

Abstract
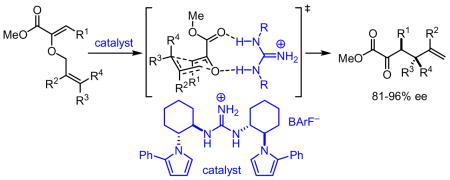
N,N′-Diphenylguanidinium ion associated with the non-coorinating BArF counterion is shown to be an effective catalyst for the [3,3]-sigmatropic rearrangement of a variety of substituted allyl vinyl ethers. Highly enantioselective catalytic Claisen rearrangements of ester-substituted allyl vinyl ethers are then documented using a new C2-symmetric guanidinium ion derivative.
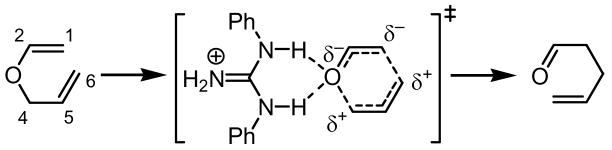
Chorismate mutases catalyze the [3,3]-sigmatropic rearrangement of chorismate to prephenate. Structural, site-directed mutagenesis, and computational studies have established the importance of several key hydrogen-bond donating residues in the active site of the enzyme. Through a combination of transition-state stabilization and restriction of the substrate in a reactive conformation, chorismate mutase is capable of achieving rate accelerations on the order of 106 relative to the thermal reaction.1 Acceleration of the Claisen rearrangement has also been observed in hydrogen-bonding solvents;2,3 using quantum mechanical computational methods, Jorgensen has advanced a model for the aqueous acceleration of the Claisen rearrangement involving H-bond interactions between two water molecules and the core heteroatom of the allyl vinyl ether in the optimized transition state structure.4 Consistent with this hypothesis, compounds capable of dual hydrogen-bonding such as ureas and thioureas have been studies in the context of Claisen rearrangement reactions and demonstrated to induce modest rate accelerations when used in stoichiometric or super-stoichiometric amounts.5 However, negligible rate accelerations have been realized under catalytic conditions. We report here catalysis of the Claisen rearrangement by simple guanidinium ions, and the development of chiral variants for highly enantioselective rearrangements of ester-substituted allyl vinyl ethers.
Approaches to the asymmetric catalysis of the Claisen rearrangement have focused on the use of Lewis acidic metal complexes based on aluminum,6a–c boron,6d–f magnesium,6g and copper;6h,i however, in almost all cases, these systems suffer from strong product inhibition or competing background pathways. To date, only one example of an asymmetric Claisen rearrangement utilizing catalytic quantities of a Lewis acid has been realized: Hiersemann’s highly enantioselective rearrangement of ester-substituted allyl vinyl ethers using copper (II) bisoxazoline complexes.6h,i
Although urea and thiourea derivatives effectively promote a broad range of important transformations via H-bonding,7 our evaluation of representative chiral and achiral catalysts of this class revealed that none provided significant rate acceleration in model Claisen rearrangement reactions. This result was predictable on the basis of earlier experimental and computational studies.5 In contrast, the simple N,N′-diphenylguanidinium salt 1 was found to induce rate enhancements in the rearrangement of a variety of substituted allyl vinyl ethers (Table 1).8 Substrates bearing electron-donating substituents at the 4- (e.g., 4) or 6-positions (e.g., 5–6), or electron-withdrawing groups at the 2-position (e.g., 8) all underwent rearrangement with significant rate acceleration in the presence of catalytic levels of 1. These observations are aligned with the proposal that transition structures with a high degree of charge separation are most susceptible to stabilization by hydrogen-bond donors.9 Claisen rearrangement of aryl ether 7 was also catalyzed by 1 to provide the corresponding ortho-prenylated phenol in high yield.
Table 1.
