot-photobleaching experiments were gradually replaced by line-scanning photobleaching protocols during the 90s (19–24). FRAP methods based on photobleaching by a scanning beam have the advantage of a freely definable bleach area, both in size and shape. Hence, since the speed of recovery is proportional to the area of the bleach region, a much larger range of diffusion coefficients is accessible within an acceptable measurement time. Also, in many spot-photobleaching experiments the fluorescence recovery is measured with the same (attenuated) stationary laser beam, resulting in a single fluorescence trace with a usually low signal/noise ratio. On a laser-scanning microscope, on the other hand, full images are acquired of the recovery phase, allowing us to integrate the recovery signal over many individual pixels and resulting in a much improved signal/noise ratio. Additionally, a reference region can be defined in the images to correct for bleaching and laser fluctuations during imaging of the recovery phase, which is not possible otherwise.
Complementary to standard confocal imaging, multiphoton microscopy has proven to be a useful tool for imaging deep into highly scattering tissues and materials (25). In multiphoton microscopy, the excitation of the fluorescent molecules is intrinsically limited to the small focal volume of the focused laser beam. Therefore, the photobleaching is also limited to the same small focal volume, contrary to single-photon FRAP, where a substantial region above and below the focal plane is bleached as well. Due to this property, multiphoton FRAP has been suggested as a method to probe the diffusion with increased axial resolution (26,27). However, multiphoton FRAP has been developed for a spot-photobleaching protocol only, and consequently has not found much application so far.
As an alternative to photobleaching, techniques based on the photoactivation of fluorophores have recently been developed to study diffusion and reaction/diffusion phenomena (28–31). Basically, photoactivatable fluorescent molecules exhibit a change in their absorption spectrum when illuminated with light of proper wavelengths, resulting in an increase of the fluorescence signal when excited with light within the absorption band. In a fluorescence redistribution after photoactivation (FRAPa) experiment, the molecules in a specified region of the sample are rapidly turned on using a highly intense laser beam. The subsequent redistribution of fluorescence due to diffusion is then recorded by timelapse imaging, similar to a classic FRAP experiment. Since photoactivatable fluorophores usually exhibit a 100–1000-fold increase of the fluorescent signal (32–35), higher signal/noise ratio redistribution curves can be obtained with a substantially lower light load compared to classic photobleaching experiments (29). However, quantitative analysis of photoactivation experiments have only been done via numerical methods so far (28,29), thus limiting the widespread use of the method.
To address both needs, we report here a versatile and easy-to-use quantitative multiphoton FRAP/FRAPa method based on the photobleaching/photoactivation of a circular area by a multiphoton laser-scanning microscope. Contrary to previously reported spot-photobleaching protocols (26), this method has the advantage of full scalability of the size of the photobleached area and thus the range of diffusion coefficients, which can be measured conveniently. Moreover, the method is compatible with low as well as high numerical aperture objective lenses and allows us to perform quantitative diffusion measurements in three-dimensional extended samples as well as in very small volumes, such as cell nuclei.
Additionally, by photobleaching/photoactivating a large area, diffusion along the optical axis can be measured separately, which is a convenient property when studying anisotropic diffusion. First, we present the rigorous derivation of the FRAP model, leading to a closed-form solution for the recovery phase which can be easily implemented in a fitting routine without the need for special programming skills. We also derive the equivalent expressions for multiphoton FRAPa experiments. Next, we examine the influence of the most important model parameters and present a thorough experimental validation of the model, both for photobleaching and photoactivation. Finally, an example experiment is provided in which the new method is applied to the diffusion of free photoactivatable green fluorescent protein in the nucleus of living mouse embryo fibroblasts NIH-3T3.
THEORY
n-photon activation by a scanning laser beam
Consider a sample with fluorescent molecules at concentration C (number of molecules per unit of volume). Each molecule has a cross section σn for n-photon absorption. If 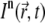 is the n-photon illumination intensity distribution with a spatial and temporal average intensity of the (pulsed) laser beam
is the n-photon illumination intensity distribution with a spatial and temporal average intensity of the (pulsed) laser beam 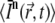 within a volume V, it follows that the number of absorbed photons per unit time is
within a volume V, it follows that the number of absorbed photons per unit time is 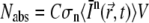 (36). Since C × V is the number of molecules within the illuminated volume, the number of photons that are absorbed per unit time and per molecule is
(36). Since C × V is the number of molecules within the illuminated volume, the number of photons that are absorbed per unit time and per molecule is 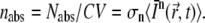 More generally, because n photons are necessary for an n-photon excitation event, the rate of absorption per molecule is
More generally, because n photons are necessary for an n-photon excitation event, the rate of absorption per molecule is 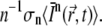 If qn is the quantum efficiency for n-photon photobleaching, the rate of photobleaching per molecule is given by
If qn is the quantum efficiency for n-photon photobleaching, the rate of photobleaching per molecule is given by 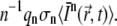 As described by Braeckmans et al. (22), assuming first-order photobleaching kinetics and a short photobleaching time (to avoid diffusion during photobleaching), it follows that the concentration of fluorophores after n-photon photobleaching of a two-dimensional geometry B(x,y) with a scanning beam can be calculated from
As described by Braeckmans et al. (22), assuming first-order photobleaching kinetics and a short photobleaching time (to avoid diffusion during photobleaching), it follows that the concentration of fluorophores after n-photon photobleaching of a two-dimensional geometry B(x,y) with a scanning beam can be calculated from
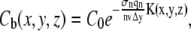 |
(1) |
where C0 is the homogeneous initial fluorophore concentration, v the line scanning speed, and Δy the distance between consecutive scanning lines. Note that the use of Eq.1 implies that Δy should be smaller than the radial resolution of the bleaching beam to avoid gaps in between the bleached lines (22). The effective bleaching intensity distribution K(x,y,z) is calculated from the convolution product of the geometry B(x,y) and the time average bleaching intensity distribution:
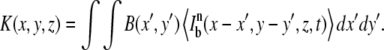 |
(2) |
Photobleaching and recovery in a large uniform disk
Let us now consider the n-photon photobleaching of a large uniform disk (see Fig. 1),
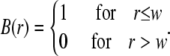 |
The n-photon excitation distribution can be effectively modeled by a three-dimensional Gaussian distribution (26),
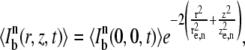 |
(3) |
where 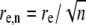 and
and 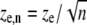 are the effective radial and axial resolution of the n-photon photobleaching beam. However, when the radius w is much larger than the radial resolution re,n, the latter can be effectively neglected and the excitation intensity distribution becomes (22)
are the effective radial and axial resolution of the n-photon photobleaching beam. However, when the radius w is much larger than the radial resolution re,n, the latter can be effectively neglected and the excitation intensity distribution becomes (22)
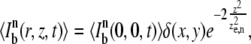 |
where δ(x,y) is the Dirac delta function. We will determine experimentally the implications of this assumption. From Eqs. 1 and 2, it immediately follows that
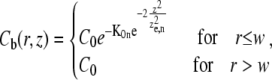 |
(4) |
where 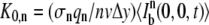 is the n-photon photobleaching parameter that determines the amount of photobleaching.
is the n-photon photobleaching parameter that determines the amount of photobleaching.
FIGURE 1.

Schematic representation of the bleaching phase of a two-photon FRAP measurement. The bleaching illumination distribution Ib(x,y,z) scans line by line the selected circular region of radius w. A high laser power is delivered on the sample when the system is scanning the inner part of the circle (dashed lines), inducing the photobleaching/photoactivation of the fluorescent molecules. An x,z view of the scanning process is also shown, where re and ze are the axial and radial 1/e2 extensions of the Gaussian bleaching illumination distribution.
