

Abstract
The mechanism of site-specific recognition of DNA by proteins has been a long-standing issue. The DNA glycosylase MutY, for instance, must find the rare 8-oxoguanine-adenine mismatches among the large number of basepairs in the DNA. This protein has a [4Fe-4S] cluster, which is highly conserved in species as diverse as Escherichia Coli and Homo sapiens. The mixed-valent nature of this cluster suggests that charge transfer may play a role in MutY's function. We have studied the energetics of the charge transfer in Bacillus stearothermophilus MutY-DNA complex using multiscale calculation including density functional theory and molecular dynamics. The [4Fe-4S] cluster in MutY is found to undergo 2+ to 3+ oxidation when coupling to DNA through hole transfer, especially when MutY is near an oxoguanine modified base (oxoG). Employing the Marcus theory for electron transfer, we find near optimal Frank-Condon factors for electron transfer from MutY to oxoguanine modified base. MutY has modest selectivity for oxoguanine over guanine due to the difference in oxidation potential. The tunneling matrix element is significantly reduced with the mutation R149W, whereas the mutation L154F reduces the tunneling matrix element as well as the Frank-Condon factor. Both L154F and R149W mutations are known to dramatically reduce or eliminate repair efficiency. We suggest a scenario where the charge transfer leads to a stabilization of the specific binding conformation, which is likely the recognition mode, thus enabling it to find the damaged site efficiently.
INTRODUCTION
Through the life of any organism, DNA is always subject to various types of damage, such as the direct misincorporation of bases occurring during genetic replication or the chemical modification of the polynucleotide from oxidative damage (1). To protect DNA from the potential deleterious and mutagenic effects of such damage, various DNA glycosylases exist in the organism to initiate base excision repair by catalyzing excision of damaged bases from DNA (2,3). The task of efficiently locating damaged bases from the overwhelming excess of native bases on the genome by the repair proteins becomes a challenging search problem (4,5). It is widely believed that proteins can search the target sites rapidly through a combination of one-dimensional diffusion along DNA segments and three-dimensional hopping among DNA segments (6). Concerning the one-dimensional diffusion process, however, the proteins need to have direct contact with the bases to recognize the damaged sites. Interrogating each basepair thoroughly could make the search process slow and inefficient. To solve the problem, a potential explanation involving two search modes with different diffusion rates during the one-dimensional diffusion process has been suggested (4,7); the protein can do fast sliding on the DNA with nonspecific binding while scanning the “normal” segments of the genome and be in the recognition mode through specific binding when near the target sites. The transition between the specific binding mode and the nonspecific-binding mode is accompanied by a conformational change (8,9). However, whether such a switch is purely stochastic or is caused by a specific trigger as the target site is approached remains unclear.
Recently, electron transfer has been hypothesized to play a role in the sensing of DNA damage by MutY (10). MutY is a DNA glycosylase in Escherichia coli that recognizes the 8-oxoguanine:adenine and G:A (to a lesser extent) mismatches (11–15) and removes adenine from the DNA. Like some other DNA base excision repair proteins, such as endonuclease III (16), MutY contains an [4Fe-4S] cluster with undetermined function. The [4Fe-4S] cluster is highly conserved in diverse species from Bacillus stearothermophilus to humans and must have biological relevance. Interestingly, the [4Fe-4S] cluster is not crucial to the stability of MutY; the protein is capable of folding without the cluster. However, the cluster is critical for DNA binding and catalysis (17). In solutions, the stable charge state of the cluster in MutY is [4Fe-4S]2+. Experiments by Barton's group showed the enhancement of the tendency for the cluster to go from the 2+ charge state to the 3+ charge state with MutY bound to DNA-modified gold electrode (10). This inspired them to propose the idea of MutY working in pairs as redox couples to sense DNA damaged sites with the DNA being an intermediate for electron transfer. For electron transfer to be relevant to DNA repair, the rate from the donor to acceptor should be faster than the diffusion rate of the protein along the DNA. This is virtually ensured if the transfer is quantum mechanically coherent.
The fact that hole transport is easier in DNA than excess electron transport and that the oxoguanine has lower oxidation energy than even guanine means that oxoguanines are likely to act as traps for any excess holes in DNA (18). Here we study the energetics of charge transfer in BsMutY-DNA complexes (19), where the trapped hole is then transferred to the MutY, using multiscale calculations including quantum mechanical (QM) calculation, molecular dynamics (MD), and the electron transfer pathway method. We find low energy barriers for hole transfer from oxoguanine to MutY, which suggests that MutY can switch from a 2+ to a 3+ charge state in the vicinity of oxoguanine with a bound hole. We also find that MutY has slight selectivity for oxoguanine over guanine due to the lower oxidation energy of oxoguanine. Also, the electron transfer rates are reduced in BsMutY mutations R149W and L154F compared to wild-type MutY, which is consistent with the observations of reduced DNA binding and glycosylase activities with these two mutations in human MutY homolog (hMYH) (20). With the [4Fe-4S] cluster in the 3+ state, MutY should be more tightly bound to DNA. This should stabilize the specific binding conformation over the nonspecific binding conformation and allow the protein to stay in a recognition mode, finding the target site until it detaches from the DNA.
THEORETICAL METHODS
Marcus theory
The theoretical framework of the electron transfer process is based upon Marcus theory (21). The electron transfer rate is given by
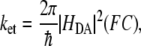 |
where the electron tunneling matrix element HDA describes the donor-acceptor interaction associated with electron tunneling. Assuming the energy functions of the initial state and the final state have quadratic dependence of the reaction coordinate and the Marcus parabolas of the two states have the same curvature (Fig. 1), the Frank-Condon (FC) factor describing the effects associated with nuclear tunneling and with the thermally activated barrier can be expressed as
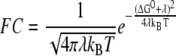 |
with ΔG0 being the free energy change, and λ being the reorganization energy associated with the nuclear relaxation.
FIGURE 1.

The schematics of energy surfaces for electron transfer in the MutY-DNA complex. In the initial state, the [4Fe-4S] cluster in MutY is in 2+ charge state, and a hole is located at the DNA base; in the final state, the [4Fe-4S] cluster is in 3+ charge state, and the DNA base is neutral. Using Marcus theory, three different regimes appear in our calculation: (a) the electron transfer from a wild-type MutY to the oxoguanine in the nonrepair position in DNA is near the optimal region; (b) the electron transfer from a wild-type MutY to the guanine in the nonrepair position is in the inverted region; and (c) the MutY mutation L154F causes the electron transfer to the oxoguanine in the nonrepair position to be in the normal region.
In our study, the tunneling matrix element HDA is estimated by the electron tunneling pathway strategy (22), which has been successful in understanding the electronic coupling in electron transfer proteins like cytochrome c2 (23,24). We calculate the FC factor with the QM treatment for the [4Fe-4S] cluster and the MD treatment for the rest of the protein, DNA, and the water environment. To minimize the wild fluctuations of the free energy calculated directly in MD, we perform free energy perturbation method (25) to calculate the free energy change and reorganization energy of the charge transfer from MutY to DNA. For the calculation of MD, we take the crystal structure of the BsMutY-DNA complex (19) (1RRQ in the Protein Data Bank) as the initial structure and carry out MD for 1 ns after energy minimization.