Claisen Rearrangements Catalyzed by 1.a
 | |||||
|---|---|---|---|---|---|
| substrate | product | time (h) | temp. (°C) | % conv.b0 mol% 1 | % conv.b20 mol% 1 |
|
3 |
|
24 | 80 | 9 | 15c |
4 |
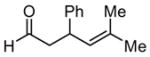
|
48 | 40 | 28 | 82 |
|
5 |
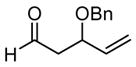
|
48 | 40 | 0 | 75 |
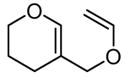
6 |
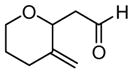
|
24 | 40 | 0 | 72 |
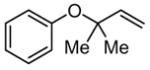
7 |
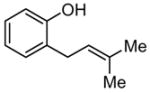
|
30 | 80 | 12 | 87 |
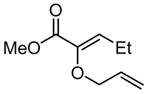
8 |
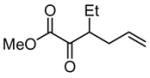
|
48 | 40 | 12 | 76 |
Reactions carried out on a 0.1 mmol scale in 0.5 mL of C6D6.
Conversions measured by 1HNMR integration relative to p-xylene as an internal standard.
Extensive decomposition observed.
Efforts to induce asymmetric rearrangement of substrates 4–6 using chiral guanidinium ion catalysts proved unsuccessful, with product formation in low-to-negligible enantioselectivities. In contrast, ester-substituted allyl vinyl ether 8 underwent reaction in moderate enantioselectivity with a variety of guanidinium catalysts.10 After extensive catalyst development, we observed that C2-symmetric guanidinium ions derived from trans-1-pyrrolo-2-aminocyclohexane were particularly effective, and fine-tuning of the pyrrole substituents led to the identification of 2 as the optimal catalyst (see Supporting Information).
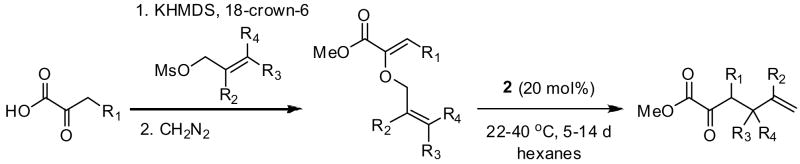
High enantioselectivities were obtained in the rearrangement of a range of substrates in reactions carried out between 22 and 40 °C over the period of several days (Table 2). Optimal rates and enantioselectivities were observed in hexanes, despite the fact that catalyst 2 is virtually insoluble in this solvent; use of dichloromethane or benzene resulted in slightly suppressed ee’s, and no catalysis was observed in ethereal solvents such as diethyl ether or TBME.
Table 2.
Enantioselective Claisen Rearrangements Catalyzed by 2a
 | ||||
|---|---|---|---|---|
| entry | substrate | product | yielda,d (%) | eee (%) |
| 1 |
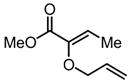
9 |
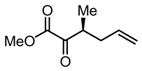
|
80 | 92 |
| 2 |
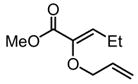
|
|
86 | 92 |
| 3 |
|
|
92
> 20:1 dr |
85 |
| 4 |
|
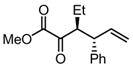
|
91
19:1 dr |
81 |
| 5 |
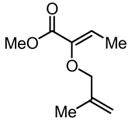
|
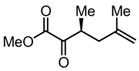
|
73 | 96 |
| 6 |
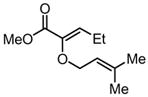
|
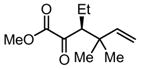
|
89 | 81 |
| 7 |
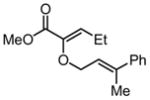
|
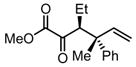
|
89
> 20.1 dr |
82 |
| 8 |
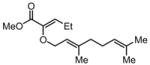
|
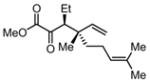
|
73
> 20.1 dr |
84 |
Reactions run on a 0.1 mmol scale in 2 mL of hexanes.
The absolute configuration for entry 2 was established by comparison to material prepared with [Cu{(S,S)-t-butylbox}](H2O)(SbF6)2 (ref. 6i); all other products assigned by analogy
Isolated yields after column chromatography.
Diastereomeric ratios determined by 1HNMR.
Enantiomeric excesses determined by GC or HPLC analysis using commercial chiral columns (see Supporting Information).
