

Abstract
Interpreting the results of plasma human immunodeficiency virus type 1 (HIV-1) genotypic drug-resistance tests is one of the most difficult tasks facing clinicians caring for HIV-1–infected patients. There are many drug-resistance mutations, and they arise in complex patterns that cause varying levels of drug resistance. In addition, HIV-1 exists in vivo as a virus population containing many genomic variants. Genotypic-resistance testing detects the drug-resistance mutations present in the most common plasma virus variants but may not detect drug-resistance mutations present in minor virus variants. Therefore, interpretation systems are necessary to determine the phenotypic and clinical significance of drug-resistance mutations found in a patient's plasma virus population. We describe the scientific principles of HIV-1 genotypic-resistance test interpretation and the most commonly used Web-based resources for clinicians ordering genotypic drug-resistance tests.
Retrospective studies have shown that the presence of HIV-1 drug resistance before starting a new antiretroviral drug treatment regimen is an independent predictor of the virologic response to that regimen. Prospective controlled studies have shown that patients whose physicians have access to drug-resistance data, particularly genotypic-resistance data, respond better to therapy than control patients of physicians without such access. The accumulation of such retrospective and prospective data has led several expert panels to recommend drug-resistance testing in the management of HIV-1–infected patients [1–3].
However, interpreting the results of HIV-1 genotypic drug-resistance tests is one of the most difficult tasks facing clinicians caring for HIV-1–infected patients because of the complex interactions among the many mutations that contribute to drug resistance; the varying levels of reduced susceptibility caused by these mutations; and the inability of drug-resistance tests to detect minor, yet clinically relevant, drug-resistant variants in a patient's virus quasispecies.
This article introduces the scientific principles underlying interpretation of genotypic-resistance test results and reviews currently available Web-based systems for interpretation of genotypic data, as well as those Web sites with clinically relevant summaries of HIV-1 drug-resistance mutations.
Scientific Basis for Interpretation of Genotypic Data
Three fundamental types of data form the basis of HIV-1 drug-resistance knowledge (table 1): (1) correlations between viral genotype and the antiretroviral treatments of persons from whom sequenced HIV-1 isolates have been obtained (genotype-treatment); (2) correlations between viral genotype and in vitro drug-susceptibility test results (genotype-phenotype); and (3) correlations between viral genotype and virological response to a new treatment regimen (genotype-outcome). Most genotypic-resistance systems are based on 1 or more of these types of data, although systems differ with regard to the relative importance that they ascribe to each of the different data types.
Table 1. Fundamental correlations underlying HIV-1 drug-resistance knowledge.
| Correlation type | Description |
|---|---|
| Genotype-treatment | These correlations link mutations to drug resistance by a Darwinian argument: mutations that are selected in vitro during drug passage experiments or in vivo in persons receiving a drug are likely to be important in conferring drug resistance. Although mutations selected during drug-passage experiments receive the most recognition, they usually are only a subset of the mutations selected by therapy in vivo. |
| Genotype-phenotype | These correlations quantify the effect of a mutation on in vitro drug susceptibility. The correlations may be derived from laboratory isolates with mutations resulting from virus passage or site-directed mutagenesis or derived from clinical isolates. These correlations are limited in their clinical significance, because (1) some mutations do not cause drug resistance but are markers for other drug-resistance mutations (e.g., transitional and sentinel mutations); (2) some mutations decrease virus replication capacity, thereby providing an advantage to continued therapy despite high-level resistance; and (3) drugs differ in their antiviral activity and pharmacokinetics: 50-fold resistance to one drug may have the same clinical significance as 5-fold resistance to another. |
| Genotype-outcome | These correlations are derived from studies in which a patient's viral genotype prior to starting a new regimen is correlated with the virologic response to a drug included in the new regimen. Such correlations are complicated by many factors, including (1) variable past treatment histories; (2) variable drugs accompanying the drug under evaluation; (3) relatively few patients per study; (4) variable definitions of what constitutes a virologic response, with follow-up ranging from 4 to 48 weeks; and (5) variability in how the baseline genotype was used to guide therapy. |
Genotype-phenotype correlations quantify the effect of HIV-1 mutations in reducing drug susceptibility. These correlations, however, are not sufficient for genotypic interpretation, because HIV-1 exists within individuals as a quasispecies of innumerable variants that are related to (but distinct from) the predominant variants detected in plasma. Many drug-resistance mutations are sentinel mutations that have a minimal effect on drug susceptibility by themselves but are markers for the presence of other mutations that are likely to emerge with continued selective drug pressure. The clinical significance of such sentinel mutations is discovered through genotype-outcome rather than through genotype-phenotype studies.
HIV-1 Drug-Resistance Summaries
Three Web sites contain comprehensive summaries of HIV-1 drug-resistance mutations (table 2). The International AIDS Society–USA drug-resistance mutation panel is maintained by a group of experts that creates a biannual summary of the mutations believed to be the most clinically relevant [4]. The HIV Sequence Database at Los Alamos National Laboratories maintains the updated review “Mutations in retroviral genes associated with drug-resistance” [5], which contains data on approved and investigational HIV-1 inhibitors, including those that target proteins other than protease, reverse-transcriptase (RT), and gp41. A complete, searchable version of the review can be found in the compendium section of the Web site (table 2). The Drug Resistance Summary section of the Stanford University HIV Drug Resistance Database contains diagrammatic summaries of HIV-1 drug-resistance mutations by drug class, summaries of the main mutations associated with each anti-retroviral drug, and a complete list of the drug-specific comments and scores used by the HIVdb drug-resistance interpretation program (table 2).
