

Abstract
Neurodegeneration observed in Alzheimer disease (AD) is believed to be related to the toxicity from reactive oxygen species (ROS) produced in the brain by the amyloid-β (Aβ) protein bound primarily to copper ions. The evidence for an oxidative stress role of Aβ-Cu redox chemistry is still incomplete. Details of the copper binding site in Aβ may be critical to the etiology of AD. Here we present the structure determined by combining x-ray absorption spectroscopy (XAS) and density functional theory analysis of Aβ peptides complexed with Cu2+ in solution under a range of buffer conditions. Phosphate-buffered saline buffer salt (NaCl) concentration does not affect the high-affinity copper binding mode but alters the second coordination sphere. The XAS spectra for truncated and full-length Aβ-Cu2+ peptides are similar. The novel distorted six-coordinated (3N3O) geometry around copper in the Aβ-Cu2+ complexes include three histidines: glutamic, or/and aspartic acid, and axial water. The structure of the high-affinity Cu2+ binding site is consistent with the hypothesis that the redox activity of the metal ion bound to Aβ can lead to the formation of dityrosine-linked dimers found in AD.
INTRODUCTION
Alzheimer disease (AD) is a progressive neurodegenerative disorder characterized by the presence of misfolded protein depositions or amyloid plaques. The major constituent of AD plaques is the amyloid-β peptide (Aβ, up to 42 amino acids: D1AEFRH6DSGY10E11VH13H14QK16LVFFAEDVGSNK28GAIIGLM35VGGVVIA42), which is cleaved from the membrane-bound amyloid precursor protein via the β/γ-secretase pathway. Current consensus in AD research is that soluble Aβ oligomer intermediates rather than the plaque burden (1,2) are the major contributors to Aβ-mediated neurodegeneration. In vitro Aβ binds to Cu, Zn, and Fe with relatively high affinity, and elevated levels of these metals are found in AD amyloid deposits (3,4). Metals can mediate oligomerization of Aβ, and the neurotoxicity of Aβ has been related to its ability to form complexes with redox-susceptible ions, Cu2+ in particular (4). The Aβ-Cu2+ complexes can be involved in extensive redox chemical reactions that produce H2O2 and other reactive oxygen species (ROS) from molecular oxygen (4,5). Summarizing these studies (6), it is evident first that the monomeric soluble full-length Aβ1−40/42 peptides and truncated Aβ1−28 and Aβ1−16 peptides bind the Cu2+ ion at a high-affinity site with the same coordination environment in a 1:1 stoichiometric ratio (7–12). Residues 1–17 are unstructured and do not participate in the β-sheet packing (13,14). The Cu2+ coordination by the hydrophobic C-terminal residues (aa 29–42), in particular by Met-35, was ruled out (15), and the voltammetric studies of three Cu2+ complexes with Aβ1−16, Aβ1−28, and Aβ1−42 were similar (9,16). Second, recent studies indicate the involvement of the three histidines His-6, His-13, and His-14 (8,11,17–19). The fourth ligand is most likely an oxygen atom donor (20). Some choices for the fourth ligand included Tyr10 (20–22), the N-terminal nitrogen, or Asp-1 carboxylate (7,8,17,19), or other carboxylate side chains. The Glu-11 residue provides the carboxylate side chain when Zn2+ is bound to human Aβ1–16 (23) and Aβ1–28 (24), and there is evidence that the Asp-1 carboxylate side chain interacts via hydrogen bonding to an axial water from the first coordination shell of Cu2+ (25). Marked 1H relaxation enhancements were also noticeable for the Glu-11 (Fig. 2 in the study by Gaggelli et al. (25)). Recent extended x-ray absorption fine structure (EXAFS) analyses of the Aβ1-40-Cu2+ (26) and Aβ1-16-Cu2+ (27) complexes with 1:1 metal/peptide ratio suggested that Cu2+ is pentacoordinated to three nitrogen atoms (from His-6, His-13, and His-14) and two oxygen atoms, with one from Tyr-10 and the other one from either a water molecule or an amino acid other than the bound histidine and tyrosine residues. Conversely, there is a compelling evidence that tyrosine (Tyr-10) is not the oxygen atom donor (7,8,11,18,19). Furthermore, in an analysis (28) of metal binding sites in metalloproteins, aspartate and glutamate were often found in the coordination sphere of Cu or Zn, whereas tyrosine binding was rare.
Native, full-length Aβ peptide forms aggregates with metastable and polydisperse structures, whereas truncated Aβ1−16 peptide does not, which is relatively hydrophilic and more soluble compared to the full-length peptide in the range of conditions compared to the full-length peptide. The discrepancy in metal binding geometry in Aβ peptides has been attributed to the differences in preparation of peptide, buffer conditions, pH, and in the mode of presentation of Cu2+ to the Aβ peptide (10,11,29). Some studies (30,31) showed that NaCl presence in buffer greatly encourages metal-mediated oligomerization of Aβ and may alter the metal binding site. The structural diversity in the aggregated structures and possibly in metal binding geometry can be interpreted in terms of the difference in the degree of the electrostatic shielding at different salt concentrations and can be explored by EXAFS spectra analysis.
Here, we present the combined multiple data (MD) multiple scattering (MS) EXAFS at 16–20°K and density functional theory (DFT) analysis of the truncated Aβ1−16 peptide complexed with Cu2+ in solution under a range of buffer conditions, in particular in phosphate-buffered saline (PBS) buffer with a range of NaCl concentrations. For comparison with the truncated Aβ1−16, we also measured EXAFS spectra for low concentration solutions of Cu2+ complexed with the full-length peptides Aβ1−42M35(O) and Aβ1−42M35V, where Met-35 has been oxidized to methionine sulfoxide and mutated to valine, respectively. The oxidation of Met-35 reduces aggregation of the peptide (32,33), which is important for the solution EXAFS, whereas the M35V mutant data should help to clarify further the role of Met-35 in copper coordination.
MATERIALS AND METHODS
Sample preparation
All human Aβ peptide samples in Table 1 except sample 4 (Aβ16-PB100) were synthesized at the University of Melbourne. Sample 4 was purchased from Auspep Pty Ltd. (Melbourne, Australia). The Aβ-Cu2+ complex samples were freshly prepared before the data collections. Known quantities of Aβ16 peptides were dissolved in phosphate buffer (1 × PB: 10 mM Na2HPO4, 2mM KH2PO4, and 1000 mL H2O; pH 7.4) with 0%, 50% (68.5 mM), 75% (103 mM), and 100% (137 mM) addition of NaCl. Sample 5 (Aβ16-PBS) of Aβ16 was dissolved in common isotonic PBS buffer. For comparison, samples of the full-length peptide Aβ1−42 with M35(O) and M35V modifications and complexed with Cu2+ were also prepared and analyzed. Both samples were synthesized as described previously (32,34) and dissolved in 0.05 M ammonia before isotonic PBS buffer was added. Freshly prepared solution of CuCl2 was added to peptide solutions to the concentration ratio 1:1 of metal/peptide. Finally, glycerol (21% for samples 1–4 and 24% for samples 5–7 in Table 1) was added to the sample solution as a cryoprotectant. Glycerol is an accepted cryoprotectant in protein crystallography and in EPR and EXAFS spectroscopy. It has been shown that inclusion of glycerol up to 50% does not affect EPR properties of Cu2+ bound to Aβ (8,17). Immediately after preparation, the samples were injected into 230 μL Teflon (DuPont, Wilmington, Delaware) cells made with two Kapton (Goodfellow Cambridge, Cambridge, UK) windows (∼4 × 25 mm) and rapidly frozen in liquid nitrogen. Aggregation of peptide complexes in the presence of NaCl from buffers was not observed. The truncated peptide Aβ1−16 does not contain the C-terminal fibrillization domain, and the time between addition of Cu2+ to the peptides and freezing of the samples was not sufficient to generate a substantial concentration of β-sheets in case of the full-length peptides Aβ1−42 with M35(O) and M35V modifications.
TABLE 1.