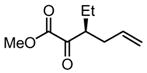
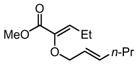
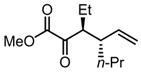
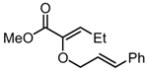
Substrates bearing methyl- or ethyl-substituents at the 1-position afforded products in high enantioselectivities (Table 2, entries 1 and 2); however, more sterically hindered isopropyl or isobutyl derivatives were less reactive and underwent rearrangement with diminished ee’s (73% and 69% ee, respectively).11 Disubstituted compounds (Table 2, entries 3 and 4) rearranged to form adjacent tertiary stereogenic centers in high enantio- and diastereoselectivity with the anti stereochemical relationship predicted by a six-membered chair-like transition state. Quaternary stereogenic centers could also be generated with good stereocontrol (Table 2, entries 7 and 8).
X-Ray structural analysis of 2, recrystallized from an isopropanol/water solvent mixture, reveals a guanidinium functionality disposed in a pseudo-C2-symmetric (Z,Z) conformation and hydrogen-bonded to two isopropanol molecules (Figure 2a). DFT studies were conducted in order to probe the mode of catalyst interaction in the transition state of the Claisen rearrangement of representative substrate 9.12 Calculations were performed using a simplified N,N′-dimethylguanidinium ion catalyst, and the lowest energy transition structure identified is depicted in Figure 2b. Transition state stabilization via hydrogen-bonding interactions between catalyst and both the ether- and ester carbonyl-derived oxygens are evident. As expected, the computed transition structure of the catalyzed reaction bears greater charge separation between the allyl and oxallyl fragments relative to the transition structure of the uncatalyzed thermal rearrangement.
Figure 2.
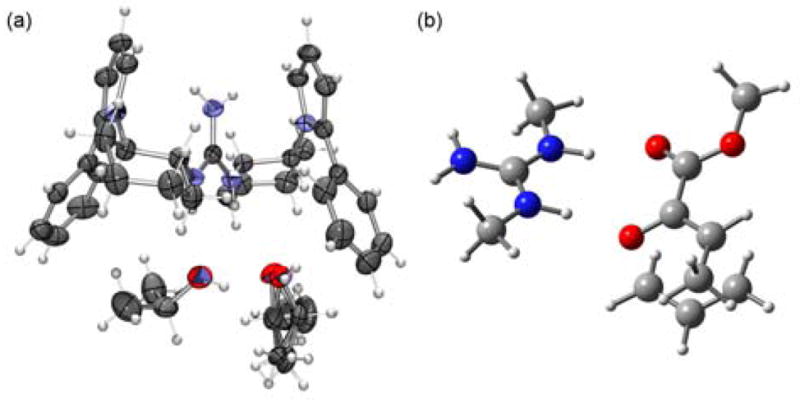
(a) ORTEP plot (50% probability ellipsoids for non-hydrogen atoms) of the guanidinium ion 2 (counterion omitted for clarity) as a complex with two isopropanol molecules. (b) Fully optimized, lowest-energy transition structure for the N,N′-dimethylguanidinium-promoted Claisen rearrangement of 9 at the B3LYP/6-31G(d) level of theory.
Although guanidinium BArF species have approximately the same equilibrium acidity as N,N′-diarylthioureas,13 they possess superior catalytic activity in all of the Claisen rearrangements we have studied to date. The basis for this unexpected difference and application of chiral guanidinium ions as hydrogen-bond donor catalysts in other reactions is the focus of ongoing studies.
Supplementary Material
Complete experimental procedures, summary of catalyst optimization studies, characterization data for new compounds, and complete ref. 12. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 1.

Guanidinium BArF catalysts.
Acknowledgments
This work was supported by the NIGMS (GM-43214). We thank Dr. Eric Ashley for helpful discussions and Dr. Douglas Ho for carrying out the X-ray structural analysis.
References
- 1.For reviews: Lee AY, Stewart JD, Clardy J, Ganem B. Chem Biol. 1995;2:195–203. doi: 10.1016/1074-5521(95)90269-4.Ganem B. Angew Chem, Int Ed. 1996;35:936–945.
- 2.Gajewski JJ, Jurayj J, Kimbrough DR, Gande ME, Ganem B, Carpenter BK. J Am Chem Soc. 1987;107:1170–1186. [Google Scholar]
- 3.For a review: Gajewski JJ. Acc Chem Res. 1997;30:219–225.
