

Abstract
Dopamine-mediated neurotransmission has been implicated in the modulation of synaptic plasticity and in the mechanisms underlying learning and memory. In the present study, we tested different forms of activity-dependent neuronal and behavioral plasticity in knockout mice for the dopamine transporter (DAT-KO), which constitute a unique genetic model of constitutive hyperdopaminergia. We report that DAT-KO mice exhibit slightly increased long-term potentiation and severely decreased long-term depression at hippocampal CA3-CA1 excitatory synapses. Mutant mice also show impaired adaptation to environmental changes in the Morris watermaze. Both the electrophysiological and behavioral phenotypes are reversed by the dopamine antagonist haloperidol, suggesting that hyperdopaminergia is involved in these deficits. These findings support the modulation by dopamine of synaptic plasticity and cognitive flexibility. The behavioral deficits seen in DAT-KO mice are reminiscent of the deficits in executive functions observed in dopamine-related neuropsychiatric disorders, suggesting that the study of DAT-KO mice can contribute to the understanding of the molecular basis of these disorders.
Keywords: Animals, Brain Diseases, Metabolic, genetics, metabolism, physiopathology, Cognition Disorders, genetics, metabolism, physiopathology, Delirium, Dementia, Amnestic, Cognitive Disorders, genetics, metabolism, physiopathology, Disease Models, Animal, Dopamine, metabolism, Dopamine Antagonists, pharmacology, Dopamine Plasma Membrane Transport Proteins, genetics, Dopamine Uptake Inhibitors, pharmacology, Female, Haloperidol, pharmacology, Hippocampus, metabolism, physiopathology, Long-Term Potentiation, genetics, Male, Maze Learning, drug effects, physiology, Memory Disorders, genetics, metabolism, physiopathology, Methylphenidate, pharmacology, Mice, Mice, Inbred C57BL, Mice, Inbred DBA, Mice, Knockout, Schizophrenia, genetics, metabolism, physiopathology
Keywords: dopamine transporter, synaptic plasticity, Morris watermaze, haloperidol, methylphenidate
INTRODUCTION
In addition to its key role in motor and reward systems, brain dopamine (DA) has also been implicated in integrative functions contributing to adaptive behaviors such as attention, learning, and memory (Nieoullon, 2002; Cools, 2006). Whereas DA or DA agonists improve memory, selective depletion, dysfunction or lesion of forebrain DA systems strongly impair acquisition and retention, as well as the ability to focus and adapt to environmental changes. In support of its role in cognitive processes, DA has been shown to exert a major modulatory input in synaptic plasticity in different brain areas, including the striatum (Calabresi et al, 1992; Arbuthnott et al, 2000), hippocampus (Lisman and Otmakhova, 2001), and prefrontal cortex (Gurden et al, 1999; Jay, 2003). However, clinical and experimental models of both excessive and deficient D00A transmission have revealed an inverted “U-shaped” relationship between DA levels and cognitive performance, and suggested that the beneficial or detrimental effects of DA depend on basal DA levels, task demands, and regional differences (Cools, 2006). Thus, the specific contribution of DA to the cognitive impairments observed in DA-related neuropsychiatric disorders such as schizophrenia, Parkinson’s disease, and attention-deficit hyperactivity disorder (ADHD) remains to be elucidated.
In the present study, we examined the role of DA in synaptic plasticity, learning and memory in mice lacking the dopamine transporter (DAT), which constitute a unique genetic model of persistent functional hyperdopaminergia (Giros et al, 1996; Gainetdinov and Caron, 2003). Mutant mice (DAT-KO) exhibit extremely high levels of DA synthesis and turnover (Jones et al, 1998), with a greatly prolonged (300-fold) extracellular lifetime of DA (Giros et al, 1996), resulting in a five- to ten-fold increase in basal extracellular DA levels in the striatum and nucleus accumbens (Jones et al, 1998; Spielewoy et al, 2000; Shen et al, 2004). Besides their obvious hyperactivity, DAT-KO mice exhibit heightened rates of stereotypies and perseverative patterns of non-goal directed behavior, distorting their motor, exploratory, and social behaviors (Gainetdinov et al, 1999; Spielewoy et al, 2000; Ralph et al, 2001; Rodriguiz et al, 2004). Based on their constitutive hyperdopaminergia, DAT-KO mice were used as a genetic model of long-term exposure to psychostimulants and were shown to exhibit an enhanced long-term potentiation (LTP) of the synaptic strength in the nucleus accumbens, a major component of the brain reward system (Yao et al, 2004).
Here, we studied whether constitutive hyperdopaminergia impaired activity-dependent synaptic plasticity in CA1 hippocampal slice preparations and learning and memory processes in the Morris watermaze.
MATERIALS AND METHODS
Animals
DAT knockout (KO) mice were obtained by homologous recombination (Giros et al, 1996) and maintained on two inbred genetic backgrounds: C57BL/6JOrl (B6) and DBA/2JOrl (D2), as previously described (Morice et al, 2004). In the present study, homozygous null mutants and their wild-type (WT) and heterozygous (HT) littermates were obtained from the mating of B6-DAT-HT females with D2-DAT-HT males in order to test the effect of the mutation on the hybrid B6xD2F1 genetic background. The genotype of the mice was determined by polymerase chain reaction (PCR) analysis as previously described (Carboni et al, 2001). Experiments were conducted during the light phase of a 12-h light/dark schedule, with lights on at 07:30 h. All behavioral and electrophysiological studies were performed on independent groups of naive animals. The studies were performed in accordance with the European Communities Council Directive (86/809/EEC) regarding the care and use of animals for experimental procedures and approved by the local ethical committee.