Samples and experimental data
| No. | Sample | Buffer, PB + NaCl% | Cu/Aβ, mM | T, K | Usable kmax, Å−1 | N scans | Scan t, min | ɛk 104* |
|---|---|---|---|---|---|---|---|---|
| 1 | Aβ16-PB: | PB | 2.2 | 16.5 | 11.8 | 13 | 40 | 0.414 |
| 2† | Aβ16-PB50 | PB+50% | 2.2 | 16.5 | 11.8 | 13 | 40 | 0.432 |
| 3 | Aβ16-PB75 | PB+75% | 2.2 | 16.5 | 11.9 | 12 | 40 | 0.543 |
| 4 | Aβ16-PB100 | PB+100% (PBS) | 2.2 | 20K | 11.7 | 15 | 40 | 0.589 |
| 5 | Aβ16-PBS | PBS | 1.1 | 20K | 11.3 | 10 | 25 | 1.303 |
| 6 | Aβ42M35V | PBS | 0.9 | 20K | 11.7 | 12 | 25 | 1.450 |
| 7‡ | Aβ42M35(O) | PBS | 0.85 | 20K | 11.8 | 12 | 25 | 2.136 |
X-ray absorption data collection
A series of Cu K-edge (8980.4 eV) x-ray absorption spectrum scans (Table 1) were obtained from each sample in a fluorescence mode at low T using a helium displex cryostat. The experiments were conducted at the Pacific Northwest Consortium Collaborative Access Team (PNC-CAT) 20BM bending magnet beamline at the Advanced Photon Source (APS) of Argonne National Laboratories, Argon, IL. The beamline optical setup was equipped with a double crystal Si(111) monochromator and a 5 mrad rhodium-coated harmonic-rejection mirror. To increase sensitivity with millimolar concentration solutions, a 12-element liquid N2-cooled Ge detector was used. The beam size was 1 × 15 mm (vertical × horizontal). The incident x-ray intensity was monitored using an ionization chamber. The stability of the monochromator energy was checked for all spectra by the simultaneous accumulation of a Cu foil spectrum by transmittance. The energy was calibrated with reference to the lowest energy inflection point of a Cu foil spectrum, which was assumed to be 8980.4 eV. X-radiation reduction of Cu2+ was monitored by comparing edge spectra (the x-ray absorption near-edge structure (XANES)) for consecutive scans. Cu2+ in aqueous solutions, especially the halogen-containing salts, can be reduced by the intense x-ray beams (35,36). After the detection of marked photoreduction of Cu2+ in sample 2 with NaCl salt (spectra scans shown in Fig. S1 in Supplementary Material, Data S1), i.e., increasing intensity ∼8984 eV and reduction of the white-line intensity typical for Cu1+ in the XANES region of the consecutive scans, an attempt was made to collect scans by moving the sample in the vertical direction in 150 μm steps within the cell window area. This movement effectively eliminated the radiation damage of the rest of the samples with halogen-containing buffers. The superposition of EXAFS spectra for all samples is shown in Fig. S2 in Data S1. The spectrum of radiation-damaged sample 2 (Aβ16-PB50) deviates noticeably from the others and produced marked changes in r space (Fourier-transformed results are not shown). These data will not be considered in further analyses. The measurement uncertainty ɛk (37) shown in Table 1 demonstrates that samples 5 (Aβ16-PBS) and 6 (Aβ42M35V-PBS), and especially sample 7 (Aβ42M35(O)-PBS), are much noisier (higher ɛk), as is expected for the lower concentration of the Cu2+ peptide complexes. Overall, features of Aβ42M35V and Aβ42M35(O) spectra are similar to those of the spectra of short peptides. Furthermore, the spectra in this study are similar (although less noisy) to those of Aβ1-40-Cu2+ (26) and Aβ1-16-Cu2+ (27), which are reported to be in the k range of ∼3–11 Å−1. Further subtle differences between the spectra measured in this study will be analyzed by comparison of energy-calibrated and normalized XANES, because the XANES region is more sensitive to small structural differences and less affected by noise.
X-ray absorption data analysis
XANES region
XANES spectra were extracted from the experimentally measured absorption coefficient using background subtraction and normalization methods implemented in the program ATHENA (38), an interface to IFEFFIT (39). For a reliable comparison of the XANES spectra, a consistent way of normalizing near-edge data has been used by matching the measured spectra to the energy dependence of the Cromer-Liberman calculations (40) for the absorption of a free atom at energies far below and above the edge (38). Fig. 1 A (see also Fig. S3 A in Data S1) shows the superimposed XANES regions of normalized absorption amplitude versus energy E for six samples, as in Table 1 (1: Aβ16-PB; 3: Aβ16-PB75; 4: Aβ16-PB100; 5: Aβ16-PBS; 6: Aβ42-M35V; and 7: Aβ42-M35(O)). Although the spectra are generally similar, they can be split in two groups: 1), three spectra for samples 1, 3, and 4 with higher concentration (∼2 mM) of complexes and 2), three spectra for samples 5, 6, and 7 with the lower concentration (∼1 mM) of the Cu2+ peptide complexes. Fig. 1 B shows superimposed spectra within the groups. The noisy spectrum for sample 7 was excluded from further analysis. The group with data 5 and 6 (Fig. 1 B, top spectra) have more pronounced shoulders B at ∼8987eV and D at 9010 eV, and slightly reduced white-line amplitude C at 8997 eV (see also Fig. S1 A in Data S1). The similarity in the series of 1, 3, and 4 measurements of the same system (Aβ1−16-Cu2+) in buffer with varying concentrations of NaCl salt suggests that the geometry of the Cu2+ coordination does not change noticeably through this series. In contrast, the similarity of spectra in the series of 5 and 6 (and, to some extent, noisy 7) measurements of different systems including the short peptide Aβ1−16-Cu2+ and the full-length peptide Aβ1−42M35V-Cu2+ (or Aβ1−42M35(O)-Cu2+) complexes suggests that the Cu2+ coordination also does not change significantly through this series and that the Met-35 modifications do not have a noticeable effect on the Cu2+ environment. The samples 5–7 from one group are in PBS buffer, which is effectively PB with 100% NaCl buffer used for the sample 4 from the other group. The only significant difference in the preparation of these two groups of samples is the concentration of the complex in solution. The concentration of the 1, 3, and 4 complexes was twice as high compared to that of the 5–7 complexes. The observed, subtle XANES changes between the two groups of data in Fig. 1 A must be primarily due to the Aβ-Cu2+ concentration in solution.
FIGURE 1.

The XANES regions of normalized absorption amplitude versus energy E for (A) six samples as in Table 1 (1) Aβ16-PB, (3) Aβ16-PB75, (4) Aβ16-PB100, (5) Aβ16-PBS, (6) Aβ42-M35V, and (7) Aβ42-M35Ox) and (B) two groups of superimposed according to similarity spectra for samples 1 (solid line), 3 (dashed line), and 4 (dotted line) in the lower group and for samples 5 (solid line) and 6 (dashed line) in the upper group. The noisy spectrum for sample 7 is not shown. Spectral features at 8979, 8987, 8997, 9005, 9010, and 9045 eV are marked as A, B, C, D, E, and F, respectively. For clarity, the spectra are stacked with offset (A) 0.2 and (B) 0.5 units along the vertical axes.
EXAFS region
The EXAFS oscillations χ(k) were extracted from the experimentally measured absorption coefficient using an automated background subtraction AUTOBK algorithm (41) implemented in the program ATHENA (38). The EXAFS oscillations χ(k) were quantitatively analyzed by the ARTEMIS (38) program, also an interface to IFEFFIT (39), using ab initio theoretical amplitude, phase, and mean-free path factors calculated by FEFF6 (42). Constrained and restrained refinement procedures were used to minimize the number of free parameters in the least-squares refinement to increase the degree of determinacy of the model and to keep the geometry of ligands (amino acids) close to those of stereochemically acceptable conformations. The metal ligands in this study were treated as idealized rigid bodies and included atoms of selected amino acids at the distance ≤5 Å from the metal atom (43). The parameters used to construct these units were taken from the averaged bond distances and angles of crystallographically characterized polypeptide structures (44). The geometry of the ligands was kept restrained within standard uncertainties given for bond lengths and angles, and its position relative to the metal atom was defined by the position of the first shell nitrogen/oxygen atom ri and the two angular parameters φi and θi (45). To further reduce the number of refined parameters, the values of the Debye-Waller factors (σ2) for the ligand (treated as rigid bodies), which represent the root mean-square variation in ri due to both static and thermal disorder, were initially assigned the same value as for the first-shell atoms. At the final stages of refinement, however, the displacement terms for higher shells of peptide atoms (at the second shell 2.3 Å < r < 4.0 Å and at the third shell r > 4.0 Å) were multiplied by estimated and fixed coefficients A2 and A3 (σ2 (second shell) = A2 σ2 (first shell refined) and σ2 (third shell) = A3 σ2 (first shell refined)) to reflect the increase of the displacement terms with the distance to the absorber (Cu). The structural parameters refined during constrained/restrained refinements included 1), the distances ri of the histidine groups (that is, the Cu-N bond) and the other low-Z ligands (for example, Cu-O bonds), 2), the angular parameters associated with the ligands, and 3), the Debye-Waller factors 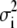 for each ligand. The coordination number Ni of the shell for each type of neighboring atom was kept fixed. In addition to the structural parameters, the photoelectron energy threshold (Fermi energy ΔE0 shift), which was treated as a single overall parameter for the multiple shell fits, was varied to correct for the simple estimate of E0 made in ATHENA. The amplitude reduction factor (core-hole factor)
for each ligand. The coordination number Ni of the shell for each type of neighboring atom was kept fixed. In addition to the structural parameters, the photoelectron energy threshold (Fermi energy ΔE0 shift), which was treated as a single overall parameter for the multiple shell fits, was varied to correct for the simple estimate of E0 made in ATHENA. The amplitude reduction factor (core-hole factor) 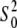 was allowed to be adjusted around the starting value of
was allowed to be adjusted around the starting value of 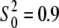 for Cu, which is accurate to within a few percent for copper compounds (46). The fitting procedure consisted of successive cycles of constrained refinement followed by restrained refinement. The Cu χ(k) data were weighted by k3 and windowed between 0 < k < kmax Å−1 using a Kaiser-Bessel window with dk = 2.0 Å−1 to minimize spectral ringing. In the course of refinement, the multiple refinements of k weights (simultaneous refinements with k1, k2, and k3, or their combinations) were also used. Different values of k weight emphasize different regions of the spectrum, reducing correlations between parameters. The fits were carried out on both the real and imaginary parts of χ(r) in the region of 0 < r < 5.0 Å using a Kaiser-Bessel window with dr = 0.2 Å. After refinement of the model with single scattering (SS) contributions from all atoms ≤5 Å from the Cu metal, all MS contributions >10% and l ≤ 4 (triple scattering paths with four legs) were analyzed and included in the refined model.
