

Abstract
To investigate the mechanism of interaction of gramicidin S-like antimicrobial peptides with biological membranes, a series of five decameric cyclic cationic β-sheet-β-turn peptides with all possible combinations of aromatic D-amino acids, Cyclo(Val-Lys-Leu-D-Ar1-Pro-Val-Lys-Leu-D-Ar2-Pro) (Ar ≡ Phe, Tyr, Trp), were synthesized. Conformations of these cyclic peptides were comparable in aqueous solutions and lipid vesicles. Isothermal titration calorimetry measurements revealed entropy-driven binding of cyclic peptides to POPC and POPE/POPG lipid vesicles. Binding of peptides to both vesicle systems was endothermic—exceptions were peptides containing the Trp-Trp and Tyr-Trp pairs with exothermic binding to POPC vesicles. Application of one- and two-site binding (partitioning) models to binding isotherms of exothermic and endothermic binding processes, respectively, resulted in determination of peptide-lipid membrane binding constants (Kb). The Kb1 and Kb2 values for endothermic two-step binding processes corresponded to high and low binding affinities (Kb1 ≥ 100 Kb2). Conformational change of cyclic peptides in transferring from buffer to lipid bilayer surfaces was estimated using fluorescence resonance energy transfer between the Tyr-Trp pair in one of the peptide constructs. The cyclic peptide conformation expands upon adsorption on lipid bilayer surface and interacts more deeply with the outer monolayer causing bilayer deformation, which may lead to formation of nonspecific transient peptide-lipid porelike zones causing membrane lysis.
INTRODUCTION
Interaction of antimicrobial peptides with cell membranes has been considered as a fundamental, if not the only, step in the mechanism of their antimicrobial activity (1,2). It has been known that helical cationic peptide antibiotics interact with cell membranes of both prokaryotic and eukaryotic cells, alter or destroy the cell membrane integrity, and cause lysis leading to the cell death (3,4). In comparison to the linear helix-forming cationic antimicrobial peptides, the mechanism of interaction of cyclic cationic antimicrobial peptides (CCAPs) with biological membranes is much less understood (5). To investigate the mechanism of interaction of small CCAPs with biological model membranes, we have designed and synthesized a series of amphipathic decameric β-sheet-β-turn cyclic peptides with different combinations of aromatic D-amino-acid (phenylalanine, tyrosine, and tryptophan) substitutions. The prototypic models for the peptides of this study are the naturally found decameric CCAPs, gramicidin S (GS), and tyrocidines of the same bacterial origin (6,7).
Gramicidin S, the principal prototype for the peptides of this study, is a CCAP from the Gram-positive bacteria Bacillus brevis that has been widely utilized as a topical antibiotic (8). The backbone structure of GS is comprised of a double-stranded antiparallel β-sheet connected by a pair of type II′ β-turns on both ends (Fig. 1) (9–11). GS is active against a broad range of both Gram-positive and Gram-negative bacteria as well as fungi, but is also highly hemolytic against mammalian erythrocytes (12). The physicochemical properties, synthetic procedures, and biological activity of GS and tyrocidines, as well as hundreds of their different analogs, have been investigated in detail (6,7,13).
FIGURE 1.

Molecular model for the amphipathic β-sheet-β-turn conformation of GS.
The sequences of GS and its five analogs are shown in Table 1. The peptides share two type II′ β-turns composed of four amino acids (-Leu-D-Xxx-Pro-Val-, where Xxx is an aromatic amino acid) on the two ends of an antiparallel β-sheet, stabilized by four backbone intramolecular hydrogen bonds. In addition to sharing the β-sheet-β-turn conformation, the peptides of Table 1 are also amphipathic. The hydrophobic residues (Val and Leu) in these peptide molecules are located on one face and Lys (Orn in the case of GS) residues are located on the opposite face. A typical structure and orientation of the amino-acid residues of the peptides of Table 1 are depicted in Fig. 1, a representation of the three-dimensional structure of GS supported by molecular modeling.
TABLE 1.
Peptide sequences, their molecular mass, retention time, and relative hydrophobicity (see legend for definitions)
| Peptide | Sequence | MW (Da): c/m | RT (min) | W-Oct ΔGWO | W-IF ΔGWIF | IF-Oct ΔΔGIFO |
|---|---|---|---|---|---|---|
| GS10WW | Cyclo-(VKLdWPVKLdWP) | 1246.76 / 1247.03 | 32.0 | −1.72 | −1.80 | +0.08 |
| GS10FW | Cyclo-(VKLdFPVKLdWP) | 1207.74 / 1207.69 | 31.7 | −1.34 | −1.08 | −0.26 |
| GS | Cyclo-(VOLdFPVOLdFP) | 1140.69 / 1140.77 | 31.4 | −0.96 | −0.36 | −0.60 |
| GS10WY | Cyclo-(VKLdWPVKLdYP) | 1223.70 / 1224.06 | 30.5 | −0.34 | −0.89 | +0.63 |
| GS10FY | Cyclo-(VKLdFPVKLdYP) | 1184.72 / 1184.17 | 30.2 | −0.04 | −0.17 | +0.13 |
| GS10YY | Cyclo-(VKLdYPVKLdYP) | 1200.74 / 1200.82 | 28.8 | +1.04 | +0.02 | +1.02 |
Peptide sequences: O represents ornithine, and d represents the D-enantiomer. Molecular mass: Calculated (c), and measured (m) molecular masses of peptides, using electrospray mass spectrometry. MW is the molecular mass. Molecular masses are measured as [M+H]+. Retention times: (RT) on the RP-HPLC C4 column. Solvents A (water + 0.05% TFA) and B (acetonitrile + 0.05% TFA) were used at a flow rate of l mL/min with a gradient from 100% A →100% B in 50 min. Hydrophobicity: Relative free energy of transfer of the peptides from water to octanol (ΔGWO), water to POPC bilayer interface (ΔGWIF), and the difference between the two (ΔΔGIFO) were calculated on the basis of whole-residue hydrophobicity scales (16). The free energy of transfer of peptide bonds to octanol and the effect of the peptide backbone structure, which are comparable for all peptides, were not considered in these calculations. Note that W stands for water; Oct stands for octanol; IF stands for interfacial; and IF-Oct stands for interfacial to octanol; free energies are in kcal/mol.
In this study, we have utilized the biophysical techniques, circular dichroism (CD), fluorescence spectroscopy, and isothermal titration calorimetry (ITC), to explore the conformation and modes of interaction of aromatic β-sheet-β-turn amphipathic CCAPs with model biological membranes and suggested a model for the mechanism of interaction of these peptides with lipid membranes.
MATERIALS AND METHODS
Chemicals
Gramicidin S was purchased from Sigma (St. Louis, MO) and further purified by RP-HPLC. All other peptides were synthesized by solid-phase procedures using tert-butyloxycarbonyl (Boc) chemistry. N-[(1H-benzotriazol-1yl)(dimethylamino)methylene]-n-methylmethanaminium hexafluorophosphate n-oxide (HBTU) and 1-hydroxybenzotriazole (HOBT) were used to activate C-terminus of the amino acids. Lys side chains were protected by formyl groups throughout the syntheses. After cleavage from resin and purification by RP-HPLC, linear peptides were cyclized in a head-to-tail manner (Pro at the C-terminus) utilizing benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate (BOP), 1-hydroxybenzotriazole, and n,n-diisopropylethylamine in n,n-dimethylformamide, and Lys residues of the cyclized peptides were then deprotected in dilute methanolic HCl at 40°C (12,14). Peptides were purified and analyzed by RP-HPLC, and their final purity was further confirmed by electrospray mass spectrometry (Table 1). Concentration of the pure peptides in aqueous stock solutions was determined by amino-acid analysis (±5% error). Phospholipids, 1-palmitoyl-2-oleoyol-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyol-sn-glycero-3-phosphoethanolamine (POPE), and 1-palmitoyl-2-oleoyol-sn-glycero-3-[phosphor-rac-1-glycerol] (sodium salt) (POPG) were from Avanti Polar Lipids (Alabaster, AL). Stock lipid solutions were in chloroform. All other chemicals were of reagent grade and used as received without further purification.
Antimicrobial and hemolytic activity assays
Microbial strains were prepared as described previously (12,14). The peptide activity against microorganisms was measured by a liquid broth assay method (12). Staphylococcus aureus (SAP0017 and K147), Staphylococcus epidermidis, Bacillus subtilis, Enterococcus faecalis, and the coryneform bacteria Corynebacterium xerosis were the Gram-positive bacterial strains. Gram-negative bacterial strains were Pseudomonas aeruginosa (K799 and Z61), Escherichia coli (UB1005 and DC2), and Salmonella typhimurium (C587 (14028S) and C610 (MS4252S)). The yeast strain employed was Candida albicans. Freshly collected human blood cells were prepared as described, and hemolytic measurements were performed in a liquid-based assay (12,14). The reported values for both antimicrobial and hemolytic assays were determined after 24 h at 37°C.
Large unilamellar vesicles for spectroscopic and ITC experiments
Chloroform solutions of the lipids POPC and POPE/POPG (7/3 molar ratio) were dried, under a mild nitrogen flush, in a round-bottomed flask to form a thin film. The film was further dried overnight under reduced pressure. The dried lipid film was then hydrated with buffer at room temperature. The resulted multilamellar vesicles were then freeze-thawed a few times and extruded through a 100-nm filter in a LiposoFast apparatus (Avestin, Ottawa, Canada) (15). The prepared large unilamellar vesicles (average diameter of ∼80 nm) were stable in dark for several days at 4°C.
Two phospholipid vesicle systems were utilized for this study. The first system contained POPC lipid bilayers, which can be considered as a model for the mammalian blood cell membranes. The second system contained POPE and POPG phospholipids ([phosphatidylethanolamine (PE)]/[phosphatidylglycerol (PG)] = 7/3) to model the negatively-charged bacterial membranes. Similar to biological membranes, the two lipid bilayer systems used for the biophysical measurements in this study were in the liquid crystalline state.