Electron tunneling pathway
The electron tunneling matrix element is determined by the tunneling energy and the geometry of the bridge between the donor and acceptor. To calculate the donor electron coupling through the bridge, all the bridge orbitals near the HOMO (the highest occupied molecular orbital) close in energy to the tunneling energy need to be included. The contribution of all these bridge states leads to an exponential decay of coupling with distance. Because of the need to include all states and the difficulty of conducting QM calculation on such large many-body systems, simple models have been developed to include the specific nature of the through-bond and through-space electronic interactions in the bridges without losing the essential physics of the tunneling problem. Based upon the through-bond and through-space decay process, the tunneling pathway strategy has been built to estimate HDA (26,27), which can be expressed as
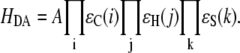 |
Here A is a prefactor that depends upon details of the interaction between the donor or acceptor with the first or the last bond of the tunneling pathway, and 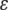 is the decay factor per unit. With the pathway strategy, any protein structure defines a network of pathway decay parameters; every pair of atoms is connected through a covalent bond (C), a hydrogen bond (H), or through space (S). The decay factors corresponding to each connection type have been determined by experiments or simple estimates (28,29) with
is the decay factor per unit. With the pathway strategy, any protein structure defines a network of pathway decay parameters; every pair of atoms is connected through a covalent bond (C), a hydrogen bond (H), or through space (S). The decay factors corresponding to each connection type have been determined by experiments or simple estimates (28,29) with 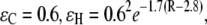 and
and 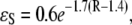 where R is the distance between atoms in units of Å. Here the decay factor through the hydrogen bond arises from approximating the hydrogen bond as two stretched covalent bonds. These values for tunneling factors have been successfully used in understanding interprotein electron transfer in proteins like cytochrome c2 (23,24). Although larger electron coupling through the hydrogen bond (0.51 for the prefactor of
where R is the distance between atoms in units of Å. Here the decay factor through the hydrogen bond arises from approximating the hydrogen bond as two stretched covalent bonds. These values for tunneling factors have been successfully used in understanding interprotein electron transfer in proteins like cytochrome c2 (23,24). Although larger electron coupling through the hydrogen bond (0.51 for the prefactor of 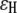 ) was found in some experiments (30), this will not change our results qualitatively but will strengthen the role of the hydrogen bond on the tunneling rate, as indicated in later discussion. Based upon the pathway strategy, we use the tunneling pathway program HARLEM (developed by I. V. Kurnikov, http://www.kurnikov.org/harlem_main.html) to find the best pathway and estimate the optimal electron tunneling rate in the MutY-DNA complex.
) was found in some experiments (30), this will not change our results qualitatively but will strengthen the role of the hydrogen bond on the tunneling rate, as indicated in later discussion. Based upon the pathway strategy, we use the tunneling pathway program HARLEM (developed by I. V. Kurnikov, http://www.kurnikov.org/harlem_main.html) to find the best pathway and estimate the optimal electron tunneling rate in the MutY-DNA complex.
QM/MD calculations
In our calculation, the system is divided into two parts, with the inner shell treated quantum mechanically and the outer shell treated classically. The inner shell includes the donor and acceptor. The donor consists of the [4Fe-4S] cluster and the side chains (-S-CH2-) of the four cysteine residues connected to the cluster, and the acceptor is the single guanine/8-oxoguanine base in DNA. There have been enormous theoretical efforts made to estimate the reduction potential of the [4Fe-4S] cluster (31,32) and single nucleobase (33–36). Here, we use the atomic-based density functional theory program SIESTA (37) to calculate the energy difference between the two charge states of the donor/acceptor. The initial structure is taken from the crystal structure, and the geometry optimization is carried out until the atomic force is <0.04 eV/Å. The calculated energy differences using SIESTA are in good agreement with experimental and other calculated results (see Table S1 in Supplementary Material, Data S1 for more details).
To incorporate the contribution of the outer shell, we obtain the atomic charges of the quantum part by fitting to the electrostatic potential (38) calculated from the QM calculation. The calculated atomic charges are then used as parts of the force field, with other parameters which were used in studies of similar structures (39–41), for the residues of the inner shell for the subsequent classical treatment of the whole protein-DNA complex. With the fixed optimized structure of the [4Fe-4S] cluster from SIESTA, we carried out the free energy calculation for the MutY-DNA complex using the MD package AMBER 8 with the PARM99 force field (42,43) and with a TIP3P water model (44). The system is equilibrated at constant pressure P = 1 atm and at a temperature T = 300 K. The periodic boundary condition is assumed, and the long-range electrostatic interactions are treated by the particle mesh Ewald method (45). The total of 1 ns of MD is simulated for the system with 365 residues in the MutY-DNA complex and 14,630 water molecules. Finally, we replace the inner shell contribution of the free energy change calculated by the MD with the QM results. In other words, the total energy difference between two charge states can be expressed as
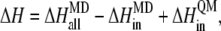 |
where 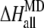 is the energy difference obtained from the MD simulation for the whole protein-DNA complex,
is the energy difference obtained from the MD simulation for the whole protein-DNA complex, 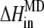 is the energy difference obtained from the MD simulation for the inner shell itself, and
is the energy difference obtained from the MD simulation for the inner shell itself, and 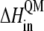 is the energy difference obtained from the QM calculation for the inner shell.
is the energy difference obtained from the QM calculation for the inner shell.
Free energy perturbation
The free energy change between two states can be obtained by the sum of the adiabatic work of transition between the two states (25). The Hamiltonian of the transition state can be expressed as
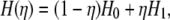 |
where 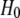 is the Hamiltonian of the initial state,
is the Hamiltonian of the initial state, 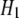 is the Hamiltonian of the final state, and
is the Hamiltonian of the final state, and 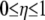 is the parameter associated with the reaction coordinate. The free energy change between the two states is then given by
is the parameter associated with the reaction coordinate. The free energy change between the two states is then given by
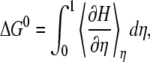 |
where 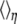 indicates the thermal average at reaction coordinate with parameter η.
indicates the thermal average at reaction coordinate with parameter η.
Assuming that the energy surface of the initial state has the same curvature of the Marcus parabolas as that of the energy surface of the final state, 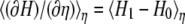 is a linear function of η. It is a good approximation to calculate for a few different transition states instead of integrating over infinitesimal steps to get the free energy change. Hence, the reorganization energy can be obtained by the calculations at the initial and final coordinates, i.e.,
is a linear function of η. It is a good approximation to calculate for a few different transition states instead of integrating over infinitesimal steps to get the free energy change. Hence, the reorganization energy can be obtained by the calculations at the initial and final coordinates, i.e., 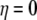 and
and 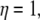 and is given by
and is given by
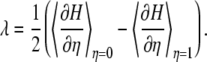 |
This relation was used in studies of the reorganization energy using the linear response approximation approach (23,46,47), and more details about this method can be found in Data S1. We also note that the thermal variations of ΔG0 and λ due to the actual fluctuations about the optimized geometry of the [4Fe-4S] cluster will cancel out as long as the fixed optimized geometry of the cluster in the MD simulation is close to the corresponding zero-point geometry.