Table 2. Web sites providing HIV-1 drug-resistance mutation summaries.
| Name, sponsoring body, URL | Comments |
|---|---|
| International AIDS Society–USA | |
| http://www.iasusa.org/resistance_mutations/mutations_figures.pdf | Expert panel summary of the drug-resistance mutations that are most clinically relevant. |
| HIV Sequence Database, Los Alamos National Laboratories | |
| http://www.hiv.lanl.gov/content/hiv-db/COMPENDIUM/2005/partI/clark.pdf | Summary of nearly all HIV-1 mutations associated with in vitro or in vivo drug resistance. |
| http://resdb.lanl.gov/Resist_DB/default.htm | Searchable form of Los Alamos National Laboratories drug summary. |
| Stanford University HIV Drug Resistance Database, Stanford University | |
| http://hivdb.stanford.edu/cgi-bin/PIResiNote.cgi | Graphical summary of PI drug-resistance mutations. |
| http://hivdb.stanford.edu/cgi-bin/NRTIResiNote.cgi | Graphical summary of NRTI drug-resistance mutations. |
| http://hivdb.stanford.edu/cgi-bin/NNRTIResiNote.cgi | Graphical summary of NNRTI drug-resistance mutations. |
| http://hivdb.stanford.edu/pages/genotype-clinical.html#ARV_Summaries | Antiretroviral drug summaries by drug. |
| http://hivdb.stanford.edu/cgi-bin/PositionPhenoSummary.cgi | Drug-resistance mutation phenotypic data. |
| http://hivdb.stanford.edu/pages/genotype-clinical.html#Summaries_of_Clinical_Studies | Summary of published clinical studies linking baseline genotype and virologic response to a new treatment regimen. |
Genotypic-Resistance Interpretation Systems
Most genotypic-resistance reports are generated by 1 of 4 types of interpretation systems: (1) systems packaged with the US Food and Drug Administration–approved genotypic-resistance testing kits TruGene (Bayer Diagnostics) [6] and ViroSeq (Celera Diagnostics) [7], which are free with each of the testing kits but are not otherwise publicly available (they are also the only systems reviewed by the US Food and Drug Administration each time they are updated); (2) systems used by large reference laboratories, which are also not publicly available; (3) the proprietary VirtualPhenotype and the VircoType systems (Virco); and (4) systems created by academic laboratories [2, 8].
Of the >10 academic systems for genotypic interpretation, 6 are maintained on freely accessible Web sites (table 3). Each of these 5 systems accepts user-submitted protease and RT mutations and/or nucleotide sequence data and reports levels of inferred resistance to each of the protease and RT inhibitors. The rules and scores underlying 3 of these systems (Agence Nationale de Recherches sur le Sida [ANRS], Rega Institute, and HIVdb) are also publicly available.
Table 3. Publicly available systems for interpretation of genotypic-resistance data.
| Name, sponsoring body, URL | Comments |
|---|---|
| Agence Nationale de Recherches sur le Sida (ANRS) system, version 13, ANRS | |
| http://www.hivfrenchresistance.org/tab2005.html | ANRS system rules. |
| http://pugliese.club.fr/index.htm | ANRS implementation for user-submitted mutations. |
| Antiretroscan, Italian Antiretroviral Resistance Cohort Analysis multicenter collaboration | |
| https://www.hivarca.net/PubblicaInglese/Index.asp | Implementation available for user-submitted sequences. |
| Geno2pheno, version 3.0, German National Reference Center | |
| http://www.Geno2pheno.org/cgi-bin/Geno2pheno.pl | Geno2pheno implementation for user-submitted sequences. |
| Stanford University HIV Drug Resistance Database (HIVdb), version 4.1, Stanford University | |
| http://hivdb6.stanford.edu/asi/deployed/xmlTools/rules_scores_hivdb.cgi?class=PI | HIVdb list of PI drug-resistance mutation penalties. |
| http://hivdb6.stanford.edu/asi/deployed/xmlTools/rules_scores_hivdb.cgi?class=NRTI | HIVdb list of NRTI drug-resistance mutation penalties. |
| http://hivdb6.stanford.edu/asi/deployed/xmlTools/rules_scores_hivdb.cgi?class=NNRTI | HIVdb list of NNRTI drug-resistance mutation penalties. |
| http://hivdb6.stanford.edu/asi/deployed/xmlTools/rules_comments_hivdb.cgi?class=PI | HIVdb list of PI drug-resistance mutation comments. |
| http://hivdb6.stanford.edu/asi/deployed/xmlTools/rules_comments_hivdb.cgi?class=NRTI | HIVdb list of NRTI drug-resistance mutation comments. |
| http://hivdb6.stanford.edu/asi/deployed/xmlTools/rules_comments_hivdb.cgi?class=NNRTI | HIVdb list of NNRTI drug-resistance mutation comments. |
| http://hivdb.stanford.edu/pages/algs/HIVdb.html | HIVdb implementation of user-submitted mutations or sequences. |
| Rega Institute System, version 6.4, Katholieke Universiteit | |
| http://www.kuleuven.be/rega/cev/links/rega_algorithm/index.htm | Rega Institute system rules. |
| http://hivdb.stanford.edu/pages/algs/HIValg.html | Rega Institute implementation of user-submitted mutations. |
The ANRS [9] and Rega Institute (Katholieke Universiteit; Leuven, Belgium) [10] systems are rules-based systems that report 3 levels of resistance: susceptible, intermediate, and resistant (table 3). Each rule consists of a Boolean expression. For example, an ANRS rule for abacavir (version 13, July 2005) states: “If there are five or more of the following RT mutations (M41L, D67N, L74V, M184V/I, L210W, T215Y/F), report resistance to abacavir.” Both systems contain interpretations for all available antiretroviral drugs, including the fusion inhibitor enfuvirtide. They are frequently updated and widely accessible.
The HIVdb system (Stanford University; Stanford, CA) is a rules-based system that consists of a list of drug penalty scores and comments for each mutation [11]. The total score for a drug is derived by adding the scores for all mutations associated with resistance to that drug to infer 1 of 5 levels of resistance: susceptible, potential low-level resistance, low-level resistance, intermediate resistance, and high-level resistance. Drug penalty scores for enfuvirtide are being developed. Figure 1 shows the 2 HIVdb input forms: one accepts lists of protease and/or RT mutations, and the other accepts nucleotide sequences. Figure 2 shows sample output for the protease and RT inhibitors.
Figure 1.