When considering the case where the fluorescence recovery after photobleaching is due to free diffusion of the fluorescent molecules, Fick's second law can be used:
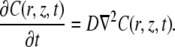 |
(5) |
To calculate the recovery in the photobleached disk, Eq. 5 has to be solved for the initial condition defined by Eq. 4. This is formally the same problem as solved by Braeckmans et al. (22), and leads to the solution
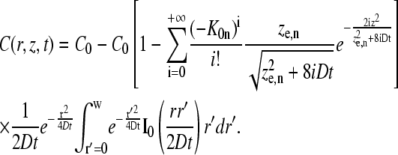 |
(6) |
The fluorescence recovery as observed by m-photon microscopy can be calculated from the convolution product of the concentration distribution in Eq. 6 with the overall microscope m-photon point-spread (PSF) function  This can be reasonably assumed as Gaussian (see Eq. 3), both in conventional one-photon excitation (provided that a small pinhole is used) and in multiphoton excitation. We allow the radial and axial extensions of the PSF, rd and zd, to be different from re and ze since previous studies have pointed out that saturation effects can increase the effective resolution of the bleaching intensity distribution (23,37,38). Again, the radial resolution of the PSF
This can be reasonably assumed as Gaussian (see Eq. 3), both in conventional one-photon excitation (provided that a small pinhole is used) and in multiphoton excitation. We allow the radial and axial extensions of the PSF, rd and zd, to be different from re and ze since previous studies have pointed out that saturation effects can increase the effective resolution of the bleaching intensity distribution (23,37,38). Again, the radial resolution of the PSF 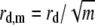 can be neglected if it is much smaller than the radius w of the bleached disk. Thus, as explained in Appendix A, the recovery of the total fluorescence inside the photobleached disk, Ftot, can be calculated from
can be neglected if it is much smaller than the radius w of the bleached disk. Thus, as explained in Appendix A, the recovery of the total fluorescence inside the photobleached disk, Ftot, can be calculated from
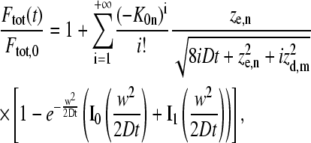 |
(7) |
where Ftot,0 is the total fluorescence inside the disk before bleaching, and I0 and I1 are the modified Bessel functions of order 0 and 1, respectively. While the infinite series may seem inconvenient, it converges quite rapidly for small values of K0n. Since in a FRAP experiment K0n is usually <2, taking five terms into account is already more than sufficient (1.5% error for K0n = 2). Note that for t = 0 the radial part of Eq. 7 is undetermined. A solution for t = 0 can be found by making use of the large argument asymptotic expansion of the modified Bessel functions,
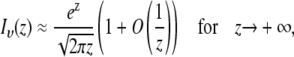 |
(8) |
from which it follows that 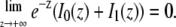 Thus we find at t = 0,
Thus we find at t = 0,
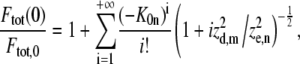 |
(9) |
which can be used to calculate the (im)mobile fraction (see Eq. 16 further on). As explained in Braeckmans et al. (22), the effective size of the bleached disk weff depends slightly on the amount of bleaching (the bleaching parameter K0n). Therefore, to obtain the most accurate results, the radius w of the bleached disk in the formulae presented here should be replaced by
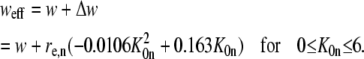 |
(10) |
Finally, we can consider some special cases of Eq. 7. Only radial diffusion 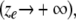
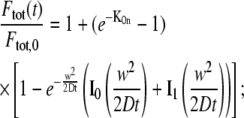 |
(11) |
and only axial diffusion 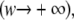
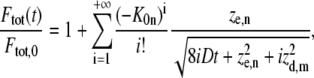 |
(12) |
which is immediately found by making use of the large argument asymptotic expansion in Eq. 8. As we will show further on, this formula can be used to selectively measure axial diffusion by photobleaching a large area and analyzing the recovery in the central part only.
Fluorescence redistribution after photoactivation of a large uniform disk
The formulae derived above can be readily converted to the case of photoactivation by an n-photon scanning beam as follows. Consider a sample of photoactivatable molecules. Let C0 be the initial concentration of nonactivated molecules and CA,0 the initial concentration of activated molecules that are present before the photoactivation step. Just as in the case of photobleaching, one can assume the photoactivation process to follow first-order kinetics, leading to (see Eq. 1)
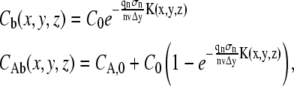 |
(13) |
where Cb and CAb represent the concentration distribution of nonactivated and activated molecules at the end of the photoactivation step. Entirely analogous to the case of photobleaching, we can calculate the observed fluorescence of both populations separately as a function of time after photoactivation. What is observed experimentally, however, is the sum of both fluorescence signals, for which we finally find
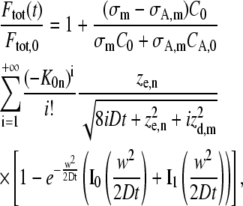 |
(14) |
where σm and σA,m are the cross sections for m-photon fluorescence emission of the nonactivated molecules and activated molecules, respectively (σm < σA,m at the wavelength that is used for imaging the activated molecules). Let it be noted that Eq. 14 is equivalent to the model derived for photobleaching (see Eq. 7), except for the factor 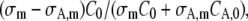 which accounts for the increase of signal associated to the activated molecules. It can easily be seen that this factor depends on the ratios C0 /CA,0 and σA,m/ σm only, which can be calculated with a linear unmixing method if the absorption spectra of the nonactivated and activated molecules are known (see Appendix B).
which accounts for the increase of signal associated to the activated molecules. It can easily be seen that this factor depends on the ratios C0 /CA,0 and σA,m/ σm only, which can be calculated with a linear unmixing method if the absorption spectra of the nonactivated and activated molecules are known (see Appendix B).
Diffusion in a volume with limited axial extent
Let us now consider the case when the photobleaching/photoactivation experiment is performed within a volume having limited axial extent. Here we assume that the focal plane is positioned at the middle of the volume, such that the walls are located at z = ±h/2. Following the method of reflection and superposition (39), we find
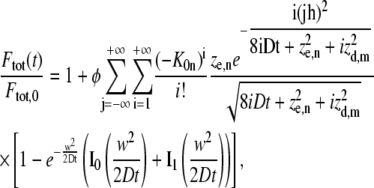 |
(15) |
where φ = 1 in case of photobleaching (see Eq. 7) and 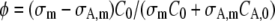 in case of photoactivation (see Eq. 14).
in case of photoactivation (see Eq. 14).
Immobile fraction
A fraction of immobile molecules inside the photobleached/photoactivated area can be taken into account by substituting any one of the Eqs. 7, 11, 12, 14, or 15 into the right-hand side of
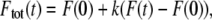 |
(16) |
where k is the fraction of mobile molecules. Even when working in samples where all molecules are mobile (k = 1), we still recommend using Eq. 16 with k as a free additional fitting parameter. In such experiments, the value of k can provide information on the quality of the experiment. For example, in case of flow in the sample, k will usually be ≫1. A value <1 could be an indication of the sample containing two (or more) populations of molecules with different diffusion coefficients.