Electrophysiological assessments
Mice were decapitated under deep halothane anesthesia, and the hippocampus was quickly removed and placed in cold artificial cerebrospinal fluid (aCSF) continuously bubbled with a 95% O2/5% CO2 gas mixture (pH 7.4). The composition of the aCSF was as follows (in mM): NaCl, 124; KCl, 3.5; MgSO4, 1.5; CaCl2, 2.3; NaHCO3, 26.2; NaHPO4, 1.2; glucose, 11. Slices (400 μm thick) were cut and placed in a holding chamber to recover. Then, a single slice was transferred on a net to the test chamber and continuously perfused with aCSF (2 ml/min). Extracellular recordings were obtained at 25–30°C from the apical dendritic layer of CA1 area using micropipettes filled with 2 M NaCl (resistance of 2–6 MΩ). Field excitatory postsynaptic potentials (fEPSPs) were evoked every 10 s by electrical stimulation (100 μs duration) of afferent fibers located in the stratum radiatum.
In order to investigate the potentiation of synaptic transmission, test stimuli were applied every 15 s and adjusted to get a fEPSP slope at 30% of the maximum response. The initial slope of three averaged fEPSPs was measured for 15 min before the delivery of a theta-burst stimulation consisting of five trains of four pulses at 100 Hz separated by 200 ms. This sequence was repeated three times with an interburst interval of 10 s. In the case of high frequency stimulation, the conditioning stimulation consisted of two trains of 100 pulses at 100 Hz separated by 20 s. Testing with single pulse was then resumed for 60 min to determine the level of long-term potentiation (LTP). Synaptic depression was induced by a low frequency stimulation consisting of 1200 pulses at 2 Hz. This conditioning stimulation was induced after 15 min of baseline recording with a current intensity adjusted to obtain a fEPSP slope at 60% of the maximum response. Testing with single pulse was then resumed for 40 min after low frequency stimulation to determine the level of long-term depression (LTD). The effects of haloperidol (Haldol®, Janssen-Cilag, 1 μM) perfused for 20 min and methylphenidate (kindly supplied by Novartis Pharma AG, 20 μM)pre-incubated for 2 h before the baseline, were investigated on LTD in WT and KO mice. In these experiments, perfusion with the drugs was maintained throughout the recording.
Locomotor activity
Naive animals were individually assessed for their horizontal activities in transparent cages (20 × 15 × 25 cm), with automatic monitoring of photocell beam breaks (Imetronic, France). The animals were first habituated to the activity boxes for 30 min, then given an injection of saline (WT and KO mice) or haloperidol (KO mice) and immediately placed back into the apparatus for 2 h. For the dose-response to methylphenidate, after an initial assessment of spontaneous activity for 30 min, mice were injected with saline or methylphenidate (30, 60, or 80 mg/kg) and locomotor activity was recorded for 2 h.
Morris watermaze
The watermaze consists of a circular stainless steel pool (150 cm diameter, 29 cm height) filled to a depth of 16 cm with water maintained at 20–22°C and made opaque using a white aqueous emulsion (Acusol® OP 301 opacifier, Rohm lhaas, France). The escape platform, made of rough stainless steel, was submerged 1 cm below the water surface. A video tracking system (View Point, France) was used to monitor activity.
Hidden platform version
During the training phase of the standard place learning version of the Morris watermaze, mice learned the fixed position of a small hidden platform (6 cm diameter), using prominent distal extra-maze cues arranged in the room around the pool. Each trial started with the mice facing the interior wall of the pool and ended when they climbed onto the platform or after a maximum searching time of 90 s. The starting position was changed pseudo-randomly between trials. Animals that did not find the platform were gently guided and placed on it for 20 s. The animals received one habituation trial on the first day and then two trials per day for five consecutive days. The mice were left undisturbed in their home cage for the 90-min intertrial interval. On the 7th day, mice were given the last training trial before the 60-s probe test in which the platform had been removed. The distance traveled in each quadrant and the number of times the animal crossed each of the four possible platform sites (annulus) was automatically calculated from the video tracking system. After the first probe trial, all mice were given a reversal test in which the hidden platform was moved to a new position. The mice were trained for seven days following the same training procedure, and then tested for the second probe trial.
Cued-platform version
During the cued version of the watermaze, an independent group of naive mice were trained to find and escape onto a wider platform (9 cm diameter), made visible to the mice by a small black ball (4.5-cm diameter) fixed 11 cm above the platform. The training procedure was identical to that of the spatial version except that: i) animals were trained for only five consecutive days and ii) both the platform’s position and the animal’s starting position were pseudo-randomized for each trial.
Cued-platform version under haloperidol or methylphenidate treatment
Naive mice were given cued training trials in the watermaze as previously described, except that KO mice were injected (i.p. in a volume of 10 ml/kg body weight) with haloperidol (Haldol, Janssen-Cilag, diluted in NaCl 0.9%, 0.075 mg/kg) or methylphenidate (dissolved in NaCl 0.9%, 80 mg/kg) and WT mice were injected with saline, 30 and 15 min respectively before the test.
Statistical analysis
Repeated measures analysis of variance (ANOVA) was performed to assess the interaction between genotypes (between factor) and time (within factor). Significant main effects were further analyzed by post-hoc comparisons of means using Newman-Keuls tests. The one-tailed two-sample Kolmogorov-Smirnov test was used to determine if the distribution of the proportion of animals succeeding in a trial differed significantly depending on genotype. Comparison of mice according to their genotypes at the end of the training phase was done using the chi-square test (or the Mantel-Haenzel chi-square test when necessary). Only significant statistical tests are reported in the text, with the significance established at a P-value < 0.05.
RESULTS
Selective and reversible impairment in hippocampal LTD
Electrophysiology experiments were conducted using extracellular recordings in ex vivo hippocampal slices from naive WT, HT, and DAT-KO mice. Our results showed that basal glutamatergic transmission, including N-methyl-D-aspartate (NMDA) and non-NMDA receptor-mediated synaptic potentials, did not differ between genotypes (data not shown). The induction of LTP by theta-burst stimulation was significantly facilitated in DAT-KO relative to their WT littermates (last 15 min: F1,27 = 4.96; P < 0.05; Figure 1a), suggesting a reduced threshold in mutant mice. The magnitude of high frequency stimulation-induced LTP during the last 15 min of recording was also enhanced in DAT-KO, but the averaged LTP during the same period was not statistically significant between both genotypes (Figure 1b). In contrast, LTD induced by low frequency stimulation was severely impaired in DAT-KO mice (Figure 1c). Low frequency stimulation induced a robust and stable NMDA receptor-dependent LTD in WT, whereas LTD was dramatically reduced in DAT-KO (last 15 min: F1,23 = 12.9, P < 0.01). HT mice showed an intermediate phenotype compared to WT and KO mice, indicating that the LTP and LTD deficit are fine-tuned by the dopaminergic tone.