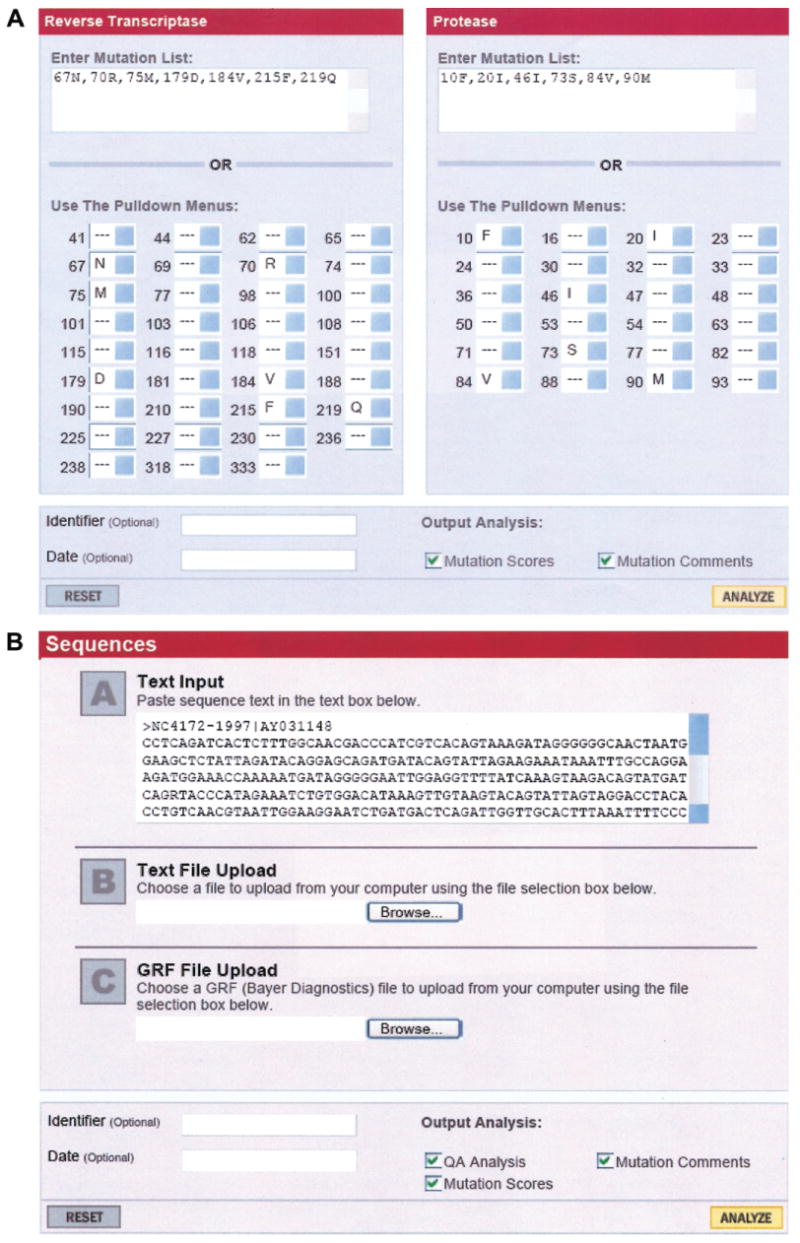
A, Screenshot of the mutation entry form of the HIVdb program available from the Stanford University HIV Drug Resistance Database [12]. Mutations can be selected from the drop down list or entered using a text box. B, Screenshot of the sequence entry form of the HIVdb program [13]. One or more sequences can be pasted into the text box or uploaded into the program. Machines can also interface with the HIVdb program to obtain scores, comments, and inferred levels of resistance using a Web service called Sierra (http://hivdb.stanford.edu/pages/webservices/).
Figure 2.

A, Screenshot of the nucleoside reverse-transcriptase inhibitor (NRTI) and nonnucleoside reverse-transcriptase inhibitor (NNRTI) interpretation of the mutations shown in figure 1A. The first part of the interpretation divides each of the mutations (i.e., each difference from consensus B) into 3 categories: NRTI-associated, NNRTI-associated, and other. The second part lists the inferred level of resistance to each of the NRTIs and NNRTIs. The third part contains comments associated with each of the drug-resistance mutations. The fourth part lists the individual scores for each mutation, with each score hyperlinked to data about the mutation and the drug. B, Screenshot of the protease inhibitor–resistance interpretation. Protease mutations are divided into 3 categories: major mutations, minor mutations, and other. The remainder of the output is similar to that for the reverse-transcriptase inhibitor interpretations.
The Geno2pheno system uses decision trees and support vector machines to infer phenotypic drug susceptibility. The statistical models were trained on a database of genotypes and phenotypes created at the Institute of Clinical and Molecular Virology of the German National Reference Center for Retro-viruses (Erlangen, Germany) [14, 15]. To facilitate the interpretation of resistance phenotype predictions, the output of Geno2pheno includes normalized resistance scores obtained from comparison of the predicted value with the distribution of fold-changes observed among untreated patients. A recently added component of the system is the software tool theo, which applies phenotypic-resistance predictions to clinical data and evaluates drug combinations on the basis of predicted changes in viral load [16].
The Retrogram is a commonly used interpretation system developed by Boucher and colleagues at Utrecht University [17]. One clinical trial showed that patients of physicians using this system had better outcomes than did control patients whose physicians did not have access to a genotypic interpretation system [17]. The AntiRetroScan was developed at the University of Sienna [18] and is maintained on the Italian Antiretroviral Resistance Cohort Analysis Web site. Three additional academic systems developed by the Detroit Medical Center, Sao Paolo University, and Centre Hospitalier de Luxembourg are available through a commercial Web site maintained by Advanced Biological Laboratories (Luxembourg).
Commercially Available Systems for Genotypic Interpretation
VirtualPhenotype and VircoType
The VirtualPhenotype system is a proprietary, commercially available, online genotypic interpretation system that accepts a virus sequence and reports the expected level of phenotypic resistance to each of the approved antiretroviral drugs (table 4). The procedure involves using Virco's database of viruses for which both genotype and phenotype testing have been performed to identify viruses with patterns of mutations matching those in the submitted sequence. Only those mutations that are believed to be pertinent to drug resistance are required to match. The phenotypes of matching sequences are then analyzed to determine the median and range in the levels of fold-resistance. In 2 prospective studies, the VirtualPhenotype system and Antivirogram (the phenotypic assay upon which the VirtualPhenotype system is based) led to similar reductions in plasma HIV-1 RNA levels following a change in treatment regimen [19, 20].