- 4.Severance DL, Jorgensen WL. J Am Chem Soc. 1992;114:10966–10969. [Google Scholar]
- 5.(a) Curran DP, Kuo LH. Tetrahedron Lett. 1995;36:6647–6650. [Google Scholar]; (b) Kirsten M, Rehbein J, Hiersemann M, Strassner T. J Org Chem. 2007;72:4001–4011. doi: 10.1021/jo062455y. [DOI] [PubMed] [Google Scholar]
- 6.(a) Maruoka K, Saito S, Yamamoto H. J Am Chem Soc. 1995;117:1165–1166. [Google Scholar]; (b) Tayama E, Saito A, Ooi T, Maruoka K. Tetrahedron. 2002;58:8307–8312. [Google Scholar]; (c) Kazmaier U, Mues H, Krebs A. Chem Eur J. 2002;8:1850–1855. doi: 10.1002/1521-3765(20020415)8:8<1850::AID-CHEM1850>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]; (d) Corey EJ, Lee D-H. J Am Chem Soc. 1991;113:4026–4028. [Google Scholar]; (e) Ito H, Sato A, Taguchi T. Tetrahedron Lett. 1997;38:4815–4818. [Google Scholar]; (f) Ito H, Sato A, Kobayashi T, Taguchi T. Chem Commun. 1998:2441–2442. [Google Scholar]; (g) Yoon TP, MacMillan DWC. J Am Chem Soc. 2001;123:2911–2912. doi: 10.1021/ja015612d. [DOI] [PubMed] [Google Scholar]; (h) Abraham L, Czerwonka R, Hiersemann M. Angew Chem, Int Ed. 2001;40:4700–4703. doi: 10.1002/1521-3773(20011217)40:24<4700::aid-anie4700>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]; (i) Abraham L, Körner M, Schwab P, Hiersemann M. Adv Synth Catal. 2004;346:1281–1294. [Google Scholar]
- 7.(a) Taylor MS, Jacobsen EN. Angew Chem, Int Ed. 2006;45:1520–1543. doi: 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]; (b) Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]
- 8.Of the salts evaluated, only guanidinium ions associated to the non-coordinating BArF counterion were found to be effective. See Supporting Information.
- 9.(a) Coates RM, Rogers BD, Hobbs SJ, Peck DR, Curran DP. J Am Chem Soc. 1987;109:1160–1170. [Google Scholar]; (b) Cramer CJ, Truhlar DG. J Am Chem Soc. 1992;114:8794–8799. [Google Scholar]; (c) Sehgal A, Shao L, Gao J. J Am Chem Soc. 1995;117:11337–11340. [Google Scholar]
- 10.For examples of asymmetric catalysis using chiral guanidines, see: Ishikawa T, Kumamoto T. Synthesis. 2006;5:737–753.Sohtome Y, Hashimoto Y, Nagasawa K. Eur J Org Chem. 2006;13:2894–2897.Shen J, Nguyen TT, Goh YP, Ye W, Fu X, Xu J, Tan CH. J Am Chem Soc. 2006;128:13692–13693. doi: 10.1021/ja064636n.Terada M, Nakano M, Ube H. J Am Chem Soc. 2006;128:16044–16045. doi: 10.1021/ja066808m.Terada M, Sorimachi K. J Am Chem Soc. 2007;129:292–293. doi: 10.1021/ja0678166.
- 11.All of the substrates for the asymmetric Claisen rearrangement were synthesized from the corresponding α-ketoacids with complete selectivity for the Z-enol ether by a simple two-step procedure involving O-alkylation and methyl ester formation.
- 12.Frisch MJ, et al. Gaussian 98. Gaussian, Inc; Pittsburgh, PA: 2002. DFT calculations were performed using Gaussian 98. [Google Scholar]
- 13.A pKa of 14.1 for 2 in DMSO was measured according to the procedure of Bordwell: Matthews WS, Bares JE, Bartmess JE, Bordwell FG, Cornforth FJ, Drucker GE, Margolin Z, McCallum RJ, McCollum GJ, Varnier NR. J Am Chem Soc. 1975;97:7006–7014.Bordwell measured a pKa of 13.5 for N, N’-diphenylthiourea in DMSO: Bordwell FG, Algrim DJ, Harrelson JA., Jr J Am Chem Soc. 1988;110:5903–5904.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete experimental procedures, summary of catalyst optimization studies, characterization data for new compounds, and complete ref. 12. This material is available free of charge via the Internet at http://pubs.acs.org.