RESULTS AND DISCUSSION
Electron tunneling pathway
Using the electron tunneling pathway method, we estimate the optimal (peak FC factor) electron tunneling rates from MutY to DNA for snapshots taken every 2 ps and average the rates over the final 600 ps in the MD run. The time dependence of the root mean-square deviation of the atomic coordinates of the backbone of the protein and DNA in the complex indicates that the system is equilibrated after 400 ps (Fig. 2). The best tunneling pathway found, as shown in Fig. 3, is from the [4Fe-4S] cluster through Cys-198, Arg-149 to the closest DNA base (A17), with the average donor-acceptor distance being 18.2 Å. The Arg-149 has a hydrogen-bonding interaction with the phosphate group of DNA, which provides a good tunneling bridge. Due to the fluctuations of the structures, the electron tunneling rate can fluctuate around the order of 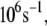 as shown in Fig. 2 b. According to the histograms of the tunneling rates in Fig. 4, most of the states have tunneling rates between
as shown in Fig. 2 b. According to the histograms of the tunneling rates in Fig. 4, most of the states have tunneling rates between 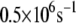 and
and 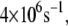 and in some occasions up to
and in some occasions up to 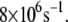 The time dependence of the optimal electron tunneling rate, however, shows bound fluctuations in the 1-ns MD simulation. Assuming the fluctuations of the complex keep bounded over a longer timescale, it is reasonable to study the dynamics of the system from the 1-ns MD simulation. Over the timescale of the electron tunneling, the fluctuations of the structures are averaged out, and the average physical values are those which become relevant. As shown in Table 1, the average optimal electron tunneling rate of the pathway from Fe to A17 in DNA is
The time dependence of the optimal electron tunneling rate, however, shows bound fluctuations in the 1-ns MD simulation. Assuming the fluctuations of the complex keep bounded over a longer timescale, it is reasonable to study the dynamics of the system from the 1-ns MD simulation. Over the timescale of the electron tunneling, the fluctuations of the structures are averaged out, and the average physical values are those which become relevant. As shown in Table 1, the average optimal electron tunneling rate of the pathway from Fe to A17 in DNA is 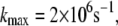 which is well within the rate of the protein sliding to the next neighbor base (48).
which is well within the rate of the protein sliding to the next neighbor base (48).
FIGURE 2.

(a) The root mean-square deviation of the atomic coordinates of the backbone in MutY-DNA complex as a function of time. The system is heated up from 0–300 K for the initial 20 ps with weak positional restraints on the DNA and strong positional restraints on the [4Fe-4S] cluster. The MD equilibration is then carried out without restraints on the DNA at constant pressure P = 1 atm and at temperature T = 300 K for 1 ns. The system is equilibrated after 400 ps. (b) The optimal (peak FC value) electron tunneling rate from the [4Fe-4S] cluster to the nearest nucleobase A17 on the DNA in the BsMutY-DNA complex as a function of time.
FIGURE 3.

The best electron tunneling pathway in the BsMutY-DNA complex after MD simulation. MutY is bound to the specific site of the oxoG-A mismatch, where the adenine (A18) is extruded from the DNA helix and is inserted into an extrahelical pocket in the catalytic domain of MutY. The best pathway (shown in yellow) is the electron transfer from the [4Fe-4S] cluster, through Cys-198, Arg-149 to A17 in the DNA with the donor-acceptor distance being 18.2 Å. The electron tunnels through two hydrogen bonds in the pathway with one between Cys-198 and Arg-149 and the other between Arg-149 and the phosphate group of the backbone of the DNA.
FIGURE 4.

The distributions of the optimal (peak FC factor) electron tunneling rate from the [4Fe-4S] cluster to the nearest nucleobase A17 on the DNA for the 300 snapshots taken every 2 ps from t = 0.4 ns to t = 1 ns in the MD simulation for the MutY-DNA complex. The upper panel is for the wild-type MutY, and the lower panel is for the mutant L154F.
TABLE 1.
Average optimal electron tunneling rate 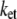 (s−1) in MutY and DNA complex
(s−1) in MutY and DNA complex
| System | S(Cys-198)-A17 | Fe-A17 | S(Cys-198)-C16 | Fe-C16 |
|---|---|---|---|---|
| MutY-DNA (Crystal) | 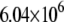 |
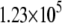 |
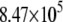 |
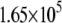 |
| R149W-DNA (Crystal) | 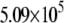 |
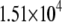 |
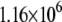 |
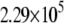 |
| MutY-DNA (MD) | 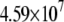 |
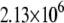 |
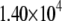 |
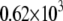 |
| L154F-DNA (MD) | 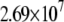 |
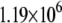 |
- | - |
A rough understanding of the variation of the tunneling matrix elements can be ascertained in a harmonic approximation for the overall fluctuations in the donor-acceptor distance assuming that the tunneling matrix element is simply proportional to exp(−βRDA/2). With the Gaussian distribution, approximately supported by Fig. 5, we obtain a mean value of 〈exp(−βRDA)〉 = exp(−β〈RDA〉) exp(β2kBT/(2K)), where K is the effective spring constant for the harmonic fluctuations of RDA. Hence the mean rate is enhanced slightly over the rate at the mean separation. We can obtain upper/lower bounds for the rate by taking the mean value times exp(±β(δRDA)), where 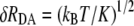 is the harmonic fluctuation induced variance in the donor-acceptor separation. From the histograms of log10(ket) plotted in Fig. 5, we can infer that β(δRDA) ≈ 0.69 (0.3 in base 10 logarithm; see also Data S1), so we expect exp(β2kBT/(2K)) ≈ exp(0.692/2) = 1.3. In fact, we can see that 〈exp(−βRDA)〉 = 1.2 exp(〈−βRDA〉). Also, exp(β(RDA)) = 2, consistent with the upper and lower bound estimates of Table 2.
is the harmonic fluctuation induced variance in the donor-acceptor separation. From the histograms of log10(ket) plotted in Fig. 5, we can infer that β(δRDA) ≈ 0.69 (0.3 in base 10 logarithm; see also Data S1), so we expect exp(β2kBT/(2K)) ≈ exp(0.692/2) = 1.3. In fact, we can see that 〈exp(−βRDA)〉 = 1.2 exp(〈−βRDA〉). Also, exp(β(RDA)) = 2, consistent with the upper and lower bound estimates of Table 2.
FIGURE 5.

The distributions of the logarithm of the optimal electron tunneling rate, 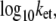 from Fe to A17 in the DNA for the 300 snapshots taken every 2 ps from t = 0.4 ns to t = 1 ns in the MD simulation for the MutY-DNA complex.
from Fe to A17 in the DNA for the 300 snapshots taken every 2 ps from t = 0.4 ns to t = 1 ns in the MD simulation for the MutY-DNA complex.
TABLE 2.