Figure 1.
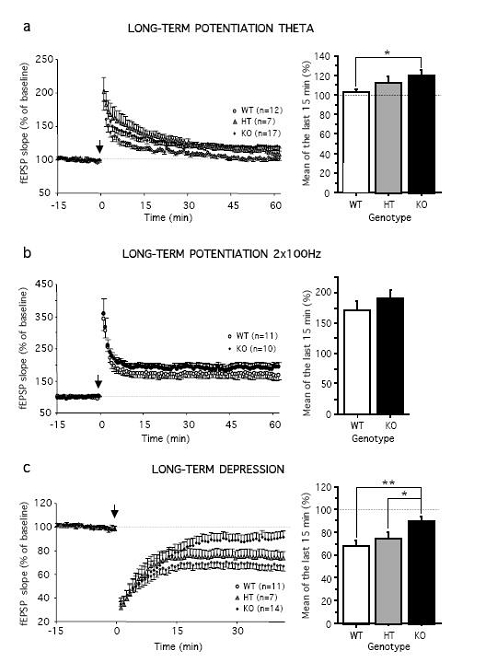
DAT-KO mice exhibit enhanced LTP but impaired LTD. (a, left) Time course of mean theta-burst stimulation-induced LTP (arrow) in hippocampal slices of WT (N = 7), HT (N = 4), and KO (N = 13) mice, (a, right) Summary histograms of the percent in fEPSP slope averaged from the last 15 min of recordings, (b, left) Time course of mean high frequency stimulation-induced LTP (arrow) in WT (N = 7) and KO (N = 6) mice, (b, right) Summary histograms of the percent in fEPSP slope averaged from the last 15 min of recordings, (c, left) Time course of mean low frequency stimulation-induced LTD (arrow) in WT (N = 8), HT (N = 5), and KO (N = 9) mice, (c, right) Summary histograms of the percentage in fEPSP slope averaged from the last 15 min of recordings. The dotted line represents percentage of baseline. Values represent means ± s.e.m. (n) represent numbers of slices per group. *P< 0.05 and **P< 0.01.
In order to assess whether enhanced DA levels at the synaptic cleft were directly responsible for the decreased LTD, we examined the effect of the DA antagonist haloperidol. In hippocampal slices of both WT and KO mice, haloperidol did not affect basal glutamatergic neurotransmission (data not shown). Interestingly, haloperidol was able to fully prevent the deficit of LTD in mutant mice. After administration of the dopaminergic antagonist, the LTD in DAT-KO mice was significantly facilitated and did not differ any longer from WT mice (Figure 2a).
Figure 2.
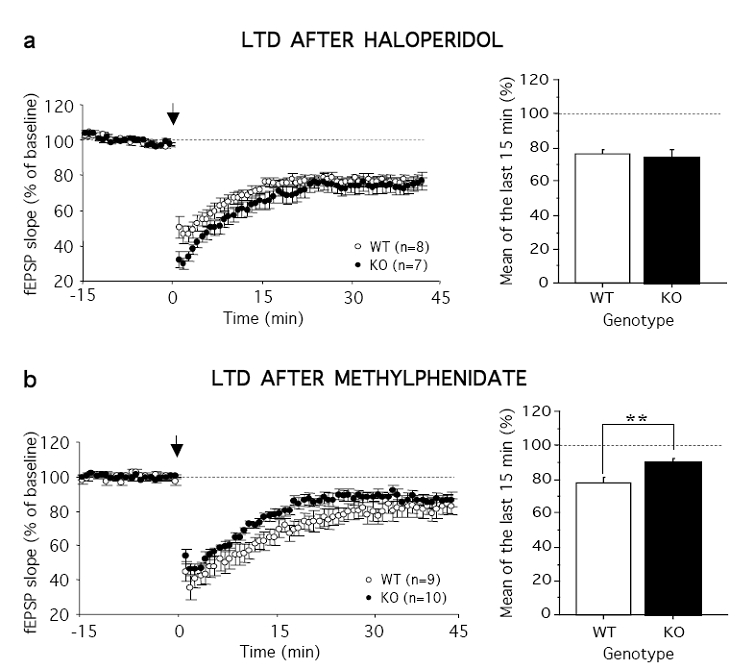
Haloperidol, but not methylphenidate, fully restores LTD in DAT-KO mice, (a, left) Time course of mean low frequency stimulation-induced LTD (arrow) calculated from slices of WT (N = 8) and KO (N = 5) mice in the presence of haloperidol. (a, right) Summary histograms of the percent in fEPSP slope averaged from the last 15 min of recordings performed in the presence of haloperidol. (b, left) Time course of mean low frequency stimulation-induced LTD (arrow) in WT (N = 6) and KO (N = 6) mice in the presence of methylphenidate. (b, right) Summary histograms of the percent in fEPSP slope averaged from the last 15 min of recordings performed in the presence of methylphenidate. The dotted line represents percentage of baseline. Values represent means ± s.e.m. (n) represent numbers of slices per group. **P< 0.01.
The role of NMDA receptor activation on synaptic plasticity was investigated after blocking NMDA receptors with the competitive antagonist DL-2-amino-5-phosphonovalerate acid (D-APV, 50 μM). D-APV completely blocked the induction of both LTP and LTD (with or without haloperidol) in DAT-KO and WT mice, indicating that these processes were NMDA receptor-dependent.