Instrumentation
Peptides were analyzed on a Waters 600E HPLC system (Waters, Franklin, MA) using a Waters C4 Symmetry (150 × 4.6 mm I.D., 5 μm particle size, 30 nm pore size) at 25°C. CD spectra were measured on an Aviv 215 spectropolarimeter (Aviv Biomedical, Lakewood, NJ). Ellipticities are reported as mean residue ellipticity, [θ]. Spectra were measured in buffer solutions composed of final concentrations of Tris(hydroxymethyl)aminomethane hydrochloride (Tris) (10 mM), NaF (100 mM), at pH 7.4, unless specified otherwise. The measurements were in quartz cells with 0.1 or 0.05 cm pathlengths, at 25°C. Sodium fluoride was used in buffer to reduce the high noise levels of chloride ions at wavelengths at <200 nm.
Fluorescence spectra were measured on a Cary Eclipse fluorescence spectrophotometer (Varian, Palo Alto, CA), using quartz cells of 1 cm pathlength. The slit-widths for both excitation and emission wavelengths were 5 nm. Spectra were measured at 25°C in buffer solutions composed of final concentrations of Tris (10 mM), NaCl (100 mM), at pH 7.4; 80% methanol; and 0.5–1 mM POPC lipid dispersions in Tris (10 mM), NaCl (100 mM) buffer, at pH 7.4.
ITC measurements were performed on a VP-ITC microcalorimeter (MicroCal, Northampton, MA) at 30°C. All solutions were degassed under reduced pressure for at least 5 min before use. The calorimeter was calibrated electronically. In all experiments, phospholipids (5 mM lipid concentration) in buffer (Tris, 10 mM and NaCl, 100 mM, at pH 7.4) were injected in 3–20 μL aliquots to the peptide buffer solutions (20–40 μM; reaction cell volume = 1.4352 mL). In the models employed for the analysis of ITC data, we have assumed that the peptides interact with the outer leaflets of vesicles and therefore, on the basis of geometric parameters for large unilamellar vesicles, an estimated 50% of the total lipid concentration was used in all calculations. Control and baseline adjustment experiments were performed in both vesicle systems by injecting vesicles in buffer in the absence of peptides. The incremental enthalpies (heat flow per injection) were corrected by subtracting the reference baselines from the measured values. Peptides were fully titrated with lipids until no change in the baseline profile was observed. Binding isotherms of the peptides with both lipid systems were derived and surface binding (partitioning) models were utilized for data analysis.
RESULTS
Antimicrobial and hemolytic activities of the cyclic peptides
Cyclic peptides of this study are listed in Table 1 in order of their retention times, on a reversed phase HPLC column. These retention times correlate well with the whole-residue scale for free energies of the transfer from water to octanol (ΔGWO) and water to POPC interface (ΔGWIF), representing the lipid bilayer hydrophobic interior and interface, respectively (16) (Table 1). GS10WW and GS10YY had the highest and lowest retention times, respectively. Biological activities of three peptides (GS, GS10YY, and GS10WW) are exhibited in Tables 2 and 3. These peptides were all hemolytic (Table 2) and their hemolytic activity (possible affinity for hydrophobic interaction with dominantly zwitterionic membrane of erythrocytes) was also in direct correlation to their retention times (affinity for the hydrophobic stationary phase of RP-HPLC columns), ΔGWO and ΔGWIF values. GS10WW was the most hemolytic peptide and had the highest retention time and greatest overall hydrophobicity. These results are in agreement with the direct relation between the hemolytic activity and hydrophobicity/amphipathicity (as measured by retention times on RP-HPLC) of other biologically active cyclic cationic peptides ((5,13,14), and references therein). Activities against certain strains of the Gram-positive bacteria were comparable for three peptides and GS had the most potent activity (Table 2). Finally, compared to GS10WW, both GS and GS10YY were more active against the tested Gram-negative bacteria (Table 3).
TABLE 2.
Biological activity of peptides against human erythrocytes, Gram-positive bacteria, and yeast after 24 h at 37°C
| Minimal inhibitory concentration (μg/mL)*
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Peptide | Hemolytic†‡ (μg/mL) | S. aureus SAP0017 | S. aureus K147 | S. epidermidis | B. subtilis | E. faecalis | C. xerosis | Geometric mean Gram-positive§ | C. albicans |
| GS | 12.5 | 1.5 | 1.5 | 1.5 | 3.1 | 1.5 | 1.5 | 1.7 | 4.0 |
| GS10YY | 25 | 6.2 | 6.2 | 4.5 | 6.2 | 3.1 | 1.5 | 4.1 | 6.2 |
| GS10WW | 3.1 | 1.5 | 3.1 | 1.5 | 3.1 | 1.5 | 1.5 | 1.9 | 6.2 |
A minimum of three experiments was performed for each biological activity assay.
Activity against Gram-positive bacteria was measured in liquid broth assays as described previously ((14) and references therein).
Freshly collected human blood cells were prepared and hemolytic liquid-based assays were performed as described previously (12,14).
Concentrations for hemolytic activity were the minimum required for complete lysis of erythrocytes.
Mean MIC values (after 24 h) against Gram-positive bacteria. Geometric mean for n numbers: 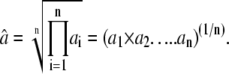
TABLE 3.
Biological activity of peptides against Gram-negative bacteria after 24 h at 37°C
| Minimal inhibitory concentration (μg/ml)*
|
|||||||
|---|---|---|---|---|---|---|---|
| Peptide | P. aeruginosa K799 (H187) | P. aeruginosa† Z61(H188) | E. coli UB1005 | E. coli† DC2 | S. typhimurium C587 | S. typhimurium† C610 | Geometric mean Gram-negative‡ |
| GS | 25 | 6.2 | 9.0 | 3.1 | 18 | 9.0 | 5.6 |
| GS10YY | 50 | 6.2 | 25 | 6.2 | 200 | 25 | 9.9 |
| GS10WW | 100 | 12.5 | 50 | 6.2 | 200 | 200 | 24.9 |
A minimum of three experiments was performed for each biological activity assay. For the definition of geometric mean, please see footnote§ in Table 2.
Activity against Gram-negative bacteria was measured in liquid broth assays as described previously ((14) and references therein).
Supersusceptible derivatives of Gram-negative bacteria with more permeable outer membranes.
Mean MIC values (after 24 h) against supersusceptible Gram-negative bacteria, P. aeruginosa Z61, E. coli DC2, and S. typhimurium C610.
CD analysis of the cyclic peptide conformations
The far-ultraviolet (UV) CD spectra of cyclic peptides in buffer and unilamellar phospholipid vesicles, supported by other structural data and molecular modeling studies, suggested comparable conformations for all peptides. The CD spectra for the six cyclic peptides in these milieus are exhibited in Fig. 2, A and B, respectively. Spectra of all peptides had double-negative maxima in the range 203–208 nm and 215–220 nm. In comparison to the CD spectra in the buffer system, in the POPC liposomes the overall shape of the spectra were changed and the negative ellipticities of all peptides were enhanced. The single positive maximum of the spectra is not shown due to increased noise detected at wavelengths <190 nm in the 0.1 cm path-length cell. In other measurements using a 0.05 cm path-length cell, the maxima for the peptides were detected between 185 and 188 nm (data not shown). Under the same conditions, the CD spectra of the six cyclic peptides in the negatively-charged POPE/POPG (7/3 molar ratio) unilamellar vesicle system, was comparable to the CD spectra in POPC vesicles (spectra not shown). Based on the structural data (NMR, x-ray diffraction, and CD) for GS and GS10YY (9–11,14) a cyclic backbone structure composed of an antiparallel β-sheet with two type II′ β-turns on both ends of the sheet has been assigned to these peptides, an example of which is shown in Fig. 1.
FIGURE 2.

CD spectra of the six peptides in Tris buffer (A), and POPC vesicles in Tris buffer (B). See Instrumentation for experimental conditions.
The concentration-dependent CD spectra of GS (as the representative of the cyclic peptides of this study) in neutrally charged POPC vesicles (Fig. 3 A) did not change in the range 1:100 to 1:10 peptide/lipid molar ratios (P/L), indicating a stable conformation with a positive maximum at 186 nm and two negative maxima at 206 and 218 nm. The negative maximum at 218 nm was shoulderlike at low P/L ratios (inset in Fig. 3 A). In contrast to the spectra in phosphatidylcholine (PC) vesicles, the concentration-dependent spectra of GS in the negatively-charged PE/PG vesicles (Fig. 3 B) indicated a decrease in ellipticity as the P/L ratio increased from 1:100 to 1:10. Increase in the P/L ratio in the PE/PG system resulted in a gradual change of the GS spectrum accompanied by a decrease in the ellipticity ratio (θ206/θ218) and an overall red shift of the negative maxima.
FIGURE 3.

CD concentration-dependence spectra of GS in POPC (A), and POPE/POPG (B) vesicles dispersed in Tris buffer. See Instrumentation for experimental conditions.
Use of fluorescence resonance energy transfer (FRET) in determining the longer dimension of the cyclic peptides
All of the peptides listed in Table 1 contain one or two types of aromatic amino-acid residues (intrinsic fluorophores: F, W, Y) in their structure. Tryptophan has the highest absorptivity and emission intensity and is the most sensitive of the three aromatic residues (F, W, Y) in that its emission maximum wavelength, in comparison to the aqueous solvents, has a conspicuous blue shift and relative increase in intensity in hydrophobic environments (17,18). In this study, we used the emission spectra of aromatic amino acids to characterize the interaction of the peptides with lipid bilayers and to estimate the distance between the two ends (two aromatic residues) of the peptides in different milieus. An example of emission spectra profile of Trp-containing cyclic peptides is exhibited in Fig. 4. The maxima of the emission (excitation at 280 nm) of GS10WY in buffer, 80% methanol, and PC vesicles are at 353, 343, and 342 nm, respectively. The intensity of emission increased in the same order from buffer to more hydrophobic milieus. Tryptophan fluorescence was dominant in these peptides and obscured the contribution of tyrosine or phenylalanine emission. Tryptophan normally displays emission maximum of ∼350 nm in aqueous environment and the emission maximum shifts to lower wavelengths in less polar hydrophobic solvents. Phenylalanine does not contribute to emission at the corresponding excitation wavelength at 280 nm. Excited at 280 nm, Trp-lacking, Tyr-containing peptides GS10YY and GS10FY had an emission maximum at 302–303 nm in buffer and 80% methanol (higher intensity in methanol) and the emission spectra of these peptides in PC vesicles had a maximum at 306 nm. All these emission maxima corresponded to tyrosine emission. The Phe-containing peptide GS did not emit at 280-nm excitation.
