

Abstract
Phospholipase A2 (PLA2), a common toxic component of snake venom, has been implicated in various pharmacological effects. Ecarpholin S, isolated from the venom of the snake Echis carinatus sochureki, is a phospholipase A2 (PLA2) belonging to the Ser49-PLA2 subgroup. It has been characterized as having low enzymatic but potent myotoxic activities. The crystal structures of native ecarpholin S and its complexes with lauric acid, and its inhibitor suramin, were elucidated. This is the first report of the structure of a member of the Ser49-PLA2 subgroup. We also examined interactions of ecarpholin S with phosphatidylglycerol and lauric acid, using surface plasmon resonance, and of suramin with isothermal titration calorimetry. Most Ca2+-dependent PLA2 enzymes have Asp in position 49, which plays a crucial role in Ca2+ binding. The three-dimensional structure of ecarpholin S reveals a unique conformation of the Ca2+-binding loop that is not favorable for Ca2+ coordination. Furthermore, the endogenously bound fatty acid (lauric acid) in the hydrophobic channel may also interrupt the catalytic cycle. These two observations may account for the low enzymatic activity of ecarpholin S, despite full retention of the catalytic machinery. These observations may also be applicable to other non-Asp49-PLA2 enzymes. The interaction of suramin in its complex with ecarpholin S is quite different from that reported for the Lys49-PLA2/suramin complex, where the interfacial recognition face (i-face), C-terminal region, and N-terminal region of ecarpholin S play important roles. This study provides significant structural and functional insights into the myotoxic activity of ecarpholin S and, in general, of non-Asp49-PLA2 enzymes.
INTRODUCTION
Phospholipase A2 (PLA2, enzyme classification number 3.1.1.4) enzymes constitute a large family of hydrolases that catalyze the hydrolysis of the sn-2 ester bond of phospholipids, producing free fatty acids and lysophospholipids (1–5). The PLA2 superfamily was classified into 11 groups (6). The PLA2 enzymes of groups I and II are major components of snake venom in the Elapidae and Viperidae families, respectively. Both groups exhibit Ca2+-dependent enzymatic activity, with Ca2+ binding to a Ca2+-binding loop and Asp49 (according to the numbering system of Renetseder et al. (7)). Although a majority of group II PLA2 enzymes have Asp at their position 49, this residue can occasionally be substituted with nonacidic residues such as Lys, Ser, Asn, and Arg (8–10). As expected, Asp49-PLA2 enzymes show high catalytic activity, whereas non-Asp49-PLA2 enzymes show low or no enzymatic activity. Interestingly, these non-Asp49-PLA2 enzymes exhibit strong myotoxic activity. Extensive research has been devoted to understanding the structure and function of Lys49-PLA2 enzymes. In Lys49-PLA2 enzymes, the Nɛ atom of Lys49 occupies the space of the essential Ca2+ ion in Asp49-PLA2 enzymes, resulting in a substantial loss of catalysis (11,12). Furthermore, some studies suggest that a fatty-acid molecule may lie in the hydrophobic channel, leading to the interruption of catalytic cycles (13,14). The myotoxic/cytolytic site of Lys49-PLA2 enzymes was identified using synthetic peptides (15–17). Subsequent studies using inhibition by heparin, chemical modification, and site-directed mutagenesis strongly supported the role of the C-terminal region in myotoxic activity, as reviewed in detail elsewhere (18). The positively charged patch on the surface of Lys49-PLA2 was suggested to play an active role in myotoxic activity (19,20). Ambrosio et al. (21) proposed that electrostatic interactions involving the C-terminal lysine residues play a role in the insertion of the hydrophobic “knuckle” induced by Lys122 into the membrane.
So far, most of the structural and functional studies of non-Asp49-PLA2 enzymes focused on Lys49-PLA2 enzymes. On the other hand, little is known about Ser49-PLA2 and other non-Asp49-PLA2 enzymes. Here we report on the enzymatic activity and myotoxic activity of a Ser49-PLA2 (ecarpholin S) from the venom of the snake Echis carinatus sochureki. We also determined crystal structures of ecarpholin S in its native form and in complexes with lauric acid, and of its myotoxic inhibitor, suramin. This is the first representative structure of the Ser49-PLA2 subgroup. Moreover, the mechanism of action of Ser49-PLA2 is elucidated, using structural data combined with biochemical and biophysical studies.
EXPERIMENTAL PROCEDURES
Materials
Ecarpholin S (belonging to the Ser49-PLA2 subgroup) was isolated from Echis carinatus sochureki snake venom (Latoxan, Rosans, France) by following the protocol of Polgar et al. (22), with minor modifications. Lyophilized venom was dissolved at 100 mg/mL in 50 mM Tris-HCl buffer, pH 7.0. Soluble components were loaded onto a Superdex 200 gel-filtration column (1.0 × 60 cm, Amersham Biosciences, Uppsala, Sweden) eluted with the same buffer. The sixth peak, estimated to have a molecular mass of ∼14 kDa, was loaded onto an SP Sepharose High Performance cation-exchange column (5 mL, Amersham Biosciences). Ecarpholin S was eluted as a main peak from this column. We isolated ScPLA2 (belonging to the Asp49-PLA2 subgroup) from E. c. sochureki venom according to the protocol of Jasti et al. (23). Both isolated proteins were confirmed by mass spectrometry and N-terminal sequencing. Suramin (8,8′-[carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-1,3,5-naphthalenetrisulfonic acid hexasodium), phosphatidyglycerol (from egg yolk), and lauric acid were obtained from Sigma Chemical Co. (St. Louis, MO). All other reagents were of analytical grade.
Phospholipase enzymatic activity
The enzymatic activity of PLA2 was measured by routine colorimetric assay, using a 1,2-dithio analog of diheptanoylphosphatidylcholine as substrate (PLA2 assay kit, Cayman Chemicals, Ann Arbor, MI). The ScPLA2 from E. c. sochureki served as positive control.
Myotoxic activity
Myotoxic activity is the damage to skeletal muscle leading to the release of cellular contents and the death of muscle cells. A number of snake venom proteins were shown to induce myotoxicity (18). Groups of five Swiss albino mice (18–22 g body weight) were injected (intramuscularly to the right gastrocnemius muscle) with 50, 75, or 100 μg of ecarpholin S in 50 μL of phosphate-buffered saline (PBS). After 3 h, the mice were anesthetized, and their blood was collected via heart puncture. The creatine kinase (CK) activity of serum was then measured using a kit (No. 2910) from Stanbio Laboratory (Boerne, Texas). One unit was defined as the amount of enzyme that produces 1 mmol of NADPH/min, and activity was expressed in units/liter. In inhibition studies, different doses of suramin were incubated with ecarpholin S (100 μg/mouse) for 15 min at 37°C before administration to animals.
Edema-inducing activity
The accumulation of fluid in the interstitial space and swelling during the inflammatory response are measured as edema. The PLA2 from snake venom is a potent mediator of edema (5,24). The edema-inducing effects of ecarpholin S were measured in mice, as described earlier (25). Different doses of ecarpholin S were dissolved in 20 μL of PBS (pH 7.5) and administered via subplantar injection into the left footpad of Swiss albino mice under anesthesia. After 1 h, while still under anesthesia, mice were sacrificed by inhalation of carbon dioxide. Both their feet (injected and uninjected) were severed just above the plantar joint, and weighed. The percentage of edema was calculated from the difference in weight between injected and uninjected feet, with PBS as the control.
Isothermal titration calorimetry (ITC)
Ecarpholin S titrations with suramin were performed on a VP-ITC calorimeter system (MicroCal LLC, Northampton, MA) at 30°C. Solutions of 0.03 mM of ecarpholin S in 50 mM Tris-HCl (pH 7.4) were titrated with 0.5 mM suramin dissolved in the same buffer. The reaction cell contained 1.5 mL of the protein solution in buffer. Suramin was added in 5-μL increments at 500-s intervals. Injections of suramin solution in the same buffer, but in the absence of protein, were used to account for the heats of mixing and dilution. Binding parameters were estimated from isotherms obtained using scripts (Origin 7.0, OriginLab, Northampton, Massachusetts) developed in our laboratory.