DAT-KO mice have been proposed as an useful model of ADHD, based on the observation that the psychostimulant-like methylphenidate, one of the most frequently prescribed treatments for ADHD, remarkably attenuates their hyperactivity (Gainetdinov et al, 1999). We thus tested whether methylphenidate could also improve the electrophysiological impairment in synaptic plasticity in DAT-KO mice. However, we found that methylphenidate did not improve LTD in KO mice (Figure 2b), which remained significantly reduced compared to WT mice (last 15 min: F1,34 = 8.90, P < 0.001).
Delayed acquisition of place navigation but intact spatial memory in the Morris watermaze
DAT-KO mice were tested for spatial learning and memory performance in the Morris watermaze. Since DAT-KO mice swam faster than their WT littermates (speed: F1,98 = 18.1, P < 10−4), distance rather than latency to find the platform was chosen as a learning variable. Compared with their WT and HT littermates, KO mice swam a longer distance to find the hidden escape platform (F2,147 = 15.24, P < 10−4, Figure 3a). However, the distance decreased during training in the three genotypes, indicating that learning had occurred (F6,147 = 18.39, P < 0.0001), and by the last two days, all mice exhibited the same level of performance. Furthermore, during the probe trial, when the platform was removed, the three genotypes showed a strong preference for the target quadrant (F3,84 = 40.12, P < 0.0001; Figure 3b) and for the place where the platform was located (annulus crossings, F3,84 = 16.64, P < 0.0001). These results demonstrate that DAT-KO mice are able to utilize a spatial strategy to learn the position of the platform.
Figure 3.
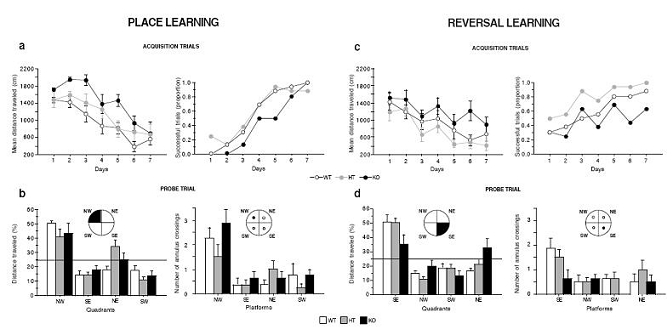
DAT-KO mice exhibit normal spatial reference memory, but show disrupted reversal learning in the Morris watermaze. Performance of WT, HT, and KO mice during acquisition trials of the place (a) and reversal (c) learning. The results are expressed as distance traveled (cm) and proportion of successful trials. Probe trial of the place (b) and reversal learning (d). Mice were scored for the percentage of distance traveled in the four quadrants: north-est (NE), south-est (SE), south-west (SW), and north-west (NW), and for the number of annulus crossings. The targeted quadrant and platform are indicated in black. The horizontal line indicates the distance traveled using a random search strategy. Values represent means ± s.e.m. (n = 8 mice per group).
Disturbed reversal learning in the Morris watermaze
We then examined learning flexibility on the reversal test, when the hidden-platform was moved to a new location. Although all groups improved with training (F6,147 = 8.72, P < 0.0001; Figure 3c), DAT-KO mice swam greater distance to find the platform (F2,147 = 12.05, P < 0.0001), and this deficit persisted until the end of the training phase (for training days 6 and 7: genotype, F2,42 = 7.65, P < 0.01, and successful trials, KO versus WT, χ22df = 14.93, P < 0.001). Moreover, during the probe trial, WT and HT mice exhibited a spatial strategy, favoring the target quadrant and the place where the platform was located, whereas DAT-KO mice swam an equal distance in all quadrants (quadrant, F3,84 = 34.87; P < 0.0001, and genotype x quadrant interaction, F6,84 = 3.48; P < 0.001; Figure 3d) and crossed the four virtual platforms as often (annulus crossings, F3,84 = 6.2, P < 0.001, and genotype, F2,84 = 2.2, P < 0.03). Therefore, DAT-KO mice fail to adapt their behavior following a change of the platform position, indicating a lack of behavioral flexibility.
The deficit of KO mice was not due to inability to shift away from the previously reinforced quadrant because during the first trial following the quadrant test, they did not visit the previous reinforced quadrant more often than the other quadrants. Instead, when they didn’t find the platform during the reversal training, their behavior became chaotic and not focused to searching the platform, as illustrated by the variability in the distance swam to reach the platform and in the number of successful trials (Figure 3c). These findings demonstrate that the disturbed reversal learning of DAT-KO mice is not the expression of a perseverative behavior in the watermaze.
Severe impairment in the cued version of the Morris watermaze
In the cued version of the watermaze, mice were trained to swim to a platform mounted with a visible cue and placed in a different spatial location on each trial. The distance traveled to escape decreased significantly with time in both WT and HT mice, whereas DAT-KO mice showed hardly any improvement over the course of training (genotype, F2,75 = 37.74, P < 0.0001; training, F4,75= 10.32, P < 0.0001; and interaction, F8,75 = 2.77, P < 0.01; Figure 4a). DAT-KO mice also succeeded in escaping on fewer trials than WT and HT mice (χ22df = 29.0, P < 0.0001 and χ22df = 14.8, P < 0.001, respectively). In order to test whether this deficit was due to an inability of DAT-KO mice to associate the cue ball to the escape platform, a new group of mice was trained in the cued version following the same training procedure except that the platform was kept in a fixed position. In this condition, we found that DAT-KO mice escaped successfully on twice as many trials as before, and their performance improved with training (KO, F4,30 = 3.8, P < 0.05; Figure 4b), demonstrating that deficits observed in the previous test were due to the lack of adaptation to rapid changes in the environment rather than to a deficit of associative learning.
Figure 4.