Table 4. Commercially available genotypic drug-resistance data interpretation systems.
| Name, company | URL | Comments |
|---|---|---|
| GeneSeq, Monogram Biosciences | http://www.virologichiv.com/assays/hcp/geneSeqHIV.aspx | GeneSeq interpretation system used by Monogram Biosciences. |
| VircoType, Virco Laboratories | http://www.vircolab.com | VircoType can be ordered over the Web. |
| ViroSeq, Celera Diagnostics/Abbott Laboratories | http://www.celeradiagnostics.com/cdx/ViroSeq | Interpretation system that is used with the Celera Diagnostics genotyping system. |
| TruGene HIV-1, Bayer Diagnostics | http://www.labnews.de/en/products/pr_truso.php | Interpretation system that is used with the Bayer Diagnostics genotyping system. |
| PhenoScript, Viralliance | http://www.viralliance.com/html/company.html | A third phenotyping method using recombinant viruses, its precision and sensitivity have not been studied. |
| ViroScore, Advanced Biological Laboratories | http://www.therapyedge.com/site/en/product_vs.html | ViroScore suite includes Agence Nationale de Recherches sur le Sida, Detroit Medical Center, Sao Paolo University, Centre Hospitalier de Luxembourg, HIVdb program, and Rega Insititute. |
Three studies have used sets of clinical sequences to compare the VirtualPhenotype system to 1 or more rules-based systems. They reported high levels of concordance, except for those nucleoside reverse-transcriptase inhibitors with a narrow range in observable phenotypic susceptibility levels; these were more likely to be considered resistant by the rules-based systems [21–23]. This is consistent with a subsequent study reporting that the Antivirogram is considerably less sensitive than the PhenoSense assay (Monogram Biosciences) at detecting decreased susceptibility to didanosine, stavudine, abacavir, and probably tenofovir [24].
The VircoType system is the VirtualPhenotype system with clinical cutoff values derived from a retrospective study of >3200 genotype-outcome correlations between genotype as determined at baseline and virological responses to a new treatment regimen as determined after 8 weeks of therapy [25]. A linear regression model was developed to adjust for confounders such as past treatment history, the likely activity of drugs used in combination with the drug under evaluation, and baseline plasma HIV-1 RNA levels. The first clinical cutoff value was defined as the VirtualPhenotype value at which a 20% loss of virological response (compared with wild-type virus) is predicted, and the second clinical cutoff value was defined as the VirtualPhenotype value at which an 80% loss of virological response is predicted. The analysis of genotype-outcome correlations in the data set used to develop the VirtualPhenotype cutoff values generated interesting preliminary results with respect to the relative antiviral activity of the boosted protease inhibitors in vivo. It has been reported that reductions in susceptibility of ∼3-fold for amprenavir-ritonavir, ∼15-fold for saquinavir-ritonavir and indinavir-ritonavir, and ∼40-fold for lopinarvir-ritonavir were associated with a 50% reduction in expected virological response [25].
Advanced Biological Laboratories
Advanced Biological Laboratories provides a commercial service called the ViroScore suite that provides simultaneous interpretations using genotypic interpretation systems developed by 6 different institutions: ANRS, Centre Hospitalier de Luxembourg, Detroit Medical Center, HIVdb, Rega Institute, and Sao Paolo University (table 4). The ViroScore does not provide the complete report (including comments and individual scores) associated with each of these 6 systems; rather, it just provides the inferred level of resistance.
Level of Concordance among Genotypic Interpretation Systems
Different interpretation systems often produce different interpretations when applied to the same virus mutations. Usually, these differences are a matter of degree. Ravela et al. [26] applied 4 algorithms (ANRS, HIVdb, Rega Institute, and TruGene) to the sequences of isolates from 2045 individuals in the United States. The results of 30,675 interpretations (2045 sequences × 15 drugs) were as follows: 4.4% were completely discordant, with at least 1 system assigning susceptible and another system assigning resistant; 29.2% were partially discordant, with at least 1 system assigning susceptible and another system assigning intermediate or 1 system assigning intermediate and another system assigning resistant; and 66.4% displayed complete concordance.
Different interpretation systems often use different criteria for inferring resistance. For example, Geno2pheno and the VirtualPhenotype are based solely on genotype-phenotype correlations. In contrast, most other systems consist of prespecified rules derived from published studies linking HIV-1 genotype to other forms of data. Most experts consider genotype-outcome correlations to be the most relevant type of data, followed by genotype-phenotype and genotype-treatment correlations. However, as noted in table 1, genotype-outcome studies involve many confounding factors and are underpowered, forcing different interpretation systems to develop different approaches for dealing with this data shortage. For example, the ANRS system bases its interpretations almost entirely on genotype-outcome studies [9], and the ANRS has published a large proportion of the studies linking genotype to virological outcome, including studies on the genotypic predictors of response to abacavir [27, 28], tenofovir [29], didanosine [30], lopinavir-ritonavir [31, 32], amprenavir-ritonavir [33], saquinavir-ritonavir [34], and atazanavir [35]. In contrast, the HIVdb system balances genotype-outcome, genotype-phenotype, and genotype-treatment correlations.
The structure of rules-based systems also contributes to discordances among interpretation systems. For example, most systems report 3 levels of resistance: susceptible, intermediate or possible resistance, and resistance. The Retrogram reports 4 levels of resistance, and the HIVdb system and AntiRetroScan each report 5 levels of resistance. Although the use of 3 levels is consistent with the approach used for other pathogenic microorganisms—for which it is usually possible to use only fully active drugs—it is often necessary to use drugs to which there is some level of resistance when treating HIV-1–infected persons for whom multiple previous treatment regimens have failed.