for Cu, which is accurate to within a few percent for copper compounds (46). The fitting procedure consisted of successive cycles of constrained refinement followed by restrained refinement. The Cu χ(k) data were weighted by k3 and windowed between 0 < k < kmax Å−1 using a Kaiser-Bessel window with dk = 2.0 Å−1 to minimize spectral ringing. In the course of refinement, the multiple refinements of k weights (simultaneous refinements with k1, k2, and k3, or their combinations) were also used. Different values of k weight emphasize different regions of the spectrum, reducing correlations between parameters. The fits were carried out on both the real and imaginary parts of χ(r) in the region of 0 < r < 5.0 Å using a Kaiser-Bessel window with dr = 0.2 Å. After refinement of the model with single scattering (SS) contributions from all atoms ≤5 Å from the Cu metal, all MS contributions >10% and l ≤ 4 (triple scattering paths with four legs) were analyzed and included in the refined model.
The determinacy of a refinement can be further improved by increasing the number of datapoints per fitted parameter. Having two groups of measurements ((1, 3, and 4) and (5 and 6)) in Table 1 with different concentrations of Aβ-Cu2+ complexes and observing that the XANES and EXAFS spectra (Fig. 1; see Fig. S2 in Data S1), and therefore the geometry of the Cu2+ coordination, did not change significantly within groups, we have attempted an MD refinement for each group. The MD refinement is similar to protein crystallographic structure determination using diffraction measurements from multiple crystals. In this case, each of the global models that are defined below was used to simultaneously fit all data sets in each group of measurements. All structural parameters and 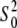 values remained the same throughout the sequence, and ΔE0 was refined for each data set separately. Best-fit parameters for models of Aβ-Cu2+ complexes are shown in Table 2 and in the Data S1.
values remained the same throughout the sequence, and ΔE0 was refined for each data set separately. Best-fit parameters for models of Aβ-Cu2+ complexes are shown in Table 2 and in the Data S1.
TABLE 2.
Best-fit EXAFS and DFT parameters of Model 1 for Aβ-Cu2+ complexes
| Model 1 “Asp-1/Glu-11” multiple EXAFS data fits | DFT | ||
|---|---|---|---|
| Samples | 1, 3, 4 | 5, 6 | |
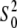 |
0.95 (0.03) | 0.92 (0.04) | |
| ΔE0 | |||
| Sample 1 | −0.7 (0.5) | ||
| Sample 3 | −0.8 (0.5) | ||
| Sample 4 | −0.6 (0.5) | ||
| Sample 5 | 0.2 (0.7) | ||
| Sample 6 | 0.3 (0.7) | ||
| r1 (His-6) | 1.981 (0.008) | 1.99 (0.02) | 2.03 |
| r2 (His-13) | 1.908 (0.007) | 1.94 (0.02) | 2.03 |
| r3 (His-14) | 2.08 (0.01) | 2.06 (0.02) | 2.07 |
| r4 (O1 carboxylate) | 1.941 (0.007) | 1.94 (0.01) | 2.02 |
| r5 (w1) | 2.03 (0.01) | 2.04 (0.02) | 2.30 |
| r6 (O2 carboxylate)* | 2.27 (0.01) | 2.26 (0.01) | 2.63 |
| r7 (“solvent”) | 4.45 (0.01) | 4.46 (0.01) | |
| φ1 (His-6) | 3.2 (1.0) | 0.4 (1.7) | −6.4 |
| φ2 (His-13) | −1.6 (8.6) | −1.8 (10.0) | −6.4 |
| φ3 (His-14) | −4.7 (1.0) | −6.4 (2.8) | 17.4 |
| φ4 (carboxylate)† | 96.5 (0.2) | 95.9 (0.4) | 104.0 |
| σ2 (1st shell) | 0.0020 (0.0005) | 0.003 (0.001) | |
| σ2 (2nd shell)‡ | 0.006 (0.008) | 0.003 (0.001) | |
| σ2 (3rd shell) ‡ | 0.008 (0.001) | 0.009 (0.002) | |
| σ2 (“solvent”) | 0.005 (0.002) | 0.003 (0.002) | |
| Nind | 111 | 72 | |
| Nvar | 16 | 15 | |
| χ2 | 4.108 | 1.406 | |
| Rall§ | 0.0134 | 0.0277 | |
| R1 | 0.0097 | ||
| R3 | 0.0185 | ||
| R4 | 0.0121 | ||
| R5 | 0.0244 | ||
| R6 | 0.0310 | ||
ΔEo is the edge position relative to the photoelectron energy threshold Eo for samples: 1:- 8992.4; 3: 8992.2; 4: 8991.9; 5: 8992.7; and 6: 8992.4 eV; ri and φi refer to the distances (in Å) and the polar angle (in degrees) for shell i; σ2 Debye-Waller terms (in Å2); Nind, Nvar, χ2,and R are defined in the main text or in Data S1; estimated standard deviation from least-squares is given in parentheses.
Calculated distance to the second Asp-1/Glu-11 carboxyl oxygen O2.
Cu-Oɛ1-Cδ (Glu-11) or Cu-Oδ1-Cγ (Asp-1) angles.
Debye-Waller terms for peptide atoms at the second shell (2.3 Å < r < 4.0Å) and at the third shell (r > 4.0 Å), respectively, adjusted by the estimated coefficients A2 and A3: σ2 (second shell) = A2 σ2 (first shell) and σ2 (third shell) = A3 σ2 (first shell).
All R factors are calculated in r-space, and Rn values are partial factors for MD sets used in refinements.
Aβ-Cu2+ starting models
The initial fittings of the XAFS data with the first shell SS theory including nitrogen and/or oxygen atoms around copper showed that the coordination number of Cu2+ is six. This suggests that the Cu2+ can be potentially octahedral coordinated with three histidines, oxygen(s) donating amino acids, and one or two water molecules. It should be noted that, because the Cu-O and Cu-N bond distances and backscattering amplitudes are somewhat similar, it is difficult to distinguish between their respective contributions in the EXAFS spectra. Considering the most probable coordination of Cu2+ to Aβ via three histidines, aspartic or/and glutamic acid, and waters, we constructed a six-coordinated model (Model 1), which includes the equatorial imidazole rings of His-6, His-13, His-14, the carboxylate side chain of Glu-11 or Asp-1, and one axial water. Because of the similar nature of the side-chain electron donor of Asp and Glu, these two amino acid residues can be considered as equivalent. Within ∼5 Å from the central Cu atom, the geometry of the Glu side chain (Cβ, Cγ, Cδ, Oɛ1, and Oɛ2) is virtually identical to the Asp side chain, including the backbone carbons (Cα, Cβ, Cγ, Oδ1, and Oδ2), and cannot be easily distinguished by the EXAFS spectra measurements. For comparison, we also constructed a six-coordinated model (Model 2), which includes the equatorial imidazole rings of His-6, His-13, His-14, the phenol ring of Tyr10, and two axial waters. DFT calculations were then used to comparatively explore the ground state electronic structures for these models, and the DFT optimized geometries were used as the starting models in EXAFS spectra fitting.