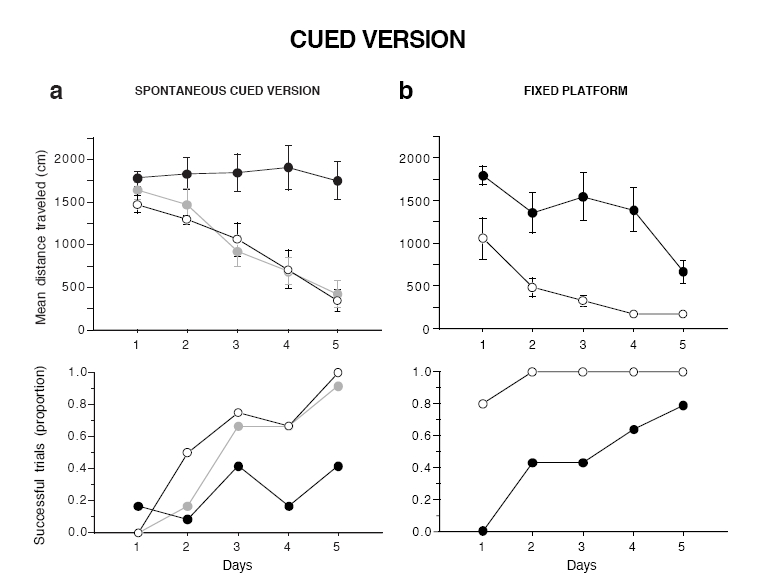
DAT-KO mice are severely impaired in the cued version of the Morris watermaze. (a) Performance of WT, HT, and KO mice on the spontaneous cued version, (b) Performance of WT and KO mice in the cued version with the platform kept at a fixed position. Results are expressed as mean distance traveled (top) and proportion of successful trials (bottom). Values represent means ± s.e.m. (n = 5–7 mice per group).
To test whether the excess of DA neurotransmission contributed to the loss of behavioral flexibility exhibited by DAT-KO mice, an independent group of animals was treated with haloperidol and tested on cued navigation. The dose of haloperidol was based on previous experiments (Spielewoy et al, 2000). The dose of 0.075 mg/kg was found to restore normal locomotor activity in DAT-KO, not only in the actimeter (data not shown), but also in the watermaze, as illustrated by the swimming speed that did not differ any longer among the KO and WT genotypes and increased in a similar way with training (F4,70 = 8.0, P < 0.0001; Figure 5a). In the watermaze, following haloperidol administration, DAT-KO became as successful as WT mice in finding the platform, and their performance markedly improved with training (KO, F1,35= 4.35; P < 0.05; Figure 5b).
Figure 5.
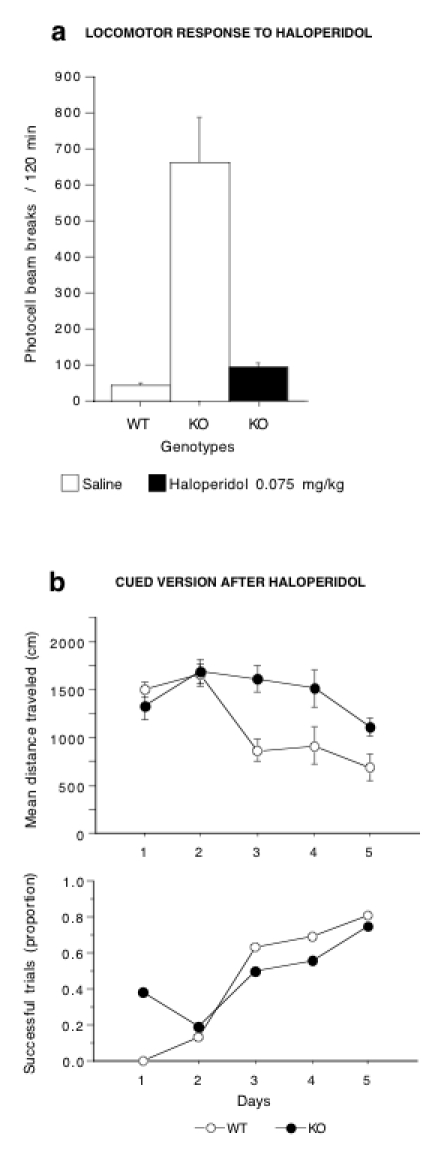
Haloperidol improves the performances of DAT-KO mice in the cued version of the Morris watermaze. (a) Total locomotor horizontal activity in WT and KO mice automatically recorded over a 2-h period after acute injection of saline or haloperidol (n = 5–8 mice per group), (b) Performance of WT and KO mice in the cued version of the watermaze. Thirty minutes before each daily session, mutant mice were injected with haloperidol (0.075 mg/kg, i.p.) and WT mice received a saline injection. Results are expressed as mean distance traveled (top) and proportion of successful trials (bottom). Values represent means ± s.e.m. (n = 8 mice per group).
We then examined whether, in addition to its calming effect (decrease in hyperlocomotion) (Gainetdinov et al, 1999), methylphenidate could improve the cognitive performance of DAT-KO mice in the cued version of the Morris watermaze. Methylphenidate exerts its psychostimulant-like effects in WT mice via the DAT, whereas its mechanism of action in DAT-KO mice is not yet understood. Thus, the purpose of this experiment was not to compare DAT-KO mice to psychostimulated animals but to test whether methylphenidate restored cognition in DAT-KO mice to the level of control animals. A dose-response experiment confirmed its inhibitory effect on horizontal activity of mutant mice and revealed that the dose of 80 mg/kg reduced the locomotion of DAT-KO mice to the level of WT animals (Figure 6a). However, in the Morris watermaze (Figure 6b), DAT-KO showed hardly any improvement with training after methylphenidate treatment (genotype, F1,84 = 17.5, P < 0.0001; training, F5,84= 4.8, P < 0.001; and interaction, F5,84 = 11.5, P < 0.0001) and escaped successfully on twice as fewer trials as their WT littermates (χ21df = 21.8, P < 0.001).
Figure 6.