MATERIALS AND METHODS
Confocal and two-photon FRAP equipment
The FRAP experiments were performed on a Leica TCS SP5 laser microscope equipped with an acousto-optic beam splitter (Leica Microsystems, Heidelberg, Germany). For conventional single-photon FRAP, the 488 nm line of the argon laser was used in combination with a 10×, 0.4 NA objective lens for both bleaching of the fluorophores and observation of the fluorescence recovery. Two-photon FRAP experiments were performed using a 63×, 1.4 NA oil immersion objective lens in combination with a Ti:sapphire tunable ultrafast Chameleon XR pulsed laser source (Coherent, Santa Clara, CA) having a pulse width of ∼140 fs at the laser beam output window and a repetition rate of 90 MHz. The laser beam is coupled in air with the Leica SP-5 scan head after having passed through an electro-optic modulator (Linos Photonics, Gottingen, Germany), which is used to control the laser power delivered to the sample. For the two-photon experiments reported here, excitation was performed by tuning the laser at 760 nm. A laser power meter (Ophir, Wilmington, MA) was used to measure the laser power coming out of the objective lens. The microscope is also equipped with an environmental chamber, allowing us to perform the experiments at a constant temperature (30.0 ± 0.2°C for the validation experiments on test solutions and 37.0 ± 0.2°C for the cell experiments). See schematic representation in Fig. 2.
FIGURE 2.

Schematic representation of the optical system. The light emitted by an argon laser is delivered by an optical fiber to the confocal head. The power delivered to the sample can be tuned via an acousto-optic tunable filter. An infrared Ti:sapphire laser is also coupled to the confocal head for two-photon experiments. In this case, the power is controlled by an electro-optic modulator. A short-pass dichroic mirror (715 nm, SP715) prevents reflected infrared light to reach the detector. Scanning is accomplished either with conventional scanning mirrors SM1 and SM2, or with a couple of resonant scanning mirrors to acquire images at a fast rate (not shown). The fluorescent light coming from the sample is discriminated from the excitation light by the acousto-optic beam splitter and brought to the detector after passing through the pinhole (in the case of confocal imaging) and through a diffractive element which allows selecting the detected wavelength range.
Test solutions
Two types of fluorescent molecules were used to test the two-photon FRAP model: fluorescein isothiocyanate dextrans (FITC-Dextrans, FD) (Sigma-Aldrich, St. Louis, MO) of different molecular weights (FD150, 1.5 × 105 g/mol; FD250, 2.5 × 105 g/mol; FD500: 5 × 105 g/mol) and photoactivatable green fluorescent protein (paGFP) (2.7 × 104 g/mol) (33). For FITC-Dextrans a concentration of 2 mg/ml was chosen which is within the linear concentration range (determined separately). To obtain a range of diffusion coefficients, aqueous solutions containing different amounts of glycerol were prepared for each of the FITC-dextrans. paGFP samples were prepared by diluting the stock solution of purified protein in PBS buffer to a final concentration of 0.5 mg/ml. Sucrose was then added to obtain solutions with different viscosities. For the FRAP experiments we added 6 μL of the solutions to a small chamber obtained by attaching a 0.12-mm-thick adhesive silicon spacer (Secure-Seal spacers, Molecular Probes, Leiden, The Netherlands) on a microscope coverslip. The chamber was then sealed with a microscope slide to eliminate any detectable flow in the sample.
NIH-3T3 cell culture and transient transfection
Embryo mouse fibroblasts NIH-3T3 were grown on microscope coverslips, in Dulbecco's modified Eagle's medium supplemented with 2 mM glutamine, 10% fetal bovine serum, and 1% penicillin-streptomycin. NIH-3T3 cells were transiently transfected using FuGene 6 reagent (Roche, Milan, Italy) according to the manufacturer's instructions with paGFP plasmid. The cells were subsequently incubated at 37°C in a 5% CO2 atmosphere to allow for paGFP expression.
Experimental FRAP protocol
FITC-dextran solutions
The validation of the multiphoton FRAP model is performed by comparison with the (single-photon) confocal disk FRAP method which has previously been described (22) and which has already been applied to various research topics (2,40–44). In agreement with the requirements of the disk FRAP model, we have photobleached in a single iteration a disk of 10-μm radius with an NA 0.4 objective lens at a pixel size of 303 nm. After photobleaching, a time series was recorded of typically 50 frames where the time between the images was between 1.4 and 15 s depending on the speed of the recovery process. Two-photon FRAP was performed by photobleaching disks of various size (see Results and Discussion) in a single photobleaching iteration with an NA 1.4 objective lens. In all experiments the focal plane was placed at ∼10 μm from the coverslip. Two-photon excitation was used for both bleaching and imaging of the recovery phase (n = m = 2). In this case we used a fully open pinhole setting to collect most of the light emitted by the molecules involved in the two-photon process. An example of a FRAP series on a FITC-dextran sample is shown in Fig. 3 A. The zoom was adjusted to obtain a pixel size of 60.2 nm, which is in agreement with the requirement of the interline distance Δy being smaller than the resolution of the photobleaching beam. Since the recovery process after two-photon photobleaching is considerably faster than for the confocal FRAP measurements, the image size was reduced to 512 × 128 pixels, resulting in a time between the images of 0.355 s at a line-scanning rate of 400 Hz. When needed, we decreased the time between the images even further to 0.185 s by scanning the sample in bidirectional mode. This shortens the photobleaching time without affecting the bleaching parameters such as the scanning speed v and the interline distance Δy.
FIGURE 3.

(A) An example is shown of a two-photon disk FRAP experiment on FD500 in an 85% (w/w) glycerol solution. Images of the sample are acquired at a regular time interval of 0.36 s. The first image shows the sample before photobleaching. The white disk (3 μm radius) in the second image comes from the photobleaching step at t = 0. Within the (user-defined) disk, the laser intensity is switched to a high value to quickly induce local photobleaching. From the third image on, the laser is switched back to a low intensity and a series of images is acquired of the recovery process at regular time intervals. The outlined circle in the first frame represents the selected reference region to account for bleaching during imaging and laser intensity fluctuations. (B) Custom image processing software is used to extract the normalized recovery curve from the images, as explained in the main text (solid dots). The diffusion coefficient D, the mobile fraction k, and the bleaching parameter K0n are calculated from a best fit of the model to the recovery data (solid line). (C and D) A corresponding two-photon FRAPa experiment is shown on paGFP in a 51% (w/w) sucrose solution. The solid dots are the experimental data and the solid line is the best fit of the model. The outlined circle in the first frame of panel C indicates the selected reference region.
paGFP solutions and cells
Validation of the two-photon FRAPa experiments on paGFP samples was done in the same way as for the FITC-dextran solutions, i.e., by comparing the two-photon results with the ones obtained with the disk FRAP method on the same samples. Confocal disk FRAPa measurements were preformed with the 488-nm line of the argon laser, which is capable of inducing a moderate photoconversion of paGFP (45). As reported earlier, paGFP can efficiently undergo photoactivation by two-photon excitation in the wavelength range between 720 nm and 860 nm (35). We therefore used the pulsed laser, tuned at 760 nm, to photoactivate the fluorophores for the two-photon FRAPa experiments. However, since we noticed that the activated form does not efficiently fluoresce when excited by 760 nm, we collected the fluorescence redistribution images using conventional confocal imaging with the 488 nm laser at low power (n = 2, m = 1). In this case, the confocal aperture was set to 1 Airy Unit to detect the in-focus fluorescence only. However, due to the large difference between the activating wavelength (760 nm) and the imaging wavelength (488 nm), we found a 1-μm difference between the focal planes for each wavelength due to chromatic aberrations. This effect was experimentally remedied by automatically adjusting the z position of the galvanometric table by 1 μm immediately after the photoactivation step. An example of a FRAPa series on a paGFP sample is provided in Fig. 3 C. Although the observation of the fluorescence redistribution is performed by confocal imaging, the benefits associated to two-photon induced perturbation are preserved, resulting in three-dimensionally defined activation volumes, together with the possibility of using high NA objectives. Due to the fast kinetics associated with paGFP diffusion, a line-scanning rate was selected of 4000 Hz resulting in a time of 140 ms between the 512 × 512 pixel images. Again a pixel size of 60.2 nm was selected here.