DFT investigation of starting models
DFT calculations were carried out in the gas phase using the B3LYP functional set (47) and the LANL2DZ basis set (48) for appropriate model systems with the Gaussian 03 suite of programs (49). The suitability of calculations at this level of theory, functional, and basis set were confirmed by computing the optimized geometry of trans-bis(L-alaninato-N,O)-copper(ii) (50), for which a high-resolution crystal structure is available in the Cambridge Structural Database (id: CUALTE01). The optimized geometry for trans-bis(L-alaninato-N,O)-copper(ii) was square planar and showed very good agreement with the experimental structure with Cu-O bond distances of 1.91 Å and 1.92 Å (1.95 Å in the crystal structure), Cu-N bond distances of 2.03 Å and 2.04 Å (1.98 Å in the crystal structure), and the N-Cu-O bond angle of 95.0° (93.8° in the crystal structure). The deviations in bond lengths from the experimental values are of the same order observed previously for Cu containing small molecules (51).
Next, Cu2+ bound to Aβ was modeled by a Cu atom centrally coordinated to three imidazole rings and a carboxylate side chain (Asp or Glu) (Model 1) or a tyrosinate (Model 2) at equatorial positions, and one (Model 1) or two (Model 2) water molecules at axial positions. The model system had a total charge of one and a spin multiplicity of two. In all geometry optimizations, the modified GDIIS (52) algorithm and tight convergence criteria were used. Stationary points were checked for true energy minima by performing frequency calculations. Setup of input files and visualization of results was carried out with GaussView v.3.09 (53).
RESULTS AND DISCUSSIONS
Quantum mechanical calculations
The DFT-optimized geometries for Cu2+ bound to glutamate/aspartate, three imidazoles, and one water molecule (Model 1) and to tyrosinate, three imidazoles, and two water molecules (Model 2) are shown in Table 2 and Table S2 in Data S1, respectively. Both optimized structures (see Fig. 3 A and Fig. S5 A in Data S1) are of Jahn-Teller-type distorted octahedral coordination about the copper ion, with the axial ligands being two waters placed asymmetrically (at elongated distances of 2.41 Å and 2.78 Å from the Cu2+ ion) in the case of Model 2. The more distal water is potentially hydrogen bonded to the tyrosinate oxygen, which is 2.00 Å from the Cu2+ ion. In the case of the carboxylate system (Model 1), the axial ligands are a water molecule at a distance of 2.30 Å from Cu2+ ion, and one of the carboxylate oxygen atoms at 2.63 Å distant from the Cu2+ ion. The other carboxylate oxygen (2.02 Å) and the three imidazole nitrogen atoms (2.03–2.07 Å) are in an approximately planar arrangement about the Cu ion. The DFT-optimized geometries for Model 1 and Model 2 were used in the EXAFS spectra data refinements as described below.
FIGURE 3.

A ball-and-stick representation of the Model 1 (carboxylate side chain from Asp-1 or Glu-11) from (A) DFT optimization and (B) MD EXAFS refinement of 1, 3, and 4 data sets. The axial oxygen atom is labeled as water W1. The two additional solvent oxygen (Osol) atoms in (B) may belong to the carboxylate group from the N-terminal amino acids such as Asp-1, which may participate in a hydrogen bonding with axial water W1 to stabilize the Cu binding site. Parts of the EXAFS structure (B) not included in the refinement are transparent.
X-ray absorption analysis
XANES region
Fig. 1 A shows the XANES regions of Aβ-Cu2+ spectra for all samples from Table 1, except for sample 2 (Aβ16-PB50), which showed significant x-radiation damage. Fig. 1 B shows a similarity between XANES spectra within the two groups, which mainly differ in the concentration of the peptide complex in solution. The measurement uncertainty ɛk shown in Table 1 demonstrates that sample 7 (Aβ42-M35Ox) spectra is much noisier than all the others (highest ɛk), and it was not included in Fig. 1 B. The series of 5, 6, and 7 samples has effectively doubled the content of salt (NaCl) per molecule of the Aβ-Cu2+ complex in solution compared to the series of 1, 3, and 4 samples. This relative salt increase may create a different hydration and salt/peptide interaction effect on the geometry of the metal binding site. At higher concentrations of salt, the Cl− anions readily form ion pairs with nitrogen-based (cationic) peptide side chains, and they are known to adsorb to nonpolar surfaces or portions of Arg, His, and Lys side chains (54). The most interesting features that show differences or similarities between the two groups of measurements are ∼ 8979, 8987, 8997, 9005, 9010, and 9045 eV. These features are marked as A, B, C, D, E, and F, respectively, in Fig. 1 A and are enlarged in Fig. S3 in Data S1.
A systematic study (55) of 19 Cu1+ and 40 Cu2+ complexes showed that the intensity and position of near-edge features are strongly correlated with the oxidation state and the coordination mode. Our recent XANES study (56) of Aβ1−16 in complex with Cu1+ and Cu2+ is in good agreement with these previous studies. The Aβ-Cu2+ complexes have very weak peaks at ∼8979 eV (labeled A in Fig. 1 B; see also Fig. S3 B in Data S1), which were assigned as originating from 1s→3d quadrupolar-allowed transitions in the Cu2+ species (55,57). It was shown (57) that the features from B to F (Fig. 1 B) depend on the geometric details of the Cu2+ coordination environment. The small shoulder at ∼8987–8988 eV (labeled B in Fig. 1 B), which stems from either a vibronically allowed 1s→4s transition or 1s→4p transitions with a metal-ligand charge-transfer shakedown, has been assigned primarily to scattering by the axial ligands, and its intensity and position has been related to the number and position of these ligands (57). Such low-energy, rising-edge features can also be due to single-electron scattering from distant (solvent) atoms (58). The B peak shown in Fig. 1 B is shifted to 8987eV and is more pronounced for the lower-concentration 5 and 6 peptide complexes relative to that at 8988 eV for the 1, 2, or 4 higher-concentration peptide complexes (see also Fig. S3 C in Data S1). This shift may reflect an increased contribution from the distant Cl− axial ligands to the Cu2+ environment for the 5 and 6 samples. The features marked C and D at 8997–9005 eV in Fig. 1 B were assigned to the main 1s→4p (or 1s→continuum) transition. The reduction of the main peak for the 5 and 6 measurements appear to be related to the increased contribution of distant ligands, such as the secondary shell axial solvents (H2O and/or Cl−) (58). The differences in amplitudes at 8985–9005 eV observed in CuBr2 solutions (59) with either an equal or higher relative concentration of NaBr were attributed to the association of the Br− and Cu2+ ions at the higher concentration of NaBr. There are other important features at E and F in the XANES region of Cu2+ spectra, which are represented by shoulders at 9010 and 9045 eV, respectively, in Fig. 1 B (see also Fig. S3 D in Data S1). These features do not change noticeably across the series of measurements and were assigned as being due to MS from the first shell ligands in the Cu2+ species (55,59); they depend, for example, on the orientation of the imidazole rings with respect to the CuN4 plane in Cu2+ imidazole complexes (57). This finding suggests that the Cu2+ species has similar imidazole environment in all peptides studied here.
EXAFS region
Figs. 2, B and D, show the EXAFS regions of Aβ-Cu2+ spectra (dashed lines) for two groups of samples from Table 1, except for sample 2 (Aβ16-PB50), which showed significant x-radiation damage, and sample 7 (Aβ42-M35Ox), which had a high level of noise. The similarity in the XANES and EXAFS regions within the two groups of measurements ((1, 3, and 4) and (5 and 6)) suggests that geometry of the Cu2+ coordination does not change significantly across the samples within the groups. Therefore, we attempted an MD EXAFS refinement in each group of measurements. Each of the two DFT-optimized global models was used to simultaneously fit the experimental information extracted from the EXAFS regions of Aβ-Cu2+ spectra of the 1, 3, and 4 and then of the 5 and 6 MD sets. All structural parameters and 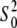 values remained the same throughout each group of data, and ΔE0 was refined for each data set separately. The data sets were weighted by the measurement uncertainties ɛk (Table 1).
values remained the same throughout each group of data, and ΔE0 was refined for each data set separately. The data sets were weighted by the measurement uncertainties ɛk (Table 1).
FIGURE 2.