Lower and upper limits of optimal electron tunneling rate 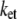 (s−1) for electron transfer from Fe to A17 in DNA
(s−1) for electron transfer from Fe to A17 in DNA
| System | Lower Limit | Mean | Upper Limit |
|---|---|---|---|
| MutY-DNA (MD) | 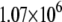 |
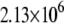 |
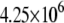 |
| L154F-DNA (MD) | 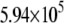 |
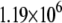 |
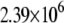 |
We note that the average optimal electron tunneling rate for the structures in MD simulation is much faster than the rate of the same pathway using the crystal structure of the complex. In the crystal structure, the rate of electron transfer through Arg-149 to A17 is about 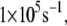 and there is another pathway, where the electron transfers through Pro-200 to another DNA base (C16), having the same order of tunneling rate. After the MD run, the tunneling rate through the latter pathway is significantly reduced to the order of
and there is another pathway, where the electron transfers through Pro-200 to another DNA base (C16), having the same order of tunneling rate. After the MD run, the tunneling rate through the latter pathway is significantly reduced to the order of 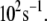 The pathway through Arg-149 to A17 emerges as the dominant pathway. The reason is that the thermal fluctuations make the space gap between Pro-200 and the phosphate of C16 increase from 3.2 Å in the crystal structure to 4.4 Å in the equilibrated MD structure. The fluctuations also reduce the density of atoms between the donor and C16, whereas the hydrogen bond between Arg-149 and the phosphate group of A17 keeps the distance between the [4Fe-4S] cluster and A17 from increasing. It is worth noting that for the MD structures, the tunneling rate for the second best tunneling pathway, other than going through Arg-149, is 10−3–10−4 of the best tunneling rate. Hence although the phases of the various electron tunneling pathways are different, the pathway interference effects will be very small; the single best tunneling pathway is sufficient for us to understand the electron tunneling process.
The pathway through Arg-149 to A17 emerges as the dominant pathway. The reason is that the thermal fluctuations make the space gap between Pro-200 and the phosphate of C16 increase from 3.2 Å in the crystal structure to 4.4 Å in the equilibrated MD structure. The fluctuations also reduce the density of atoms between the donor and C16, whereas the hydrogen bond between Arg-149 and the phosphate group of A17 keeps the distance between the [4Fe-4S] cluster and A17 from increasing. It is worth noting that for the MD structures, the tunneling rate for the second best tunneling pathway, other than going through Arg-149, is 10−3–10−4 of the best tunneling rate. Hence although the phases of the various electron tunneling pathways are different, the pathway interference effects will be very small; the single best tunneling pathway is sufficient for us to understand the electron tunneling process.
MutY mutation R149W
The importance of Arg-149 can be highlighted by the tunneling rate, which is reduced due to the mutation. With the substitution of Arg-149 with tryptophan in the crystal structure of MutY, the coherent charge tunneling rate of the dominant tunneling pathway is reduced by a factor of 10 mainly due to the missing hydrogen bonds between Arg-149 and DNA and between Arg-149 and Cys-198, as shown in Fig. 6. The hydrogen bond provides a good bridge for electron tunneling as it can be simply approximated as two stretched covalent bonds. If the electron coupling through the hydrogen bond is actually larger than the simple approximation, as found in some experiments (30), it will further enhance the ratio by a factor of four in our estimated tunneling rates between the wild-type MutY and the mutation. We note that we did not consider the rate of the incoherent charge transfer through the intervening tryptophan for the mutation. It is possible that the tryptophan interacts with DNA radicals and results in a hopping process on a relevant timescale. However, the missing hydrogen bonds in the tunneling pathway may also make the donor-acceptor distance in R149W-DNA unstable against the thermal fluctuations, which could decrease the charge transfer rate. The decreased transfer rate due to the mutation R149W may explain the reduced efficiency of the recognition of the damaged sites on DNA by MutY. Furthermore, the absence of the hydrogen-bonding interaction between R149 and DNA reduces the binding affinity, which can also affect the ability of recognition.
FIGURE 6.

The best tunneling pathway for electron transfer from MutY mutation R149W to the DNA. There are two through-space tunnelings in this pathway with one between Cys-198 and Trp-149 and the other between Trp-149 and the phosphate group of A17 in the DNA.
Frank-Condon factor
The free energy change and reorganization energy of the charge transfer process in the MutY-DNA complex are calculated using the free energy perturbation method, as shown in Table 3. To investigate if there is selectivity for oxoguanine over guanine in the charge transfer process, we first study the case where the acceptor is set as the guanine (G8), which is two basepairs away from the 8-oxoguanine (OG6) in the repair position, as shown in Fig. 3. Since the protein is expected to switch modes before reaching the damaged site, the acceptor must be located in the nonrepair position when the charge transfer occurs. The energetics of transferring an electron to the oxoguanine in the nonrepair position can be estimated by adding a correction due to the difference in oxidation energy to 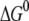 of the case where the acceptor is G8, assuming the reorganization energy does not change when replacing G8 by an oxoguanine. In the MD simulation, we calculate the average of
of the case where the acceptor is G8, assuming the reorganization energy does not change when replacing G8 by an oxoguanine. In the MD simulation, we calculate the average of 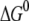 and λ over every 100 ps after the system is equilibrated. We note that the standard deviation of the FC factors obtained by these samplings is within 5%, which is a reasonably small deviation.
and λ over every 100 ps after the system is equilibrated. We note that the standard deviation of the FC factors obtained by these samplings is within 5%, which is a reasonably small deviation.
TABLE 3.
Calculated energies and FC factor in the electron transfer process in MutY-DNA complex
| System |
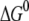 (eV) (eV) |
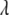 (eV) (eV) |
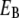 (eV) (eV) |
FC (1/eV) |
|---|---|---|---|---|
| MutY-G (nonrepair position) | −2.83 | 2.65 | 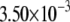 |
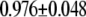 |
| L154F-G (nonrepair position) | −2.76 | 2.72 | 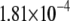 |
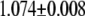 |
| MutY-OxoG (nonrepair position) | −2.56 | 2.65 | 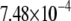 |
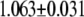 |
| L154F-OxoG (nonrepair position) | −2.50 | 2.72 | 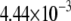 |
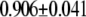 |
| MutY-OxoG (repair position) | −2.61 | 2.38 | 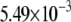 |
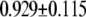 |
| L154F-OxoG (repair position) | −2.67 | 2.47 | 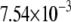 |
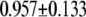 |
We see that the magnitudes of 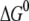 and λ are both ∼2–3 eV, which makes small energy barriers. This suggests that the oxidation of MutY can take place in the vicinity of a hole in DNA, consistent with the observations in recent experiments (49,50). The small difference (0.09 eV) between the magnitude of
and λ are both ∼2–3 eV, which makes small energy barriers. This suggests that the oxidation of MutY can take place in the vicinity of a hole in DNA, consistent with the observations in recent experiments (49,50). The small difference (0.09 eV) between the magnitude of 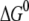 and λ for charge transfer to the oxoguanine in nonrepair position indicates that it is near the optimal region of the charge transfer, whereas in the case of charge transfer to the guanine in the same position, the magnitude of
and λ for charge transfer to the oxoguanine in nonrepair position indicates that it is near the optimal region of the charge transfer, whereas in the case of charge transfer to the guanine in the same position, the magnitude of 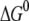 is larger than λ by 0.18 eV, indicating the system shifts toward the inverted region, as shown in Fig. 1. The energy barrier for charge transfer to the oxoguanine in nonrepair position is fivefold smaller than that for transfer to the guanine in the same position, which suggests there is selectivity for oxoguanine over guanine. However, since the energy barriers are both small compared to
is larger than λ by 0.18 eV, indicating the system shifts toward the inverted region, as shown in Fig. 1. The energy barrier for charge transfer to the oxoguanine in nonrepair position is fivefold smaller than that for transfer to the guanine in the same position, which suggests there is selectivity for oxoguanine over guanine. However, since the energy barriers are both small compared to 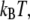 the difference in the values of the FC factor is modest. MutY has slight selectivity of oxoguanine over guanine if there is a hole at the nucleobase. Nevertheless, the lower oxidation energy of oxoguanine makes the hole more likely to be trapped in the oxoguanine than the guanine. The difference of the oxidation energy between the oxoguanine modified base (oxoG) and G in our calculation is ∼0.3 eV (see Table S1 in Data S1), which results in
the difference in the values of the FC factor is modest. MutY has slight selectivity of oxoguanine over guanine if there is a hole at the nucleobase. Nevertheless, the lower oxidation energy of oxoguanine makes the hole more likely to be trapped in the oxoguanine than the guanine. The difference of the oxidation energy between the oxoguanine modified base (oxoG) and G in our calculation is ∼0.3 eV (see Table S1 in Data S1), which results in 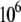 times larger probability for a hole to be trapped in oxoG than G.