Surface plasmon resonance
Surface plasmon resonance (SPR) studies were performed with Biacore 3000 (Biacore AB, Uppsala, Sweden). We prepared phosphatidyglycerol (PG) liposomes by sonication, as previously described (26). Briefly, an appropriate amount of PG (2 mg) was dissolved in 2 mL of a chloroform solution, and the organic solvent was subsequently removed by vacuum desiccation. The resultant dried lipid film was rehydrated with PBS (pH 7.4) at the appropriate concentration at 25°C for 2 h. The resulting aqueous lipid mixture was sonicated until the solution appeared optically transparent in white light. A homogeneous liposomal suspension of approximately uniformly sized unilamellar vesicles with an average diameter (from dynamic light-scattering measurement) of 45 ± 7 nm was obtained. We performed immobilization of the PG liposome on the L1 chip, as described by the manufacturer. Briefly, PBS (pH 6.8) was used as the running buffer, the washing solution was 40 mM N-octyl β-D-glucopyranoside, and the regenerating solution was 10 mM NaOH. Experiments were performed at 25°C. Phosphatidyglycerol unilamellar vesicles (100 μL, 1 mM) were applied to the chip surface at a low flow rate (2 μL/min). To remove any multilamellar structures from the lipid surface, we injected NaOH (50 μL, 10 mM) at a flow rate of 50 μL/min, which resulted in a stable baseline corresponding to the lipid bilayer linked to the chip surface. The negative-control bovine serum albumin was injected (50 μL, 0.1 mg/μL in PBS) to confirm complete coverage of nonspecific binding sites. Ecarpholin S was diluted into a suitable final concentration, using the running buffer. All sensorgrams were processed by using automatic correction for nonspecific bulk refractive index effects. The equilibrium constant (KD) was determined by the 1:1 Langmuir binding fitting model provided by the Biacore 3000 instrument software.
To study the interaction between ecarpholin S and lauric acid, ecarpholin S was immobilized on the sensor chip CM5 (Biacore AB) by the standard primary amine-coupling reaction. The equilibration of baseline was completed by the continuous flow of HBS-EP running buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, and 0.005% (v/v) surfactant P20, pH 7.4) through the chip. One of the four serial flow cells was activated for 10 min by injecting a 1:1 fresh mixture of 0.2 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 50 mM N-hydroxysuccinimide (NHS) at 25°C. Ecarpholin S was diluted with 10 mM HEPES buffer (pH 7.5) at a concentration of 50 μg/mL, and immobilized to the surface of sensor chip CM5. Finally, unreacted groups were blocked by injecting 1 M ethanolamine-HCl at pH 8.5 for 7 min, resulting in immobilized densities of 3500 RU. After stabilizing the baseline, Biacore data were collected at 25°C at a constant flow of 20 μL/min. Lauric acid was diluted into a suitable final concentration, using the running buffer. We used 70% ethanol as regeneration buffer. The equilibrium constant (KD) was determined by the 1:1 Langmuir binding fitting model.
Crystallization and data collection
Ecarpholin S was crystallized by hanging-drop vapor-diffusion method using screens from Hampton Research (Aliso Viejo, CA). All crystallization experiments were performed at room temperature, with drops containing equal volumes (1 μL) of reservoir and protein solution. The protein was kept in a buffer of 50 mM Tris-HCl, pH 7.4, at a concentration of 10 mg/mL. The apo ecarpholin S crystal belonged to the space group P212121 and was grown from a condition of 20% w/v polyethylene glycol (PEG) 3350, 20% v/v iso-propanol, and 0.1 M sodium citrate, pH 5.6. Crystals of natural ecarpholin S/fatty-acid complex were crystallized in the presence of 2 M ammonium sulfate, 0.1 M Tris-HCl, pH 8.5, and belonged to the space group P3121. Ecarpholin S/suramin complex crystals were obtained using 10% w/v PEG3350, 10% v/v iso-propanol, and 0.1 M sodium citrate, pH 5.6, with a molar ratio of suramin/protein at 3:2. These crystals belonged to the space group P21. Before the collection of x-ray diffraction data, crystals were soaked in increasing steps of glycerol concentration (5% increase in each step; 30 min per step), and the final glycerol concentration was ∼20% before flash-freezing using liquid N2. The x-ray diffraction data for the apo and suramin complex were collected using an R-axis IV2+ image plate detector mounted on a RU-H3RHB rotating anode generator (Rigaku Corp., Tokyo, Japan). The data for the fatty-acid complex were collected at the X29 beamline (National Synchrotron Light Source (NSLS), Brookhaven National Laboratory, New York), using a Q315 CCD detector (Area Detector Systems Corp., Poway, CA). All data sets were processed using the HKL2000 suite (27). Crystallographic statistics are shown in Table 1.
TABLE 1.
X-ray data collection and refinement statistics
| Apo-ecarpholin S | Ecarpholin S-lauric acid complex | Ecarpholin S-suramin complex | |
|---|---|---|---|
| Data collection | |||
| Cell parameters (Å) | a = 34.23, b = 39.48, c = 68.94 | a = b = 62.09, c = 124.25 | a = 51.45, b = 132.25, c = 86.10; β = 99.32 |
| Space group | P212121 | P3121 | P21 |
| Molecules/asymmetry unit | 1 | 2 | 8 |
| Resolution range (Å) | 50–2.0 | 50–1.95 | 50–2.2 |
| Wavelength (Å) | 1.5418 | 0.97 | 1.5418 |
| Observed reflections | 32,276 | 214,445 | 186,658 |
| Unique reflections | 6,463 | 20,851 | 55.923 |
| Completeness (%) | 95.7 (87.4) | 99.7 (100) | 97.5 (86.6) |
| Rsym (%)* | 0.051 | 0.050 | 0.089 |
| I/σ (I) | 24.7 (9.1) | 21.5 (2.4) | 10.8 (2.3) |
| Refinement and quality | |||
| Resolution range (Å) | 20–2.0 | 30–1.95 | 30–2.3 |
| Rwork (%)† | 19.2 | 22.8 | 21.7 |
| Rfree (%)‡ | 25.5 | 26.8 | 27.5 |
| rmsd bond lengths (Å) | 0.005 | 0.006 | 0.013 |
| rmsd bond angles(deg) | 1.3 | 1.3 | 1.6 |
| Average B-factors (Å2)§ | 30.5 | 29.1 | 33.7 |
| Main-chain | 28.4 | 26.3 | 31.9 |
| Side-chains | 31.3 | 30.0 | 33.2 |
| Waters | 38.0 | 35.4 | 32.0 |
| Lauric acid | 50.2 | ||
| Suramin chains | 36.4 | ||
| Ramachandran plot (%) | |||
| Most favored regions | 90.4 | 89.4 | 85.9 |
| Additional allowed regions | 9.6 | 10.6 | 13.9 |
| Generously allowed regions | 0 | 0 | 0.1 |
| Disallowed regions | 0 | 0 | 0 |
| Number of protein atoms | 959 | 1918 | 7672 |
| Number of ligand atoms | 0 | 28 | 1031 |
| Number of waters | 96 | 243 | 370 |
| Protein Data Bank code | 2QHE | 2QHD | 3BJW |
Rsym = ∑|Ii − 〈I〉|/∑|I|, where Ii is the intensity of the ith measurement, and 〈I〉 is the mean intensity for that reflection.
Rwork = ∑|Fobs–Fcalc|/∑|Fobs |, where Fcalc and Fobs are the calculated and observed structure factor amplitudes, respectively.
Rfree = Rwork, but for 10% of the total reflections randomly selected and omitted from refinement.
Individual B-factor refinement was performed.
Structural determination and refinement
Initial phases for apo ecarpholin S were obtained by the molecular replacement method with Molrep (28), using the Asp49-PLA2 structure from Agkistrodon halys Pallas as a search model (Protein Data Bank code 1JIA; sequence identity, ∼56%). The initial Rwork was 52%, and subsequent auto-model building was performed with ARP/wARP (29). The resultant model with the electron-density map was examined, and the model was built with the O program (30). Structures of the lauric acid and suramin complexes were determined using the apo ecarpholin S structure as the initial search model for the molecular replacement method, using the program Molrep (28). Omit maps were calculated for positioning the ligand. All three model-buildings and refinements were performed using O (30) and CNS (31) programs, with appropriate entries in their respective dictionaries. The overall geometry of the final models was analyzed by PROCHECK (32). Refinement statistics are given in Table 1.
RESULTS
Characterization of ecarpholin S
Ammodytin L, the first known Ser49-PLA2 isolated from the snake venom of Vipera ammodytes, was initially reported to exhibit low enzyme activity (33). However, this activity was later attributed to contamination with ammodytoxins (34). Polgar et al. (22) reported that Ser49-PLA2 ecarpholin S had 1.5–2.9 times more enzymatic activity than the recombinant human group II PLA2. Our study shows that ecarpholin S has enzymatic activity (0.1 μmol/min/mg). However, compared with the activity of scPLA2, an Asp49-PLA2 from the same venom, ecarpholin S shows only 0.5% activity (24.5 μmol/min/mg; see Fig. S1 in Supplementary Material, Data S1). Thus it is tempting to conclude that the activity of ecarpholin S is similar to that of some Lys49-PLA2 enzymes, which have low catalytic ability (35–41).