Model 1 (Asp-1/Glu-11 carboxylate) best fits of (B and D) the Cu K-edge k3-weighted EXAFS and (A and C) the corresponding Fourier-transformed data for five Aβ-Cu2+ samples used in two separate MD refinements: (A and B) for samples (1) Aβ16-PB, (3) Aβ16-PB75, and (4) Aβ16-PB100, and (C and D) for samples (5) Aβ16-PBS and (6) Aβ42M35V-PBS. The solid lines are the theory; the dashed lines correspond to experimental data. The spectra are stacked with offset of (A) 2.5, (B) 5, (C) 2, and (D) 10 units along the vertical axes.
After the thorough MS MD refinement of the DFT-optimized Model 1, further significant improvement of the fitting of the 1, 3 and 4 MD were achieved with the addition of the SS contribution from oxygen atoms (or atoms with occupancy 2) at the distance of 4.4 Å. With two additional refined parameters, the R-factor and χ2 decreased from 2.01% to 1.34% and from 5.980 to 4.108, respectively (Table 2). The significance of this improvement is supported by the F-test (see Data S1), which shows that the calculated F2,95 = 21.646 is seven times the tabulated values of F-distribution, F2,95,0.05 = 3.092, at the common significance level of α = 5%. This finding justified adding two oxygen atoms to the refined Model 1 for the 1, 3, and 4 MD fit. These oxygen atoms are called solvent in Table 2. We can hypothesize that these two oxygen atoms may belong to the carboxylate group from the N-terminal amino acids, such as Asp-1, which may participate in hydrogen bonding with axial water W1 to stabilize the Cu binding site. This observation is consistent with a recent electron paramagnetic resonance spectroscopic analysis (19), where it was proposed that Asp-1 can be involved in metal binding through hydrogen bonding interactions and not via direct equatorial ligation to Cu2+. In this model, the direct coordination of Cu2+ is made by three histidines (His-6, His-13, and His-14), glutamic acid (Glu-11), and axial water, which is involved in hydrogen bonding with the Asp-1 carboxylate.
In the 5 and 6 MD refinement, the Debye-Waller factors for the two solvent oxygen atoms were refined to very low values, suggesting that the coordination number of solvent should be increased or heavier solvent ions should be included. Best fits were obtained with either four oxygen or three chlorine atoms at approximately the same distance of 4.5 Å from the central Cu2+ ion. Given the XANES analysis described above, it is reasonable to suppose that, at the higher content of NaCl per molecule of complex in solution, the Cl− anions can interact with the nonpolar portions of His side chains (54) and/or replace oxygen atoms from water or Asp-1 modeled at ∼4.4 Å in the 1, 3, and 4 MD refinement. For example, it was reported that a Cl− replaced the Asp carboxylate when the Asp of an Asp-Arg salt link in a phosphate-binding protein was mutated to a Gly or Thr (60). The final refinement in Table 2 for samples 5 and 6 included three chlorine atoms at the same distance. The significance of this model improvement is supported by the F-test. With two additional refined parameters, the R-factor and χ2 decreased from 5.980 to 4.108 and from 2.191 to 1.406, respectively (Table 2). The calculated F2,57 = 15.912 is five times the tabulated values of F-distribution, F2,57,0.05 = 3.158, respectively, at the significance level of α = 5%. The best fits of the Model 1 to the EXAFS MD sets are shown in Fig. 2. Fig. 2, B and D, show the k3-weighted χ(k) data within the usable k range ≤12 Å−1 (Table 1). Figs. 1, A and C, show the χ(R) results or the magnitude of the Fourier-transformed χ(k) data. The solid lines represent the theoretical fits to the experiment data, and values for fitted parameters are shown in Table 2.
The best fits obtained with the Cu2+ coordination number of six were also supported by the values of the amplitude reduction factor 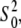 which strongly correlates with the coordination number of the model. The
which strongly correlates with the coordination number of the model. The 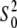 values in Table 2 are within a standard deviation from that expected for the Cu atom (46). The refinement of the five-coordinate model with the omission of w1 (Table 2) produced unphysical
values in Table 2 are within a standard deviation from that expected for the Cu atom (46). The refinement of the five-coordinate model with the omission of w1 (Table 2) produced unphysical 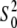 ∼1.5 and significantly higher R factors. The Debye-Waller parameters in Table 2 are in agreement with the values obtained by x-ray absorption for other Cu2+-imidazole systems (43) and those obtained by ab initio calculations for Zn2+-imidazole systems (61,62). The geometries of the ligands were kept restrained within standard uncertainties given for bond lengths and angles and their positions relative to the metal ion, as defined by the positions of the first shell nitrogen/oxygen atom ri and the two angular parameters φi and θi. The θi angles contributed insignificantly to the refinements and were fixed at zero. The EXAFS angles φi and distances ri for histidine residues in Table 2 suggest that these ligands are somewhat rotated within the plane of imidazole rings and that they tend to be close to the Cu2+ ion compared to the DFT-optimized parameters in vacuo for side chains of selected residues unrestricted by the peptide backbone.
∼1.5 and significantly higher R factors. The Debye-Waller parameters in Table 2 are in agreement with the values obtained by x-ray absorption for other Cu2+-imidazole systems (43) and those obtained by ab initio calculations for Zn2+-imidazole systems (61,62). The geometries of the ligands were kept restrained within standard uncertainties given for bond lengths and angles and their positions relative to the metal ion, as defined by the positions of the first shell nitrogen/oxygen atom ri and the two angular parameters φi and θi. The θi angles contributed insignificantly to the refinements and were fixed at zero. The EXAFS angles φi and distances ri for histidine residues in Table 2 suggest that these ligands are somewhat rotated within the plane of imidazole rings and that they tend to be close to the Cu2+ ion compared to the DFT-optimized parameters in vacuo for side chains of selected residues unrestricted by the peptide backbone.
The structural geometry for Model 1 based on the MD refinement of the 1, 3, and 4 data sets is shown in Fig. 3 B. The distorted six-coordinated Cu2+ environment can be viewed as a dissymmetric square pyramidal arrangement. This geometry includes an axially elongated square pyramid formed by three imidazole nitrogen atoms at 1.91–2.06 Å and one carboxylate oxygen atom at 1.94 Å in the near equatorial plane and an axially localized second carboxylate oxygen atom at 2.27 Å. The water molecule is transverse-axially localized at a short distance of 2.04 Å, and the second shell of solvent atoms is at 4.5 Å. The structural geometry does not contradict the XANES features discussed above, even though this structure is at variance with the generally expected Jahn-Teller distortion octahedral model, which was suggested by DFT optimization with two axial oxygen atoms at longer distances. It should be noted that the DFT optimization did not include the second shell effects from the peptide backbone, potentially hydrogen bonded ligands, and distant solvent atoms. The Jahn-Teller distortion in Cu2+ complexes is a dynamic process (63), which suggests that the geometry of Aβ-Cu2+ complexes should be somewhat plastic (64). The obtained structure in solution is in accordance with the recently proposed dynamical view based in particular on XAS, where different six-, five-, and four-coordinated structures can coexist and exchange among themselves in solutions of Cu2+ aqueous complexes, and the classical view of the Jahn-Teller distortion is no longer supported (65,66). The observed distorted Cu2+ environment can be related to the Aβ peptide multifunctional properties including chelating Cu, Zn, and Fe metals with a relatively high affinity.
The EXAFS distances shown in Table 2 are slightly greater than those obtained from the Aβ1-40-Cu2+ (26) and Aβ1-16-Cu2+ (27) EXAFS for pentacoordinated Cu2+ with three nitrogen atoms at 1.85–1.95 Å from the same three histidines (His-6, His-13, and His-14) and two oxygen atoms, one at 2.00–2.06 Å from Tyr-10 and the other one at 1.91–1.95 Å from either a water molecule or from some other amino acid residue. It should be noted that the position of the potential water molecule is even closer to the Cu2+ ion than the water localization (2.04 Å) in our model. Despite the different Cu2+ coordination model suggested in other studies (26,27), the EXAFS spectra for Aβ1-16-Cu2+ and Aβ1-40-Cu2+ are similar to those presented here. However, the statistical significance of the simulations, that is, the R factors = 28–32% (even though they were calculated in k-space including high-frequency noise components) (26,27), and the overall determinacy of the refinement (ratio of independent datapoints to the number of refined parameters of 17/9 (26)) may indicate a somewhat limited accuracy of the proposed model involving tyrosine. In addition, the lack of a proper alignment of the theory to the measured EXAFS spectra (the energy shift parameter ΔE0 ∼10 eV (27)) may result in erroneous minima in the refinement (67).