FIGURE 4.

Emission spectra (excitation at 280 nm) of GS10WY in Tris buffer, 80% methanol, and POPC vesicles dispersed in Tris buffer. See Instrumentation for experimental conditions.
Resonance energy transfer between Tyr and Trp in GS10WY was utilized to estimate the distance between the fluorophores (17–19). This distance corresponds to the longer dimension (length) of the cyclic peptide estimated by measuring the distance between the Cα carbons of the D-Phe residues (or any two aromatic residues) located symmetrically in the peptide molecule (Fig. 1 and Fig. 5 A).
FIGURE 5.

Top views of molecular models of GS: top view of the GS in Fig. 1 with omitted amino-acid side chains (A), and top view of GS bound to a bacterial proteinase (22) (only GS is shown) (B).
Tyrosine in GS10WY was the donor (D) and tryptophan was the acceptor (A) of Tyr energy. To complete the calculations for the efficiency of the energy transfer and the Förster distance of the Tyr-Trp pair in GS10WY, the spectroscopic properties of two other conformationally equivalent peptides in the series, GS10FY (the donor-only peptide) and GS10FW (the acceptor-only peptide) were also determined. At the excitation wavelengths used for measurements (280 and 295 nm), the energy transfer between FY and FW fluorophore pairs in the latter peptides is minimal and not detectable.
Förster distance, R0, is the donor-acceptor distance at 50% FRET efficiency and measured by the equation (17)
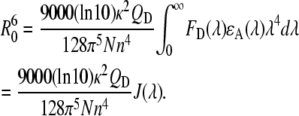 |
(1) |
In the above equation, FD (λ) is the corrected fluorescence intensity of the donor (area under the curve in the λ to λ + Δλ range normalized to unity); QD is the quantum yield of the donor in the absence of acceptor; ɛA(λ) is the molar extinction coefficient of the acceptor; n is the refractive index in aqueous solutions (1.33–1.4 for biomolecules (19), n =1.35 in these calculations); N is the Avogadro number; and 0 ≤ κ2 ≤ 4 is a term denoting the relative orientations of the donor and acceptor transition dipoles in space. Assuming the free rotations of donor and acceptor fluorophores in space, κ2 is normally considered to be equal to 2/3, which is an appropriate average value for the calculation of R0 without a significant error unless κ2 reaches zero (perpendicular transition dipoles) (17). The integral 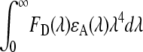 in Eq. 1 is equivalent to the term J(λ), which is the overlap integral between the emission of the donor (Tyr) and absorption of the acceptor (Trp). Considering the known experimental values and constants, Eq. 1 can be simplified as
in Eq. 1 is equivalent to the term J(λ), which is the overlap integral between the emission of the donor (Tyr) and absorption of the acceptor (Trp). Considering the known experimental values and constants, Eq. 1 can be simplified as
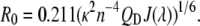 |
(2) |
In the above equations, λ and J(λ) are in nm and M−1 cm−1 (nm)4 units, respectively. Förster distance thus calculated is in Å units. The overlap integral J(λ) or JDA (DA stands for donor-acceptor) was determined experimentally by calculating the area under the overlap spectrum between the corrected and normalized emission of GS10FY (the donor without acceptor construct) and the absorption of GS10FW (the acceptor without donor construct) spectra. The overlap integral JDA can be replaced for J(λ) in Eq.2 to cover the whole range of overlap wavelengths:
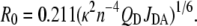 |
(3) |
The relative quantum yield of the donor fluorophore (QD) in GS10FY construct is calculated by the following equation:
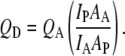 |
(4) |
In this equation QA is the quantum yield of the standard amino acid (0.14 for Tyr in water) (20); IP and IA are integrated emission (fluorescence intensity) for the peptide and standard amino acid, respectively; and AP and AA are the maximum absorption (optical density at ∼280 nm) for the peptide and standard amino acid, respectively. The QD values for the donor peptide, GS10FY, are shown in Table 4. These values correspond to the range of Tyr quantum yields (0.03–0.05) observed in proteins (19). Since the absorption spectrum was not attainable for the peptide in POPC vesicles due to high scattering, assuming that the peptide is located at the interface of lipid membranes, the average value of the absorption of peptide in buffer and 80% methanol was taken for AP in POPC vesicles.
TABLE 4.
FRET parameters for the decameric cyclic cationic peptide GS10WY at 25°C
| Environment | QD | JDA (M−1 cm−1 nm4) | R0 (Å) | r (Å) | FRET efficiency |
|---|---|---|---|---|---|
| Buffer | 0.039 | 3.034 × 1012 | 11.4 | 7.2 | 0.94 |
| 80% Methanol | 0.027 | 4.067 × 1012 | 11.2 | 12.6 | 0.33 |
| POPC vesicles | 0.033 | 2.072 × 1012 | 10.7 | 13.3 | 0.21 |
For description and calculation of parameters, see the Materials and Methods and Results sections. Peptide concentrations for FRET measurements were 10 μM. The P/L ratio for the cyclic peptides in POPC lipid dispersions was 1:100; at this ratio, all peptides were bound to vesicles, as verified by CD concentration-dependent experiments (e.g., Fig. 3 A).
The energy transfer efficiencies (E) in the last column in Table 4 were experimentally determined on the basis of
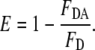 |
(5) |
FDA and FD are the fluorescence intensities of the donor in the presence and absence of the acceptor, respectively. The E and R0 values in Table 4 were used to calculate the distance between Tyr and Trp (r) in GS10WY in three different environments (buffer, 80% methanol, and POPC vesicles) (Table 4) employing
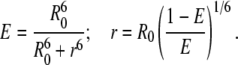 |
(6) |
The r values are in the range of 0.63R0 to 1.24R0, which is acceptable for FRET-based estimations of molecular distances (0.5–2R0 range). In comparison with the r value in buffers, the r values in 80% methanol and POPC vesicles were increased 75% and 85%, respectively (Table 4). The DA (r) distance derived for the peptide molecules in 80% methanol and PC vesicles (12.6 and 13.3 Å, respectively) was in a good correspondence with the distance shown for the longer dimension of the extended peptide conformation in Fig. 5 A (13.2 Å: distance between the Cα carbons of the aromatic residues), the crystal structure of the bis-Boc-tetra-n-methyl derivative of GS (12.8 Å) (21), as well as the longer dimension of the high resolution crystal structure of GS bound to the active domain of a bacterial Subtilisin saviase proteinase molecule shown in Fig. 5 B (13.9 Å) (note that the crystal structure of a complex formed between alkaline proteinase savinase and gramicidin S at 1.5 Å resolution has been deposited at the Protein Data Bank under PDB ID: 1tk2 (22)).
ITC measurements
Amphipathic cyclic peptides of this study have comparable conformations and overall charge (+2), therefore, application of surface binding (partitioning) models can provide an appropriate relative scale to evaluate the thermodynamics of peptide-lipid interactions. For this purpose, binding isotherms of peptide-lipid interactions were determined by calorimetric measurements of titration of lipid dispersions into peptide solutions. On the basis of these binding isotherms, it was assumed that peptide molecules bind to the surface and partly partition into the interior of lipid bilayers.
Representative isothermal calorimetric data for the titration of lipids into peptide solutions are exhibited in Fig. 6 (in POPC vesicles) and Fig. 7 (in POPE/POPG vesicles). These data represent all examples of the binding profiles observed for the cyclic peptide series of Table 1. Fig. 6 A exhibits the heat flow profile of the titration of PC vesicles into GS solutions (top), as well as the total heat per injection at different lipid/peptide molar ratios (bottom). At the initial peptide and lipid concentrations (5 μL aliquots of 5 mM lipid vesicles were injected into 20 μM peptide solutions), the binding of GS to PC vesicles was endothermic and resolved into two components as shown in the inset of Fig. 6 A. In contrast, binding of the Trp-containing peptide GS10WW to PC vesicles was exothermic with no clear resolution into components, as exhibited in Fig. 6 B. The heat flow profile of the binding of the rest of the cyclic peptides to PC vesicles was endothermic (GS10YY, GS10FW, and GS10FY) or exothermic (GS10WY). Partial resolution of the single injection heat flow was also observed in other peptides such as GS10FY (Fig. 6 C). Total heat per injection versus lipid/peptide molar ratio graphs are shown in the inset of Fig. 6, B and C.
FIGURE 6.

Heat of binding of peptides to POPC vesicles: GS (A), GS10WW (B), and GS10FY (C). See Instrumentation for experimental conditions. The “molar ratios” in the figures are lipid/peptide molar ratios.
FIGURE 7.

Heat of binding of peptides to POPE/POPG vesicles: GS (A), GS10WW (B), and GS10FY (C). See Instrumentation for experimental conditions. The “molar ratios” in the figures are lipid/peptide molar ratios.
The binding of GS to PE/PG vesicles was also endothermic but not clearly resolved into two components (Fig. 7 A). Another unique feature in this heat flow profile of GS in the PE/PG lipid system (not observed in other cyclic peptides under similar conditions) is the appearance of a chaotic pattern after the 9th injection of lipids. The heat flow diminished to baseline at L/P ≥ 14. The data points between 9th and 14th injections were not included in the curve-fitting for calculating the thermodynamic parameters to exclude the processes that could be related to vesicle fusion and other nonspecific interactions, therefore unrelated to peptide binding (Fig. 7 A, inset). We have repeated this experiment several times with independent preparations and observed similar heat flow patterns. Binding of the rest of cyclic peptides to PE/PG vesicles was also endothermic as exhibited in Fig. 7, B and C, for GS10WW and GS10FY, respectively. Total heat per injection versus L/P molar ratio graphs are shown in the inset of Fig. 7, B and C.