times larger probability for a hole to be trapped in oxoG than G.
The large value of 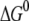 with negative sign, however, indicates that the electron transfer from 2+ MutY to DNA with a bound hole is a downhill process and there is a huge energy barrier for the back electron transfer. The electron transfer process is not reversible. Once MutY switches from a 2+ to a 3+ charge state, it remains in the 3+ state until finding the damaged site. We also study the case where OG6 in the repair position is set as the acceptor. We see that the FC factor is ∼87% of that in the case where the oxoguanine is in the nonrepair position. This result indicates that the electron transfer is more likely to happen before MutY reaches the damaged site. In addition, the energy barrier for the back electron transfer is larger when the acceptor is in the repair position, which makes the protein more stuck in the recognition mode at the damaged site to carry out the base excision. We note that the structure we study here is MutY bound specifically to the target site on DNA. Different conformations of MutY on DNA may lead to different electron transfer rates between MutY and DNA. Energetics and reversibility of the electron transfer, when MutY is in the nonspecific binding conformation, remains to be studied.
with negative sign, however, indicates that the electron transfer from 2+ MutY to DNA with a bound hole is a downhill process and there is a huge energy barrier for the back electron transfer. The electron transfer process is not reversible. Once MutY switches from a 2+ to a 3+ charge state, it remains in the 3+ state until finding the damaged site. We also study the case where OG6 in the repair position is set as the acceptor. We see that the FC factor is ∼87% of that in the case where the oxoguanine is in the nonrepair position. This result indicates that the electron transfer is more likely to happen before MutY reaches the damaged site. In addition, the energy barrier for the back electron transfer is larger when the acceptor is in the repair position, which makes the protein more stuck in the recognition mode at the damaged site to carry out the base excision. We note that the structure we study here is MutY bound specifically to the target site on DNA. Different conformations of MutY on DNA may lead to different electron transfer rates between MutY and DNA. Energetics and reversibility of the electron transfer, when MutY is in the nonspecific binding conformation, remains to be studied.
MutY mutation L154F
We also study the influence of the mutation L154F on the charge transfer rate. The range of the most probable tunneling rate for L154F is noticeably smaller than that in the wild-type MutY. The histograms of the optimal electron tunneling rates in Fig. 4 show that most of the states have tunneling rates between 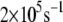 and
and 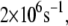 and the largest tunneling rate is
and the largest tunneling rate is 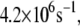 whereas the most probable rate for the wild-type MutY ranges from
whereas the most probable rate for the wild-type MutY ranges from 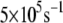 to
to 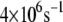 and can be as large as
and can be as large as 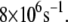 On average, the optimal electron tunneling rate decreases by 50% with the substitution of Leu-154 with phenylalanine, as shown in Table 2. The average donor-acceptor distance in L154F is found to be larger than that in wild-type MutY by ∼0.3 Å. Using the decay exponent
On average, the optimal electron tunneling rate decreases by 50% with the substitution of Leu-154 with phenylalanine, as shown in Table 2. The average donor-acceptor distance in L154F is found to be larger than that in wild-type MutY by ∼0.3 Å. Using the decay exponent 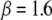 Å−1, this can justify the decrease in the tunneling rate with the mutation. The increase in the donor-acceptor distance may result from the ring-like side chain of the phenylalanine, which takes more space than leucine does.
Å−1, this can justify the decrease in the tunneling rate with the mutation. The increase in the donor-acceptor distance may result from the ring-like side chain of the phenylalanine, which takes more space than leucine does.
As shown in Fig. 7, the side chain of Phe-154 points away from DNA and the [4Fe-4S] cluster, and it can push neighbor residues away, which eventually affects the [4Fe-4S] loop. The effect on the loop may not be significant enough to change the binding affinity to DNA, but such a slight increase in donor-acceptor distance is able to reduce the tunneling matrix element noticeably, due to the exponential decay with distance. As to the FC factor, with the substitution of Leu-154 with phenylalanine, we find that for the electron transfer from MutY to an oxoguanine in the nonrepair position on DNA the magnitude of 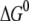 decreases by ∼0.06 eV, whereas λ increases by 0.07 eV, compared to the wild-type case. This shifts the system toward the normal region
decreases by ∼0.06 eV, whereas λ increases by 0.07 eV, compared to the wild-type case. This shifts the system toward the normal region 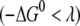 of charge transfer, as shown in Fig. 1. Consequently, the energy barrier is increased by one order of magnitude, and the FC factor is reduced by 10%. The combination of decrease in the electron tunneling matrix element and the modest decrease in the FC factor due to the mutation can reduce the possibility of transition from a 2+ to a 3+ state for MutY and affect its efficiency of recognition of the mismatches.
of charge transfer, as shown in Fig. 1. Consequently, the energy barrier is increased by one order of magnitude, and the FC factor is reduced by 10%. The combination of decrease in the electron tunneling matrix element and the modest decrease in the FC factor due to the mutation can reduce the possibility of transition from a 2+ to a 3+ state for MutY and affect its efficiency of recognition of the mismatches.
FIGURE 7.

The comparison of the structures of MutY mutation L154F and wild-type MutY-DNA complexes after MD simulation. The two structures are aligned along the backbone of Arg-149. The thin lines represent wild-type MutY, and the thick sticks represent the structure of L154F. Both are the average structures over the final 600 ps after equilibration in MD simulation.
Implications for DNA repair
A rapid search of specific DNA targets by proteins involves a combination of one-dimensional diffusion along the DNA and three-dimensional diffusion among DNA segments. The diffusion rate along DNA depends upon the activation barrier that the protein needs to overcome to slide to the adjacent site. Specific binding to DNA results in slow diffusion with a large activation barrier for translocation, whereas nonspecific binding allows fast sliding along DNA. The fast sliding rates have been measured for DNA glycosylases hOgg1 and MutM recently (48). Switching from nonspecific binding to specific binding is believed to be accompanied by a conformational change. The binding of a protein to DNA typically covers up to 10 basepairs. Presumably the sliding diffusion constant for MutY is similar to the measured diffusion constant for MutM, i.e., 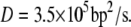 Hence, the time that MutY spends on scanning 10 basepairs is ∼0.15 ms. There may not be enough time for MutY to complete the full process of conformational switch to specific binding while in contact with the damaged site, if the switching initiates only after it reaches the damaged site.