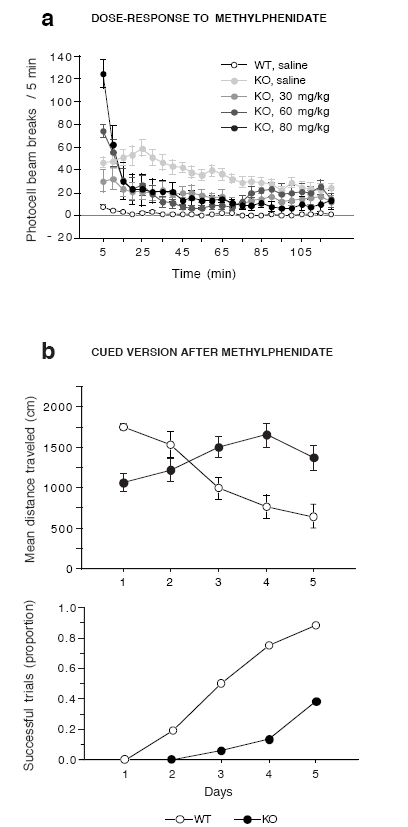
Methylphenidate does not improve the performance of DAT-KO mice in the cued version of the Morris watermaze. (a) Locomotor dose-response to administration of methylphenidate in DAT-KO mice over a 2-h period (n = 5–15 mice per group), (b) Performance of the WT and KO in the cued version following methylphenidate treatment. Fifteen minutes before each daily session, mutant mice were injected with methylphenidate (80 mg/kg, i.p.), and WT mice received a saline injection. All results are expressed as mean distance traveled (top) and proportion of successful trials (bottom). Values represent means ± s.e.m. (n = 8 mice per group).
DISCUSSION
The present study illustrates a profound modification of various forms of activity-dependent plasticity in DAT-KO mice, highlighted by enhanced LTP, markedly impaired LTD, and disturbed cognitive adaptation to environmental changes in the watermaze. Notably, both impaired LTD and cued-learning are reversed by haloperidol. This result stongly suggests that the hyperdopaminergic tone during the test, rather than compensatory mechanisms occurring during development, is directly involved in these deficits, although other mechanisms in DAT-KO mice, such as a direct effect of haloperidol cannot be excluded (Arvanov et al, 1997; Gemperle et al, 2003). In addition, this parallel loss/rescue strongly supports the hypothesis of common mechanisms underlying both forms of cellular and behavioral plasticity (Malenka and Bear, 2004).
In vitro electrophysiological studies have provided clear evidence that DA acts as a major modulator of fast glutamatergic neurotransmission (Jay, 2003). The modulatory role of DA in synaptic plasticity is further supported by in vivo studies showing that repeated systemic exposure to psychostimulants facilitates LTP induction in the midbrain (Liu et al, 2005), reduces LTD in the nucleus accumbens (Thomas et al, 2001), and converts LTD into LTP in the striatum (Nishioku et al, 1999). Interestingly, enhanced LTP of the cortico-accumbal synapses was recently reported in DAT-KO mice, used as a genetic model of long-term exposure to psychostimulants (Yao et al, 2004). Altogether, these data and ours, demonstrating that both LTP and LTD are altered in hippocampal CA1 synapses, suggest that hyperdopaminergia mediates a loss of bidirectional synaptic plasticity in high-order pathways and strengthen the emerging consensus that drug addiction and memory processes may rely on similar cellular mechanisms (Nestler et al, 1993; Berke and Hyman, 2000; Hyman and Malenka, 2001; Malenka and Bear, 2004).
Our results indicate that the hyperdopaminergia of DAT-KO mice produces cognitive deficits similar to those observed following lesion or pharmacologically-induced hypodopaminergia of the mesocorticolimbic DA system (Cools, 2006), and are thus in agreement with the observation that both excessive and insufficient DA levels impair cognitive performances (Arnsten, 1998). The hyperdopaminergia of DAT-KO mice does not suppress the ability to perform behavior, they are not debilitated and they interact socially with their littermates (Spielewoy et al, 2000). Moreover, they do not exhibit a general defect in learning. In a self-administration paradigm, where food is used as a reinforcer, DAT-KO mice are able to learn an operant behavior within the same training sessions as controls (Rocha et al, 1998). Similarly, KO mice are able to discriminate olfactory stimuli, and show no deficits in associative learning as tested in the place preference test (Sora et al, 1998; Rodriguiz et al, 2004). Here, we show that their spatial navigation and motivation are spared: DAT-KO mice actively search for the platform, climb on it as soon as they find it, and do not exhibit any floating behavior. Rather, they appear dramatically impaired in their capacity to adapt their behavior to environmental changes, as illustrated by their disturbed reversal learning, impaired learning in the cued version, and improved performance only when the cued platform was kept at a fixed position. Together with previous studies showing their perseverative patterns of social behaviors and their greater distractibility (Ralph et al, 2001; Rodriguiz et al, 2004), these data support a permissive rather than a mnemonic role of central DA (Whishaw and Dunnett, 1985).
Complex cognitive functions, referred to as executive functions, which contribute to the continuous orchestration of well-adapted and goal-directed behaviors, rely on both prefrontal cortex and striatal DA transmission (Cools, 2006). Several lines of evidence suggest that the cognitive deficits observed in DAT-KO mice are more likely to depend on striatal DA rather than on prefrontal cortex DA. First, whereas the DAT is a major determinant of the intensity and duration of DA neurotransmission in the striatum, microdialysis experiments demonstrate a less prominent role of the DAT in DA clearance in the prefrontal cortex (Cass and Gerhardt, 1995), which correlates with the relative sparse distribution of the DAT in the prefrontal cortex (Sesack et al, 1998), as well as with several studies showing heterologous DA uptake by the norepinephrine transporter in this region (Carboni et al, 1990; Tanda et al, 1997). Second, in DAT-KO mice, dialysate DA is approximately 10-fold higher in both the striatum and nucleus accumbens, whereas the basal level of extracellular DA is not affected in the prefrontal cortex (Shen et al, 2004). Furthermore, the rate of DA uptake in the prefrontal cortex has also been shown to be normal in DAT-KO mice (Mundorf et al, 2001).