Three-dimensional point spread function measurements
The FRAP method presented here requires the knowledge of the effective PSF during both the perturbation and imaging phase. The photoactivation/photobleaching PSF was evaluated via a calibration FRAPa/FRAP experiment as described below. The overall microscope imaging PSF was measured by acquiring three-dimensional images of fluorescent polystyrene microspheres of 170 nm in diameter (Molecular Probes) at the very same laser power used to observe the fluorescence redistribution after the perturbation pulse. Thus we obtained zd = 0.92 μm and rd = 0.42 μm under multiphoton excitation at 760 nm, and zd = 0.64 μm and rd = 0.28 μm for confocal imaging operating at Airy disk 1 pinhole at 488 nm.
Data analysis and fitting
Custom image analysis software was written in MATLAB (The MathWorks, Natick, MA) to extract the experimental recovery curves from the timecourse microscopy images. First, the position of the photobleached/photoactivated circular region is determined and the absence of flow is verified using a center-of-mass algorithm. Secondly, the total intensity inside the disk as a function of time is calculated. For each time point, the total fluorescence intensity within the disk is normalized to the total intensity in a reference region far away from the perturbed area, to account for laser fluctuations and photobleaching during imaging. The recovery data points are then normalized to the prebleach value. Finally, the experimental parameters are determined by a least-squares fit of any one of Eqs. 7, 12, 14, or 15 in combination with Eqs. 10 and 16 to the experimental recovery data. Exemplary normalized curves obtained from photobleaching of FITC-dextrans and photoactivation of paGFP are provided in Fig. 3, B and D, respectively.
RESULTS AND DISCUSSION
First we will examine the influence of the different model parameters on the calculated diffusion coefficient. In particular, we will concentrate on the role of the perturbation laser power and the size of the perturbed disk. Next, having established the influence of those parameters, we will validate the two-photon disk model by performing experiments on solutions with known diffusion coefficients. Finally we will show the application of the method to diffusion measurements of free paGFP in the nucleus of NIH-3T3 cells.
Influence of the perturbation laser power
Recently we have demonstrated that, due to saturation effects at high laser powers, the effective extension of the photobleaching PSF generally depends on the photon flux of the photobleaching beam, both in single photon (23,37) and two-photon photobleaching experiments (38), as well as in photoactivation experiments (46). In particular we have shown that, while the size of the effective PSF under high power photobleaching conditions can increase substantially, the shape of the photobleaching PSF can still be approximated by a three-dimensional Gaussian distribution as in Eq. 3. For a particular zoom setting (i.e., line-scanning speed), the axial and radial resolution, ze and re, of the effective perturbation PSF, depend on the laser power. The precise dependency of ze and re on the laser power has to be determined experimentally for a particular fluorophore. As explained in Theory, we assume a perturbation disk whose diameter is much larger than the radial resolution re of the effective perturbation PSF. Under those conditions the redistribution is independent of re and we only have to take ze into account (see Eq. 7). To examine the effect of the perturbation laser power on ze, we performed two-photon FRAP experiments at different photobleaching powers on a single solution of fluorophores whose diffusion coefficient D is known from an independent measurement with the conventional single photon disk FRAP method. The value ze can be calculated from a best fit of Eq. 7 to the experimental recovery curve (with D fixed). This was done for an FD500 solution (85% glycerol, D = 0.362 ± 0.016 μm2/s), the results of which are shown in Fig. 4 A. Consistent with our previous studies, again we find a substantial increase of the effective photobleaching PSF with an average value of ze = 2.64 μm compared to zd = 0.93 μm for the overall imaging PSF. Interestingly, ze varies only little with increasing laser power, similar to what we have found for single photon photobleaching experiments on FITC dextrans (23). For our further experiments we have selected a photobleaching power between 60 and 75 mW. In addition, the bleaching parameter K0 values as calculated from these experiments are plotted versus laser power in Fig. 4 B. While theoretically one would expect a quadratic power law (see Eq. 4), instead we find a slope of 2.7. This is in agreement with previous findings where it was reported that photobleaching under two-photon excitation might involve higher-order photon processes as well (47,48).
FIGURE 4.

(A) Two-photon FRAP experiments were performed on FD500 in an 85% (w/w) glycerol solution by bleaching a disk of 3 μm in radius with different laser powers to evaluate the axial extension ze of the effective photobleaching PSF. The axial bleaching resolution was determined by fitting of the two-photon disk FRAP model to the experimental recovery curves. Every data point is the average of 10 measurements. The error bars are the corresponding standard deviations (SDs). (B) The corresponding K0 values are shown as a function of the bleaching laser power in a log-log plot. The slope of the linear fit is 2.7. (C) The same measurement was performed on paGFP in a solution containing 56% (w/w) of sucrose. The horizontal solid line represents the average of the ze values corresponding to a laser power ≤25 mW. The dashed lines indicate the corresponding SD. (D) A log-log plot of K0 as a function of the photoactivation laser power is shown. The solid line is a linear fit to the data at the left of the vertical dashed line, having a slope of 1.8.
The same type of experiment was repeated on a solution of paGFP (in 56% w/w sucrose) having a diffusion coefficient D = 2.27 ± 0.13 μm2/s. The results from the two-photon experiments are shown in Fig. 4 C. We note that less laser power was required for the photoactivation of paGFP while a stronger dependency of ze on the laser power is observed. For our further experiments we have used activation powers between 10 and 25 mW, corresponding to an average value of ze = 2.7 μm. Interestingly, for the photoactivation experiments we do find a quadratic relation between K0 and the laser power (the slope of the linear fit in Fig. 4 D is 1.8).
These experiments confirm that two-photon activation of paGFP requires a lower total light dose as compared to fluorescein photobleaching. A similar conclusion has been drawn by comparing photoactivation of paGFP to green fluorescent protein photobleaching (29). In our experiments, the power of the photoactivating beam was typically five-times less than the photobleaching beam, while the scanning speed was 10 times more. This results in a noteworthy difference of a factor of 50 in total light dose during the perturbation phase. Therefore, photoactivation might be preferred when observing fast diffusing molecules as well as when photodamage could be an issue.
Influence of the size of the perturbed disk w
In the mathematical derivation of the model we have made the assumption of the radius of the bleached disk being much larger than the effective radial resolution re of the bleaching beam. We will now examine what is an acceptable minimal radius w by performing two-photon FRAP experiments on the same solution for various w between 0.75 μm and 3 μm. The resulting diffusion coefficients can then be compared to the one obtained by a conventional single photon disk FRAP measurement on the same solution. The measurements were repeated for two different solutions of FITC-dextrans (FD150 in 90% w/w glycerol and FD500 in 85% w/w glycerol) and the results are shown in Fig. 5, A and B, respectively. Noteworthy is that, contrary to what we have found for single photon FRAP (22), correct diffusion coefficients are obtained for all sizes of the photobleached disk, even for the very small ones. This is most likely because of a substantial contribution by axial diffusion in case of two-photon FRAP, while in single-photon FRAP with a low NA lens the recovery is due to radial diffusion only. Hence, in practice no restrictions apply to the disk size in case of multiphoton FRAP experiments in three-dimensional extended samples. This is a convenient property since it allows us to tune the recovery time—which is proportional to the square of the radius of the perturbed disk—depending on the diffusion coefficient. In addition, the possibility for us to freely scale the size of the perturbation region would be a real benefit for the measurement of anomalous diffusion. Indeed, anomalous diffusion can be detected by the less than linear scaling of the recovery time with the area of the perturbation region (49). Finally we would like to note that we are currently working on a FRAP/FRAPa method that allows continuous scaling of the size of the disk for two-dimensional diffusion as well (e.g., diffusion in plasma membrane).