The total molar enthalpy of binding of peptides to lipid systems (ΔHb) was calculated as 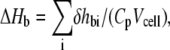 where δhbi is the heat of binding for injection i, Cp is the total peptide molar concentration, and Vcell is the volume of the sample cell of calorimeter. The experimental values for δhbi and ΔHb (Table 5) were used to calculate the binding isotherms (23,24). The ratio of the heat of binding of the first injection to the total enthalpy of binding (δhb1 /ΔHb) suggested a range for the amount of peptide bound to lipids. For example, in the first lipid injection, close to 25% of the total GS and 16% of the total GS10WW were bound to both lipid systems, whereas 15 and 10% of GS10FY was bound to PC and PE/PG lipids, respectively.
where δhbi is the heat of binding for injection i, Cp is the total peptide molar concentration, and Vcell is the volume of the sample cell of calorimeter. The experimental values for δhbi and ΔHb (Table 5) were used to calculate the binding isotherms (23,24). The ratio of the heat of binding of the first injection to the total enthalpy of binding (δhb1 /ΔHb) suggested a range for the amount of peptide bound to lipids. For example, in the first lipid injection, close to 25% of the total GS and 16% of the total GS10WW were bound to both lipid systems, whereas 15 and 10% of GS10FY was bound to PC and PE/PG lipids, respectively.
TABLE 5.
Thermodynamic parameters of the peptide-lipid interactions
| Peptide | ΔHb × 103 (total) (cal.mol−1) | Keq (M−1) | Bmax (mol/mol) | Kb (M−1) | ΔHb × 103 (cal.mol−1) | ΔSb (cal.mol−1.K−1) | ΔGb × 103 (cal.mol−1) | ΔGb × 103 (total) (cal.mol−1) |
|---|---|---|---|---|---|---|---|---|
| GS | ||||||||
| PC | 1.3 | Keq (15.1 μM) 3.7 × 104 | Bmax1 0.36 | Kb1 7.0 × 107 | ΔHb1 0.9 | ΔSb1 46.7 | ΔGb1 −13.3 | −12.1 |
| Keq (1.7 μM) 2.5 × 105 | Bmax2 0.35 | Kb2 1.5 × 105 | ΔHb2 0.4 | ΔSb2 32.8 | ΔGb2 −9.5 | |||
| PE/PG | 5.2 | Keq (15.6 μM) 3.2 × 104 | Bmax1 0.29 | Kb1 9.0 × 108 | ΔHb1 3.4 | ΔSb1 60.0 | ΔGb1 −14.8 | −13.1 |
| Keq (1.1 μM) 3.2 × 105 | Bmax2 0.26 | Kb2 2.8 × 105 | ΔHb2 1.8 | ΔSb2 38.8 | ΔGb2 −9.9 | |||
| GS10YY | ||||||||
| PC | 0.9 | Keq (22.4 μM) 2.2 × 104 | Bmax1 0.27 | Kb1 108 | ΔHb1 0.6 | ΔSb1 46.5 | ΔGb1 −13.5 | −12.1 |
| Keq (4.2 μM) 7.9 × 104 | Bmax2 0.36 | Kb2 8.5 × 104 | ΔHb2 0.3 | ΔSb2 30.5 | ΔGb2 −9.2 | |||
| PE/PG | 5.1 | Keq (16.5 μM) 1.0 × 104 | Bmax1 0.11 | Kb1 4.0 × 108 | ΔHb1 4.1 | ΔSb1 60.1 | ΔGb1 −14.2 | −13.3 |
| Keq (0.6 μM) 2.0 × 105 | Bmax2 0.13 | Kb2 1.5 × 105 | ΔHb2 1.0 | ΔSb2 34.9 | ΔGb2 −9.6 | |||
| GS10WW | ||||||||
| PC | −2.5 | Keq (16.7 μM) 3.7 × 104 | Bmax 0.74 | Kb 3.2 × 105 | ΔHb −2.5 | ΔSb 24.9 | ΔGb −10.0 | −10.0 |
| Keq (1.0 μM) 2.2 × 105 | ||||||||
| PE/PG | 2.2 | Keq (16.7 μM) 2.3 × 104 | Bmax1 0.26 | Kb1 6.9 × 107 | ΔHb1 1.5 | ΔSb1 48.6 | ΔGb1 −13.2 | −12.2 |
| Keq (1.2μM) 2.4 × 105 | Bmax2 0.16 | Kb2 4.2 × 105 | ΔHb2 0.7 | ΔSb2 36.1 | ΔGb2 −10.2 | |||
| GS10FW | ||||||||
| PC | 0.25 | Keq (25.7 μM) 6.4 × 104 | Bmax1 0.82 | Kb1 108 | ΔHb1 0.15 | ΔSb1 44.9 | ΔGb1 −13.5 | −11.9 |
| Keq (3.1 μM) 3.3 × 105 | Bmax2 1.2 | Kb2 8.3 × 104 | ΔHb2 0.1 | ΔSb2 30.7 | ΔGb2 −9.5 | |||
| PE/PG | 4.2 | Keq (15.8 μM) 1.5 × 104 | Bmax1 0.10 | Kb1 5.3 × 108 | ΔHb1 2.3 | ΔSb1 55.3 | ΔGb1 −14.5 | −12.2 |
| Keq (1.3μM) 1.0 × 105 | Bmax2 0.20 | Kb2 1.4 × 105 | ΔHb2 1.9 | ΔSb2 37.7 | ΔGb2 −9.5 | |||
| GS10WY | ||||||||
| PC | −1.5 | Keq (16.6 μM) 2.4 × 104 | Bmax 0.70 | Kb 8.7 × 104 | ΔHb −1.5 | ΔSb 25.4 | ΔGb −9.2 | −9.2 |
| Keq (1.7 μM) 7.6 × 104 | ||||||||
| PE/PG | 3.8 | Keq (17.7 μM) 1.5 × 104 | Bmax1 0.17 | Kb1 2.9 × 108 | ΔHb1 2.7 | ΔSb1 55.2 | ΔGb1 −14.0 | −12.8 |
| Keq (1.1μM) 1.8 × 105 | Bmax2 0.12 | Kb2 2.2 × 105 | ΔHb2 1.1 | ΔSb2 36.0 | ΔGb2 −9.8 | |||
| GS10FY | ||||||||
| PC | 0.9 | Keq (34.1 μM) 2.0 × 104 | Bmax1 0.35 | Kb1 8.7 × 108 | ΔHb1 0.5 | ΔSb1 50.3 | ΔGb1 −13.3 | −11.6 |
| Keq (1.3 μM) 3.0 × 105 | Bmax2 0.50 | Kb2 1.3 × 105 | ΔHb2 0.4 | ΔSb2 32.6 | ΔGb2 −9.5 | |||
| PE/PG | 7.4 | Keq (18.2 μM) 1.1 × 104 | Bmax1 0.09 | Kb1 1.1 × 109 | ΔHb1 4.0 | ΔSb1 62.1 | ΔGb1 −14.8 | −12.5 |
| Keq (1.2 μM) 1.0 × 105 | Bmax2 0.14 | Kb2 2.3 × 105 | ΔHb2 3.4 | ΔSb2 43.5 | ΔGb2 −9.8 | |||
The reported data are the average of 2–4 independent measurements. See the Results and Discussion sections for details of one- and two-site binding models. In ITC experiments, all peptides were in the sample cell and titrated with 3–5 μL of 5 mM lipid dispersions per single injection. In POPC titrations, GS10YY, GS10FW, and GS10FY were at 40 μM concentrations; peptide concentrations in all other titrations with lipid dispersions were 20 μM.
Binding isotherms for the interaction of three peptides (GS, GS10WW, and GS10FY) with two lipid systems are exhibited in Fig. 8. Each data point is derived from a single injection of lipids into peptide solutions. Binding isotherms were also obtained for the rest of the cyclic peptides. In Fig. 8, Xb represents the degree of peptide binding defined as the moles of peptide bound per moles of lipid (Xb = npb/nl), and Cpf is the concentration of free peptide in solution. An equilibrium condition is assumed in the binding of peptides to lipid vesicles and on the basis of this assumption and the binding isotherms, Xb is a function of the free peptide concentration: Xb = f(Cpf). The binding isotherms derived for the cyclic peptides (Fig. 8) suggest that the relation between Xb and Cpf is not linear and Keq (an apparent partition equilibrium constant independent of peptide concentration: Xb = KeqCpf ) varies with the peptide concentration. The Keq for GS in the PC lipid system varies between 3.7 × 104 M−1 (Cpf = 15.1 μM) and 2.5 × 105 M−1(Cpf = 1.7 μM). The values of Keq for the cyclic peptides, in a broad range of free peptide concentrations, are tabulated in Table 5. To calculate the binding constants for the cyclic peptides to the lipid systems under equilibrium conditions, we have applied simple one- and two-site binding models to the binding isotherms, using the equations
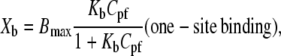 |
(7) |
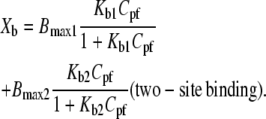 |
(8) |
In these equations, the Bmax, Bmax1, and Bmax2 are the maximum binding capacity (in mol/mol) of the peptides to lipid vesicles, and Kb, Kb1, and Kb2 are the equilibrium binding (surface partitioning) constants. One- and two-site models were fit to the data by Levenberg-Marquardt nonlinear least-square algorithms to obtain the values for the Bmax and Kb. Calculation of Kb values resulted in the calculation of free energies of binding (ΔGb) of peptides using the equation
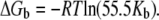 |
(9) |
In this equation, R is the universal molar gas constant and T is the absolute temperature. The factor 55.5 is the molar concentration of water and in this way the cratic contribution to the free energy (∼2.4 kcal/mol at 30°C) is taken into consideration (25). Finally, entropy of binding (ΔSb) was calculated as ΔSb = (ΔHb−ΔGb)/T.