Hence, the time that MutY spends on scanning 10 basepairs is ∼0.15 ms. There may not be enough time for MutY to complete the full process of conformational switch to specific binding while in contact with the damaged site, if the switching initiates only after it reaches the damaged site.
The redox process for MutY provides a way to slow down the diffusion along DNA. MutY spends most of its time in the stable 2+ charge state while in the cycle of searching along DNA, dissociating from DNA, and translocating to other segments of DNA to scan along DNA again. The charge transfer takes place when MutY is in the vicinity of a hole in DNA. The low oxidation potential of 8-oxoguanine makes it easy to trap the hole. The sacrificial role of oxoguanine in the protection of other DNA bases from oxidative damage has also been suggested (51). The stability of the hole bound on the oxoguanine is relevant for the charge transfer process to complete. The lifetime of a hole trapped on an isolated oxoguanine has been shown to be in the timescale of milliseconds to seconds (52), which is much longer than the charge transfer time we compute here.
A recent study (53) showed a lifetime as large as 15–20 s for the oxoguanine radical in a double-stranded DNA in the presence of superoxide dismutases, which are important in vivo to deactivate superoxide radicals to the less reactive H2O2 and O2 molecules. Such a long lifetime may actually be stable enough for the MutY to complete the entire process of DNA scanning, which can be estimated to be <10 s for E. coli with 5 × 106 DNA basepairs and 30 MutY molecules (54) in a cell, using the three-dimensional/one-dimensional diffusion model (4). Our findings that MutY can switch from the 2+ state to a 3+ state through hole transfer, especially in the vicinity of the oxoguanine with a bound hole, suggest that MutY switches to the slow diffusion mode most likely when it gets close to the damaged site. It is possible that a hole can transfer over longer distances in DNA (55). The fast coherent charge transfer makes MutY activatable within 5–10 basepairs from the damaged site even though it is outside the optimal region of the nonrepair position in our calculation.
Our study with the structure of MutY bound specifically to the target site does not imply that the charge transfer triggers the switch from the nonspecific binding mode to the specific binding mode. Rather, it implies that switching from a 2+ to a 3+ state for MutY stabilizes the specific binding conformation. It has been postulated that proteins can continuously and stochastically probe DNA bases with the conformation changing between nonspecific and specific binding modes, but the specific binding conformation is not stable during the fast searching process (4). The transition from a 2+ to a 3+ state of MutY in the vicinity of the oxoguanine with a bound hole helps stabilize the specific binding conformation. The stronger DNA binding affinity for 3+ MutY (which is at least 103 times greater than the affinity for 2+ MutY, as indicated from the shift in oxidation potential observed when MutY binds to DNA (56)) slows down the diffusion rate and increases its binding time on DNA, which allows MutY to recognize the damaged site before dissociation.
Regarding the competition between electron transfer and diffusion for MutY, the time needed to slide for one basepair along DNA is 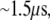 which is long enough for the electron transfer to take place. Electron transfer from the [4Fe-4S] cluster to the nearest nucleobase (A17) takes
which is long enough for the electron transfer to take place. Electron transfer from the [4Fe-4S] cluster to the nearest nucleobase (A17) takes 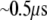 in our calculation. The strong covalence between Fe and S atoms of the cysteine residues connected to the [4Fe-4S] cluster can even shorten the transfer time to 20 ns since the sulfur atom of Cys-198 becomes the edge of the donor. Considering that the electron has to tunnel farther through the hydrogen bonds to the paired base (T7) in the opposite DNA strand to transfer to the damaged site with a bound hole through base stacking, the time needed remains within 1
in our calculation. The strong covalence between Fe and S atoms of the cysteine residues connected to the [4Fe-4S] cluster can even shorten the transfer time to 20 ns since the sulfur atom of Cys-198 becomes the edge of the donor. Considering that the electron has to tunnel farther through the hydrogen bonds to the paired base (T7) in the opposite DNA strand to transfer to the damaged site with a bound hole through base stacking, the time needed remains within 1 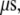 which is still able to occur before the protein diffuses away.
which is still able to occur before the protein diffuses away.
As for the mutants, the missing hydrogen bonds between MutY and DNA in the best tunneling pathway for the mutation R149W not only increase the tunneling time by one order of magnitude but also make the equilibrated donor-acceptor distance larger due to the lack of the ability to keep Trp-149 and the phosphate group of DNA together. The reduction in the optimal electron tunneling rate for R149W can explain the observed severe defect in oxoG-A binding and glycosylase activities (20); though in this case, the deficiency in repair ability could come from the reduced binding affinity as well. The effect on the tunneling rate induced by the mutation L154F, where the mutated residue is not on the best tunneling pathway, however, strengthens the case for the role of electron tunneling in DNA repair. In this case, the slight increase in the donor-acceptor distance has a greater impact on the tunneling rate, due to exponential decay with distance, than on the binding affinity. This is consistent with the observation that the repair efficiency is partially reduced due to the corresponding mutation V232F of hMYH (20).
We note that our study of charge transfer in the DNA-MutY complex is based upon the crystal structure of the specifically bound complex. When the protein changes from specific binding to nonspecific binding conformation, the most significant changes involve those residues which have direct contacts with DNA bases. The electrostatic interactions and hydrogen bonds between DNA phosphates and protein side chains in the specific binding mode remain preserved in the nonspecific complex (9,57). Based upon the facts, the best electron tunneling pathway involved with the hydrogen bond between Arg-149 in MutY and DNA phosphate should remain unchanged in the nonspecific binding mode. Although the DNA contacting protein side chains may have more mobility in the nonspecific complex with motions on the microsecond to millisecond timescale (9), the high flexibility of the complex may make efficient searching of the fast tunneling pathway, and MutY can still be activated when Arg-149 comes close to the DNA backbone. The electron tunneling rate calculated using the crystal structure should be similar to that in nonspecific binding mode.
We have elaborated here the DNA site-specific search process by MutY before the recognition of the damaged bases. For MutY to switch from the 3+ state in the recognition mode back to the 2+ state after excising the adenine base from the oxogG-A mismatch, it has to gain back an electron. Since the charge transfer is not reversible, this might happen in the following ways. MutY might accept an electron during the process of catalyzing the excision of the adenine. It may also cooperate with other repair proteins or replication enzymes (58) after initiating the repair process and accept an electron from them; MutY may be a mark on the target site for other polymerases. A much simpler possibility is that the 3+ MutY can still dissociate from DNA after catalyzing base excision and obtain the electron in the solution, where the 2+ state is more stable. In addition to coupling with DNA, it is possible that MutY may be activated independent of DNA in cellular conditions. The redox activity of the Fe-S complex may be used by the cell to preemptively prepare itself for DNA oxidative damage, especially when cellular conditions become ripe for damage due to oxidative stress. Such possibilities of protecting DNA from potential oxidative damage for MutY or other DNA repair proteins consisting of the Fe-S complex remain to be explored in the future.