Ecarpholin S is a basic PLA2 (calculated pI = 8.4), with several conserved, positively charged residues (particularly lysine) that are thought to be important in the myotoxicity of low molecular mass PLA2 enzymes. Hence ecarpholin S was predicted to have myotoxic activity (22,42). Our results confirmed that ecarpholin S does exhibit myotoxic activity. Intramuscular injections of ecarpholin S increased serum creatine kinase (CK) levels in a dose-dependent manner (Fig. 1). The injection of ecarpholin S at a dose of 100 μg/mouse induced a significant increase in serum CK activity from 160 (± 40) units/L (mean ± SD, n = 5) to 1900 (± 400) units/L (n = 5), compared with physiological saline solution (PBS, pH 7.3). Injecting ecarpholin S into the footpad of mice induced mild edema. At 20 μg/mouse, ecarpholin S induced ∼20% edema compared with PBS-injected control mice (Fig. S2 in Data S1).
FIGURE 1.

Effects of ecarpholin S on levels of creatine kinase (CK) in mouse serum. Various doses of ecarpholin S (0 μg of ecarpholin S represents 50 μL of PBS buffer), with or without suramin, were injected into gastrocnemius muscles of mice, 3 h before their blood was collected. Released CK in plasma was measured, and results are presented as average ± SD (n = 5).
Structure of apo ecarpholin S
Although structures of a number of Asp49 and Lys49 PLA2 enzymes are known, to date, no structure has been available for Ser49 PLA2 enzymes. Therefore, we determined the structure of ecarpholin S. The structure of its apo-form was solved by the molecular replacement method, using the Asp49-PLA2 structure from Agkistrodon halys Pallas as a starting model, and refined to an Rwork of 19.2% (Rfree = 25.4%) up to 2.0-Å resolution. The model was refined with good stereochemical parameters (Table 1). Statistics for the Ramachandran plot from an analysis using PROCHECK (32) gave 90.4% of nonglycine residues in the most favored region, and no residues in the disallowed region.
The overall structure of ecarpholin S resembles that of classic group II PLA2 enzymes. The superimposition of ecarpholin S with a classic group II PLA2 (ScPLA2; Protein Data Bank code 1OZ6) gave a root mean-square deviation (rmsd) of 1.2 Å for 120 Cα atoms. Thus we will provide only a general overview of the structure and the subtle differences between ecarpholin S and other PLA2 enzymes. Ecarpholin S consists of an N-terminal α-helix (α1), a loop equivalent to the Ca2+-binding loop, two long antiparallel α-helices (α2 and α3), the short two-stranded antiparallel β-sheet (β-wing), and the C-terminal loop region. The sequence of ecarpholin S contains 14 cysteine residues, forming seven disulfide bonds. The pattern of disulfide bridges is identical to that of classic group II PLA2 (5). Other main structural features include highly conserved catalytic residues (His48, Tyr52, and Asp99) and a hydrophobic channel (formed by residues such as Val2, Leu5, Ile9, Pro18, Tyr22, and Gly30).
The Ca2+-binding loop in Asp49-PLA2 is highly conserved (Tyr28, Gly30, Gly32, and Asp49; Fig. 2). In Lys49-PLA2, the Nɛ of Lys49 was shown to occupy the position of catalytically essential calcium, and thus reduces catalytic activity (12). In ecarpholin S, Asp49 is replaced by Ser, and Tyr28 in the Ca2+-binding loop is replaced by Phe. Although ecarpholin S partially retains its enzymatic activity, this activity is significantly lower than that of Asp49-PLA2. Fig. 3 shows the superimposition of the Ca2+-binding loop of ecarpholin S with scPLA2, an Asp49-PLA2 from the same venom, and myotoxin II, a Lys49-PLA2 from Bothrops asper venom. The ScPLA2 is a classic Asp49-PLA2, with Ca2+-binding at the highly conserved Ca2+-binding loop. In the case of myotoxin II, the long side-chain of Lys49 extends into the Ca2+-binding loop and destabilizes Ca2+-binding. The previous homology modeling studies of ecarpholin S suggested that serine is a potential candidate for replacing Asp49 without significantly affecting the Ca2+-binding capacity (22). But this model failed to predict the unique conformation of the Ca2+-binding loop (see below). However, in the ecarpholin S structure (Fig. 3), no calcium ion was found in the putative Ca2+-binding loop. A stretch of three residues (Gly30-Trp31-Gly32) of ScPLA2 essentially bends to form a coordination bond with Ca2+. In the case of ecarpholin S, these three residues (Gly30-Gly31-Gly32) are linearly extended, and cause the carbonyl O of Gly32 to be moved away (∼5 Å) from the putative Ca2+-binding position. In addition, the whole loop moves closer to the Ser49 residue, and the Nα of Gly31 nearly occupies the putative Ca2+-binding position (1.1 Å away from the Gly31 N). The position of Oγ of Ser49 of ecarpholin S does not occupy the position of Oδ of Asp49 of Asp49-PLA2 enzymes. The side-chain of the Ser49 residue of ecarpholin S is too short to extend into the Ca2+-binding site (Oγ of Ser49 is 3.8 Å away from the putative Ca2+-binding site), unlike the Nɛ of Lys49 in Lys49 PLA2 (Fig. 3). Furthermore, no water molecule was observed near the putative Ca2+-binding site of ecarpholin S. The nearest water molecule is ∼3 Å away from the Ca2+-binding site, and thus no alternative electron density can be modeled as Ca2+. These observations strongly suggest that the conformation of the Ca2+-binding loop in ecarpholin S is different from that of Asp49-PLA2 enzymes and Lys49-PLA2 enzymes (Fig. 3), and the putative Ca2+-binding loop in ecarpholin S is not favorably placed to coordinate the Ca2+ ion, which partially explains its lower enzymatic activity than that of Asp49-PLA2. An attempt to cocrystallize ecarpholin S with 100 mM calcium chloride did not result in the incorporation of calcium ions in the Ca2+-binding loop. Binding studies using ITC showed that Ca2+ did not bind to ecarpholin S (Fig. S3 in Data S1). No calcium ion was found at the putative Ca2+-binding loops of lauric acid and suramin complexes of ecarpholin S (see below). Thus Ser49 and the putative Ca2+-binding loop of ecarpholin S do not support the binding of Ca2+ ions.
FIGURE 2.

Sequence alignment for PLA2 and chemical structure of suramin. (A) Sequence comparison of ecarpholin S and other PLA2 enzymes from snake venom. Identical residues are in red, nonsimilar residues are in black, and conserved residues are in blue. (B) Chemical structure of suramin.
FIGURE 3.

Stereo diagram of superposition of Ca2+-binding loop of PLA2 enzymes. ScPLA2 (Protein Data Bank code, 1OZ6; blue), ecarpholin S (red), and myotoxin II (Protein Data Bank code, 1CLP; yellow) were superimposed. Conformational differences of three different Ca2+-binding loops are shown. Three residues Gly30-Trp31-Gly32 of scPLA2 bend to form a coordination bond with Ca2+, whereas three residues Gly30-Gly31-Gly32 of ecarpholin S extend linearly, causing Gly32 carbonyl O to move away from possible Ca2+ area (∼5 Å). The whole loop moves closer to Ser49 residue, and Nα of Gly31 nearly occupies putative Ca2+-binding position (1.1 Å away from Gly31 N), whereas in myotoxin II, Nɛ of Lys49 residue occupies Ca2+ position. The side-chain of Ser49 residue of ecapholin S is too short to extend into Ca2+-binding site (Oγ of Ser49 is 3.8 Å away from putative Ca2+-binding site). Image was prepared using the program Pymol (59).
It is important to note that Ward et al. (43) carried out a mutational study of Lys49 back to Asp, using Lys49-PLA2 bothropstoxin I. This mutant was still catalytically inactive, demonstrating that the single Asp49 replacement with lysine alone was not important for the lack of enzymatic activity of this protein. Other features may also be important. In the case of ecarpholin S, Ser49 is not ideal in terms of coordinating with the calcium ion. Moreover, the whole Ca2+-binding loop moves closer to the Ser49 residue, and the Gly31 N nearly occupies the putative Ca2+ binding position. In the case of the Asp49-PLA2 subgroup, such as ScPLA2, the Tyr28 side-chain O hydrogen bonded with the Gly35 backbone nitrogen atom (Fig. S4 in Data S1). In ecarpholin S, Tyr28 was replaced by Phe, and no hydrogen bond was observed between Gly35 and Phe28. In the Asp49-PLA2 subgroup, Tyr28 is highly conserved, and this hydrogen bond appears to play an important role in stabilizing the Ca2+-binding loop. In addition, Gly33 is a highly conserved residue in Asp49-PLA2, with a few exceptions, such as Lys33 in CRV-W6D49 from Callosellasma rhodostoma (44), Asn33 in textilotoxin subunit C, and Ser33 in the D subunit of textilotoxin from Pseudonaja textilis (45). Interestingly, all of these exceptions show no enzyme activity. Thus Gly33 could be another critical residue for PLA2 enzyme activity, although it is not directly involved with the coordination bonding with Ca2+. In scPLA2, Gly30-Trp31-Gly32 bends into a loop to form coordination bonds with Ca2+, and Gly33 moves closer to the Ca2+ binding site. Replacing Gly33 with a residue with a side chain, such as Lys in the case of CRV-W6D49 and Ser in textilotoxin, could abolish the movement of these residues toward the Ca2+ binding site because of steric clashes. In ecarpholin S, Glu33 occupies the Gly33 position. Thus we speculate that the point mutation of Ser49Asp alone in ecarpholin S may not restore its enzyme activity.