FIGURE 5.

The capability of the model to provide correct estimates of D depending on the size of the bleached region has been tested by performing FRAP experiments for different radii of the disk between 0.75 μm and 3 μm. The experiments have been performed on FD150 in 90% w/w glycerol (A) and on FD500 in 85% w/w glycerol (B). Each value is the average of 10 measurements and the error bars are the corresponding SDs. The horizontal solid line represents the value of the diffusion coefficient as measured by conventional confocal FRAP.
Validation of the model
Having determined the influence of the laser perturbation power and the size of the perturbation disk, we will now examine whether the model can measure absolute diffusion coefficients correctly. To obtain a variety of diffusion coefficients, for each of the FITC-dextrans (150 kDa, 250 kDa, and 500 kDa), we have prepared a series of aqueous solutions with different viscosities by adding different amounts of glycerol (ranging from 70% w/w to 95% w/w). Validation of the new multiphoton FRAP method is performed by comparing the calculated diffusion coefficients with the ones measured by the conventional disk FRAP method on the same samples. While the conventional disk FRAP method demands for a low NA lens to be used, this new two-photon FRAP model does not impose any restriction on the numerical aperture of the objective. We therefore used a 63× 1.4 NA oil immersion objective lens. The radius w of the bleached disk was set to 3 μm and a bleaching power was chosen of 75 mW, which corresponds to an effective axial extension ze of the bleaching PSF of 2.8 μm (see Fig. 4 A). As is clear from the results in Fig. 6, A–C, we find a good correspondence between both methods within the experimental accuracy. These results also show that, while the ze value was obtained for FD500, the same value can be used for FITC-dextrans of other molecular weights, again in agreement with our single-photon results (23,37).
FIGURE 6.

(A–C) The diffusion coefficients of three different FITC-dextrans probes in solutions of different viscosities are measured with the multiphoton FRAP method (10 measurements for each sample) and with the conventional confocal FRAP method (five measurements for each sample): (A) FD150, (B) FD250, and (C) FD500. Within the experimental error a good correspondence is found between both measurements. (D) The diffusion coefficients of paGFP solutions of different viscosities are measured with the two-photon FRAPa method (10 measurements for each sample) and with the confocal FRAPa method (five measurement for each sample). Again, a good correspondence is found between both measurements.
The same experiment was repeated for the photoactivation of paGFP as well. In this case, solutions with different amounts of sucrose were prepared (35%, 41%, 47%, 51%, and 56% w/w). Again single-photon and two-photon disk FRAPa experiments were performed on each solution. As explained in Materials and Methods, single photon excitation was used for imaging the fluorescence redistribution of activated paGFP molecules in both cases. The normalized experimental data were, therefore, fitted with Eq. 14 with n =2, m = 1 for the two-photon data. A two-photon photoactivation power of 20 mW was used, corresponding to ze = 2.7 μm. As is clear from the results in Fig. 6 D, again we found a good correspondence between both types of measurements.
The same method of two-photon photobleaching/photoactivation can be used to measure axial diffusion (i.e., parallel to the optical axis) separately by photobleaching a very large disk and analyzing the recovery data in the central part only (to exclude any contribution from radial diffusion). We performed experiments on solutions of FD500 by inducing two-photon photobleaching in a large region of 15 μm in radius. The recovery curve was then calculated from the fluorescence signal in a region of 4 μm in radius at the center of the bleached region. The diffusion coefficients as calculated from Eq. 12 are shown in Table 1 and are in agreement with the results obtained by conventional confocal FRAP experiments on the same solutions.
TABLE 1.
Measurement of axial diffusion with multiphoton FRAP
| Glycerol (% w/w) | Dconfocal (μm2/s) | Dtwo-photon (μm2/s) |
|---|---|---|
| 85% | 0.33 ± 0.04 | 0.36 ± 0.04 |
| 80% | 0.70 ± 0.08 | 0.60 ± 0.03 |
| 75% | 0.97 ± 0.04 | 0.97 ± 0.14 |
The diffusion coefficients of FD500 solutions as measured by conventional confocal FRAP and two-photon FRAP in case of axial diffusion only. The latter situation was obtained by photobleaching a very large area (15 μm radius) and analyzing the recovery data only in the central part of the bleached area.
One might notice from Fig. 6 that the standard deviations (SDs) of the diffusion coefficients measured under two-photon perturbation are usually larger (∼10–15%) than for the values obtained by confocal FRAP (typically ∼5%). This is due to the fast scanning (and consequently a worse S/N), which is required for imaging the fast recovery in case of a two-photon experiment. On the other hand, compared to single-photon FRAP, two-photon FRAP provides the possibility of faster measurements (or the measurement of slower diffusion) since the volume that undergoes the perturbation is much smaller than in confocal FRAP. For the same reason two-photon FRAP allows us to perform much more localized diffusion measurements as well, also in very small volumes such as cell nuclei. Another important property of the multiphoton method presented here is the possibility to select a perturbation disk of any size (see Fig. 5), thus bringing a wide range of diffusion coefficients within reach. The largest diffusion coefficient that can be measured is limited by two factors, both related to the rate of recovery, which is usually referred to in terms of the characteristic recovery times 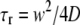 and
and 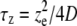 for radial and axial diffusion, respectively. First, the scan rate must be high enough to collect a sufficient number of recovery data points. In case of large bleach regions (w ≫ ze) for probing axial diffusion, it is our experience that a time between the images of τz/2 is usually sufficient to calculate a correct diffusion coefficient. For example, modern confocal scanheads have the possibility to acquire up to 15 images per second. Since ze was close to 3 μm in our experiments, diffusion coefficients up to 30 μm2/s are accessible. In case of smaller disks, radial diffusion will start to play a significant role as well, resulting in faster recovery and a smaller maximum diffusion coefficient. Secondly, the photobleaching or photoactivation step must be performed as quickly as possible in relation to the characteristic recovery time to avoid diffusion during this phase (9). As a rule of thumb, one usually assumes that the photobleaching time should be <10% of the characteristic recovery time (9). At a power of 75 mW, in our experiments we were only just able to induce sufficient photobleaching at a line scanning rate of 1600 Hz or lower. Bleaching a disk consisting of, e.g., 100 lines at this rate, takes 60 ms. This means that τz should be at least 0.6 s, corresponding to a maximum diffusion coefficient of ∼4 μm2/s. On the other hand, we have found that it is possible to obtain significant photoactivation of paGFP when scanning at line scanning rates up to 16,000 Hz, which can be achieved by using the resonant galvanometric mirrors available on the Leica SP-5 in bidirectional scan mode. In this case, diffusion coefficients can be measured up to ∼40 μm2/s. However, as explained above, this will rather be limited to 30 μm2/s due to the limited frame rate.
for radial and axial diffusion, respectively. First, the scan rate must be high enough to collect a sufficient number of recovery data points. In case of large bleach regions (w ≫ ze) for probing axial diffusion, it is our experience that a time between the images of τz/2 is usually sufficient to calculate a correct diffusion coefficient. For example, modern confocal scanheads have the possibility to acquire up to 15 images per second. Since ze was close to 3 μm in our experiments, diffusion coefficients up to 30 μm2/s are accessible. In case of smaller disks, radial diffusion will start to play a significant role as well, resulting in faster recovery and a smaller maximum diffusion coefficient. Secondly, the photobleaching or photoactivation step must be performed as quickly as possible in relation to the characteristic recovery time to avoid diffusion during this phase (9). As a rule of thumb, one usually assumes that the photobleaching time should be <10% of the characteristic recovery time (9). At a power of 75 mW, in our experiments we were only just able to induce sufficient photobleaching at a line scanning rate of 1600 Hz or lower. Bleaching a disk consisting of, e.g., 100 lines at this rate, takes 60 ms. This means that τz should be at least 0.6 s, corresponding to a maximum diffusion coefficient of ∼4 μm2/s. On the other hand, we have found that it is possible to obtain significant photoactivation of paGFP when scanning at line scanning rates up to 16,000 Hz, which can be achieved by using the resonant galvanometric mirrors available on the Leica SP-5 in bidirectional scan mode. In this case, diffusion coefficients can be measured up to ∼40 μm2/s. However, as explained above, this will rather be limited to 30 μm2/s due to the limited frame rate.