CONCLUSION
In this work, we have addressed the role of MutY's [4Fe-4S] complex in the protein's function of DNA damage recognition. Our calculations strongly support the possibility of charge transfer between MutY and DNA, especially as it nears its intended target site containing an oxoguanine. Using a combined QM/molecular mechanics calculation, we have shown that there can be a rapid electron transfer from the complex to an oxoguanine with an extra hole in the process of changing the complex from a 2+ to a 3+ state. We suggest that this can slow down the search process by stabilizing the more tightly bound conformation. This allows the protein enough time to interrogate the specific bases and recognize the damaged sites. We have shown that two mutations, L154F and R149W, which are known to impair the repair efficacy, affect the charge transfer process negatively, thus strengthening the case for charge transfer in the recognition process. We hope our work will stimulate further work on the role of charge transfer between proteins and DNA in the broader context of the recognition of specific DNA sites by DNA binding proteins.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank J. N. Onuchic and O. Miyashita for discussions on the electron tunneling pathway strategy.
This work was supported by the U.S. Department of Energy Office of Basic Energy Sciences, Division of Materials Research and by the Center for Theoretical and Biological Physics through National Science Foundation grant no. PHY0216576.
Editor: Steven D. Schwartz.
References
- 1.Cooke, M. S., M. D. Evans, M. Dizdaroglu, and J. Lunec. 2003. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 17:1195–1214. [DOI] [PubMed] [Google Scholar]
- 2.Michaels, M. L., and J. H. Miller. 1992. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J. Bacteriol. 174:6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tchou, J., and A. P. Grollman. 1993. Repair of DNA containing the oxidatively-damaged base, 8-oxoguanine. Mutat. Res. 299:277–287. [DOI] [PubMed] [Google Scholar]
- 4.Slutsky, M., and L. A. Mirny. 2004. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophys. J. 87:4021–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zharkov, D. O., and A. P. Grollman. 2005. The DNA trackwalkers: principles of lesion search and recognition by DNA glycosylases. Mutat. Res. 577:24–54. [DOI] [PubMed] [Google Scholar]
- 6.von Hippel, P. H., and O. G. Berg. 1989. Facilitated target location in biological systems. J. Biol. Chem. 264:675–678. [PubMed] [Google Scholar]
- 7.Winter, R. B., O. G. Berg, and P. H. von Hippel. 1981. Diffusion-driven mechanisms of protein translocation on nucleic acids. 3. The Escherichia coli lac repressor-operator interaction: kinetic measurements and conclusions. Biochemistry. 20:6961–6977. [DOI] [PubMed] [Google Scholar]
- 8.Spolar, R. S., and M. T. Record. 1994. Coupling of local folding to site-specific binding of proteins to DNA. Science. 263:777–784. [DOI] [PubMed] [Google Scholar]
- 9.Kalodimos, C. G., N. Biris, A. M. J. J. Bonvin, M. M. Levandoski, M. Guennuegues, R. Boelens, and R. Kaptein. 2004. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science. 305:386–389. [DOI] [PubMed] [Google Scholar]
- 10.Boon, E. M., A. L. Livingston, N. H. Chmiel, S. S. David, and J. K. Barton. 2003. DNA-mediated charge transport for DNA repair. Proc. Natl. Acad. Sci. USA. 100:12543–12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Au, K. G., M. Cabrera, J. H. Miller, and P. Modrich. 1988. Escherichia coli MutY gene product is required for specific A-G to C-G mismatch correction. Proc. Natl. Acad. Sci. USA. 85:9163–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Au, K. G., S. Clark, J. H. Miller, and P. Modrich. 1989. Escherichia coli MutY gene encodes an adenine glycosylase active on G-A mispairs. Proc. Natl. Acad. Sci. USA. 86:8877–8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michaels, M. L., C. Cruz, A. P. Grollman, and J. H. Miller. 1992. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl. Acad. Sci. USA. 89:7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michaels, M. L., J. Tchou, A. P. Grollman, and J. H. Miller. 1992. A repair system for 8-oxo-7,8-dihydrodeoxyguanine. Biochemistry. 31:10964–10968. [DOI] [PubMed] [Google Scholar]
- 15.Porello, S. L., A. E. Leyes, and S. S. David. 1988. Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry. 37:14756–14764. [DOI] [PubMed] [Google Scholar]
- 16.Kuo, C. F., D. E. McRee, C. L. Fischer, S. F. O'Handley, R. P. Cunningham, and J. A. Tainer. 1992. Atomic structure of the DNA repair [4Fe-4S] enzyme endonuclease III. Science. 258:434–440. [DOI] [PubMed] [Google Scholar]
- 17.Porello, S. L., M. J. Cannon, and S. S. David. 1988. A substrate recognition role for the [4Fe-4S]2+ cluster of the DNA repair glycosylase MutY. Biochemistry. 37:6465–6475. [DOI] [PubMed] [Google Scholar]
- 18.Steenken, S., S. V. Jovanovic, M. Bietti, and K. Bernhard. 2000. The trap depth (in DNA) of 8-oxo-7,8-dihydro-2′ deoxyguanosine as derived from electron-transfer equilibria in aqueous solution. J. Am. Chem. Soc. 122:2373–2374. [Google Scholar]
- 19.Fromme, J. C., A. Banerjee, S. J. Huang, and G. L. Verdine. 2004. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 427:652–656. [DOI] [PubMed] [Google Scholar]
- 20.Bai, H. B., S. Jones, X. Guan, T. M. Wilson, J. R. Sampson, J. P. Cheadle, and A. L. Lu. 2005. Functional characterization of two human MutY homolog (hMYH) missense mutations (R227W and V232F) that lie within the putative hMSH6 binding domain and are associated with hMYH polyposis. Nucleic Acids Res. 33:597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcus, R. A., and N. Sutin. 1985. Electron transfer in chemistry and biology. Biochim. Biophys. Acta. 811:265–322. [Google Scholar]
- 22.Beratan, D. N., and J. N. Onuchic. 1996. The protein bridge between redox centers. In Protein Electron Transfer. D. S. Bendall, editor. BIOS Scientific Publishers, Oxford, UK. 23–42.