FIGURE 8.

Binding isotherms for GS, GS10WW and GS10FY in POPC (solid symbols) and POPE/POPG (open symbols) lipid systems.
Table 5 provides a relative scale for comparison of the thermodynamic parameters of peptide-lipid interactions. The one-site binding (partitioning) model was only used for the exothermic interactions of GS10WW and GS10WY with PC vesicles. Two-site binding (partitioning) model was used for all other endothermic peptide-lipid interactions. In the two-site models, the thermodynamic parameters were resolved for each binding event. Both binding models had excellent fits to the binding isotherms as is shown by simulations (solid lines) in Fig. 8.
The ΔHb values were variable in different peptides. In the PC lipid system, ΔHb was small and positive (endothermic interaction) for GS, GS10YY, GS10FW, and GS10FY, and negative (exothermic interaction) for GS10WW and GS10WY (Table 5). In the PE/PG lipid system, ΔHb was positive (endothermic interaction) for all six peptides, and had higher values in comparison to the PC system (Table 5). In the two-step binding processes, the first binding constant (Kb1) was 102−103 times larger than the second binding constant (Kb2), which suggested a much higher affinity for the first step of the binding process. In the two-step processes, ΔGb1 in PC and PE/PG systems were comparable and in the range of −13.2 to −14.8 kcal/mol. In the same processes, ΔGb2 in PC and PE/PG systems were in the range of −9.2 to −10.2 kcal/mol. The latter free energies, as well as the Kb2 values, were comparable to the corresponding parameters, ΔGb and Kb, in the one-step binding processes (GS10WW and GS10WY binding to PC). In the two-step processes, both ΔSb1 and ΔSb2 in PE/PG systems were larger than their corresponding values in PC. In the one-step binding processes, ΔSb of GS10WW and GS10WY to PC were <ΔSb2 in the two-step binding processes (Table 5). Overall free energies of binding of the peptides to the two lipid systems are reported in the last column of Table 5. These values are calculated on the basis of the contribution of enthalpy of binding, which is proportional to the moles of bound peptide, to each binding process. Compared with the PC system, the total ΔGb of peptides are slightly lower in the PE/PG system. The lowest free energies of binding in the PC system are those of GS10WW and GS10WY (2–3 kcal/mol less than other peptides), which participate in the one-site binding process. The total ΔGb of peptides in the PE/PG system are comparable (−12.2 to −13.1 kcal/mol). Total ΔGb of GS for binding to POPC (−12.1 kcal/mol at 30°C) is lower in comparison with the previously reported ΔGb for GS (−11.0 kcal/mol at 25°C) (26).
DISCUSSION
Conformation of the cyclic peptides
All decameric cyclic cationic peptides of this study (Table 1) share common structural features and their difference is in their aromatic amino acids (Fig. 1 and Fig. 5 A). The difference in retention times of peptides could be attributed to the difference in the hydrophobicity of aromatic amino acids with Trp being the most and Tyr the least hydrophobic amino acids (16,27). Equivalent β-sheet-β-turn backbone conformations of the cyclic cationic peptides can be further verified by comparing their CD spectra in aqueous and lipid vesicles. The difference in the shape and ellipticities of these spectra can be attributed to the differences in their aromatic amino-acid composition—Trp and Tyr can interfere with the CD spectra of the backbone conformation in the 225–240 nm range—and their tendencies to interact with lipid vesicles. In the last column in Table 1, ΔΔGIFO values can be used to estimate the tendencies of the peptides to interact with the hydrophobic core versus the interface of POPC bilayers. In this scale, GS and GS10YY have the highest tendency to interact with the bilayer core and bilayer interface, respectively. GS10WW has a comparable affinity for both environments. Other peptides with different aromatic residues are located in between GS and GS10YY. Different tendencies of peptide aromatic side chains to interact with different regions of POPC bilayers could therefore influence their CD spectra. The common feature of the CD spectra of cyclic peptides in lipid vesicles was their substantial enhancement of negative ellipticity in comparison to the spectra in buffer. The enhancement of ellipticity in lipid vesicles suggests strong interactions of these peptides with lipid vesicle surfaces as well as their limited conformational change in the lipid environments. Moreover, the similarity of CD spectra of cyclic cationic peptides in zwitterionic and negatively-charged vesicles implies that despite the role of electrostatic interaction in attracting peptides to the surface of membranes, hydrophobic interaction is the main factor in stabilizing the relatively inflexible structures of peptides in lipid membranes (28).
The conformation of GS is comparable and stable at different P/L ratios in PC vesicles; however, at higher P/L ratios (1:15 and 1:10), the negative maximum at 218 nm was more pronounced and replaced a shoulderlike negative maximum at lower P/L ratios (Fig. 3 A and inset). The enhancement of ellipticity at this negative maximum can be attributed to a conformational change caused by intermolecular interactions of peptides and/or peptides with lipids. Conversely, the concentration-dependent spectra of GS in the negatively-charged PE/PG vesicles (Fig. 3 B) indicated a decrease in ellipticity as the P/L ratio increased from 1:100 to 1:10. Increase in the P/L ratio in the PE/PG system resulted in a gradual change of the GS spectrum accompanied by a decrease in the ratio of the negative maxima from 1.2 (θ206/θ218) in the 1:100 to 1 (θ208/θ220) in the 1:10 P/L ratios. The shift in the shape of CD spectra in the PE/PG system implies a conformational change that can be caused by peptide association and/or specific electrostatic interaction between the cationic peptides and anionic lipids at lower P/L ratios (5). The change in the CD spectra at higher L/P ratios in the PE/PG system can be also attributed to the change of optical properties of the peptide/vesicle dispersion caused by the appearance of larger fused vesicles induced by higher peptide concentrations ((5), and M. Jelokhani-Niaraki, unpublished results).
The comparative fluorescence spectra of cyclic peptides in buffer, 80% methanol, and PC vesicles (Fig. 4) support the CD data in confirming the interaction of the cyclic peptides with membranes. Three conformationally equivalent cyclic peptides of the series (GS10WY (donor-acceptor), GS10FY (donor-only), and GS10FW (acceptor-only)) (Fig. 2) were employed to estimate the longer dimension of the cyclic peptides (17,29). Table 4 exhibits the main parameters obtained from the FRET experiments. Förster distances were between 10.7 and 11.4 Å for GS10WY. FRET efficiency was the highest for the peptide in Tris buffer (0.94) and lowest for the peptide in the PC system, which directly corresponded to the shortest (7.2 Å) and longest (13.3 Å) DA distances, respectively. The DA distance in 80% methanol (12.6 Å) is very close to the DA distance in the PC vesicles. The DA distances (r), in Table 4, suggest that the cyclic β-sheet-β-turn construct of peptides undergoes a conformational change from aqueous environment to less polar alcoholic and lipid membrane environments. As with the CD spectra of the peptides, this conformational change was attributed to stabilization of the β-sheet-β-turn backbone structure in less polar environments. Overall, from the results of fluorescence studies, interaction of the cyclic peptides with lipid membranes was further confirmed. Consequently, we can assume two main conformations for the β-sheet-β-turn peptide constructs in aqueous and less polar milieus: a contracted tense β-sheet-β-turn in aqueous environment (short interaromatic distance, 7.2 Å, before interaction with lipid membrane surface) and an expanded relaxed β-sheet-β-turn in alcoholic and PC vesicles (long interaromatic distance, 12.6–13.3 Å, after interaction with lipid membrane surface).
Thermodynamics of interaction of the cyclic peptides with lipid membranes
The interactions of GS with different phospholipid systems PC, PE/PG, and PC/PG (26,30) were endothermic. In comparison to the zwitterionic PC vesicles, interaction of the peptides with the negatively-charged lipid vesicles, PE/PG and PC/PG (Fig. 7 A and (30), respectively), was more endothermic. For example, the enthalpy of binding of GS in binding to PE/PG vesicles was four times its enthalpy of binding to PC vesicles (Table 5).
Under our experimental conditions, the heat flow profile for the binding of GS to POPC lipid vesicles was resolved into two components (Fig. 6 A and its inset). This phenomenon has not been reported in a previous publication on the POPC binding to GS using the ITC technique (26). The two-site binding model used to analyze the binding isotherms of GS (Fig. 8) suggests a consistent qualitative and quantitative interpretation for this experimental observation. The first sharp peak in the heat flow profile of GS can be related to the fast and high affinity binding of free GS peptides to the added peptide-free POPC vesicles (Kb1 = 7 × 107 M−1) (Table 5), whereas the second peak exhibits slower processes that implies a second binding (low affinity, Kb2<Kb1/100) step involving further peptide-lipid and peptide-peptide interactions. As an interpretation, in a two-step binding (partitioning) process, initially a population of peptides binds to free lipid vesicles (fast, high affinity), and then a second population of peptides binds to these peptide-bound vesicles (slow, low affinity). Binding of the first population of positively-charged peptides generates a positive charge density and membrane potential on the lipid vesicle surface, and therefore reduces the binding affinity of the second population of peptides for the membrane surface. Both partitioning steps, particularly the second step, may also involve other exothermic or endothermic interactions such as peptide association processes, penetration of hydrophobic regions of peptides into the bilayer hydrophobic core, and rearrangement (association/dissociation) of water molecules caused by the increase in the peptide concentration at the bilayer surface and disruption of bilayer surface. It is noteworthy to mention that concentration-dependent CD and UV measurements of GS in POPC vesicles excluded vesicle fusion (Fig. 3 A and M. Jelokhani-Niaraki, unpublished results) and its possible contribution to the endothermic process in the second binding step. In both binding steps, hydrophobic effects are the main binding driving force as supported by large positive entropy values (ΔSb1 = 46.7 and ΔSb2 = 32.8 cal/mol.K). A two-step binding model was also applied to the binding isotherm of GS in the PE/PG system and the thermodynamic parameters were resolved (higher Kb values, Table 5). It is worth noting that we also applied a surface binding (partition) model considering electrostatic effects to the binding isotherms of GS and its aromatic analogs (24,31). In this case, a two-site binding model was also applied to resolve the derived nonlinear binding isotherms. For both lipid systems, membrane potential was always positive under our experimental conditions and by considering the electrostatic effects, the binding constants (in Table 5) were increased accordingly (up to two orders of magnitude). These results further supported the main role of hydrophobic effect in the binding of GS and its aromatic analogs with lipid membranes. Since there was no qualitative difference between the binding isotherms in the two models, and the partitioning equilibrium constant (Kp) in the electrostatic model was not independent of peptide concentration (24), we chose the above-mentioned simple one- and two-site binding (partitioning) models (Eqs. 7 and 8) for the interpretation of our experimental data.