APPLICATION TO INTRACELLULAR DIFFUSION
As an example, we show the application of the photoactivation multiphoton FRAP method to the measurement of the diffusion of free paGFP in the nuclei of living NIH-3T3 cells. We selected a region inside the nucleus of 2.5 μm in radius to be activated by 30 mW of the 760-nm infrared pulsed laser. A corresponding axial resolution of ze = 3.4 μm was used (see Fig. 4 B for reference) for analyzing the redistribution data. A representative experiment on a living cell is shown in Fig. 7. Seven similar experiments were performed on different cells and the resulting recovery curves were normalized as described in Materials and Methods. An average redistribution curve was calculated from the seven individual measurements to improve the signal/noise ratio (see Fig. 7 C). Fitting the recovery curve with Eq. 14 for diffusion in an infinite volume leads to an underestimated diffusion coefficient D = 11.9 μm2/s. Since the nuclei of NIH-3T3 typically have a radius of 7.5–15 μm and a thickness of 7–10 μm, Eq. 15 should rather be used to account for the limited axial extent. Using an average thickness of h = 8 μm, a best fit of Eq. 15 to the experimental recovery curve (solid line in Fig. 7 C) leads to a diffusion coefficient D = (19 ± 4) μm2/s, in good agreement with what has been reported for line scanning FRAP or FCS measurements (23,50,51).
FIGURE 7.

(A and B) Two-photon FRAP experiment on paGFP diffusing in the nucleus of a mouse embryo fibroblast. The first image shows the sample before photoactivation in the selected region (2.5 μm in radius). The subsequent images show the fluorescence redistribution after photoactivation of the selected region. The scale bar in (A) is 5 μm. (C) The redistribution curve is the average of seven experiments in different cells to obtain a smoother curve. The thickness of the nuclei was h = 8 μm on average. This value was used to fit Eq. 15 to the experimental data, from which a diffusion coefficient was obtained of (19 ± 4) μm2/s. As expected, all molecules were found to be mobile (k = 1.002 ± 0.007).
CONCLUSIONS
We have presented, to our knowledge here, a new multiphoton FRAP method for three-dimensionally resolved diffusion measurements based on the photobleaching of a disk-shaped area by a scanning laser beam. In particular it was our aim to develop a versatile quantitative method that is straightforward to carry out on a regular multiphoton laser scanning microscope. Quantitative analysis of the FRAP data is equally straightforward since we have been able to derive a closed-form solution describing the recovery phase. The diffusion coefficient and mobile fraction are readily obtained by a best fit of the model to the (properly normalized) recovery data. In addition, we have extended the model to photoactivation experiments, for which we have used the acronym FRAPa. In Table 2, a comparison is given of the benefits of multiphoton photobleaching versus photoactivation. Photobleaching might be preferred when diffusion is probed in thick or turbid samples, since both the perturbation and the observation of fluorescence recovery can be performed under multiphoton excitation. On the other hand, despite having to use regular (single-photon) confocal microscopy for imaging the redistribution phase, FRAPa experiments are attractive since generally less laser irradiation is necessary for photoactivation compared to photobleaching. Not only does this allow us to shorten the photoperturbation phase, it also reduces the chance of phototoxic effects in living cells. We have shown extensively that the multiphoton FRAP and FRAPa methods are both capable of obtaining correct absolute diffusion coefficients in three-dimensional extended samples. Importantly, we have shown that the size of the photobleached disk can be adjusted over a wide range, thus making it possible to measure a wide range of diffusion coefficients as well. Measurements inside micron-sized volumes are equally possible by making the disk very small and taking into account the (possibly) limited thickness of the sample. Additionally, we have demonstrated that axial diffusion can be measured separately by analyzing the central part of a large photobleached/photoactivated region. Finally, the FRAPa method was successfully applied to intranucleic diffusion measurements in living cells expressing free paGFP. Considering the ease-of-use and the versatility of the multiphoton FRAP/FRAPa method, we expect that it will make quantitative diffusion measurements much more accessible in the life sciences.
TABLE 2.
Comparison of the benefits of FRAP versus FRAPa
| Multiphoton photobleaching | Multiphoton photoactivation |
|---|---|
| Pro: Can be performed with virtually any fluorescent probe. | Con: Requires photoactivatable probes. |
| Pro: Allows two-photon observation of the fluorescence recovery. | Con: For practical reasons, generally requires one-photon observation of the fluorescence recovery. |
| Con: Higher light dosage delivered on the sample. | Pro: Lower light dosage delivered on the sample. |
| Con: Lower scanning speed required to induce significant photobleaching. | Pro: Photoactivation can be performed with higher scanning speeds. |
Acknowledgments
We are grateful to George Patterson and Jennifer Lippincott-Schwartz for the purified paGFP, and to Mario Faretta and Sara Barozzi for the paGFP plasmid.
Davide Mazza, Francesca Cella, Ilaria Testa, and Alberto Diaspro's research activity for this work was supported by Ministero Istruzione Università e Ricerca-Italian Research Program of National Interest (grant No. PRIN 2006-2006028909). Kevin Braeckmans is a Postdoctoral Fellow of the Fund for Scientific Research, Flanders (Belgium).
APPENDIX A: FLUORESCENCE RECOVERY AFTER n-PHOTON PHOTOBLEACHING OF A UNIFORM DISK
It is possible to calculate the fluorescence recovery as observed by m-photon microscopy from the convolution product of the concentration distribution in Eq. 6 with the microscope's overall PSF 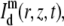
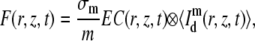 |
(17) |
where σm is the cross section for m-photon fluorescence emission and E the overall efficiency of the detection system. Again, the radial resolution of the PSF can be effectively neglected if it is much smaller than the radius w of the bleached disk:
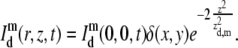 |
(18) |
By combining Eqs. 6 and 18 into Eq. 17 we find
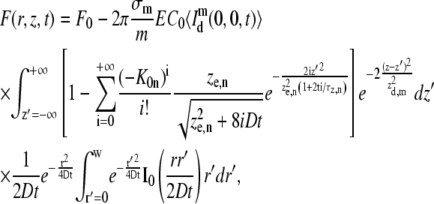 |
(19) |
where F0 is the observed fluorescence before photobleaching. While the first integral can be readily solved, the second one has no analytical solution. However, following the method outlined by Soumpasis (52), a solution in terms of modified Bessel functions can be found if the total fluorescence inside the disk of radius w is calculated at the focal plane (i.e., z = 0), finally leading to the solution in Eq. 7.