All catalytic residues are conserved in ecarpholin S. Critical residues of the catalytic triad of ecarpholin S (His48, Tyr52, and Asp99) are anchored tightly to the scaffold composed of two α helices, and superimpose with corresponding residues of two structural homologues from Asp49 and Lys49-PLA2 enzymes with nearly identical conformations (rmsd of 0.5 Å) (Fig. S4 in Data S1). The hydrophobic channel that provides access to the catalytic site is also highly conserved. Therefore, we propose that the nonideal conformation of the Ca2+-binding loop is responsible for the low enzyme activity of ecarpholin S.
The structure of ecarpholin S shows several positively charged patches on the surface of the molecule (Fig. S5 in Data S1) that may interact with negatively charged counterparts on the membrane, to initiate the process of interrupting the integrity of the cell. For example, a Lys49-PLA2 such as myotoxin I was shown to interrupt negatively charged liposomes in vitro (46). To understand these interactions further, we studied the binding between PG and ecarpholin S, using SPR (Fig. 4). Phosphatidyglycerol was immobilized to the surface of the L1 chip in the experiment, and ecarpholin S was the analyte. Sensorgrams show that PG binds to ecarpholin S in a dose-dependent manner. The dissociation constant (KD) calculated from these sensorgrams was 0.5 μM.
FIGURE 4.

Studies of binding of phophatidyglycerol (PG) and lauric acid to ecarpholin S, using SPR. Concentration-dependent binding and real-time binding affinity measurement of phophatidyglycerol (A) and lauric acid (B) to ecarpholin S used Biacore 3000. Analyte was injected for 3 min, and dissociation was monitored for more than 15 min. Reference and blank data are subtracted. (A) Analyte concentrations were 20 nM, 38 nM, 75 nM, 150 nM, and 300 nM. (B) Analyte concentrations were 10 μM, 20 μM, 30 μM, 40 μM, and 50 μM.
Structure of ecarpholin S/lauric acid complex
Our crystallization experiments showed that ecarpholin S can be crystallized in different conditions. The structure of ecarpholin S in P3121 crystal form was determined at a 1.95-Å resolution (Rwork = 22.8%, and Rfree = 26.8%). The model was refined with good stereochemical parameters (Table 1). During structural refinement, the hydrophobic channel of ecarpholin S showed an extra electron density to accommodate ∼12 atoms. Because the crystallization conditions did not contain polyethylene glycol, and considering the extra electron density location, size, and geometry, this electron density was modeled as lauric acid (Fig. 5 A), although no lauric acid was added during the experiments. However, no direct evidence is available to confirm the presence of lauric acid, and the assignment of the electron density to this molecule must be regarded with caution. We do not rule out the possibility of the binding of some other metabolite at this site. Moreover, in the presence of isopropanol (10–20%) in the crystallization conditions of the apo and suramin ecarpholin S complex (see below), no lauric acid was evident. Probably the naturally bound lauric acid was removed by the organic solvent present in the crystallization conditions, whereas the lauric-acid complex was crystallized with 2 M ammonium sulfate, and because of the high hydrophobic interactions, the lauric acid was retained in the hydrophobic channel of ecarpholin S.
FIGURE 5.

Fo-Fc map shows ligand in ecarpholin S structure. (A) Fo-Fc map in hydrophobic channel of ecarpholin S. Bound lauric-acid molecules were omitted before refinement and map calculation. Map was contoured at a level of 2σ. (B) Fo-Fc map for suramin complex. Inhibitor suramin molecules were omitted before refinement and map calculation. Map was contoured at a level of 2σ. All suramin molecules of asymmetric unit have the equivalent quality of electron density; one representative molecule is shown here. Images were prepared using the program Pymol (59).
The lauric acid was well-defined in the electron-density map (Fig. 5 A). No calcium ion was identified at the putative Ca2+-binding loop. The overall structure of ecarpholin S in the complex was similar to that of apo-ecarpholin S in P212121 crystal form. The bound ligand undergoes hydrophobic interactions with several residues of the hydrophobic channel such as Val2, Leu5, Gly6, Phe18, Cys45, and Tyr52. The acidic head is stabilized by hydrogen-bonding contact with the Nδ1 atom of His48, whereas the tail of the ligand extends into the hydrophobic channel. The presence of such a natural fatty-acid molecule was previously reported in the crystal structures of three different Lys49-PLA2 enzymes (13,14,21).
The superposition of the ecarpholin S/lauric acid complex on the known fatty-acid complexes indicates that all ligands are in the same region of PLA2 enzymes (Fig. 6), whereas the conformation of the alkyl tail is different. The alkyl tail of lauric acid of the ecarpholin S complex is close to the i-face, whereas the tails of the other three fatty acids take different orientations, closer to the N-terminal region. These three fatty acids are longer, and are in generally extended conformations. The observed electron density from Fo-Fc maps and simulated annealing omit maps did not permit us to model our ligand with an extended conformation. The ecarpholin S-bound lauric acid could be the result of phospholipid hydrolysis by ecarpholin S, followed by a failure of product release. Alternatively, it could be a transient fatty acyl intermediate in the catalytic cycle that leads to the release of fatty acid in the next step.
FIGURE 6.

Superimposition of lauric acid in hydrophobic channel of ecarpholin S and three Lys49-PLA2 enzymes. Structure of MjTX-II/stearic acid (Protein Data Bank code, 1XXS, red), PrTX-II/fatty acid (Protein Data Bank code, 1QLL, blue), ACL/lauric acid (Protein Data Bank code, 1S8G, yellow), and ecarpholin S/lauric acid (cyan) complexes were superimposed. Lauric acid molecules are shown as lines. Image was prepared using the program Pymol (59).
The Fo-Fc and omit maps of the apo and ecarpholin S/suramin complex (see below) were carefully examined for the presence of any PEG or fatty-acid molecules in the hydrophobic channel, because both conditions used for crystallization contained PEG3350. In these structures, no electron densities could be modeled as either a fatty acid or a PEG molecule. It is important to note that the crystals for both the apo-enzyme and the lauric acid complex were obtained from the same batch of protein; the only difference involved their crystallization conditions.
Watanabe et al. (47) obtained the MjTX-II/stearic acid complex from a cocrystallization experiment. In the hydrophobic channel of MjTX-II, a second stearic acid was observed in the vicinity of the first stearic acid, as shown in Fig. 6. In the case of the ecarpholin S/lauric acid complex, no electron density was observed to model the second fatty acid or other polymeric molecules in the corresponding location. This result further suggests that lauric acid could be a natural ligand for ecarpholin S, specifically binding at one location of the hydrophobic channel. Thus, this complex appears to be different from the in vitro MjTX-II/stearic acid complex (47).
To understand these interactions further, we studied the interaction between lauric acid and ecarpholin S, using SPR (Fig. 4). Sensorgrams showed that lauric acid binds to ecarpholin S in a dose-dependent manner, exhibiting characteristic slow-binding and slow-dissociation curves. The dissociation constant (KD) calculated from these sensorgrams was 4.2 μM.
Structure of ecarpholin S/suramin complex
Suramin, a highly charged polysulfonated compound, inhibits the myonecrosis caused by some snake venoms and Lys49-PLA2 enzymes (20,48–50). Similarly, suramin significantly inhibits the myotoxic activity of ecarpholin S (Fig. 1). To understand these interactions, we determined the crystal structure of the ecarpholin S/suramin complex. Data for this complex were collected at a 2.3-Å resolution, and the structure was solved and refined to an Rwork of 21.7% (Rfree = 27.5%). The model was refined with good stereochemical parameters (Table 1). Suramin molecules were well-defined in the electron-density map (Fig. 5 B). No calcium ion was identified at the putative Ca2+-binding loop. The asymmetric unit consists of eight ecarpholin S monomers, with 12 suramin molecules (3:2 inhibitor/protein ratio). The eight monomers could be divided into four dimers, each sandwiching three suramin molecules (Fig. 7). The conformations of all eight monomers were nearly identical. The superimposition of apo ecarpholin S and the ecarpholin S/suramin complex gave an rmsd in the range of 0. 9–1.0 Å for all chains. Similarly, the rmsd for the lauric-acid complex molecule A (or B) and suramin complex was in the range of 0.6–0.8 Å for each molecule. Thus, the complex formation with suramin did not result in any significant conformational changes in the protein.
FIGURE 7.

Stereo diagram of comparison of suramin bound to ecarpholin S and Basp-II. (A) Three suramin molecules are sandwiched between monomer A and monomer B of ecarpholin S. (B) In Basp-II/suramin complex (Protein Data Bank code, 1Y4L), only one suramin molecule is located between the two monomers. Image was prepared using the program Pymol (59).