The two-site binding model proved to be the best experimental model for analysis of the endothermic interaction of the cyclic peptides with both lipid systems employed in this study. A two-step binding seemed to be obvious from some of the heat flow profiles of peptides in the PC lipid system. However, in the PE/PG lipid system the single injection heat flow profiles were not clearly resolved. Applying the two-step model has therefore revealed some of the subtleties of cyclic peptide-lipid interactions, which were not otherwise observable in the heat flow trends and have not been reported previously. In the case of exothermic interactions of the two Trp-containing peptides (GS10WW and GS10WY) with PC vesicles, the single-step binding model gave the best fit (Fig. 6 B, Table 5). This observation can be related to the strong affinity of Trp for lipid vesicle membrane surfaces as well as its high intrinsic hydrophobicity, which enhances the interaction with the lipid vesicle hydrophobic core. A more detailed analysis of the ITC data considering the affinity of the aromatic amino acids for POPC bilayers using the whole-residue hydrophobicity scales (16) may lead to a better understanding of the interaction of GS and its conformationally equivalent aromatic analogs with lipid bilayers. As exhibited in Table 1, the calculated water-to-octanol relative free energies of partitioning of the peptides (ΔGWO), as a measure of their affinity for the bilayer hydrophobic interior, follow the order of their retention times and the intrinsic hydrophilicity/hydrophobicity scale (27), with GS10WW and GS10YY being the most and least hydrophobic peptides, respectively. The place of GS10WY and GS is switched in water-to-interface partitioning relative free energies (ΔGWIF) of peptides. The free energies of the transfer of peptides from bilayer interface to bilayer hydrophobic core (ΔΔGIFO) increase in the order of GS>GS10FW>GS10WW>GS10FY>GS10WY>GS10YY. By considering the ΔΔGIFO values and the thermodynamic parameters in Table 5, cyclic peptides can be categorized on the basis of their affinity for different regions of POPC bilayers—interface-water layers, polar headgroups, and hydrophobic interior. The ΔΔGIFO values imply that GS has the highest affinity for the membrane core (ΔΔGIFO = −0.6 kcal/mol), while GS10YY strongly prefers the membrane interface (ΔΔGIFO = +1.02 kcal/mol). On the other hand, GS10WW has an equal preference for membrane core and interface (ΔΔGIFO = +0.08 kcal/mol). In this way, on the basis of the relative affinity of their aromatic residues for different regions of the lipid bilayers, as shown in Fig. 9 A, the cyclic peptides can be categorized into three groups: 1), peptides with more affinity for the bilayer hydrophobic core (GS); 2), peptides with comparable affinity for the bilayer core and interface (GS10WW); and 3), peptides with more affinity for the bilayer interface (GS10YY). Fig. 9 B, as well as Tables 1 and 5, exhibit a direct relation between the total enthalpy of binding (ΔHb) in each group of peptides and their ΔΔGIFO. Single and double substitutions of the F residues in GS to W or Y, GS→GS10FW→GS10WW or GS→GS10FY→→GS10YY, result in the decrease in affinity of peptide for the bilayer core and ΔHb. Single and double substitutions of the Y residues in GS10YY to W, GS10YY→GS10WY→GS10WW, result in the decrease in affinity for the bilayer interface and ΔHb. While the dominant driving force for the binding of the cyclic peptides to bilayers seems to be the increase in the entropy of the system through classical hydrophobic effect causing the exclusion of nonpolar surfaces from water, the contribution of enthalpy to binding should not be underestimated. Contribution of enthalpy to the overall ΔGb of Trp-containing cyclic peptides stresses the additional role of nonclassical hydrophobic effect in the interaction of these peptides with lipid membranes. GS10WW and GS10WY have the least favorable entropy and the most favorable enthalpy of binding among the cyclic peptides. Tryptophan has an affinity for the lipid bilayer surfaces in membrane-interacting proteins, and the interactions of its indole derivatives with POPC membranes are exothermic; moreover, dipole interactions with the bilayer polar headgroups and close van der Waals interactions with the bilayer hydrophobic core may also contribute to the exothermic interaction of Trp-containing cyclic peptides with membranes (16). Phe-containing peptides GS and GS10FY strongly interact with the bilayer hydrophobic core and have higher positive entropies of binding. In addition to disruption of the membrane surface water molecules, these peptides can greatly disrupt the polar headgroups to reach bilayer interiors. The Tyr-containing peptide GS10YY interacts mainly with the interface-water layers and bilayer polar headgroups and seems to be the peptide with the least penetration into the bilayer hydrophobic core. A schematic view of the interaction of the cyclic peptides with different regions of the lipid bilayers is exhibited in Fig. 9 A. Overall, it seems that in these peptides and similar constructs, changing the balance between the aromatic amino acids (for example, Phe versus Trp) can modulate their mode of interaction with PC and PC-containing (30) lipid systems.
FIGURE 9.

(A) Scheme for preferential binding of the cyclic aromatic peptides to different regions of bilayer depending on their aromatic amino-acid composition (peptides have similar backbone structures); (B) comparison between the enthalpies and affinities of binding of peptides to the bilayer hydrophobic core versus bilayer interface (Tables 1 and 5).
As mentioned above, in comparison with the enthalpy values for the PC lipid system, the enthalpy of binding in PE/PG lipid system was noticeably more positive (less favorable) and the entropy of binding was generally increased (more favorable) (Table 5). This difference can be caused by the structure and degree of hydration of the membrane surfaces as well as the packing of the lipid headgroups and surface charge density of the vesicle systems. In comparison to PC lipids, pure PE lipids with one or two unsaturated hydrocarbon chains form bilayers with an inherent negative curvature and prefer to switch to the hexagonal HII phase. Addition of 30% of a bilayer-forming lipid, such as PG, to PE lipids stabilizes the bilayer form and reduces the tendency of PE to form the hexagonal HII phase (32). PE lipids are also less hydrated than PC lipids and therefore less energy is required to access the dehydrated surface of these lipids; however, addition of PG lipids disrupts the PE packing, and by increasing the water-accessible surface area and hydration of the bilayer surface, causes an increase in the overall dehydration energy and positive enthalpy of binding (33). These factors may count for the different profile of peptide-binding to PE/PG vesicles compared to PC or PC/PG vesicles. Another notable feature of peptide-binding to PE/PG vesicles was observed in the heat flow profile of GS (Fig. 7 A). In this profile, the stepwise decreasing heat flow of the titration of GS with PE/PG vesicles was interrupted with a chaotic pattern. The origin of this phenomenon could be the inherent fusogenic properties of unsaturated PE lipids induced by GS. It is plausible that after the first 8–9 titrations of GS with PE/PG vesicles almost all of the positively charged peptides were bound to the lipid vesicles and the negatively-charged PG lipids on the surface of vesicles were shielded and neutralized (the total positive charge of the peptides in the sample cell can be neutralized by addition of ∼40 μL of lipids). In further additions, free PE/PG vesicles did not experience any strong electrostatic repulsion or organized hydration repulsive force from the neutralized peptide-bound vesicles in the sample cell and could therefore approach the peptide-bound vesicles. At the same time the peptide-bound vesicle surfaces were destabilized by both the increase in surface tension and fusogenic properties of PE lipids. The chaotic pattern observed in the titration of GS with PE/PG vesicles could be therefore attributed to nonspecific vesicle fusion and other nonspecific events. This interpretation was supported by the concentration-dependent light scattering UV absorption experiments at 600 nm (data not shown).
The binding affinities of cyclic peptides for lipid systems can be also related to their biological activities (Tables 1–3 and 5). For interaction with POPC vesicles, a model of erythrocyte membranes, the Kb, for GS10WW could be compared to the Kb2 values of GS and GS10YY. Among these peptides, GS10WW was the most hydrophobic and hemolytic, and also had the highest Kb and ΔGWO value. In the POPE/POPG system, a model for bacterial membranes, there was a relation between the Kb2 values and their activity against Gram-positive bacteria; the higher Kb2 values corresponded to higher antimicrobial activity. In the same lipid system, activity against Gram-negative bacteria could be also directly related to the Kb1 values—peptides with higher Kb1 were more potent against Gram-negative bacteria.
The total ΔGb for the interaction of the peptides with lipids can be separated into different terms:
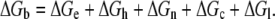 |
ΔGe is the free energy of electrostatic (includes charge and dipole interactions) interactions; ΔGh is the contribution of hydrophobic effect to binding (includes the dehydration and rehydration of peptide and lipid molecules and rearrangement of water molecules close to peptide and bilayer surfaces); ΔGn is the free energy of the interaction of peptide side chains with the nonpolar regions of the bilayer; ΔGc is the free energy related to the conformational changes of peptide upon interaction with membranes; and ΔGl is the free energy related to lipid bilayer deformation and structural change. Each component contributes to the total free energy in a subtle way that cannot be easily quantified experimentally. ΔGe has the most contribution in the interaction of peptides with the anionic PE/PG membranes and the interaction of Trp- and Tyr dipoles with bilayer headgroup dipoles or charges. Of all terms contributing to the total ΔGb, ΔGh is the main determinant in the entropy-driven binding of peptides to both lipid systems. ΔGn also contributes to the binding of all peptides, especially the Trp- and Phe-containing peptides. ΔGc could be comparable for all peptides and ΔGl contributes most in the binding of peptides such as GS and GS10FW that perturb the membrane structure most (Fig. 9, A and B). The ΔGb values reported in this study included all of the above-mentioned free energy components. The binding energies of the cationic cyclic peptides (Table 1) to lipid membranes (ΔGb = ΔHb − TΔSb) were all negative (Table 5). The TΔSb term was positive and much larger than the ΔHb term, which implies that the binding of peptides to lipid membranes were entropy-driven. Favorable contribution to the TΔSb term in the equation for the free energy of binding can be attributed to the rearrangement of hydration water molecules around the peptide and lipid bilayer surface caused by the binding of peptides, the change of the conformation of peptides (elongation), and the deformation and expansion of lipid bilayers. Electrostatic interaction between the membrane surface and the peptide charge (+2) or dipoles (including the aromatic Trp or Tyr side-chain dipoles), van der Waals interaction of the peptide side chains with bilayer hydrophobic cores, and the hydration of bilayer surfaces are the favorable contributors to the ΔHb.