APPENDIX B: LINEAR UNMIXING OF THE ACTIVATED AND NONACTIVATED POPULATIONS IN FRAPa EXPERIMENTS
Provided that the excitation spectra of the nonactivated and activated forms of the fluorophore are known, the factor 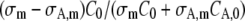 in Eq. 14 can be calculated from two images acquired before the FRAPa experiment at different excitation wavelengths under conventional one-photon excitation. It can be easily seen that this factor only depends on the ratios C0/CA,0 and σA,m/σm. The ratio σA,m/σm is readily obtained from the relative heights of the absorption spectra at the excitation wavelength used for observing the fluorescence redistribution (488 nm in our experiments, m = 1). The ratio C0/CA,0 can be calculated from two images of the sample at different excitation wavelengths. In our case, for example, we acquired one image by exciting at 488 nm and a second one at 405 nm. Laser intensities should be used that are low enough to avoid any significant photoactivation at this stage. Assuming a uniform initial concentration of both activated and nonactivated molecules, it follows from Eqs. 17 and 18 that the combined fluorescence signal from both populations as measured in a circular region for each excitation wavelength is equal to
in Eq. 14 can be calculated from two images acquired before the FRAPa experiment at different excitation wavelengths under conventional one-photon excitation. It can be easily seen that this factor only depends on the ratios C0/CA,0 and σA,m/σm. The ratio σA,m/σm is readily obtained from the relative heights of the absorption spectra at the excitation wavelength used for observing the fluorescence redistribution (488 nm in our experiments, m = 1). The ratio C0/CA,0 can be calculated from two images of the sample at different excitation wavelengths. In our case, for example, we acquired one image by exciting at 488 nm and a second one at 405 nm. Laser intensities should be used that are low enough to avoid any significant photoactivation at this stage. Assuming a uniform initial concentration of both activated and nonactivated molecules, it follows from Eqs. 17 and 18 that the combined fluorescence signal from both populations as measured in a circular region for each excitation wavelength is equal to
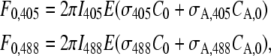 |
(20) |
where σ405 and σA,405 are the cross sections for the fluorescence emission of the nonactivated and the activated form of the protein when exciting at 405 nm, while σ488 and σA,488 are the corresponding fluorescence emission cross sections at 488 nm. I405 and I488 are the laser intensities at the sample for both wavelengths. From this set of linear equations it immediately follows that
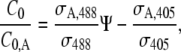 |
(21) |
where
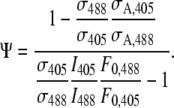 |
(22) |
Again, the cross-section ratios are readily obtained from the excitation spectra. The ratio I488/I405 can be determined by measuring the laser power coming out of the objective lens at both wavelengths. The ratio F0,488/F0,405 is obtained directly from the images. For the experiments on paGFP in this study, we have measured the absorption spectra of the activated and nonactivated form of the protein with a spectrofluorimeter. We obtained a ratio σA,m/σm = 120 at 488 nm, and a ratio CA,0/C0 between 0.01 and 0.03, leading to a factor 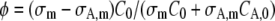 between −25 and −60.
between −25 and −60.
Davide Mazza and Kevin Braeckmans contributed equally to this work.
Editor: Michael Edidin.
References
- 1.Sanders, N. N., S. C. De Smedt, and J. Demeester. 2000. The physical properties of biogels and their permeability for macromolecular drugs and colloidal drug carriers. J. Pharm. Sci. 89:835–849. [DOI] [PubMed] [Google Scholar]
- 2.Remaut, K., N. N. Sanders, B. G. De Geest, K. Braeckmans, J. Demeester, and S. C. De Smedt. 2007. Nucleic acid delivery: where material sciences and bio-sciences meet. Mater. Sci. Eng. Rep. 58:117–161. [Google Scholar]
- 3.Chen, Y., B. C. Lagerholm, B. Yang, and K. Jacobson. 2006. Methods to measure the lateral diffusion of membrane lipids and proteins. Methods. 39:147–153. [DOI] [PubMed] [Google Scholar]
- 4.De Smedt, S. C., K. Remaut, B. Lucas, K. Braeckmans, N. N. Sanders, and J. Demeester. 2005. Studying biophysical barriers to DNA delivery by advanced light microscopy. Adv. Drug Deliv. Rev. 57:191–210. [DOI] [PubMed] [Google Scholar]
- 5.Gosch, M., and R. Rigler. 2005. Fluorescence correlation spectroscopy of molecular motions and kinetics. Adv. Drug Deliv. Rev. 57:169–190. [DOI] [PubMed] [Google Scholar]
- 6.Saxton, M. J., and K. Jacobson. 1997. Single-particle tracking: applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct. 26:373–399. [DOI] [PubMed] [Google Scholar]
- 7.Suh, J., M. Dawson, and J. Hanes. 2005. Real-time multiple-particle tracking: applications to drug and gene delivery. Adv. Drug Deliv. Rev. 57:63–78. [DOI] [PubMed] [Google Scholar]
- 8.McNally, J. G., and C. L. Smith. 2002. Photobleaching by confocal microscopy. In Confocal and Two-Photon Microscopy. Foundations, Applications and Advances. A. Diaspro, editor. Wiley-Liss, New York.
- 9.Meyvis, T. K., S. C. D. Smedt, P. Van Oostveldt, and J. Demeester. 1999. Fluorescence recovery after photobleaching: a versatile tool for mobility and interaction measurements in pharmaceutical research. Pharm. Res. 16:1153–1162. [DOI] [PubMed] [Google Scholar]
- 10.Sprague, B. L., and J. G. McNally. 2005. FRAP analysis of binding: proper and fitting. Trends Cell Biol. 15:84–91. [DOI] [PubMed] [Google Scholar]
- 11.Braga, J., J. M. Desterro, and M. Carmo-Fonseca. 2004. Intracellular macromolecular mobility measured by fluorescence recovery after photobleaching with confocal laser scanning microscopes. Mol. Biol. Cell. 15:4749–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elsner, M., H. Hashimoto, J. C. Simpson, D. Cassel, T. Nilsson, and M. Weiss. 2003. Spatiotemporal dynamics of the COPI vesicle machinery. EMBO Rep. 4:1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karpova, T. S., T. Y. Chen, B. L. Sprague, and J. G. McNally. 2004. Dynamic interactions of a transcription factor with DNA are accelerated by a chromatin remodeler. EMBO Rep. 5:1064–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verkman, A. S. 2003. Diffusion in cells measured by fluorescence recovery after photobleaching. In Biophotonics, Part A: Methods in Enzymology. G. Marriott and I. Parker, editors. Academic Press, New York. [DOI] [PubMed]
- 15.Peters, R., J. Peters, K. H. Tews, and W. Bahr. 1974. A microfluorimetric study of translational diffusion in erythrocyte membranes. Biochim. Biophys. Acta. 367:282–294. [DOI] [PubMed] [Google Scholar]
- 16.Axelrod, D., D. E. Koppel, J. Schlessinger, E. Elson, and W. W. Webb. 1976. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 16:1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez, A., L. Dupou, A. Altibelli, J. Trotard, and J. F. Tocanne. 1988. Fluorescence recovery after photobleaching (FRAP) experiments under conditions of uniform disk illumination. Critical comparison of analytical solutions, and a new mathematical method for calculation of diffusion coefficient D. Biophys. J. 53:963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blonk, J. C. G., A. Don, H. V. Aalst, and J. J. Birmingham. 1992. Fluorescence photobleaching recovery in the confocal scanning light microscope. J. Microsc. 169:363–374. [Google Scholar]
- 19.Wedekind, P., U. Kubitscheck, O. Heinrich, and R. Peters. 1996. Line-scanning microphotolysis for diffraction-limited measurements of lateral diffusion. Biophys. J. 71:1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wedekind, P., U. Kubitscheck, and R. Peters. 1994. Scanning microphotolysis: a new photobleaching technique based on fast intensity modulation of a scanned laser beam and confocal imaging. J. Microsc. 176:23–33. [DOI] [PubMed] [Google Scholar]
- 21.Gribbon, P., B. C. Heng, and T. E. Hardingham. 1999. The molecular basis of the solution properties of hyaluronan investigated by confocal fluorescence recovery after photobleaching. Biophys. J. 77:2210–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braeckmans, K., L. Peeters, N. N. Sanders, S. C. D. Smedt, and J. Demeester. 2003. Three-dimensional fluorescence recovery after photobleaching with the confocal scanning laser microscope. Biophys. J. 85:2240–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braeckmans, K., K. Remaut, R. E. Vandenbroucke, B. Lucas, S. C. D. Smedt, and J. Demeester. 2007. Line FRAP with the confocal laser scanning microscope for diffusion measurements in small regions of 3D samples. Biophys. J. 92:2172–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cutts, L. S., P. A. Robberts, J. Adler, M. C. Davies, and C. D. Melia. 1995. Determination of localized diffusion-coefficients in gels using confocal scanning laser microscopy. J. Microsc. 180:131–139. [Google Scholar]
- 25.Diaspro, A. 2002. Confocal and Two-Photon Microscopy: Foundations, Applications and Advances. Wiley-Liss, New York.