In the ecarpholin S/suramin complex, the entrance of the hydrophobic channel of monomers A and B of the dimer are occupied by three suramin molecules through hydrogen-bonding and hydrophobic interactions. Most residues that form hydrogen bonds with suramin are located in the i-face (Fig. 8 A). Suramin also interacts with the C-terminal region of ecarpholin S. In addition, Asn114, Lys115, and Lys116 of monomer A form several hydrogen-bonding contacts with suramin (Fig. 8 B). Suramin forms interactions with the N-terminal helix of ecarpholin S (Fig. 8 C). Overall, the suramin-recognition sites of ecarpholin S are mainly from the i-face, C-terminal, and N-terminal regions through hydrogen-bonding and hydrophobic interactions. In contrast, in the Lys49-PLA2 Basp-II/suramin complex, suramin-recognition sites are located near the putative Ca2+-binding loop or on the side of the i-face (Arg34, Lys53, and Lys69) (Figs. 7 and 8 D). In this case, suramin interacts with Basp-II, with a stoichiometric ratio of 1:1. Thus, in terms of the stoichiometric ratio and the interaction site, the ecarpholin S/suramin complex is different from that of the Basp-II/suramin complex.
FIGURE 8.

Comparison of interaction region of suramin with ecarpholin S and Basp-II. (A) Suramin interacts with i-face region of ecarpholin S. (B) Suramin interacts with C-terminal region of ecarpholin S. (C) Suramin interacts with N-terminal region of ecarpholin S. (D) Suramin interacts with putative Ca2+-binding loop region of Basp-II (Protein Data Bank code, 1Y4L). Hydrogen bonds are shown in green (dashed lines). Images were prepared using the program Pymol (59).
To test whether this observed difference is attributable to the ratios between suramin and ecarpholin S, we crystallized the complex at various suramin/protein ratios. The crystal complex structure remained the same at the 3:2 ratios (inhibitor/protein). In all crystal structures obtained, suramin molecules were fully occupied in all positions in the asymmetric unit described above. We think that the surplus protein present in the solution did not affect the crystallization of the ecarpholin S /suramin complex.
In the crystal structure, the ecarpholin S/suramin complex forms dimers as the minimal unit, and four dimers (octamers) are found in the asymmetric unit. This is consistent with gel-filtration experiments. In the gel filtration, the ecarpholin and suramin mixture eluted as a dimer (Fig. 9), whereas ecarpholin S alone eluted as a monomer. This shows that suramin functions as a linker molecule between the monomers, and induces the dimerization of ecarpholin S, as revealed by the crystal structure.
FIGURE 9.

Gel-filtration studies on oligomerization of ecarpholin S induced by suramin. (A) Elution profiles of ecarpholin S without suramin. Column is Superdex 200 (Amersham Biosciences). (B) Elution profiles of ecarpholin S with suramin (ecarpholin S/suramin = 1:1, molar ratio). (C) Elution profiles of ecarpholin S with suramin. (ecarpholin S/suramin = 1:2, molar ratio). (D) Column calibrated with gel-filtration standard (1.35–670 kDa from Bio-Rad Laboratories, Hercules, California.
The interaction of suramin with ecarpholin S was further examined by preliminary ITC experiments. The binding isotherm and nonlinear least-squares analysis of the data are shown in Fig. 10. Fig. 10, top shows the heat effect associated with successive injections until saturation is reached. Fig. 10, bottom shows the integrated heat effects associated with each injection. In the analysis, the concentration of ecarpholin S was considered on the basis of monomers. The titration was nonmonotonic, exhibiting a biphasic shape that does not correspond with stoichiometries for independent binding sites. This biphasic shape is not considered an artifact, because it always appeared and can be accounted for with the binding model used in the analysis. Moreover, blank titrations that injected ligand into the buffer yielded constant heat effects.
FIGURE 10.

Interaction between ecarpholin S and suramin, using ITC. Raw thermal power signal (top) and plot of integrated heat versus ligand/protein molar ratio (bottom). Conditions: 0.03 mM ecarpholin S titrated with 5-μL injections of 0.5 mM suramin. Both protein and suramin solutions were in 50 mM Tris-HCl at pH 7.0 and 30°C. In the analysis, protein concentration was considered on a monomer basis (0.03 mM). Titration curve was analyzed using a fully cooperative model in which n ligand molecules (n = 2, 3) bind to a protein dimer. Thermodynamic binding parameters obtained are: K = (2.1 ± 0.6) × 1020 (M−4), ΔH = −27 ± 1 kcal/mol, N = 0.69 ± 0.05 for n = 3 (dashed line); and K = (1.9 ± 0.3) × 1014 (M−3), ΔH = −26.7 ± 0.8 kcal/mol, N = 1.09 ± 0.05 for n = 2 (solid line).
Because the gel-filtration and x-ray diffraction results indicated that suramin promotes dimerization, and the stoichiometry is 3:1 (suramin/dimer), the simplest way to analyze titration is to consider a fully cooperative two-state model in which n ligand molecules bind to a dimer at once. Then, only two states are present: a free protein monomer, and a dimer with n bound ligands. The chemical equilibrium scheme for this model, as been outlined in the Appendix, is:
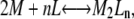 |
(1) |
where the dimerization energetics are implicitly included in the binding energetics, and no intermediate states appear. Thus, the global-equilibrium constant will be a combination of the equilibrium constant for ligand binding, KL, and the equilibrium constant for dimerization, Kd:
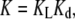 |
(2) |
and the global enthalpy will be a combination of the ligand-binding enthalpy, ΔHL, and the dimerization enthalpy, ΔHd:
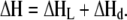 |
(3) |
If it is assumed that an ecarpholin dimer binds three ligand molecules (n = 3), the nonlinear analysis yields the following values: K = (2.1 ± 0.6) × 1020 (M−4), ΔH = −27 ± 1 kcal/mol, N = 0.69 ± 0.05. The theoretical curve is able to reproduce the experimental curve reasonably well, including the initial part of the titration where the heat effect slightly increases. Because the parameter N corrects for the effective concentration of protein, there are two possible explanations for N not being equal to unity: (1), assuming that the suramin concentration is accurately determined, only 70% of the protein is binding-competent; or (2), less than three suramin molecules bind per dimer under the conditions and concentrations used in the ITC experiments.
If it is assumed that an ecarpholin dimer binds two ligand molecules (n = 2), the nonlinear analysis yields the following values: K = (1.9 ± 0.3) × 1014 (M−3), ΔH = −26.7 ± 0.8 kcal/mol, N = 1.09 ± 0.05. Again, the theoretical curve is able to reproduce the experimental curve reasonably well, including the initial part of the titration where the heat effect slightly increases, and the fit is significantly better than the previous one with three ligand molecules bound per dimer.
The titration experiments cannot be analyzed with a model considering two or three independent binding sites (51), because the thermodynamic parameters obtained, and in particular the stoichiometry values, are not realistic (i.e., N values much lower than 1).
DISCUSSION
An explanation for the mechanism of lipid hydrolysis by Asp49-PLA2 enzymes was proposed by Verheij et al. (52), and was subsequently modified, based on the crystal structure of PLA2 from Naja atra venom complexed with a transition-state analogue (53,54). In those models, the roles of active sites, the Ca2+-binding loop, the hydrophobic channel, and the i-face were established. A structural characterization of PLA2 isoforms with low or no enzymatic activity will help us further understand the mechanism of catalysis. In a number of group II PLA2 enzymes, one of the critical residues for Ca2+-binding, Asp49, is replaced by nonacidic residues. As expected, despite the conserved Ca2+-binding loop, hydrophobic channel, and i-face, these isoforms have low or no activity. However, crystal structures of PLA2 enzymes in which Asp49 is replaced by nonacidic residues are not available, with the exception of Lys49-PLA2. Hence, we investigated whether the low enzymatic activity of these PLA2 enzymes could be explained by the same factors as with Lys49-PLA2. As shown here, the conformation of the Ca2+-binding loop of ecarpholin S, a Ser49-PLA2, is quite different (Fig. 6). The Asp49-PLA2 enzymes have a canonical Ca2+-binding loop, with Ca2+ in the binding pocket. In Lys49-PLA2 enzymes, the Nɛ of the Lys49 residue occupies the position of the Ca2+ ion, and displaces it. In ecarpholin S, the putative Ca2+-binding loop is distorted, and its conformation is not suitable for calcium binding. This inability to bind Ca2+ is further supported by ITC experiments (Fig. S3 in Data S1). This new Ca2+-binding loop conformation was not observed in any other PLA2 structures.