Not surprisingly, the dramatic molecular alterations of both pre- and post-synaptic homeostasis following inactivation of the DAT have been extensively described in the striatum (Giros et al, 1996; Rocha et al, 1998; Jones et al, 1999; Shen et al, 2004), whereas no data are available in these mutant mice concerning the mesencephalo-hippocampal DA projection. We show here that synaptic plasticity is clearly impaired at the Schaffer collateral-CA1 synapses. Even though we demonstrated that hippocampal-dependent spatial navigation is clearly preserved in DAT-KO mice, impaired DA transmission in the hippocampus could be involved in their initial delayed acquisition. The potential implication of the hippocampus in the observed deficits is further supported by a growing body of evidence demonstrating that both striatal and hippocampal systems interact intimately in various cognitive functions (Poldrack and Packard, 2003; Voermans et al, 2004), and that the hippocampus and the midbrain form a DA-dependent loop that contributes to the role of the hippocampus in novelty detection (Lisman and Grace, 2005). However, further molecular characterization of DA homeostasis within the hippocampal region of DAT-KO mice remains to be carried out.
It was previously reported that ADHD patients suffer primarily from an inability to inhibit their behavioral response, suggesting that their impaired cognitive function could be secondary to the hyperkinesia (Barkley, 1997). The observation that psychostimulants exerted a calming effect in DAT-KO mice suggested that similar mechanisms may exist in DAT-KO mice and they were thus proposed as a model of ADHD (Gainetdinov et al, 1999). The present finding demonstrating that methylphenidate, despite its efficient attenuation of the hyperactivity in DAT-KO mice, neither improved cued-learning nor restored LTD, challenges this hypothesis. Our results suggest that the serotoninergic transmission, shown to mediate the calming effect of methylphenidate on locomotor activity of DAT-KO mice (Gainetdinov et al, 1999), is not likely to be involved in their cognitive deficit.
Mental rigidity, perseveration, inability to shift, to adapt and to adjust behaviors to the context are common features of DA-related disorders such as schizophrenia, Parkinson’s disease, and ADHD. We show here that similar behavioral phenotypes are present in DAT-KO mice, as the result of genetically-induced hyperdopaminergia. Thus, DAT-KO mice offer an unprecedented animal model to study the neural and molecular mechanisms underlying these transnosographic symptoms for which no satisfactory treatment is yet available.
Acknowledgments
We thank S. Davis and S. Cooke for helpful comments on the manuscript. We also thank L. Hillard for animal care. This work was supported by INSERM and Fondation pour la Recherche Médicale. E.M. was supported by a fellowship from the Ministere de I’Education Nationale, de la Recherche et de la Technologie.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- Arbuthnott GW, Ingham CA, Wickens JR. Dopamine and synaptic plasticity in the neostriatum. J Anat. 2000;196(Pt 4):587–596. doi: 10.1046/j.1469-7580.2000.19640587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT. Catecholamine modulation of prefrontal cortical cognitive function. Trends in Cognitive Sciences. 1998;2:436–446. doi: 10.1016/s1364-6613(98)01240-6. [DOI] [PubMed] [Google Scholar]
- Arvanov VL, Liang X, Schwartz J, Grossman S, Wang RY. Clozapine and haloperidol modulate N-methyl-D-aspartate- and non-N-methyl-D-aspartate receptor-mediated neurotransmission in rat prefrontal cortical neurons in vitro. J Pharmacol Exp Ther. 1997;283:226–234. [PubMed] [Google Scholar]
- Barkley RA. Behavioral inhibition, sustained attention, and executive functions: constructing a unifying theory of ADHD. Psychol Bull. 1997;121:65–94. doi: 10.1037/0033-2909.121.1.65. [DOI] [PubMed] [Google Scholar]
- Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboni E, Tanda GL, Frau R, Di Chiara G. Blockade of the noradrenaline carrier increases extracellular dopamine concentrations in the prefrontal cortex: evidence that dopamine is taken up in vivo by noradrenergic terminals. J Neurochem. 1990;55:1067–1070. doi: 10.1111/j.1471-4159.1990.tb04599.x. [DOI] [PubMed] [Google Scholar]
- Carboni E, Spielewoy C, Vacca C, Nosten-Bertrand M, Giros B, Di Chiara G. Cocaine and amphetamine increase extracellular dopamine in the nucleus accumbens of mice lacking the dopamine transporter gene. J Neurosci. 2001;21(RC141):141–144. doi: 10.1523/JNEUROSCI.21-09-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass WA, Gerhardt GA. In vivo assessment of dopamine uptake in rat medial prefrontal cortex: comparison with dorsal striatum and nucleus accumbens. J Neurochem. 1995;65:201–207. doi: 10.1046/j.1471-4159.1995.65010201.x. [DOI] [PubMed] [Google Scholar]
- Cools R. Dopaminergic modulation of cognitive function-implications for L-DOPA treatment in Parkinson’s disease. Neurosci Biobehav Rev. 2006;30:1–23. doi: 10.1016/j.neubiorev.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG. Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science. 1999;283:397–401. doi: 10.1126/science.283.5400.397. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Caron MG. Monoamine transporters: from genes to behavior. Annu Rev Pharmacol Toxicol. 2003;43:261–284. doi: 10.1146/annurev.pharmtox.43.050802.112309. Epub 2002 Sep 2017. [DOI] [PubMed] [Google Scholar]
- Gemperle AY, Enz A, Pozza MF, Luthi A, Olpe HR. Effects of clozapine, haloperidol and iloperidone on neurotransmission and synaptic plasticity in prefrontal cortex and their accumulation in brain tissue: an in vitro study. Neuroscience. 2003;117:681–695. doi: 10.1016/s0306-4522(02)00769-8. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Gurden H, Tassin JP, Jay TM. Integrity of the mesocortical dopaminergic system is necessary for complete expression of in vivo hippocampal-prefrontal cortex long-term potentiation. Neuroscience. 1999;94:1019–1027. doi: 10.1016/s0306-4522(99)00395-4. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC. Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci. 2001;2:695–703. doi: 10.1038/35094560. [DOI] [PubMed] [Google Scholar]