Interpretation systems involving Boolean expressions (e.g., those developed by ANRS and Rega Institute) consist of rules designed for common combinations of drug-resistance mutations supplemented by rules designed to provide reasonable interpretations for the large number of remaining possible mutation combinations. Score-based systems, such as HIVdb, consist of a list of drug-specific penalty scores for each mutation, which, in effect, creates a rule (adding up the mutation penalties) for every possible combination of mutations.
Score-based systems are prone to producing more-pessimistic interpretations than are Boolean expressions, because every drug-resistance mutation is assigned a penalty In contrast, Boolean expressions are more likely to have a threshold at which resistance is not assigned unless certain combinations of mutations are present. Boolean expressions may be better predictors of short-term virological response, because in the absence of multiple mutations, the predominant plasma virus population may be only minimally resistant to a new antiretroviral drug. Scores and rules, however, are not mutually exclusive and can be combined, as in the AntiRetroScan system [18].
Comparisons of Genotypic Interpretation Systems
De Luca et al. [36] studied the performance of 11 interpretation systems on 261 patients with treatment-change episodes. Each drug used in the salvage regimen was assigned a genotypic sensitivity score (GSS) of 0 for drugs to which the virus was considered to be resistant, 0.5 for drugs to which the virus was considered to be partially resistant, and 1 for drugs to which the virus was considered to have no resistance. The mean GSS of the salvage regimens ranged from 1.4 to 2.2. The median change in plasma HIV-1 RNA level from baseline was −0.88 log10 copies/mL after 3 months and −0.52 log10 copies/mL after 6 months. The OR for achieving a virologic response (defined as plasma HIV-1 RNA levels of <500 copies/mL) was determined for each unit increase in GSS.
All analyzed interpretation systems were significantly predictive of virologic response, with ORs of 1.35–2.04 at 3 months and 1.44–2.10 at 6 months. However, in a multivariable model that included additional patient characteristics, only the ANRS, TruGene, and Rega Institute systems were independently predictive at 3 months, and only the TruGene, HIVdb, and the Retrogram systems were independently predictive at 6 months.
De Luca and colleagues also studied the performance of 13 interpretation systems for 415 previously untreated patients, 42 of whom had viruses with ≥ 1 major drug-resistance mutation. In a multivariate analysis, only 1 system (Rega Institute) significantly predicted virologic response, and only 2 systems (HIVdb and Retrogram) showed a trend towards significantly predicting response [37]. Torti et al. [23] examined the predictive accuracy of the Retrogram, TruGene, and the Virtual-Phenotype systems on 188 patients and reported that the interpretations of the rules-based systems, but not the Virtual-Phenotype system, were significantly associated with achieving plasma HIV-1 RNA levels of <400 copies/mL. One study that examined the virological response to abacavir found that the ANRS system had the greatest predictive accuracy [38].
Although it is logical to use comparative studies to understand performance variations among genotypic-interpretation systems, these studies are confounded by many factors: (1) genotypic-interpretation systems do not take into account past treatment history; (2) different systems report different gradations of resistance, and not all comparative studies adjust the GSS accordingly; (3) some systems may be better for short-term virological outcomes (e.g., after 4–8 weeks of therapy), and others may be better for longer-term outcomes; (4) GSSs that assign a score of 1 to all drugs that are presumed to be fully active are oversimplifications, because different drugs have different potencies; (5) drugs often provide a virological advantage even in the presence of high-level drug resistance, either by maintaining selection for a replication-impaired virus variant or through residual antiretroviral activity [39–41]; and (6) as genotypic testing has become routine, the genotypic interpretations themselves influence treatment selection and confound retrospective analyses.
Comparative studies of genotypic-resistance interpretation studies are important nonetheless. Indeed, the researchers performing these comparisons are aware of potential confounders and attempt to control for them by using multivariate statistical models. However, the clinical data sets used in these studies must be 1–2 orders of magnitude larger than those described to have enough statistical power to overcome these obstacles. They also must be publicly available to allow pooling of data from multiple studies and to allow researchers developing new systems to determine which rules (rather than which overall systems) work the best.
HIV-1 Subtyping
Although several groups have reported differences in the mechanisms of resistance among different subtypes [42–47], subtype-specific genotypic-resistance interpretation systems do not exist and may not be necessary. Nonetheless, many clinicians are interested in knowing the subtype of sequences that have undergone genotypic-resistance testing. Although the protease and RT genes are among the most highly conserved parts of the HIV-1 genome, they contain enough phylogenetic information (particularly the larger RT gene) to make pol subtyping possible [48, 49].
Table 5 lists the Web sites that provide reliable tools for determining the HIV-1 subtype of a submitted sequence. Each system accepts a user-submitted sequence and compares it with reference sequences for each of the pure subtypes (A, B, C, D, F, G, H, J, and K) and many of the common circulating recombinant forms (CRFs). Each system is also designed to detect non-CRF recombinants. The National Center for Biotechnology Information subtyping program can be used for several viruses in addition to HIV-1 [50]. The Los Alamos HIV Sequence Database [51] has 2 subtyping programs: one that uses distances and another that uses phylogenetic trees to assign an HIV-1 subtype.
Table 5. Web-based programs for HIV-1 subtyping.