Mechanism of interaction of the cyclic cationic peptides with biological membranes
Two general models have been proposed for the mechanism of GS interaction with lipid bilayers. In one model, GS strongly interacts with the outer surface of membranes, resides on the surface (34), and destabilizes the overall membrane structure through membrane-thinning effect (35) and peptide-lipid interactions causing membrane lysis (36). In this model, GS does not associate to form organized pores (37) and only generates nonspecific leakage through perturbation of membrane structure. In the second model, GS molecules associate to generate channels by forming supramolecular double-stranded spirals in membranes that can transverse the bilayer. This model is based on the supramolecular crystal structures of GS complexes in aqueous and organic solvents (38,39). Preliminary electrophysiological data have been reported on the GS channel-like activity in the lipid bilayers, but the published data did not represent stable channels (40,41). Moreover, in these reports, the analysis of conductance events was not conclusive and electrophysiological properties of the channels such as open-close analysis and voltage-dependence were not characterized.
The two-state model for the action of antimicrobial peptides, proposed by Huang (37,42), is a physical model based on elasticity theory of lipid membranes. In this model the peptide has two different equilibrium states in binding to membranes. At low P/L ratios, the peptide adsorbs on the lipid headgroup area and, once the P/L ratios increase and passes a threshold value P/L*, the peptide associates to form a multi-pore state, transverse the membrane, and cause leakage. The P/L* ratio is different for each peptide and each lipid membrane system. In this model, the pore formation is considered to be a spontaneous cooperative event (37). However, when the two-state model was applied to a cyclic cationic octadecapeptide (θ-defensin), despite the experimental evidence for the existence of the adsorbed state, no evidence for a pore-forming structure or vertical insertion of this peptide was found (43).
Based on the experimental results of this study and our previous studies (5), and by considering the concepts developed in the above-mentioned two general mechanistic models, a qualitative model is suggested for the entropy-driven interaction of GS and GS-like peptides with biological membranes as outlined in Fig. 10. At the concentrations used for this study, peptides are monomeric, and are in their tense conformation in the aqueous environment (28). Initially, peptides approach the bilayer surface in their tense conformations (short interaromatic length, 7.2 Å) (Fig. 10, step 0; Fig. 2 A; and Table 4). In the next step (Fig. 10, step 1), adsorption of peptide on lipid bilayer surface is accompanied by the rearrangement of water molecules on bilayer and peptide surfaces (mostly an endothermic reaction, high Kb and ΔSb; see Table 5). Electrostatic attractive forces between the positively-charged peptide and membrane surface negative charges (in case of PE/PG bilayers) also contribute to and enhance the adsorption of peptides (higher ΔHb and ΔSb compared with PC bilayers; see Table 5). In the next stage (Fig. 10, step 2), after initial interaction of peptides with bilayer surfaces, peptides expand their conformation from tense to relaxed state (long interaromatic length, 13.3 Å) (stabilization of intramolecular H-bonds, increase in ΔS) and interact more favorably with the bilayer surface (Fig. 2 B, Table 4). At this stage, the bilayer structure at the surface is perturbed and the area of the bilayer surface expands in proportion to the cross-sectional area of the peptide (e.g., the cross-sectional area of GS, ∼258 Å2 (44), roughly equals the cross-sectional area of four PC lipid headgroups). Bilayer surface expansion (increase in ΔS) results in the increase in surface tension and thinning of the bilayer (37,45,46). Cyclic peptides can compensate for this increase in the surface expansion by reducing their cross-sectional area (tilting or inversing their orientation) through deeper interaction with the bilayer surface and hydrophobic regions.
FIGURE 10.

A mechanism for the binding of GS and its aromatic analogs to lipid bilayers: (0) Monomeric cyclic peptides are attracted to and approach the bilayer surface in their tense conformation. (1) Cyclic peptides adsorb on the bilayer surface and bind to surface through hydrophobic effect and electrostatic interactions; tense conformation of cyclic peptides changes toward relaxed conformation. (2) Cyclic peptides adopt the relaxed conformation, insert deeper into bilayer, deform the membrane structure without forming peptide-peptide associations, and induce leakage (low P/L ratios). (3) Cyclic peptide-lipid complexes form nonspecific transient porelike zones enhancing the leakage through membranes (high P/L ratios).
At high concentrations, peptide interaction with membranes could result in further perturbation of bilayer structure (increase in ΔS and hydrophobic interaction) and may lead to generation of transient lipid-peptide zones of nonspecific size in equilibrium with the surrounding environment, enhancing the leakage through membranes (Fig. 10, step 3). Concentration-dependent dye-leakage experiments with GS and GS10YY, has been in support of this assumption (5). Our experimental data in this study strongly support the first two stages of peptide interaction with membrane surfaces (Fig. 10, steps 1 and 2). Further investigations are required for experimental characterization of lipid-peptide porelike zones. Finally, despite their transition from tense to relaxed state upon interaction with bilayer surfaces, depending on their aromatic amino-acid residue composition, cyclic aromatic peptides preferentially interact with different regions of the bilayer and disrupt the membrane surface to different degrees (Fig. 9 A). GS prefers to penetrate into the bilayer hydrophobic interior, GS10WW prefers the bilayer interface, and GS10YY interacts mostly with the polar headgroups of the bilayer and penetrates least into the hydrophobic core. Interestingly, a solid-state 19F-NMR study on the DMPC membrane-bound structure of a 19F-labeled GS (L/P: 80:1 mol/mol), in which both leucine residues were replaced with 4F-phenylglycine, indicated that the backbone of the 19F-GS analog lay flat at the membrane surface, with its hydrophobic face buried in the bilayer hydrophobic core and its hydrophilic face interacting with the bilayer polar headgroups (34). The results of this structural study are therefore in agreement with the scheme for GS in Fig. 9 A and the second stage (Fig. 10, step 2) of our proposed mechanism. Overall, the proposed qualitative model (Fig. 10) considers the general modes of interaction of all peptides with lipid bilayers, which includes binding, conformational change, and disruption of membrane interface and structure.
In conclusion, interaction with the phospholipid components of microbial membranes may not be the only mechanism for the biological activity of GS-like antimicrobial peptides. The mechanism of biological activity of antimicrobial peptides can include specific inhibitory interactions with the components of metabolic pathways (including membrane protein receptors) and immune systems of organisms (1,47). By understanding the mechanism of interaction of GS-like molecules with biological membranes and considering interactions of these cyclic peptides with other biomolecules, such as carbohydrates (48) and components of metabolic pathways (49), a comprehensive picture of their biological activity is anticipated in future.
Acknowledgments
We thank Mr. Marc Genest for his contribution to peptide synthesis.
This study was supported by the Canada Foundation for Innovation (CFI grant No. 6786) and the Natural Sciences and Engineering Research Council of Canada (NSERC grant No. 250119) to M.J.-N., and the Protein Engineering Network of Centers of Excellence grant to R.S.H..
Editor: Paul H. Axelsen.
References
- 1.Brodgen, K. A. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238–250. [DOI] [PubMed] [Google Scholar]
- 2.Kleinkauf, H., and H. von Döhren, editors. 1990. Biochemistry of Peptide Antibiotics. Walter de Gruyter, Berlin.
- 3.Bechinger, B. 1999. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional NMR spectroscopy. Biochim. Biophys. Acta. 1462:157–184. [DOI] [PubMed] [Google Scholar]
- 4.Shai, Y. 2005. Mechanism of membrane permeation and pore formation by antimicrobial peptides. In Protein-Lipid Interactions. L. K. Tamm, editor. Wiley-VCH, Weinheim, Germany.
- 5.Jelokhani-Niaraki, M., E. J. Prenner, C. M. Kay, R. N. McElhaney, and R. S. Hodges. 2002. Conformation and interaction of the cyclic cationic antimicrobial peptides in lipid bilayers. J. Pept. Res. 60:23–36. [DOI] [PubMed] [Google Scholar]
- 6.Izumiya, N., T. Kato, H. Aoyagi, M. Waki, and M. Kondo. 1979. Synthetic Aspects of Biologically Active Cyclic Peptides—Gramicidin S and Tyrocidines. Halstead (Wiley), New York.
- 7.Waki, M., and N. Izumiya. 1990. Chemical synthesis and bioactivity of gramicidin S and related peptides. In Biochemistry of Peptide Antibiotics. H. Kleinkauf and H. von Döhren, editors. Walter de Gruyter, Berlin.