- Jay TM. Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog Neurobiol. 2003;69:375–390. doi: 10.1016/s0301-0082(03)00085-6. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci USA. 1998;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Hu XT, Cooper DC, Wightman RM, White FJ, Caron MG. Loss of autoreceptor functions in mice lacking the dopamine transporter. Nat Neurosci. 1999;2:649–655. doi: 10.1038/10204. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Otmakhova NA. Storage, recall, and novelty detection of sequences by the hippocampus: elaborating on the SOCRATIC model to account for normal and aberrant effects of dopamine. Hippocampus. 2001;11:551–568. doi: 10.1002/hipo.1071. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Morice E, Denis C, Giros B, Nosten-Bertrand M. Phenotypic expression of the targeted null-mutation in the dopamine transporter gene varies as a function of the genetic background. Eur J Neurosci. 2004;20:120–126. doi: 10.1111/j.1460-9568.2004.03465.x. [DOI] [PubMed] [Google Scholar]
- Mundorf ML, Joseph JD, Austin CM, Caron MG, Wightman RM. Catecholamine release and uptake in the mouse prefrontal cortex. J Neurochem. 2001;79:130–142. doi: 10.1046/j.1471-4159.2001.00554.x. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Hope BT, Widnell KL. Drug addiction: a model for the molecular basis of neural plasticity. Neuron. 1993;11:995–1006. doi: 10.1016/0896-6273(93)90213-b. [DOI] [PubMed] [Google Scholar]
- Nieoullon A. Dopamine and the regulation of cognition and attention. Prog Neurobiol. 2002;67:53–83. doi: 10.1016/s0301-0082(02)00011-4. [DOI] [PubMed] [Google Scholar]
- Nishioku T, Shimazoe T, Yamamoto Y, Nakanishi H, Watanabe S. Expression of long-term potentiation of the striatum in methamphetamine-sensitized rats. Neurosci Lett. 1999;268:81–84. doi: 10.1016/s0304-3940(99)00386-9. [DOI] [PubMed] [Google Scholar]
- Poldrack RA, Packard MG. Competition among multiple memory systems: converging evidence from animal and human brain studies. Neuropsychologia. 2003;41:245–251. doi: 10.1016/s0028-3932(02)00157-4. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, Paulus MP, Fumagalli F, Caron MG, Geyer MA. Prepulse inhibition deficits and perseverative motor patterns in dopamine transporter knock-out mice: differential effects of D1 and D2 receptor antagonists. J Neurosci. 2001;21:305–313. doi: 10.1523/JNEUROSCI.21-01-00305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B, Miller GW, Caron MG. Cocaine self-administration in dopamine-transporter knockout mice. Nat Neurosci. 1998;1:132–137. doi: 10.1038/381. [see comments] [published erratum appears in Nat Neurosci 1998 Aug;1(4):330] [DOI] [PubMed] [Google Scholar]
- Rodriguiz RM, Chu R, Caron MG, Wetsel WC. Aberrant responses in social interaction of dopamine transporter knockout mice. Behav Brain Res. 2004;148:185–198. doi: 10.1016/s0166-4328(03)00187-6. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Hawrylak VA, Matus C, Guido MA, Levey Al. Dopamine axon varicosities in the prelimbic division of the rat prefrontal cortex exhibit sparse immunoreactivity for the dopamine transporter. J Neurosci. 1998;18:2697–2708. doi: 10.1523/JNEUROSCI.18-07-02697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HW, Hagino Y, Kobayashi H, Shinohara-Tanaka K, Ikeda K, Yamamoto H, Yamamoto T, Lesch KP, Murphy DL, Hall FS, Uhl GR, Sora I. Regional differences in extracellular dopamine and serotonin assessed by in vivo microdialysis in mice lacking dopamine and/or serotonin transporters. Neuropsychopharmacology. 2004;29:1790–1799. doi: 10.1038/sj.npp.1300476. [DOI] [PubMed] [Google Scholar]
- Sora I, Wichems C, Takahashi N, Li XF, Zeng Z, Revay R, Lesch KP, Murphy DL, Uhl GR. Cocaine reward models: conditioned place preference can be established in dopamine- and in serotonin-transporter knockout mice. Proc Natl Acad Sci U S A. 1998;95:7699–7704. doi: 10.1073/pnas.95.13.7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielewoy C, Gonon F, Roubert C, Fauchey V, Jaber M, Caron MG, Roques BP, Hamon M, Betancur C, Maldonado R, Giros B. Increased rewarding properties of morphine in dopamine-transporter knockout mice. Eur J Neurosci. 2000;12:1827–1837. doi: 10.1046/j.1460-9568.2000.00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielewoy C, Roubert C, Hamon M, Nosten-Bertrand M, Betancur C, Giros B. Behavioural disturbances associated with hyperdopaminergia in dopamine-transporter knockout mice. Behav Pharmacol. 2000;11:279–290. doi: 10.1097/00008877-200006000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanda G, Pontieri FE, Frau R, Di Chiara G. Contribution of blockade of the noradrenaline carrier to the increase of extracellular dopamine in the rat prefrontal cortex by amphetamine and cocaine. Eur J Neurosci. 1997;9:2077–2085. doi: 10.1111/j.1460-9568.1997.tb01375.x. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Voermans NC, Petersson KM, Daudey L, Weber B, Van Spaendonck KP, Kremer HP, Fernandez G. Interaction between the human hippocampus and the caudate nucleus during route recognition. Neuron. 2004;43:427–435. doi: 10.1016/j.neuron.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Whishaw IQ, Dunnett SB. Dopamine depletion, stimulation or blockade in the rat disrupts spatial navigation and locomotion dependent upon beacon or distal cues. Behav Brain Res. 1985;18:11–29. doi: 10.1016/0166-4328(85)90165-2. [DOI] [PubMed] [Google Scholar]
- Yao WD, Gainetdinov RR, Arbuckle Ml, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, Caron MG. Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron. 2004;41:625–638. doi: 10.1016/s0896-6273(04)00048-0. [DOI] [PubMed] [Google Scholar]