| Name, sponsoring body | URL | Comments |
|---|---|---|
| Virus subtyping tool, National Center for Biotechnology Information (NCBI) retroviruses resources | http://www.ncbi.nih.gov/projects/genotyping/formpage.cgi | Generic subtyping tool for HIV-1 and other viruses. Sequences are scanned by a user-specified window size and compared with a list of reference sequences. The output consists of a graphical summary of the closest matching subtypes by window [50]. |
| Recombinant Identification Program (RIP), Los Alamos National Laboritories (LANL) HIV Sequence Database | http://hiv-web.lanl.gov/content/hiv-db/RIPPER/RIP.html | RIP is similar to the NCBI subtyping tool, in that sequences are scanned by a user-specified window size and compared with a list of reference sequences. |
| Subtyping Distance Tool (SUDI), LANL HIV Sequence Database | http://hiv-web.lanl.gov/content/hiv-db/SUDI/sudi.html | SUDI uses a phylogenetic approach to classify a sequence using user-specified reference sequences. |
| Rega Institute HIV-1 Automated Subtyping Tool, Rega Institute, Katholieke Universitiet | http://jose.med.kuleuven.be/subtypetool/html/index.html | Automated subtyping tool that combines phylogenetic analyses and boot-scanning to identify pure subtypes and the known circulating recombinant forms with a high degree of certainty [52]. |
| Subtype Analyzer, UC London | http://www.vgb.ucl.ac.uk/starn.shtml | Position-specific scoring matrices determine HIV-1 subtype as well as a statistical measure of confidence in the subtype assignment [53]. |
The Rega Institute HIV-1 Automated Subtyping Tool [52] combines multiple programs to provide the most definitive identification of sequences matching known subtypes or CRFs. It provides users with both the underlying analysis and its interpretation, which is important when a sequence does not unequivocally match one of the known subtypes or CRFs. The Subtype Analyzer uses position-specific scoring matrices over protease- and RT-sequence windows to determine HIV-1 subtype. It also returns a statistical measure of confidence in the subtype assignment [53]. Although the HIVdb system reports subtypes, it is not as accurate as the systems described above; because the HIVdb system does not do bootscanning, it does not detect many of the CRFs and non-CRF recombinants.
Conclusion
Systems for genotypic-resistance interpretation help clinicians to understand the results of genotypic-resistance testing by providing targeted information on the phenotypic and clinical significance of mutations present in their patients' plasma virus populations. When combined with a sound understanding of the principles of antiretroviral therapy, these systems and Web-based drug-resistance summaries provide clinicians with data that help them to make the most-informed treatment decisions for their patients.
However, because these systems to do not integrate the results of other relevant clinical data, such as previous drug-resistance test results, antiretroviral treatment history, plasma HIV-1 RNA levels, and CD4 cell counts, they do not have the logical power to instruct clinicians on which antiretroviral drugs should be used when constructing a salvage therapy regimen. Moreover, clinicians must still be knowledgeable about antiretroviral cross-resistance patterns to avoid prescribing drugs that may be rendered ineffective by mutations that were selected during previous treatments but are no longer detected in a genotypic test.
Additional genotype-phenotype and genotype-outcome correlations are required to improve current interpretation systems and to develop a standard, highly predictive, publicly available system that will evolve as new drugs are developed and new studies are published. Given sufficient publicly analyzable genotype-outcome correlations, it should also be possible to develop a transparent and robust genotypic interpretation system that would assimilate antiretroviral treatment history, laboratory data, and published treatment guidelines to generate treatment recommendations on the basis of the likely success of different antiretroviral drug combinations.
Acknowledgments
Potential conflicts of interest. R.S. has received consulting fees from Bayer Diagnostics and Celera Diagnostics and research support from Celera Diagnostics. The HIVdb program has been licensed by Stanford University for commercial use to Roche-Virodec and Advanced Biological Laboratories and for academic use at no cost to Johns Hopkins University. All other authors: no conflicts.
References
- 1.Hirsch MS, Brun-Vezinet F, Clotet B, et al. Antiretroviral drug resistance testing in adults infected with human immunodeficiency virus type 1: 2003 recommendations of an International AIDS Society-USA Panel. Clin Infect Dis. 2003;37:113–28. doi: 10.1086/375597. [DOI] [PubMed] [Google Scholar]
- 2.Vandamme AM, Sonnerborg A, Ait-Khaled M, et al. Updated European recommendations for the clinical use of HIV drug resistance testing. Antivir Ther. 2004;9:829–48. [PubMed] [Google Scholar]
- 3.US Department of Health and Human Services Panel on Clinical Practices for Treatment of HIV Infection A. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. [25 April 2006]; doi: 10.1310/hct.2000.1.1.008. Available at: http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. [DOI] [PubMed]
- 4.Johnson VA, Brun-Vezinet F, Clotet B, et al. Update of the drug resistance mutations in HIV-1: 2005. Top HIV Med. 2005;13:51–7. [PubMed] [Google Scholar]
- 5.Clark S, Calef C, Mellors J. Mutations in retroviral genes associated with drug resistance. In: Leitner T, Foley B, Hahn BH, et al., editors. HIV sequence compendium 2005. Los Alamos, NM: Los Alamos National Laboratory; 2005. pp. 80–175. [Google Scholar]
- 6.Grant RM, Kuritzkes DR, Johnson VA, et al. Accuracy of the TRUGENE HIV-1 genotyping kit. J Clin Microbiol. 2003;41:1586–93. doi: 10.1128/JCM.41.4.1586-1593.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eshleman SH, Crutcher G, Petrauskene O, et al. Sensitivity and specificity of the ViroSeq human immunodeficiency virus type 1 (HIV-1) genotyping system for detection of HIV-1 drug resistance mutations by use of an ABI PRISM 3100 genetic analyzer. J Clin Microbiol. 2005;43:813–7. doi: 10.1128/JCM.43.2.813-817.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Luca A, Perno CF. Impact of different HIV resistance interpretation by distinct systems on clinical utility of resistance testing. Curr Opin Infect Dis. 2003;16:573–80. doi: 10.1097/00001432-200312000-00010. [DOI] [PubMed] [Google Scholar]
- 9.Brun-Vezinet F, Costagliola D, Khaled MA, et al. Clinically validated genotype analysis: guiding principles and statistical concerns. Antivir Ther. 2004;9:465–78. [PubMed] [Google Scholar]
- 10.Van Laethem K, De Luca A, Antinori A, Cingolani A, Perno CF, Vandamme AM. A genotypic drug resistance interpretation algorithm that significantly predicts therapy response in HIV-1–infected patients. Antivir Ther. 2002;7:123–9. [PubMed] [Google Scholar]
- 11.Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003;31:298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.HIVdb program: mutation list analysis. [25 April 2006]; Available at: http://hivdb6.stanford.edu/asi/deployed/hiv_central.pl?program=hivdb&action=showMutationForm.
- 13.HIVdb program: sequence analysis. [25 April 2006]; Available at: http://hivdb6.stanford.edu/asi/deployed/hiv_central.pl?program=hivdb&action=showSequenceForm.