Although Lys49, Ser49, and other non-Asp49-PLA2 enzymes show limited or no enzymatic activity, they exhibit strong myotoxic activities (including ecarpholin S). The mechanism of their action remains interesting, because it is believed to be independent of phospholipid hydrolysis activity. Many proposals were put forward to explain Lys49-PLA2 myotoxic activity, and they were summarized in reviews (18,19). These myotoxins should have a specific “functional site” responsible for their potent myotoxic activities. Synthetic peptides (KKYRYYLKPLCKK and KKYKAYFKLKCKK) from the C-terminal region K115–K129 of B. asper myotoxin II and A. p. piscivorus Lys49 PLA2, respectively, induce cytolytic effects and play a critical role in myotoxicity (15–17,55). This region was also identified as a binding site for heparin (a myotoxin inhibitor) in rat class IIA secreted PLA2 (51). Treatment with cyanogen bromide leads to cleavage of the N-terminal octapeptide of B. asper myotoxin II, resulting in a significant decrease in its membrane-damaging activities, such as liposome disruption and myotoxicity (56–58). Thus, the N-terminal region is also responsible for myotoxicity. The ecarpholin S/suramin complex shows that it simultaneously binds to the C-terminal, N-terminal, and i-face. These findings strongly suggest that one or more of these three regions could be functionally relevant regions in myotoxicity. The comparison of C-terminal residues Lys115-Gly129 (KKYTYYPNFWCKG) of ecarpholin S with the corresponding segment of B. asper myotoxin II and A. p. piscivorus Lys49-PLA2 reveals that they possess combinations of positively charged and hydrophobic residues. In the ecarpholin S/suramin complex, suramin is mainly held together by a combination of both hydrogen-bonding and hydrophobic interactions with residues, such as Lys115, Lys116, Phe124, and Trp125. Because Lys115 and Lys116 are highly conserved among all three PLA2 enzymes, they may play a more crucial role in the activity of myotoxic PLA2 enzymes.
Furthermore, our SPR experiments suggest that negatively charged phospholipids, such as PG, interact with ecarpholin S. Based on our studies, we propose a hypothesis to explain the myotoxic activity of ecarpholin S: positively charged residues, located in the i-face of the protein, initiate binding with the anionic acceptor(s) (phospholipids) on the membrane, leading to a further strengthening of this interaction by hydrophobic and aromatic residues located at the entrance of the hydrophobic channel. The C-terminal (mainly Lys115 and Lys116) and N-terminal positive patches interact further with the membrane, perturbing the membrane's integrity and inducing cell death. This hypothesis is highly consistent with evidence from noncrystallographic studies involving chemical modifications, synthetic peptides, inhibitors, and site-directed mutagenesis (15–17,55–58).
Our preliminary studies to characterize the thermodynamics of the ecarpholin/suramin interaction are in agreement with suramin promoting the dimerization of ecarpholin. The cooperative model, in which only two states are considered (free protein monomer and ligand-bound protein dimer), is able to describe qualitatively and quantitatively the calorimetric titrations reasonably well. According to our results, the ecarpholin dimer might bind two suramin molecules under the conditions and concentrations used for ITC experiments. Although three suramin molecules bound to an ecarpholin dimer are observed in the crystallographic structure, one of the three suramin molecules in every dimer has higher a B-factor, suggesting the possibility of partial occupancy. In addition, the protein and ligand concentrations are significantly higher in the crystallization setup. If two suramin molecules bind to a protein dimer, the global Gibbs energy is −19.8 kcal/mol, whereas if three suramin molecules bind to a protein dimer, the global Gibbs energy is −28.2 kcal/mol. This global Gibbs energy corresponds to the Gibbs energy for the binding of two or three ligand molecules plus the Gibbs energy for ecarpholin dimerization. The enthalpy values obtained from the analysis are observed values with a possible influence on the ionization enthalpy of the buffer. Additional experiments would be required to assess the extent of possible protonation events coupled to ligand binding, and to estimate the buffer-independent binding enthalpies.
The fully cooperative two-state model for suramin binding can reproduce the shape of the experimental titrations, which supports the hypotheses that suramin promotes dimerization and the population of ligand-free dimers is not significant, as observed using gel-filtration chromatography. A more realistic model would include the ligand-free monomer-dimer equilibrium. A more detailed discussion about the adequate binding model would be appropriate in future work. The influence and explicit consideration of the protein monomer-dimer equilibrium, modulated by ligand binding, would require additional information from ultracentrifugation experiments or binding experiments at different concentrations of protein, or by performing reverse titrations in which the protein is injected into the ligand solution.
In conclusion, the structure of the first representative member of the Ser49-PLA2 subgroup of enzymes, ecarpholin S and its complexes with fatty acid and suramin, is quite different from the structures of Lys49-PLA2 and Asp49-PLA2 enzymes. First, the Ca2+-binding loop of ecarpholin S has a unique conformation which is not ideal for coordinating with Ca2+. Secondly, the ecarpholin S/suramin complex shows that, in addition to the i-face, suramin interacts with both the C-terminal and N-terminal regions of ecarpholin S. Such interactions were not observed in the previous report of the Lys49-PLA2/suramin complex (20). These findings broaden the understanding of the action of non-Asp49-PLA2 enzymes, and should be helpful in the development of new drugs for the treatment of myonecrosis.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
Some of the x-ray data for this study were measured at beamline X12C of the National Synchrotron Light Source, Brookhaven National Laboratory. We acknowledge Dr. Anand Saxena for assistance during data collection. We also thank Mr. Shashikant Joshi from the Protein and Proteomic Centre of Department of Biological Sciences, National University of Singapore, for providing research facilities, and for useful discussions.
Financial support came principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the United States Department of Energy, and from the National Center for Research Resources of the National Institutes of Health. A.V.C. acknowledges funding from the Spanish Ministry of Education and Science (grant SAF2004-07722 and Ramon y Cajal Research Contract), and from Fundacion Aragon I + D (Diputacion General de Aragon). This work was also supported by the Academic Research Fund, Ministry of Education, Singapore (grant R-154-000-245-112).
APPENDIX
The procedure for the analysis of ITC experiments, according to the cooperative model (Eq. 1), is rather straightforward, starting from the conservation equations for protein and ligand:
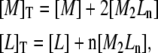 |
(4) |
where M and L represents the protein monomer and the ligand molecule, respectively. The global-equilibrium constant, K, governs the partition into bound and free species:
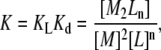 |
(5) |
where KL is the equilibrium constant for ligand binding, and Kd is the dimerization constant for the protein. Therefore, Eq. 4 can be written in terms of free concentrations:
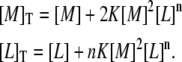 |
(6) |
The free concentration of protein, [M], can be calculated by solving analytically the first equation of the previous set:
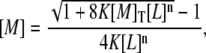 |
(7) |
and it can be introduced into the second equation of the previous set, to calculate the free-ligand concentration, [L]:
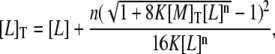 |
(8) |
which can be solved numerically (e.g., by the Newton-Raphson method). Once the free-ligand and free-protein concentrations are known, the concentration of the complex can be calculated from Eq. 5:
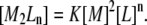 |
(9) |
The concentration of total ligand and total protein in the calorimetric cell after each injection i is calculated as:
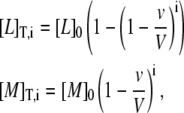 |
(10) |
where [L]0 is the concentration of ligand in the syringe, [M]0 is the initial concentration of protein in the cell, and v and V are the injection volume and the cell volume, respectively. The factor (1 − v/V) is the dilution factor after each injection.
Given the total concentration of protein and ligand in the calorimetric cell before and after each injection (Eq. 10), and assuming a certain value for the equilibrium constant K, the concentration of free ligand and free protein, before and after each injection, can be calculated by solving Eqs. 7 and 8, and the concentration of the complex can be calculated from Eq. 9. Then the heat qi associated with the injection i is calculated for the sequential ligand-binding model:
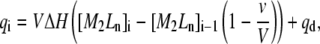 |
(11) |
where ΔH is the global enthalpy (Eq. 3), and qd is the so-called “dilution heat” (constant background heat effect due to unspecific phenomena such as ligand dilution upon injection and injection turbulence). Nonlinear least-square regression, using Eq. 11, will provide K and ΔH as adjustable parameters. It is customary to include an additional parameter N (i.e., using N[M]T instead of [M]T in all previous equations) to account for a different stoichiometry than the one expected or a mismatch between the measured concentrations and the actual ones.
Editor: Kathleen B. Hall.
References
- 1.Davidson, F. F., and E. A. Dennis. 1990. Evolutionary relationships and implications for the regulation of phospholipase A2 from snake venom to human secreted forms. J. Mol. Evol. 31:228–238. [DOI] [PubMed] [Google Scholar]
- 2.Dennis, E. A. 1994. Diversity of group types, regulation, and function of phospholipase A2. J. Biol. Chem. 269:13057–13060. [PubMed] [Google Scholar]
- 3.Dennis, E. A. 1997. The growing phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem. Sci. 22:1–2. [DOI] [PubMed] [Google Scholar]
- 4.Balsinde, J., M. A. Balboa, P. A. Insel, and E. A. Dennis. 1999. Regulation and inhibition of phospholipase A2. Annu. Rev. Pharmacol. Toxicol. 39:175–189. [DOI] [PubMed] [Google Scholar]
- 5.Kini, R. M. 1997. Venom Phospholipase A2 Enzymes: Structure, Function and Mechanism. John Wiley & Sons, Chichester.