For comparison with the refined Model 1 (Table 2), we also fitted the DFT-optimized Model 2, which includes Tyr10 residue, to the same EXAFS data sets (see Table S1 and Fig. S5 B in Data S1). The structures also had a distorted six-coordinated copper ion with three imidazole nitrogen atoms (1.91–2.10 Å) and one tyrosinate (1.94–1.97 Å) oxygen atom in an approximately equatorial arrangement. The axial ligands are two water molecules placed asymmetrically at the distances 1.99–2.04 and 2.26–2.32 Å, again shorter than expected for the Jahn-Teller distorted octahedral coordination from the DFT-optimized model (see Table S1 in Data S1). The additional solvent atoms at 4.4 Å were also included in the refinement of Model 2. Although Model 2 has one extra parameter, its refinement resulted in higher values of R factors (1.92% (data 1, 3, and 4) and 3.35% (data 5 and 6)) and χ2 (5.790 (data 1, 3, and 4) and 1.748 (data 5 and 6)) (see Table S1 in Data S1) than those for Model 1 (Table 2). The calculated statistical F-test value of F1,94 = 27.307 for 1, 2 and 4 MD fit is four times greater than the tabulated value of F-distribution (F1,94,0.05 = 3.940) at the common significance level of α = 5%, whereas the F-test value of F1,56 = 10.956 for 5 and 6 MD fit is 2.7 times greater than the tabulated F1,56,0.05 = 4.010 at the same significant level of α = 5% or four times greater of tabulated F1,56,0.1 = 2.739 at the significance level of α = 10% (see Data S1). In other words, the chances that random errors in the obtained data would cause Model 2 (Tyr-10) to fit better are zero for the 1, 3, and 4 MD and ∼0.1% in the case of the lower concentration of Cu2+ peptide complexes and noisier 5 and 6 MD. All these factors indicate that Model 1 is significantly better than Model 2. In addition, Model 1 is consistent with strong evidence that tyrosine (Tyr10) is not the oxygen atom donor in the Aβ-Cu2+ complex (7,8,11,18,19) and the observation that it is a rare ligand in other copper-proteins (28).
CONCLUSIONS
We studied the coordination environment of the Aβ1−16-Cu2+ complex by using low-temperature (16–20 K) MD sets and MS XAS analysis in a range of Aβ-Cu2+ complex solutions. We attempted to address the problems related to the XAS analysis of diluted biologic samples (46), such as low photon counts and high noise at high k values, limited useful k range, and sensitivity to the first few coordination shells (∼5 Å from the Cu atom) due to the low (millimolar) concentration of the soluble Aβ-Cu2+ in solution and relatively weak scattering centers (C, N, and O). The reliability of the MS fitting procedure in EXAFS region analysis was improved by restrained refinements of the DFT-optimized models and by simultaneously fitting MD sets measured at low temperatures. The XANES investigation allowed the refinements of the MD sets to be justified. The Cu2+ reduction by the Aβ peptide was not detected, except at high doses of x-radiation as in the case of sample 2. This finding is consistent with recent cyclic voltammetry measurements showing that the oxidation state of Cu is +2 for all of the complexes Aβ1−16, Aβ1−28, and Aβ1−42 (9,16). The buffer salt (NaCl) concentration did not significantly affect the primary copper binding mode and did not promote the Aβ-Cu2+ complex association in the cases of short Aβ1−16 peptide. In the case of relatively low (1 mM) concentration of the peptide complex in PBS buffer, the secondary coordination sphere involving ether solvents or hydrogen bonded N-terminal residue (for example, Asp-1) was altered by the chaotropic Cl− anions. Neither the additional histidine-bridged Cu2+ ion nor significant distortions of the first Cu2+ ion environment by bridged histidine (as may be expected for dimeric species (20,29)) were observed. However, dimerization can be a specific concentration-dependent phenomenon, and the tendency for the full-length Aβ1−42 peptides to form aggregates may facilitate cross-linking under induced redox chemistry. The structure of the high-affinity Cu2+ binding site is consistent with the hypothesis that the redox activity of the metal ion bound to Aβ can lead to the formation of dityrosine-linked dimers found in AD (68). It is likely that soluble Aβ1–42 initially forms a 1:1 complex with Cu2+, as is seen for shorter Aβ1−16 forms (16). Similar x-ray absorption spectra were observed for the Cu2+ complexed with full-length (Aβ1−42M35(O) and Aβ1−42M35V), truncated (Aβ1−16) peptides, and independently measured (however differently interpreted) spectra for Aβ1−16-Cu2+ and Aβ1−40-Cu2+ (26,27). The oxidized form of Met-35(O) does not provide the oxygen ligand, even though this residue appears to be related to the Aβ toxicity and the ROS production (32).
Contrary to the report of a pentacoordinated structure with Tyr-10 involved (26,27), we found that, under the conditions of our experiments, the preferred Cu2+ binding site in Aβ is a distorted six-coordinated (dissymmetric square pyramidal arrangement) with three histidine (His-6, His-13 and His-14) residues and a carboxylate oxygen (from either Glu-11 or Asp-1 residue) in an approximately equatorial planar arrangement, with the axial ligands consisting of a water molecule and the other carboxylate oxygen also from Glu-11 or Asp-1. It is also possible that that the carboxylate side chain of N-terminal Asp-1 participates in hydrogen bonding via the axial water molecule to stabilize the Cu2+ binding site and Glu-11 coordinates directly the Cu2+ ion along with three histidines. This N-terminus involvement, however, can be compromised by the Cl− anions in high-salt buffers. The water involvement in Cu2+ coordination supports the view that hydration is important in metal binding to the Aβ peptide and that copper binding causes less dehydration of the Aβ peptide compared to zinc (69). The participation of the Aβ N-terminus in Cu2+ coordination also suggests that the pyro-Glu-3 Aβ found in AD plaques (68) should have a different mode of copper binding.
The novel distorted six-coordination mode (3N3O) including three histidines, glutamic or/and aspartic acid, and water obtained from these EXAFS studies reflects the plasticity of the Cu2+ environment in complex with the multifunctional Aβ peptide, which binds Cu, Zn, and Fe metals with a relatively high affinity.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at www.biophysj.org.
Supplementary Material
Acknowledgments
The authors thank Keyla Perez, The University of Melbourne, for the preparation of some Aβ peptides; Dr Steve Heald, the Pacific Northwest Consortium Collaborative Access Team (PNC-CAT); the Argonne National Laboratory, for help in data collections; and Advance Photon Source (APS) synchrotron of Argonne National Laboratory, Argonne, IL, for access to the PNC-CAT 20BM beamline.
The use of APS was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences (contract No. DE-AC02-06CH11357). APS synchrotron access was supported by the Australian Synchrotron Research Program, which is funded by the Commonwealth of Australia under the Major National Research Facilities Program. K.J.B. was partially funded by a grant from the National Health and Medical Research Council of Australia.
Editor: Jill Trewhella.