- 8.Gause, G. F., and M. G. Brazhnikova. 1944. Gramicidin S and its use in the treatment of infected wounds. Nature. 154:703b. [Google Scholar]
- 9.Schmidt, G. M. J., D. Crowfoot-Hodgkin, and B. M. Oughton. 1957. A crystallographic study of some derivatives of gramicidin S. Biochem. J. 65:744–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stern, A., W. A. Gibbons, and L. C. Craig. 1968. A conformational analysis of gramicidin S-A by nuclear magnetic resonance. Proc. Natl. Acad. Sci. USA. 61:734–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hull, S. E., R. Karlsson, P. Main, M. M. Woolfson, and E. J. Dodson. 1978. The crystal structure of a hydrated gramicidin S-urea complex. Nature. 275:206–207. [Google Scholar]
- 12.Kondejewski, L. H., S. W. Farmer, D. S. Wishart, R. E. W. Hancock, and R. H. Hodges. 1996. Gramicidin S is active against both Gram-positive and Gram-negative bacteria. Int. J. Pept. Protein Res. 47:460–466. [DOI] [PubMed] [Google Scholar]
- 13.Lee, D.L., and R.S. Hodges. 2003. Structure-activity relationships of de novo designed cyclic antimicrobial peptides based on Gramicidin S. Biopolym. Peptide Sci. 71:28–48. [DOI] [PubMed] [Google Scholar]
- 14.Jelokhani-Niaraki, M., L. H. Kondejewski, S. W. Farmer, R. E. W. Hancock, C. M. Kay, and R. S. Hodges. 2000. Diastereomeric analogues of gramicidin S: structure, biological activity and interaction with lipid bilayers. Biochem. J. 349:747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacDonald, R. C., R. I. MacDonald, B. Ph. M. Menco, K. Takeshita, N. K. Subbarao, and L.-R. Hu. 1991. Small-volume extrusion apparatus for preparation of large unilamellar vesicles. Biochim. Biophys. Acta. 1061:297–303. [DOI] [PubMed] [Google Scholar]
- 16.White, S. H., and W. C. Wimley. 1999. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28:319–365. [DOI] [PubMed] [Google Scholar]
- 17.Lakowicz, J. R. 2006. Principles of Fluorescence Spectroscopy, 3rd Ed. Springer, New York.
- 18.Eftink, M. R. 1991. Fluorescence techniques for studying protein structure. In Methods for Biochemical Analysis. C. H. Suelter, editor. Wiley, New York. [DOI] [PubMed]
- 19.dos Remedios, C. G., and P. Mones. 1999. Resonance energy transfer in proteins. In Resonance Energy Transfer. D. L. Andrews and A. A. Demidov, editors. Wiley, New York.
- 20.Chen, R. F. 1967. Fluorescence quantum yields of tryptophan and tyrosine. Anal. Lett. 1:35–42. [Google Scholar]
- 21.Yamada, K., M. Unno, K. Kobayashi, H. Oku, H. Yamamura, S. Araki, H. Matsumoto, R. Katakai, and M. Kawai. 2002. Stereochemistry of protected ornithine side chains of gramicidin S derivatives: x-ray crystal structure of the bis-Boc-tetra-n-methyl derivative of gramicidin S. J. Am. Chem. Soc. 124:12684–12688. [DOI] [PubMed] [Google Scholar]
- 22.Bhatt, V.S., P. Kaur, S. Klupsch, S. Betzel, S. Brenner, and T.P. Singh. 2004. Crystal structure of a complex formed between alkaline proteinase savinase and gramicidin S at 1.5 Å resolution. PDB ID: 1tk2. http://www.rcsb.org/pdb/home/home.do.
- 23.Wiseman, T., S. Williston, J. F. Brandts, and L.-N. Lin. 1989. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179:131–137. [DOI] [PubMed] [Google Scholar]
- 24.Wieprecht, T., and J. Seelig. 2002. Isothermal titration calorimetry for studying interactions between peptides and lipid membranes. In Peptide-Lipid Interactions. S. A. Simon and T. J. McIntosh, editors. Academic Press, San Diego, CA.
- 25.Cantor, C. R., and P. R. Schimmel. 1980. Biophysical Chemistry Part I. Freeman, New York.
- 26.Abraham, T., R. N. A. H. Lewis, R. S. Hodges, and R. N. McElhaney. 2005. Isothermal titration calorimetry studies of the binding of the antimicrobial peptide gramicidin S to phospholipid bilayer membranes. Biochemistry. 44:11279–11285. [DOI] [PubMed] [Google Scholar]
- 27.Kovacs, J.M., C.T. Mant, and R.S. Hodges. 2006. Determination of intrinsic hydrophilicity/hydrophobicity of amino acid side chains in peptides in the absence of nearest neighbor or conformational effects. Biopolym. Peptide Sci. 84:283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jelokhani-Niaraki, M., E. J. Prenner, L. H. Kondejewski, C. M. Kay, R. N. McElhaney, and R. S. Hodges. 2001. Conformation and other biophysical properties of the cyclic antimicrobial peptides in aqueous solutions. J. Pept. Res. 58:293–306. [DOI] [PubMed] [Google Scholar]
- 29.Schiller, P. W. 1985. Application of fluorescence techniques in studies of peptide conformations and interactions. In The Peptides. V. Hruby, editor. Academic Press, New York.
- 30.Jelokhani-Niaraki, M., E. J. Prenner, and R. S. Hodges. 2005. Biophysical studies of cyclic cationic antimicrobial peptides. In Peptide Science 2004. Y. Shimohigashi, editor. Japanese Peptide Society, Osaka, Japan.
- 31.Seelig, J. 2004. Thermodynamics of lipid-peptide interaction. Biochim. Biophys. Acta. 1666:40–50. [DOI] [PubMed] [Google Scholar]
- 32.Bailey, A. L., and P. R. Cullis. 1997. Liposome fusion. In Current Topics in Membranes. R. M. Epand, editor. Academic Press, New York.
- 33.Rand, R. P., and V. A. Parsegian. 1992. The forces between interacting bilayer membranes and the hydration of phospholipid assemblies. In The Structure of Biological Membranes. P. Yeagle, editor. CRC Press, Boca Raton, FL.
- 34.Salgado, J., S. L. Grage, L. H. Kondejewski, R. S. Hodges, R. N. McElhaney, and A. S. Ulrich. 2001. Membrane-bound structure and alignment of the antimicrobial β-sheet peptide gramicidin S derived from angular and distance constraints by solid-state 19 F-NMR. J. Biomol. NMR. 21:191–208. [DOI] [PubMed] [Google Scholar]
- 35.Heller, T. H., A. J. Waring, R. I. Lehrer, T. A. Harroun, T. M. Weiss, L. Yang, and H. W. Huang. 2000. Membrane thinning effect of β-sheet antimicrobial protegrin. Biochemistry. 39:139–145. [DOI] [PubMed] [Google Scholar]
- 36.Katsu, T., M. Kuroko, T. Morikawa, K. Sanchika, Y. Fujita, H. Yamamura, and M. Uda. 1989. Mechanism of membrane damage induced by amphipathic peptides gramicidin S and melittin. Biochim. Biophys. Acta. 983:135–141. [DOI] [PubMed] [Google Scholar]
- 37.Huang, H. W. 2006. Molecular mechanism of antimicrobial peptides: the origin of cooperativity. Biochim. Biophys. Acta. 1758:1292–1302. [DOI] [PubMed] [Google Scholar]
- 38.Tishchenko, G. N., V. I. Andrianov, B. K. Vainstein, M. M. Woolfson, and E. Dodson. 1997. Channels in gramicidin S-with-urea structure and their possible relation to transmembrane ion transport. Acta Crystallogr. D. 53:151–159. [DOI] [PubMed] [Google Scholar]
- 39.Llamas-Saiz, A. L., G. M. Grotenberg, M. Overhand, and M. J. van Raaij. 2007. Double-stranded helical twisted β-sheet channels in crystals of gramicidin S grown in the presence of trifluoroacetic and hydrochloric acids. Acta Crystallogr. D. 63:401–407. [DOI] [PubMed] [Google Scholar]
- 40.Heitz, F., F. Kaddari, N. Van Nau, F. Verducci, R. H. Seheno, and R. L. Lazaro. 1989. Ionic pores formed by cyclic peptides. Biochimie. 71:71–76. [DOI] [PubMed] [Google Scholar]
- 41.Wu, M., E. Maier, R. Benz, and R. E. W. Hancock. 1999. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with cytoplasmic membrane of Escherichia coli. Biochemistry. 38:7235–7242. [DOI] [PubMed] [Google Scholar]
- 42.Huang, H. W. 2000. Action of antimicrobial peptides: two-state model. Biochemistry. 39:8347–8352. [DOI] [PubMed] [Google Scholar]
- 43.Weiss, T. M., L. Yang, L. Ding, A. J. Waring, R. I. Lehrer, and H. W. Huang. 2002. Two states of cyclic antimicrobial peptide RTD-1 in lipid bilayers. Biochemistry. 41:10070–10076. [DOI] [PubMed] [Google Scholar]
- 44.Mihailescu, D., and J. C. Smith. 2000. Atomic detail peptide-membrane interactions: molecular dynamics simulation of gramicidin S in a DMPC bilayer. Biophys. J. 79:1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Longo, M. L., A. J. Waring, L. M. Gordon, and D. A. Hammer. 1998. Area expansion and permeation of phospholipid bilayers by influenza fusion peptides and melittin. Langmuir. 14:2385–2395. [Google Scholar]
- 46.Brochard-Wyart, F., P. G. de Gennes, and O. Sandre. 2000. Transient pores in stretched vesicles: role of leak-out. Physica A. 278:32–51. [Google Scholar]
- 47.Yount, N. Y., and M. R. Yeaman. 2005. Immunocontinuum: perspectives in antimicrobial mechanisms of action and resistance. Protein Pept. Lett. 12:49–76. [DOI] [PubMed] [Google Scholar]
- 48.Niidome, T., H. Murakami, M. Kawazoe, T. Hatakeyama, Y. Kobashigawa, M. Matsushita, Y. Kumaki, M. Demura, K. Nitta, and H. Aoyagi. 2001. Carbohydrate recognition of gramicidin S analogs in aqueous medium. Bioorg. Med. Chem. Lett. 11:1893–1896. [DOI] [PubMed] [Google Scholar]
- 49.Mogi, T., H. Ui, K. Shiomi, S. Omura, and K. Kita. 2008. Gramicidin S identified as a potent inhibitor for cytochrome bd-type quinol oxidase. FEBS Lett. 582:2299–2302. [DOI] [PubMed] [Google Scholar]