- 14.Beerenwinkel N, Schmidt B, Walter H, et al. Diversity and complexity of HIV-1 drug resistance: a bioinformatics approach to predicting phenotype from genotype. Proc Natl Acad Sci U S A. 2002;99:8271–6. doi: 10.1073/pnas.112177799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beerenwinkel N, Daumer M, Oette M, et al. Geno2pheno: estimating phenotypic drug resistance from HIV-1 genotypes. Nucleic Acids Res. 2003;31:3850–5. doi: 10.1093/nar/gkg575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beerenwinkel N, Sing T, Lengauer T, et al. Computational methods for the design of effective therapies against drug resistant HIV strains. Bioinformatics. 2005;21:3943–50. doi: 10.1093/bioinformatics/bti654. [DOI] [PubMed] [Google Scholar]
- 17.Tural C, Ruiz L, Holtzer C, et al. Clinical utility of HIV-1 genotyping and expert advice: the Havana trial. AIDS. 2002;16:209–18. doi: 10.1097/00002030-200201250-00010. [DOI] [PubMed] [Google Scholar]
- 18.Zazzi M, Romano L, Venturi G, et al. Comparative evaluation of three computerized algorithms for prediction of antiretroviral susceptibility from HIV type 1 genotype. J Antimicrob Chemother. 2004;53:356–60. doi: 10.1093/jac/dkh021. [DOI] [PubMed] [Google Scholar]
- 19.Perez-Elias MJ, Garcia-Arota I, Munoz V, et al. Phenotype or virtual phenotype for choosing antiretroviral therapy after failure: a prospective, randomized study. Antivir Ther. 2003;8:577–84. [PubMed] [Google Scholar]
- 20.Mazzotta F, Lo Caputo S, Torti C, et al. Real versus virtual phenotype to guide treatment in heavily pretreated patients: 48-week follow-up of the Genotipo-Fenotipo di Resistenza (GenPheRex) trial. J Acquir Immune Defic Syndr. 2003;32:268–80. doi: 10.1097/00126334-200303010-00005. [DOI] [PubMed] [Google Scholar]
- 21.Puchhammer-Stockl E, Steininger C, Geringer E, Heinz FX. Comparison of virtual phenotype and HIV-SEQ program (Stanford) interpretation for predicting drug resistance of HIV strains. HIV Med. 2002;3:200–6. doi: 10.1046/j.1468-1293.2002.00116.x. [DOI] [PubMed] [Google Scholar]
- 22.Kijak GH, Rubio AE, Pampuro SE, et al. Discrepant results in the interpretation of HIV-1 drug-resistance genotypic data among widely used algorithms. HIV Med. 2003;4:72–78. doi: 10.1046/j.1468-1293.2003.00131.x. [DOI] [PubMed] [Google Scholar]
- 23.Torti C, Quiros-Roldan E, Keulen W, et al. Comparison between rules-based human immunodeficiency virus type 1 genotype interpretations and real or virtual phenotype: concordance analysis and correlation with clinical outcome in heavily treated patients. J Infect Dis. 2003;188:194–201. doi: 10.1086/376512. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Rhee SY, Taylor J, Shafer RW. Comparison of the precision and sensitivity of the Antivirogram and PhenoSense HIV drug susceptibility assays. J Acquir Immune Defic Syndr. 2005;38:439–44. doi: 10.1097/01.qai.0000147526.64863.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bacheler L, Winters B, Harrigan PR, et al. Estimation of phenotypic clinical cutoffs for VircoTYPE through meta analyses of clinical trial and cohort data [abstract H-1133]. Program and abstracts of the 44th Interscience Conference on Antimicrobial Agents and Chemotherapy; 30 October–2 November 2004; Washington, DC. 2004. [Google Scholar]
- 26.Ravela J, Betts BJ, Brun-Vezinet F, et al. HIV-1 protease and reverse transcriptase mutation patterns responsible for discordances between genotypic drug resistance interpretation algorithms. J Acquir Immune Defic Syndr. 2003;33:8–14. doi: 10.1097/00126334-200305010-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katlama C, Clotet B, Plettenberg A, et al. The role of abacavir (ABC, 1592) in antiretroviral therapy-experienced patients: results from a randomized, double-blind, trial. CNA3002 European Study Team. AIDS. 2000;14:781–9. doi: 10.1097/00002030-200005050-00003. [DOI] [PubMed] [Google Scholar]
- 28.Brun-Vezinet F, Descamps D, Ruffault A, et al. Clinically relevant interpretation of genotype for resistance to abacavir. AIDS. 2003;17:1795–802. doi: 10.1097/00002030-200308150-00008. [DOI] [PubMed] [Google Scholar]
- 29.Masquelier B, Tamalet C, Montes B, et al. Genotypic determinants of the virological response to tenofovir disoproxil fumarate in nucleoside reverse transcriptase inhibitor-experienced patients. Antivir Ther. 2004;9:315–23. [PubMed] [Google Scholar]
- 30.Molina JM, Marcelin AG, Pavie J, et al. Didanosine in HIV-1–infected patients experiencing failure of antiretroviral therapy: a randomized placebo-controlled trial. J Infect Dis. 2005;191:840–7. doi: 10.1086/428094. [DOI] [PubMed] [Google Scholar]
- 31.Masquelier B, Breilh D, Neau D, et al. Human immunodeficiency virus type 1 genotypic and pharmacokinetic determinants of the virological response to lopinavir-ritonavir–containing therapy in protease inhibitor–experienced patients. Antimicrob Agents Chemother. 2002;46:2926–32. doi: 10.1128/AAC.46.9.2926-2932.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delaugerre C, Teglas JP, Treluyer JM, et al. Predictive factors of virologic success in HIV-1–infected children treated with lopinavir/ritonavir. J Acquir Immune Defic Syndr. 2004;37:1269–75. doi: 10.1097/01.qai.0000137408.78031.37. [DOI] [PubMed] [Google Scholar]