References
- 1.McLean, C. A., R. A. Cherny, F. W. Fraser, S. J. Fuller, M. J. Smith, K. Beyreuther, A. I. Bush, and C. L. Masters. 1999. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46:860–866. [DOI] [PubMed] [Google Scholar]
- 2.Crouch, P. J., S. M. Harding, A. R. White, J. Camakaris, A. I. Bush, and C. L. Masters. 2008. Mechanisms of Aβ mediated neurodegeneration in Alzheimer's disease. Int. J. Biochem. Cell Biol. 40:181–198. [DOI] [PubMed] [Google Scholar]
- 3.Smith, M. A., P. L. Harris, L. M. Sayre, and G. Perry. 1997. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA. 94:9866–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnham, K. J., C. L. Masters, and A. I. Bush. 2004. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 3:205–214. [DOI] [PubMed] [Google Scholar]
- 5.Smith, D. G., R. Cappai, and K. J. Barnham. 2007. The redox chemistry of the Alzheimer's disease amyloid β peptide. Biochim. Biophys. Acta. 1768:1976–1990. [DOI] [PubMed] [Google Scholar]
- 6.Streltsov, V. 2008. X-ray absorption and diffraction studies of the metal binding sites in amyloid β-peptide. Eur. Biophys. J. 37:257–263. [DOI] [PubMed] [Google Scholar]
- 7.Kowalik-Jankowska, T., M. Ruta, K. Wisniewska, and L. Lankiewicz. 2003. Coordination abilities of the 1–16 and 1–28 fragments of β-amyloid peptide towards copper(II) ions: a combined potentiometric and spectroscopic study. J. Inorg. Biochem. 95:270–282. [DOI] [PubMed] [Google Scholar]
- 8.Karr, J. W., H. Akintoye, L. J. Kaupp, and V. A. Szalai. 2005. N-Terminal deletions modify the Cu2+ binding site in amyloid-β. Biochemistry. 44:5478–5487. [DOI] [PubMed] [Google Scholar]
- 9.Jiang, D., L. Men, J. Wang, Y. Zhang, S. Chickenyen, Y. Wang, and F. Zhou. 2007. Redox reactions of copper complexes formed with different β-amyloid peptides and their neuropathological relevance. Biochemistry. 46:9270–9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma, Q.-F., J. Hu, W.-H. Wu, H.-D. Liu, J.-T. Du, Y. Fu, Y.-W. Wu, P. Lei, Y.-F. Zhao, and Y.-M. Li. 2006. Characterization of copper binding to the peptide amyloid-β(1–16) associated with Alzheimer's disease. Biopolymers. 83:20–31. [DOI] [PubMed] [Google Scholar]
- 11.Syme, C. D., R. C. Nadal, S. E. J. Rigby, and J. H. Viles. 2004. Copper binding to the amyloid-β (Aβ) peptide associated with Alzheimer's disease. J. Biol. Chem. 279:18169–18177. [DOI] [PubMed] [Google Scholar]
- 12.Danielsson, J., R. Pierattelli, L. Banci, and A. Graslund. 2007. High-resolution NMR studies of the zinc-binding site of the Alzheimer's amyloid β-peptide. FEBS J. 274:46–59. [DOI] [PubMed] [Google Scholar]
- 13.Luhrs, T., C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert, and R. Riek. 2005. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 102:17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato, T., P. Kienlen-Campard, M. Ahmed, W. Liu, H. Li, J. I. Elliott, S. Aimoto, S. N. Constantinescu, J. N. Octave, and S. O. Smith. 2006. Inhibitors of amyloid toxicity based on β-sheet packing of Aβ40 and Aβ42. Biochemistry. 45:5503–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miura, T., K. Suzuki, N. Kohata, and H. Takeuchi. 2000. Metal binding modes of Alzheimer's amyloid β-peptide in insoluble aggregates and soluble complexes. Biochemistry. 39:7024–7031. [DOI] [PubMed] [Google Scholar]
- 16.Guilloreau, L., S. Combalbert, A. Sournia-Saquet, H. Mazarguil, and P. Faller. 2007. Redox chemistry of copper-amyloid-β: the generation of hydroxyl radical in the presence of ascorbate is linked to redox-potentials and aggregation state. ChemBioChem. 8:1317–1325. [DOI] [PubMed] [Google Scholar]
- 17.Karr, J. W., L. J. Kaupp, and V. A. Szalai. 2004. Amyloid-β binds Cu2+ in a mononuclear metal ion binding site. J. Am. Chem. Soc. 126:13534–13538. [DOI] [PubMed] [Google Scholar]
- 18.Guilloreau, L., L. Damian, Y. Coppel, H. Mazarguil, M. Winterhalter, and P. Faller. 2006. Structural and thermodynamical properties of CuII amyloid-β16/28 complexes associated with Alzheimer's disease. J. Biol. Inorg. Chem. 11:1024–1038. [DOI] [PubMed] [Google Scholar]
- 19.Karr, J. W., and V. A. Szalai. 2007. Role of aspartate-1 in Cu(II) binding to the amyloid-β peptide of Alzheimer's disease. J. Am. Chem. Soc. 129:3796–3797. [DOI] [PubMed] [Google Scholar]
- 20.Curtain, C. C., F. Ali, I. Volitakis, R. A. Cherny, R. S. Norton, K. Beyreuther, C. J. Barrow, C. L. Masters, A. I. Bush, and K. J. Barnham. 2001. Alzheimer's disease amyloid-β binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J. Biol. Chem. 276:20466–20473. [DOI] [PubMed] [Google Scholar]
- 21.Curtain, C. C., F. E. Ali, D. G. Smith, A. I. Bush, C. L. Masters, and K. J. Barnham. 2003. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer's disease amyloid-β peptide with membrane lipid. J. Biol. Chem. 278:2977–2982. [DOI] [PubMed] [Google Scholar]
- 22.Tickler, A. K., D. G. Smith, G. D. Ciccotosto, D. J. Tew, C. C. Curtain, D. Carrington, C. L. Masters, A. I. Bush, R. A. Cherny, R. Cappai, J. D. Wade, and K. J. Barnham. 2005. Methylation of the imidazole side chains of the Alzheimer disease amyloid-β peptide results in abolition of superoxide dismutase-like structures and inhibition of neurotoxicity. J. Biol. Chem. 280:13355–13363. [DOI] [PubMed] [Google Scholar]
- 23.Zirah, S., S. A. Kozin, A. K. Mazur, A. Blond, M. Cheminant, I. Segalas-Milazzo, P. Debey, and S. Rebuffat. 2006. Structural changes of region 1–16 of the Alzheimer disease amyloid β-peptide upon zinc binding and in vitro aging. J. Biol. Chem. 281:2151–2161. [DOI] [PubMed] [Google Scholar]
- 24.Gaggelli, E., A. Janicka-Klos, E. Jankowska, H. Kozlowski, C. Migliorini, E. Molteni, D. Valensin, G. Valensin, and E. Wieczerzak. 2008. NMR studies of the Zn2+ interactions with rat and human β-amyloid (1–28) peptides in water-micelle environment. J. Phys. Chem. B. 112:100–109. [DOI] [PubMed] [Google Scholar]
- 25.Gaggelli, E., Z. Grzonka, H. Kozlowski, C. Migliorini, E. Molteni, D. Valensin, and G. Valensin. 2008. Structural features of the Cu(II) complex with the rat Aβ(1–28) fragment. Chem. Commun.341–343. [DOI] [PubMed]
- 26.Stellato, F., G. Menestrina, M. D. Serra, C. Potrich, R. Tomazzolli, W. Meyer-Klaucke, and S. Morante. 2006. Metal binding in amyloid β-peptides shows intra- and inter-peptide coordination modes. Eur. Biophys. J. 35:340–351. [DOI] [PubMed] [Google Scholar]
- 27.Minicozzi, V., F. Stellato, M. Comai, M. D. Serra, C. Potrich, W. Meyer-Klaucke, and S. Morante. 2008. Identifying the minimal copper- and zinc-binding site sequence in amyloid-β peptides. J. Biol. Chem. 283:10784–10792. [DOI] [PubMed] [Google Scholar]
- 28.Dokmanić, I., M. Šikić, and S. Tomić. 2008. Metals in proteins: correlation between the metal-ion type, coordination number and the amino-acid residues involved in the coordination. Acta Crystallogr. D. 64:257–263. [DOI] [PubMed] [Google Scholar]
- 29.Smith, D. P., D. G. Smith, C. C. Curtain, J. F. Boas, J. R. Pilbrow, G. D. Ciccotosto, T. L. Lau, D. J. Tew, K. Perez, J. D. Wade, A. I. Bush, S. C. Drew, F. Separovic, C. L. Masters, R. Cappai, and K. J. Barnham. 2006. Copper-mediated amyloid-β toxicity is associated with an intermolecular histidine bridge. J. Biol. Chem. 281:15145–15154. [DOI] [PubMed] [Google Scholar]
- 30.Huang, X., C. S. Atwood, R. D. Moir, M. A. Hartshorn, J. P. Vonsattel, R. E. Tanzi, and A. I. Bush. 1997. Zinc-induced Alzheimer's Aβ1–40 aggregation is mediated by conformational factors. J. Biol. Chem. 272:26464–26470. [DOI] [PubMed] [Google Scholar]
- 31.Narayanan, S., and B. Reif. 2005. Characterization of chemical exchange between soluble and aggregated states of β-amyloid by solution-state NMR upon variation of salt conditions. Biochemistry. 44:1444–1452. [DOI] [PubMed] [Google Scholar]