- 26.Brown, E. B., E. S. Wu, W. Zipfel, and W. W. Webb. 1999. Measurement of molecular diffusion in solution by multiphoton fluorescence photobleaching recovery. Biophys. J. 77:2837–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waharte, F., C. M. Brown, S. Coscoy, E. Coudrier, and F. Amblard. 2005. A two-photon FRAP analysis of the cytoskeleton dynamics in the microvilli of intestinal cells. Biophys. J. 88:1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beaudouin, J., F. Mora-Bermudez, T. Klee, N. Daigle, and J. Ellenberg. 2006. Dissecting the contribution of diffusion and interactions to the mobility of nuclear proteins. Biophys. J. 90:1878–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calvert, P. D., J. A. Peet, A. Bragin, W. E. Schiesser, and E. N. Pugh, Jr. 2007. Fluorescence relaxation in 3D from diffraction-limited sources of PAGFP or sinks of EGFP created by multiphoton photoconversion. J. Microsc. 225:49–71. [DOI] [PubMed] [Google Scholar]
- 30.Chudakov, D. M., S. Lukyanov, and K. A. Lukyanov. 2007. Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat. Protocols. 2:2024–2032. [DOI] [PubMed] [Google Scholar]
- 31.Lippincott-Schwartz, J., N. Altan-Bonnet, and G. H. Patterson. 2003. Photobleaching and photoactivation: following protein dynamics in living cells. Nat. Cell Biol. Suppl:S7–S14. [PubMed]
- 32.Lippincott-Schwartz, J., J. F. Presley, K. J. Zaal, K. Hirschberg, C. D. Miller, and J. Ellenberg. 1999. Monitoring the dynamics and mobility of membrane proteins tagged with green fluorescent protein. Methods Cell Biol. 58:261–281. [DOI] [PubMed] [Google Scholar]
- 33.Patterson, G. H., and J. Lippincott-Schwartz. 2002. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 297:1873–1877. [DOI] [PubMed] [Google Scholar]
- 34.Schafer, S. P., P. S. Dittrich, E. P. Petrov, and P. Schwille. 2006. Single molecule fluorescence imaging of the photoinduced conversion and bleaching behavior of the fluorescent protein Kaede. Microsc. Res. Tech. 69:210–219. [DOI] [PubMed] [Google Scholar]
- 35.Schneider, M., S. Barozzi, I. Testa, M. Faretta, and A. Diaspro. 2005. Two-photon activation and excitation properties of PA-GFP in the 720–920-nm region. Biophys. J. 89:1346–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu, C. 2002. Cross-sections of fluorescence molecules in multiphoton microscopy. In Confocal and Two-Photon Microscopy. Foundations, Applications and Advances. A. Diaspro, editor. Wiley-Liss, New York.
- 37.Braeckmans, K., B. G. Stubbe, K. Remaut, J. Demeester, and S. C. D. Smedt. 2006. Anomalous photobleaching in fluorescence recovery after photobleaching measurements due to excitation saturation—a case study for fluorescein. J. Biomed. Opt. 11:044013. [DOI] [PubMed] [Google Scholar]
- 38.Mazza, D., F. Cella, G. Vicidomini, S. Krol, and A. Diaspro. 2007. Role of three-dimensional bleach distribution in confocal and two-photon fluorescence recovery after photobleaching experiments. Appl. Opt. 46:7401–7411. [DOI] [PubMed] [Google Scholar]
- 39.Crank, J. 1975. The Mathematics of Diffusion. Oxford University Press, London.
- 40.de Monvel, J. B., W. E. Brownell, and M. Ulfendahl. 2006. Lateral diffusion anisotropy and membrane lipid/skeleton interaction in outer hair cells. Biophys. J. 91:364–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sonesson, A. W., H. Brismar, T. H. Callisen, and U. M. Elofsson. 2007. Mobility of Thermomyces lanuginosa lipase on a trimyristin substrate surface. Langmuir. 23:2706–2713. [DOI] [PubMed] [Google Scholar]
- 42.Sonesson, A. W., T. H. Callisen, H. Brismar, and U. M. Elofsson. 2005. Lipase surface diffusion studied by fluorescence recovery after photobleaching. Langmuir. 21:11949–11956. [DOI] [PubMed] [Google Scholar]
- 43.Alvarez-Mancenido, F., K. Braeckmans, S. C. De Smedt, J. Demeester, M. Landin, and R. Martinez-Pacheco. 2006. Characterization of diffusion of macromolecules in Konjac glucomannan solutions and gels by fluorescence recovery after photobleaching technique. Int. J. Pharm. 316:37–46. [DOI] [PubMed] [Google Scholar]
- 44.Peeters, L., N. N. Sanders, K. Braeckmans, K. Boussery, J. Van de Voorde, S. C. De Smedt, and J. Demeester. 2005. Vitreous: a barrier to nonviral ocular gene therapy. Invest. Ophthalmol. Vis. Sci. 46:3553–3561. [DOI] [PubMed] [Google Scholar]
- 45.Testa, I., D. Mazza, S. Barozzi, M. Faretta, and A. Diaspro. 2007. Blue-light (488 nm)-irradiation-induced photoactivation of the photoactivatable green fluorescent protein. Appl. Phys. Lett. 91:133902. [Google Scholar]
- 46.Testa, I., D. Parazzoli, S. Barozzi, M. Garrè, M. Faretta, and A. Diaspro. 2008. Spatial control of pa-GFP photoactivation in living cells. J. Microsc. 230:48–60. [DOI] [PubMed] [Google Scholar]
- 47.Patterson, G. H., and D. W. Piston. 2000. Photobleaching in two-photon excitation microscopy. Biophys. J. 78:2159–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cannone, F., M. Caccia, S. Bologna, A. Diaspro, and G. Chirico. 2004. Single molecule spectroscopic characterization of GFP-MUT2 mutant for two-photon microscopy applications. Microsc. Res. Tech. 65:186–193. [DOI] [PubMed] [Google Scholar]
- 49.Saxton, M. J. 2001. Anomalous subdiffusion in fluorescence photobleaching recovery: a Monte Carlo study. Biophys. J. 81:2226–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maertens, G., J. Vercammen, Z. Debyser, and Y. Engelborghs. 2005. Measuring protein-protein interactions inside living cells using single color fluorescence correlation spectroscopy. Application to human immunodeficiency virus type I integrase and LEDGF/p75. FASEB J. 19:1039–1041. [DOI] [PubMed] [Google Scholar]
- 51.Chen, Y., J. D. Muller, Q. Ruan, and E. Gratton. 2002. Molecular brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys. J. 82:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soumpasis, D. M. 1983. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys. J. 41:95–97. [DOI] [PMC free article] [PubMed] [Google Scholar]