- 6.Six, D. A., and E. A. Dennis. 2000. The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim. Biophys. Acta. 1488:1–19. [DOI] [PubMed] [Google Scholar]
- 7.Renetseder, R., S. Brunie, B. W. Dijkstra, J. Drenth, and P. B. Sigler. 1985. A comparison of the crystal structures of phospholipase A2 from bovine pancreas and Crotalus atrox venom. J. Biol. Chem. 260:11627–11634. [PubMed] [Google Scholar]
- 8.Magro, A. J., A. M. Soares, J. R. Giglio, and M. R. Fontes. 2003. Crystal structures of BnSP-7 and BnSP-6, two Lys49-phospholipases A(2): quaternary structure and inhibition mechanism insights. Biochem. Biophys. Res. Commun. 311:713–720. [DOI] [PubMed] [Google Scholar]
- 9.Wei, J. F., X. L. Wei, Q. Y. Chen, T. Huang, L. Y. Qiao, W. Y. Wang, Y. L. Xiong, and S. H. He. 2006. N49 phospholipase A2, a unique subgroup of snake venom group II phospholipase A2. Biochim. Biophys. Acta. 1760:462–471. [DOI] [PubMed] [Google Scholar]
- 10.Wei, J. F., T. Li, X. L. Wei, Q. Y. Sun, F. M. Yang, Q. Y. Chen, W. Y. Wang, Y. L. Xiong, and S. H. He. 2006. Purification, characterization and cytokine release function of a novel Arg-49 phospholipase A(2) from the venom of Protobothrops mucrosquamatus. Biochimie. 88:1331–1342. [DOI] [PubMed] [Google Scholar]
- 11.Holland, D. R., L. L. Clancy, S. W. Muchmore, T. J. Ryde, H. M. Einspahr, B. C. Finzel, R. L. Heinrikson, and K. D. Watenpaugh. 1990. The crystal structure of a lysine 49 phospholipase A2 from the venom of the cottonmouth snake at 2.0-Å resolution. J. Biol. Chem. 265:17649–17656. [DOI] [PubMed] [Google Scholar]
- 12.Scott, D. L., A. Achari, J. C. Vidal, and P. B. Sigler. 1992. Crystallographic and biochemical studies of the (inactive) Lys-49 phospholipase A2 from the venom of Agkistridon piscivorus piscivorus. J. Biol. Chem. 267:22645–22657. [PubMed] [Google Scholar]
- 13.de Azevedo, W. F., Jr., R. J. Ward, J. M. Gutierrez, and R. K. Arni. 1999. Structure of a Lys49-phospholipase A2 homologue isolated from the venom of Bothrops nummifer (jumping viper). Toxicon. 37:371–384. [DOI] [PubMed] [Google Scholar]
- 14.Lee, W. H., M. T. da Silva Giotto, S. Marangoni, M. H. Toyama, I. Polikarpov, and R. C. Garratt. 2001. Structural basis for low catalytic activity in Lys49 phospholipases A2–a hypothesis: the crystal structure of piratoxin II complexed to fatty acid. Biochemistry. 40:28–36. [DOI] [PubMed] [Google Scholar]
- 15.Lomonte, B., E. Moreno, A. Tarkowski, L. A. Hanson, and M. Maccarana. 1994. Neutralizing interaction between heparins and myotoxin II, a lysine 49 phospholipase A2 from Bothrops asper snake venom. Identification of a heparin-binding and cytolytic toxin region by the use of synthetic peptides and molecular modeling. J. Biol. Chem. 269:29867–29873. [PubMed] [Google Scholar]
- 16.Calderon, L., and B. Lomonte. 1998. Immunochemical characterization and role in toxic activities of region 115–129 of myotoxin II, a Lys49 phospholipase A2 from Bothrops asper snake venom. Arch. Biochem. Biophys. 358:343–350. [DOI] [PubMed] [Google Scholar]
- 17.Calderon, L., and B. Lomonte. 1999. Inhibition of the myotoxic activity of Bothrops asper myotoxin II in mice by immunization with its synthetic 13-mer peptide 115–129. Toxicon. 37:683–687. [DOI] [PubMed] [Google Scholar]
- 18.Lomonte, B., Y. Angulo, and L. Calderon. 2003. An overview of lysine-49 phospholipase A2 myotoxins from crotalid snake venoms and their structural determinants of myotoxic action. Toxicon. 42:885–901. [DOI] [PubMed] [Google Scholar]
- 19.Murakami, M. T., and R. K. Arni. 2003. A structure based model for liposome disruption and the role of catalytic activity in myotoxic phospholipase A2s. Toxicon. 42:903–913. [DOI] [PubMed] [Google Scholar]
- 20.Murakami, M. T., E. Z. Arruda, P. A. Melo, A. B. Martinez, S. Calil-Elias, M. A. Tomaz, B. Lomonte, J. M. Gutierrez, and R. K. Arni. 2005. Inhibition of myotoxic activity of Bothrops asper myotoxin II by the anti-trypanosomal drug suramin. J. Mol. Biol. 350:416–426. [DOI] [PubMed] [Google Scholar]
- 21.Ambrosio, A. L., M. C. Nonato, H. S. de Araujo, R. Arni, R. J. Ward, C. L. Ownby, D. H. de Souza, and R. C. Garratt. 2005. A molecular mechanism for Lys49-phospholipase A2 activity based on ligand-induced conformational change. J. Biol. Chem. 280:7326–7335. [DOI] [PubMed] [Google Scholar]
- 22.Polgar, J., E. M. Magnenat, M. C. Peitsch, T. N. Wells, and K. J. Clemetson. 1996. Asp-49 is not an absolute prerequisite for the enzymic activity of low-M(r) phospholipases A2: purification, characterization and computer modelling of an enzymically active Ser-49 phospholipase A2, ecarpholin S, from the venom of Echis carinatus sochureki (saw-scaled viper). Biochem. J. 319:961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jasti, J., M. Paramasivam, A. Srinivasan, and T. P. Singh. 2004. Structure of an acidic phospholipase A2 from Indian saw-scaled viper (Echis carinatus) at 2.6 Å resolution reveals a novel intermolecular interaction. Acta Crystallogr. D Biol. Crystallogr. 60:66–72. [DOI] [PubMed] [Google Scholar]
- 24.Ownby, C. L., H. S. Selistre de Araujo, S. P. White, and J. E. Fletcher. 1999. Lysine 49 phospholipase A2 proteins. Toxicon. 37:411–445. [DOI] [PubMed] [Google Scholar]
- 25.Joseph, J. S., S. Thirumangalathu, F. Tsang, F. W. Wong, and R. M. Kini. 2003. Trocarin, a blood coagulation factor Xa homologue from snake venom, causes inflammation and mitogenesis. Toxicon. 42:769–776. [DOI] [PubMed] [Google Scholar]
- 26.Bordi, F., C. Cametti, S. Sennato, and M. Diociaiuti. 2006. Direct evidence of multicompartment aggregates in polyelectrolyte-charged liposome complexes. Biophys. J. 91:1513–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology. Carter, C.W., and Jr. & R. M. Sweet, editors. Academic Press, New York. 307–326. [DOI] [PubMed]
- 28.Vagin, A., and A. Teplyakov. 1997. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 30:1022–1025. [Google Scholar]
- 29.Perrakis, A., R. Morris, and V. S. Lamzin. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6:458–463. [DOI] [PubMed] [Google Scholar]
- 30.Jones, T. A., J. Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 47:110–119. [DOI] [PubMed] [Google Scholar]
- 31.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. 54:905–921. [DOI] [PubMed] [Google Scholar]
- 32.Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. Procheck: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26:283–291. [Google Scholar]
- 33.Gubensek, F., T. R. Pattabhiraman, and F. E. Russell. 1980. Phospholipase A2 activity of some crotalid snake venoms and fractions. Toxicon. 18:699–701. [DOI] [PubMed] [Google Scholar]
- 34.Krizaj, I., A. L. Bieber, A. Ritonja, and F. Gubensek. 1991. The primary structure of ammodytin L, a myotoxic phospholipase A2 homologue from Vipera ammodytes venom. Eur. J. Biochem. 202:1165–1168. [DOI] [PubMed] [Google Scholar]
- 35.Yoshizumi, K., S. Y. Liu, T. Miyata, S. Saita, M. Ohno, S. Iwanaga, and H. Kihara. 1990. Purification and amino acid sequence of basic protein I, a lysine-49-phospholipase A2 with low activity, from the venom of Trimeresurus flavoviridis (habu snake). Toxicon. 28:43–54. [DOI] [PubMed] [Google Scholar]