- 32.Barnham, K. J., G. D. Ciccotosto, A. K. Tickler, F. E. Ali, D. G. Smith, N. A. Williamson, Y. H. Lam, D. Carrington, D. Tew, G. Kocak, I. Volitakis, F. Separovic, C. J. Barrow, J. D. Wade, C. L. Masters, R. A. Cherny, C. C. Curtain, A. I. Bush, and R. Cappai. 2003. Neurotoxic, redox-competent Alzheimer's β-amyloid is released from lipid membrane by methionine oxidation. J. Biol. Chem. 278:42959–42965. [DOI] [PubMed] [Google Scholar]
- 33.Johansson, A. S., J. Bergquist, C. Volbracht, A. Paivio, M. Leist, L. Lannfelt, and A. Westlind-Danielsson. 2007. Attenuated amyloid-β aggregation and neurotoxicity owing to methionine oxidation. Neuroreport. 18:559–563. [DOI] [PubMed] [Google Scholar]
- 34.Ciccotosto, G. D., D. Tew, C. C. Curtain, D. Smith, D. Carrington, C. L. Masters, A. I. Bush, R. A. Cherny, R. Cappai, and K. J. Barnham. 2004. Enhanced toxicity and cellular binding of a modified amyloid β peptide with a methionine to valine substitution. J. Biol. Chem. 279:42528–42534. [DOI] [PubMed] [Google Scholar]
- 35.Mesu, J. G., F. M. F. de Groot, A. M. Beale, and B. M. Weckhuysen. 2006. Probing the influence of x rays on aqueous copper solutions using time-resolved in situ combined video/x-ray absorption near-edge/ultraviolet-visible spectroscopy. J. Phys. Chem. B. 110:17671–17677. [DOI] [PubMed] [Google Scholar]
- 36.Mesu, J. G., A. M. J. van der Eerden, F. M. F. de Groot, and B. M. Weckhuysen. 2005. Synchrotron radiation effects on catalytic systems as probed with a combined in-situ UV-Vis/XAFS spectroscopic setup. J. Phys. Chem. B. 109:4042–4047. [DOI] [PubMed] [Google Scholar]
- 37.Newville, M., B. I. Boyanov, and D. E. Sayers. 1999. Estimation of uncertainties in XAFS data. J. Synchr. Rad. 6:264–265. [DOI] [PubMed] [Google Scholar]
- 38.Ravel, B., and M. Newville. 2005. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for x-ray absorption spectroscopy using IFEFFIT. J. Synchrotron. Radiat. 12:537–541. [DOI] [PubMed] [Google Scholar]
- 39.Newville, M. 2001. EXAFS analysis using FEFF and FEFFIT. J. Synchroton. Radiat. 8:96–100. [DOI] [PubMed] [Google Scholar]
- 40.Cromer, D. T., and D. Liberman. 1970. Relativistic calculation of anomalous scattering factors for x rays. J. Chem. Phys. 53:1891–1898. [Google Scholar]
- 41.Newville, M., P. Līviņš, Y. Yacoby, J. J. Rehr, and E. A. Stern. 1993. Near-edge x-ray-absorption fine structure of Pb: a comparison of theory and experiment. Phys. Rev. B. 47:14126–14131. [DOI] [PubMed] [Google Scholar]
- 42.Zabinsky, S. I., J. J. Rehr, A. Ankudinov, R. C. Albers, and M. J. Eller. 1995. Multiple-scattering calculations of x-ray-absorption spectra. Phys. Rev. B. 52:2995–3009. [DOI] [PubMed] [Google Scholar]
- 43.Binsted, N., R. W. Strange, and S. S. Hasnain. 1992. Constrained and restrained refinement in EXAFS data analysis with curved wave theory. Biochemistry. 31:12117–12125. [DOI] [PubMed] [Google Scholar]
- 44.Engh, R. A., and R. Huber. 1991. Accurate bind and angle parameters for x-ray protein structure refinement. Acta Crystallogr. A. 47:392–400. [Google Scholar]
- 45.Blackburn, N. J., S. S. Hasnain, T. M. Pettingill, and R. W. Strange. 1991. Copper K-extended x-ray absorption fine structure studies of oxidized and reduced dopamine β-hydroxylase. Confirmation of a sulfur ligand to copper(I) in the reduced enzyme. J. Biol. Chem. 266:23120–23127. [PubMed] [Google Scholar]
- 46.Levina, A., R. S. Armstrong, and P. A. Lay. 2005. Three-dimensional structure determination using multiple-scattering analysis of XAFS: applications to metalloproteins and coordination chemistry. Coord. Chem. Rev. 249:141–160. [Google Scholar]
- 47.Becke, A. D. 1993. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98:5648–5652. [Google Scholar]
- 48.Hay, P. J., and W. R. Wadt. 1985. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 82:270–283. [Google Scholar]
- 49.Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, and J. A. Pople. 2004. Gaussian 03, Revision C.02. Gaussian, Inc., Wallingford, CT.
- 50.Hitchman, M. A., L. Kwan, L. M. Englehardt, and A. H. White. 1987. Electron spin resonance and electronic spectra and crystal and molecular structures of copper(II) amino acid complexes. J. Chem. Soc., Dalton Trans.457–465.
- 51.Legge, F. S., G. L. Nyberg, and J. B. Peel. 2001. DFT calculations for Cu-, Ag-, and Au-containing molecules. J. Phys. Chem. A. 105:7905–7916. [Google Scholar]
- 52.Csaszar, P., and P. Pulay. 1984. Geometry optimization by direct inversion in the iterative sub-space. J. Mol. Struct. 114:31–34. [Google Scholar]
- 53.Dennington II, R., T. Keith, J. Millam, K. Eppinnett, W. L. Hovell, and R. Gilliland. 2003. GaussView, Version 3.09. Semichem, Inc., Shawnee Mission, KS.
- 54.Collins, K. D. 1997. Charge density-dependent strength of hydration and biological structure. Biophys. J. 72:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kau, L. S., D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson, and E. I. Solomon. 1987. X-ray absorption edge determination of the oxidation state and coordination number of copper. Application to the type 3 site in Rhus vernicifera laccase and its reaction with oxygen. J. Am. Chem. Soc. 109:6433–6442. [Google Scholar]
- 56.Streltsov, V. A., and J. N. Varghese. 2008. Substrate mediated reduction of copper-amyloid-β complex in Alzheimer's disease. Chem. Comm. 3169–3171. [DOI] [PubMed]
- 57.Strange, R. W., L. Alagna, P. Durham, and S. S. Hasnain. 1990. An understanding of the x-ray absorption near-edge structure of copper(II) imidazole complexes. J. Am. Chem. Soc. 112:4265–4268. [Google Scholar]
- 58.Frank, P., M. Benfatto, B. Hedman, and K. O. Hodgson. 2008. Solution [Cu(amm)]2+ is a strongly solvated square pyramid: a full account of the copper K-edge XAS spectrum within single-electron theory. Inorg. Chem. 47:4126–4139. [DOI] [PubMed] [Google Scholar]
- 59.Fulton, J. L., M. M. Hoffmann, J. G. Darab, B. J. Palmer, and E. A. Stern. 2000. Copper(I) and copper(II) coordination structure under hydrothermal conditions at 325°C: an x-ray absorption fine structure and molecular dynamics study. J. Phys. Chem. A. 104:11651–11663. [Google Scholar]
- 60.Yao, N., P. S. Ledvina, A. Choudhary, and F. A. Quiocho. 1996. Modulation of a salt link does not affect binding of phosphate to its specific active transport receptor. Biochemistry. 35:2079–2085. [DOI] [PubMed] [Google Scholar]
- 61.Poiarkova, A. V., and J. J. Rehr. 1999. Multiple-scattering x-ray-absorption fine-structure Debye-Waller factor calculations. Phys. Rev. B. 59:948–957. [Google Scholar]
- 62.Dimakis, N., and G. Bunker. 2002. Group-fitted ab initio single- and multiple-scattering EXAFS Debye-Waller factors. Phys. Rev. B. 65:201103–201104. [Google Scholar]
- 63.Bersuker, I. B. 2001. Modern aspects of the Jahn-Teller effect theory and applications to molecular problems. Chem. Rev. 101:1067–1114. [DOI] [PubMed] [Google Scholar]
- 64.Rorabacher, D. B. 2004. Electron transfer by copper centers. Chem. Rev. 104:651–698. [DOI] [PubMed] [Google Scholar]
- 65.Chaboy, J., A. Munoz-Paez, F. Carrera, P. J. Merkling, and E. S. Marcos. 2005. Ab initio x-ray absorption study of copper K-edge XANES spectra in Cu(II) compounds. Phys. Rev. B. 71:134208–134215. [Google Scholar]
- 66.Chaboy, J., A. Munoz-Paez, P. J. Merkling, and E. S. Marcos. 2006. The hydration of Cu2+: Can the Jahn-Teller effect be detected in liquid solution? J. Chem. Phys. 124:64509–64518. [DOI] [PubMed] [Google Scholar]
- 67.Shelly, D. K., and R. Bruce. 2007. EXAFS energy shift and structural parameters. AIP Conf. Proc. 882:132–134. [Google Scholar]
- 68.Naylor, R., A. Hill, and K. Barnham. 2008. Neurotoxicity in Alzheimer's disease: is covalently crosslinked Aβ responsible? Eur. Biophys. J. 37:265–268. [DOI] [PubMed] [Google Scholar]
- 69.Yu, H., J. Ren, and X. Qu. 2008. Different hydration changes accompanying copper and zinc binding to amyloid β-peptide: water contribution to metal binding. ChemBioChem. 9:879–882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.