- 23.Miyashita, O., M. Y. Okamura, and J. N. Onuchic. 2003. Theoretical understanding of the interprotein electron transfer between cytochrome c(2) and the photosynthetic reaction center. J. Phys. Chem. B. 107:1230–1241. [Google Scholar]
- 24.Miyashita, O., M. Y. Okamura, and J. N. Onuchic. 2005. Interprotein electron transfer from cytochrome c(2) to photosynthetic reaction center: tunneling across an aqueous interface. Proc. Natl. Acad. Sci. USA. 102:3558–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kollman, P. A. 1993. Free energy calculations: applications to chemical and biochemical phenomena. Chem. Rev. 93:2395–2417. [Google Scholar]
- 26.Beratan, D. N., J. N. Onuchic, and J. J. Hopfield. 1987. Electron tunneling through covalent and noncovalent pathways in proteins. J. Chem. Phys. 86:4488–4498. [Google Scholar]
- 27.Beratan, D. N., J. N. Betts, and J. N. Onuchic. 1992. Tunneling pathway and redox-state-dependent electronic couplings at nearly fixed distance in electron transfer proteins. J. Phys. Chem. 96:2852–2855. [Google Scholar]
- 28.Closs, G. L., L. T. Calcaterra, N. J. Green, K. W. Penfield, and J. R. Miller. 1986. Distance, stereoelectronic effects, and the Marcus inverted region in intramolecular electron transfer in organic radical anions. J. Phys. Chem. 90:3673–3683. [Google Scholar]
- 29.Onuchic, J. N., and D. N. Beratan. 1990. A predictive theoretical model for electron tunneling pathways in proteins. J. Chem. Phys. 92:722–733. [Google Scholar]
- 30.de Rege, P. J. F., S. A. Williams, and J. T. Michael. 1995. Direct evaluation of electronic coupling mediated by hydrogen bonds: implications for biological electron transfer. Science. 269:1409–1413. [DOI] [PubMed] [Google Scholar]
- 31.Mouesca, J., J. L. Chen, L. Noodleman, D. Bashford, and D. A. Case. 1994. Density functional/Poisson-Boltzmann calculations of redox potentials for iron-sulfur clusters. J. Am. Chem. Soc. 116:11898–11914. [Google Scholar]
- 32.Torres, R. A., T. Lovell, L. Noodleman, and D. A. Case. 2003. Density functional-and reduction potential calculations of Fe4S4 clusters. J. Am. Chem. Soc. 125:1923–1936. [DOI] [PubMed] [Google Scholar]
- 33.Russo, N., M. Toscano, and A. Grand. 2000. Theoretical determination of electron affinity and ionization potential of DNA and RNA bases. J. Comput. Chem. 21:1243–1250. [Google Scholar]
- 34.Baik, M. H., J. S. Silverman, I. V. Yang, P. A. Ropp, V. A. Szalai, W. T. Yang, and H. H. Thorp. 2001. Using density functional theory to design DNA base analogues with low oxidation potentials. J. Phys. Chem. B. 105:6437–6444. [Google Scholar]
- 35.Preuss, M., W. G. Schmidt, K. Seino, J. Furthmüller, and F. Bechstedt. 2004. Ground- and excited-state properties of DNA base molecules from plane-wave calculations using ultrasoft pseudopotentials. J. Comput. Chem. 25:112–122. [DOI] [PubMed] [Google Scholar]
- 36.Crespo-Hernández, C. E., R. Arce, Y. Ishikawa, L. Gorb, J. Leszczynski, and D. M. Close. 2004. Ab initio ionization energy thresholds of DNA and RNA bases in gas phase and in aqueous solution. J. Phys. Chem. A. 108:6373–6377. [Google Scholar]
- 37.Soler, J. M., E. Artacho, J. D. Gale, A. Garcia, J. Junquera, P. Ordejón, and D. Sánchez-Portal. 2002. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter. 14:2745–2779. [DOI] [PubMed] [Google Scholar]
- 38.Bayly, C. I., P. Cieplak, W. D. Cornell, and P. A. Kollman. 1993. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97:10269–10280. [Google Scholar]
- 39.Meuwly, M., and M. Karplus. 2003. Theoretical investigations of Ferredoxin I: The possible role of internal water molecules on the coupled electron proton transfer reaction. Faraday Discuss. 124:297–313. [DOI] [PubMed] [Google Scholar]
- 40.Beck, B. W., J. B. Koerner, and T. Ichiye. 1999. Ab initio quantum mechanical study of metal substitution in analogues of rubredoxin: implications for redox potential control. J. Phys. Chem. B. 103:8006–8015. [Google Scholar]
- 41.Miller, J. H., C. C. P. Fan-Chiang, T. P. Straatsma, and M. A. Kennedy. 2003. 8-Oxoguanine enhances bending of DNA that favors binding to glycosylases. J. Am. Chem. Soc. 125:6331–6336. [DOI] [PubMed] [Google Scholar]
- 42.Pearlman, D. A., D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham, S. DeBolt, D. Ferguson, G. Seibel, and P. Kollman. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91:1–41. [Google Scholar]
- 43.Wang, J., P. Cieplak, and P. A. Kollman. 2000. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21:1049–1074. [Google Scholar]
- 44.Jorgensen, W. L., J. Chandrasekhar, J. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 45.Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 46.Lee, F. S., Z. T. Chu, M. B. Bolger, and A. Warshel. 1992. Calculations of antibody-antigen interactions: microscopic and semi-microscopic evaluation of the free energies of binding of phosphorylcholine analogs to McPC603. Protein Eng. 5:215–228. [DOI] [PubMed] [Google Scholar]
- 47.Aqvist, J., C. Medina, and J. E. Samuelsson. 1994. New method for predicting binding-affinity in computer-aided drug design. Protein Eng. 7:385–391. [DOI] [PubMed] [Google Scholar]
- 48.Blainey, P. C., A. M. van Oijent, A. Banerjee, G. L. Verdine, and X. S. Xie. 2006. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. USA. 103:5752–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yavin, E., A. K. Boal, E. D. A. Stemp, E. M. Boon, A. L. Livingston, V. L. O'Shea, S. S. David, and J. K. Barton. 2005. Protein-DNA charge transport: redox activation of a DNA repair protein by guanine radical. Proc. Natl. Acad. Sci. USA. 102:3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yavin, E., E. D. A. Stemp, V. L. O'Shea, S. S. David, and J. K. Barton. 2006. Electron trap for DNA-bound repair enzymes: a strategy for DNA-mediated signaling. Proc. Natl. Acad. Sci. USA. 103:3610–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanvah, S., and G. B. Schuster. 2005. The sacrificial role of easily oxidizable sites in the protection of DNA from damage. Nucleic Acids Res. 33:5133–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brett, A. M. O., J. A. P. Piedade, and S. H. P. Serrano. 2000. Electrochemical oxidation of 8-oxoguanine. Electroanalysis. 12:969–973. [Google Scholar]
- 53.Misiaszek, R., Y. Uvaydov, C. Crean, N. E. Geacintov, and V. Shafirovich. 2005. Combination reactions of superoxide with 8-oxo-7,8-dihyroguanine radicals in DNA. J. Biol. Chem. 280:6293–6300. [DOI] [PubMed] [Google Scholar]
- 54.Demple, B., and L. Harrison. 1994. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 63:915–948. [DOI] [PubMed] [Google Scholar]
- 55.Delaney, S., and J. K. Barton. 2003. Long-range DNA charge transport. J. Org. Chem. 68:6475–6483. [DOI] [PubMed] [Google Scholar]
- 56.Boal, A. K., E. Yavin, and J. K. Barton. 2007. DNA repair glycosylases with a [4Fe-4S] cluster: a redox cofactor for DNA-mediated charge transport? J. Inorg. Biochem. 101:1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iwahara, J., M. Zweckstetter, and G. M. Clore. 2006. NMR structural and kinetic characterization of a homeodomain diffusing and hopping on nonspecific DNA. Proc. Natl. Acad. Sci. USA. 103:15062–15067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu, Y. S., A. Parker, T. M. Wilson, H. B. Bai, D. Y. Chang, and A. L. Lu. 2003. Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J. Biol. Chem. 277:11135–11142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.