- 36.Liu, S. Y., K. Yoshizumi, N. Oda, M. Ohno, F. Tokunaga, S. Iwanaga, and H. Kihara. 1990. Purification and amino acid sequence of basic protein II, a lysine-49-phospholipase A2 with low activity, from Trimeresurus flavoviridis venom. J. Biochem. (Tokyo). 107:400–408. [DOI] [PubMed] [Google Scholar]
- 37.Shimohigashi, Y., A. Tani, H. Matsumoto, K. Nakashima, and Y. Yamaguchi. 1995. Lysine-49-phospholipases A2 from Trimeresurus flavoviridis venom are membrane-acting enzymes. J. Biochem. (Tokyo). 118:1037–1044. [DOI] [PubMed] [Google Scholar]
- 38.Rodrigues-Simioni, L., J. Prado-Franceschi, A. C. Cintra, J. R. Giglio, M. S. Jiang, and J. E. Fletcher. 1995. No role for enzymatic activity or dantrolene-sensitive Ca2+ stores in the muscular effects of bothropstoxin, a Lys49 phospholipase A2 myotoxin. Toxicon. 33:1479–1489. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi, Y., Y. Shimohigashi, T. Chiwata, A. Tani, T. Chijiwa, B. Lomonte, and M. Ohno. 1997. Lys-49-phospholipases A2 as active enzyme for beta-arachidonoyl phospholipid bilayer membranes. Biochem. Mol. Biol. Int. 43:19–26. [DOI] [PubMed] [Google Scholar]
- 40.Mancin, A. C., A. M. Soares, C. A. Giglio, S. H. Andriao-Escarso, C. A. Vieira, and J. R. Giglio. 1997. The histamine releasers crotamine, protamine and compound 48/80 activate specific proteases and phospholipases A2. Biochem. Mol. Biol. Int. 42:1171–1177. [DOI] [PubMed] [Google Scholar]
- 41.Soares, A. M., Y. Oshima-Franco, C. A. Vieira, G. B. Leite, J. E. Fletcher, M. S. Jiang, A. C. Cintra, J. R. Giglio, and L. Rodrigues-Simioni. 2002. Mn2+ ions reduce the enzymatic and pharmacological activities of bothropstoxin-I, a myotoxic Lys49 phospholipase A(2) homologue from Bothrops jararacussu snake venom. Int. J. Biochem. Cell Biol. 34:668–677. [DOI] [PubMed] [Google Scholar]
- 42.Selistre de Araujo, H. S., S. P. White, and C. L. Ownby. 1996. cDNA cloning and sequence analysis of a lysine-49 phospholipase A2 myotoxin from Agkistrodon contortrix laticinctus snake venom. Arch. Biochem. Biophys. 326:21–30. [DOI] [PubMed] [Google Scholar]
- 43.Ward, R. J., L. Chioato, A. H. de Oliveira, R. Ruller, and J. M. Sa. 2002. Active-site mutagenesis of a Lys49-phospholipase A2: biological and membrane-disrupting activities in the absence of catalysis. Biochem. J. 362:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsai, I. H., Y. M. Wang, L. C. Au, T. P. Ko, Y. H. Chen, and Y. F. Chu. 2000. Phospholipases A2 from Callosellasma rhodostoma venom gland cloning and sequencing of 10 of the cDNAs, three-dimensional modelling and chemical modification of the major isozyme. Eur. J. Biochem. 267:6684–6691. [DOI] [PubMed] [Google Scholar]
- 45.Tyler, M. I., D. Barnett, P. Nicholson, I. Spence, and M. E. Howden. 1987. Studies on the subunit structure of textilotoxin, a potent neurotoxin from the venom of the Australian common brown snake (Pseudonaja textilis). Biochim. Biophys. Acta. 915:210–216. [DOI] [PubMed] [Google Scholar]
- 46.Soares, A. M., S. H. Andriao-Escarso, Y. Angulo, B. Lomonte, J. M. Gutierrez, S. Marangoni, M. H. Toyama, R. K. Arni, and J. R. Giglio. 2000. Structural and functional characterization of myotoxin I, a Lys49 phospholipase A(2) homologue from Bothrops moojeni (Caissaca) snake venom. Arch. Biochem. Biophys. 373:7–15. [DOI] [PubMed] [Google Scholar]
- 47.Watanabe, L., A. M. Soares, R. J. Ward, M. R. Fontes, and R. K. Arni. 2005. Structural insights for fatty acid binding in a Lys49-phospholipase A2: crystal structure of myotoxin II from Bothrops moojeni complexed with stearic acid. Biochimie. 87:161–167. [DOI] [PubMed] [Google Scholar]
- 48.Arruda, E. Z., N. M. Silva, R. A. Moraes, and P. A. Melo. 2002. Effect of suramin on myotoxicity of some crotalid snake venoms. Braz. J. Med. Biol. Res. 35:723–726. [DOI] [PubMed] [Google Scholar]
- 49.de Oliveira, M., W. L. Cavalcante, E. Z. Arruda, P. A. Melo, M. Dal-Pai Silva, and M. Gallacci. 2003. Antagonism of myotoxic and paralyzing activities of bothropstoxin-I by suramin. Toxicon. 42:373–379. [DOI] [PubMed] [Google Scholar]
- 50.Murakami, M. T., M. M. Vicoti, J. R. Abrego, M. R. Lourenzoni, A. C. Cintra, E. Z. Arruda, M. A. Tomaz, P. A. Melo, and R. K. Arni. 2007. Interfacial surface charge and free accessibility to the PLA2-active site-like region are essential requirements for the activity of Lys49 PLA2 homologues. Toxicon. 49:378–387. [DOI] [PubMed] [Google Scholar]
- 51.Murakami, M., Y. Nakatani, and I. Kudo. 1996. Type II secretory phospholipase A2 associated with cell surfaces via C-terminal heparin-binding lysine residues augments stimulus-initiated delayed prostaglandin generation. J. Biol. Chem. 271:30041–30051. [DOI] [PubMed] [Google Scholar]
- 52.Verheij, H. M., J. J. Volwerk, E. H. Jansen, W. C. Puyk, B. W. Dijkstra, J. Drenth, and G. H. de Haas. 1980. Methylation of histidine-48 in pancreatic phospholipase A2. Role of histidine and calcium ion in the catalytic mechanism. Biochemistry. 19:743–750. [DOI] [PubMed] [Google Scholar]
- 53.White, S. P., D. L. Scott, Z. Otwinowski, M. H. Gelb, and P. B. Sigler. 1990. Crystal structure of cobra-venom phospholipase A2 in a complex with a transition-state analogue. Science. 250:1560–1563. [DOI] [PubMed] [Google Scholar]
- 54.Scott, D. L., S. P. White, Z. Otwinowski, W. Yuan, M. H. Gelb, and P. B. Sigler. 1990. Interfacial catalysis: the mechanism of phospholipase A2. Science. 250:1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nunez, C. E., Y. Angulo, and B. Lomonte. 2001. Identification of the myotoxic site of the Lys49 phospholipase A(2) from Agkistrodon piscivorus piscivorus snake venom: synthetic C-terminal peptides from Lys49, but not from Asp49 myotoxins, exert membrane-damaging activities. Toxicon. 39:1587–1594. [DOI] [PubMed] [Google Scholar]
- 56.Diaz, C., A. Alape, B. Lomonte, T. Olamendi, and J. M. Gutierrez. 1994. Cleavage of the NH2-terminal octapeptide of Bothrops asper myotoxic lysine-49 phospholipase A2 reduces its membrane-destabilizing effect. Arch. Biochem. Biophys. 312:336–339. [DOI] [PubMed] [Google Scholar]
- 57.Soares, A. M., R. Guerra-Sá, C. R. Borja-Oliveira, V. M. Rodrigues, L. Rodrigues-Simioni, V. Rodrigues, M. R. Fontes, B. Lomonte, J. M. Gutiérrez, and J. R. Giglio. 2000. Structural and functional characterization of BnSP-7, a Lys49 myotoxic phospholipase A(2) homologue from Bothrops neuwiedi pauloensis venom. Arch. Biochem. Biophys. 378:201–209. [DOI] [PubMed] [Google Scholar]
- 58.Soares, A. M., S. H. Andriao-Escarso, R. K. Bortoleto, L. Rodrigues-Simioni, R. K. Arni, R. J. Ward, J. M. Gutierrez, and J. R. Giglio. 2001. Dissociation of enzymatic and pharmacological properties of piratoxins-I and -III, two myotoxic phospholipases A2 from Bothrops pirajai snake venom. Arch. Biochem. Biophys. 387:188–196. [DOI] [PubMed] [Google Scholar]
- 59.DeLano, W. S. 2002. The PyMOL Molecular Graphics System. In: http://www.pymol.org. Accessed April 7, 2007.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.