- 33.Marcelin AG, Lamotte C, Delaugerre C, et al. Genotypic inhibitory quotient as predictor of virological response to ritonavir-amprenavir in human immunodeficiency virus type 1 protease inhibitor–experienced patients. Antimicrob Agents Chemother. 2003;47:594–600. doi: 10.1128/AAC.47.2.594-600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcelin AG, Dalban C, Peytavin G, et al. Clinically relevant interpretation of genotype and relationship to plasma drug concentrations for resistance to saquinavir-ritonavir in human immunodeficiency virus type 1 protease inhibitor–experienced patients. Antimicrob Agents Chemother. 2004;48:4687–92. doi: 10.1128/AAC.48.12.4687-4692.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vora S, Marcelin AG, Gunthard HF, et al. Clinical validation of atazanavir/ritonavir genotypic resistance score in protease inhibitor-experienced patients. AIDS. 2006;20:35–40. doi: 10.1097/01.aids.0000196179.11293.fc. [DOI] [PubMed] [Google Scholar]
- 36.De Luca A, Cingolani A, Di Giambenedetto S, et al. Variable prediction of antiretroviral treatment outcome by different systems for interpreting genotypic human immunodeficiency virus type 1 drug resistance. J Infect Dis. 2003;187:1934–43. doi: 10.1086/375355. [DOI] [PubMed] [Google Scholar]
- 37.De Luca A, Cozzi-Lepri A, Perno CF, et al. Variability in the interpretation of transmitted genotypic HIV-1 drug resistance and prediction of virological outcomes of the initial HAART by distinct systems. Antivir Ther. 2004;9:743–52. [PubMed] [Google Scholar]
- 38.Cabrera C, Cozzi Lepri A, Phillips A, et al. Baseline resistance and virological outcome in patients with virological failure who start a regimen containing abacavir: EuroSIDA study. Antivir Ther. 2004;9:787–800. [PubMed] [Google Scholar]
- 39.Lawrence J, Mayers DL, Hullsiek KH, et al. Structured treatment interruption in patients with multidrug-resistant human immunodeficiency virus. N Engl J Med. 2003;349:837–46. doi: 10.1056/NEJMoa035103. [DOI] [PubMed] [Google Scholar]
- 40.Deeks SG, Hoh R, Neilands TB, et al. Interruption of treatment with individual therapeutic drug classes in adults with multidrug-resistant HIV-1 infection. J Infect Dis. 2005;192:1537–44. doi: 10.1086/496892. [DOI] [PubMed] [Google Scholar]
- 41.Campbell TB, Shulman NS, Johnson SC, et al. Antiviral activity of lamivudine in salvage therapy for multidrug-resistant HIV-1 infection. Clin Infect Dis. 2005;41:236–42. doi: 10.1086/430709. [DOI] [PubMed] [Google Scholar]
- 42.Cane PA, de Ruiter A, Rice PA, Wiselka M, Fox R, Pillay D. Resistance-associated mutations in the human immunodeficiency virus type 1 subtype c protease gene from treated and untreated patients in the united kingdom. J Clin Microbiol. 2001;39:2652–4. doi: 10.1128/JCM.39.7.2652-2654.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brenner B, Turner D, Oliveira M, et al. A V106M mutation in HIV-1 clade C viruses exposed to efavirenz confers cross-resistance to non-nucleoside reverse transcriptase inhibitors. AIDS. 2003;17:F1–5. doi: 10.1097/00002030-200301030-00001. [DOI] [PubMed] [Google Scholar]
- 44.Grossman Z, Istomin V, Averbuch D, et al. Genetic variation at NNRTI resistance-associated positions in patients infected with HIV-1 subtype C. AIDS. 2004;18:909–15. doi: 10.1097/00002030-200404090-00008. [DOI] [PubMed] [Google Scholar]
- 45.Grossman Z, Paxinos EE, Averbuch D, et al. Mutation D30N is not preferentially selected by human immunodeficiency virus type 1 subtype C in the development of resistance to nelfinavir. Antimicrob Agents Chemother. 2004;48:2159–65. doi: 10.1128/AAC.48.6.2159-2165.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Camacho R, Godinho AR, Gomes P, et al. Different substitutions under drug pressure at protease codon 82 in HIV-1 subtype G compared to subtype B infected individuals including a novel I82M resistance mutation [abstract 138] Antivir Ther. 2005;10:S151. [Google Scholar]
- 47.Abecasis AB, Deforche K, Snoeck J, et al. Protease mutation M89I/V is linked to therapy failure in patients infected with the HIV-1 non-B subtypes C, F or G. AIDS. 2005;19:1799–806. doi: 10.1097/01.aids.0000188422.95162.b7. [DOI] [PubMed] [Google Scholar]
- 48.Gonzales MJ, Machekano RN, Shafer RW. Human immunodeficiency virus type 1 reverse-transcriptase and protease subtypes: classification, amino acid mutation patterns, and prevalence in a northern California clinic-based population. J Infect Dis. 2001;184:998–1006. doi: 10.1086/323601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hue S, Clewley JP, Cane PA, Pillay D. HIV-1 pol gene variation is sufficient for reconstruction of transmissions in the era of antiretroviral therapy. AIDS. 2004;18:719–28. doi: 10.1097/00002030-200403260-00002. [DOI] [PubMed] [Google Scholar]
- 50.Rozanov M, Plikat U, Chappey C, Kochergin A, Tatusova T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004;32:W654–9. doi: 10.1093/nar/gkh419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leitner T, Foley B, Hahn B, et al. HIV Sequence Compendium, 2003. Los Alamos, NM: Los Alamos National Laboratory; 2003. [Google Scholar]
- 52.de Oliveira T, Deforche K, Cassol S, et al. An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics. 2005;21:3797–800. doi: 10.1093/bioinformatics/bti607. [DOI] [PubMed] [Google Scholar]
- 53.Myers RE, Gale CV, Harrison A, Takeuchi Y, Kellam P. A statistical model for HIV-1 sequence classification using the subtype analyser (STAR) Bioinformatics. 2005;21:3535–40. doi: 10.1093/bioinformatics/bti569. [DOI] [PubMed] [Google Scholar]