

Abstract
A genome scan was previously performed and pointed to chromosome 6q21 as a candidate region for autism. This region contains the glutamate receptor 6 (GluR6 or GRIK2) gene, a functional candidate for the syndrome. Glutamate is the principal excitatory neurotransmitter in the brain and is directly involved in cognitive functions such as memory and learning. We used two different approaches, the affected sib-pair (ASP) method and the transmission disequilibrium test (TDT), to investigate the linkage and association between GluR6 and autism. The ASP method, conducted with additional markers on the 51 original families and in 8 new sibling pairs, showed a significant excess of allele sharing, generating an elevated multipoint maximum LOD score (ASPEX MLS = 3.28). TDT analysis, performed in the ASP families and in an independent data set of 107 parent-offspring trios, indicated a significant maternal transmission disequilibrium (TDTall P = 0.0004). Furthermore, TDT analysis (with only one affected proband per family) showed significant association between GluR6 and autism (TDT association P = 0.008). In contrast to maternal transmission, paternal transmission of GluR6 alleles was as expected in the absence of linkage, suggesting a maternal effect such as imprinting. Mutation screening was performed in 33 affected individuals, revealing several nucleotide polymorphisms (SNPs), including one amino acid change (M867I) in a highly conserved domain of the intracytoplasmic C-terminal region of the protein. This change is found in 8% of the autistic subjects and in 4% of the control population and seems to be more maternally transmitted than expected to autistic males (P = 0.007). Taken together, these data suggest that GluR6 is in linkage disequilibrium with autism.
Keywords: Amino Acid Sequence, Autistic Disorder, genetics, Brain, physiopathology, Child, Chromosome Mapping, Chromosomes, Human, Pair 6, Exons, Family, Female, Genetic Markers, Genotype, Glutamic Acid, physiology, Humans, Linkage (Genetics), Male, Molecular Sequence Data, Open Reading Frames, Receptors, Kainic Acid, genetics, Restriction Mapping
Keywords: autistic disorder, GluR6, GRIK2, mutation screening, affected sib-pair method, TDT, linkage disequilibrium, single nucleotide polymorphism, editing, isoforms
Introduction
Autism (MIM 209850) is a pervasive developmental disorder (PDD) characterised by impairments in reciprocal social interaction and communication as well as restricted and stereotyped patterns of interests and activities.1,2 The symptoms are usually apparent during the first three years of life and the population prevalence is 1/1000,3 with a sex ratio of 4:1 in favour of males.4 Since the original reports by Kanner,1 many studies have supported a genetic aetiology for autism. Familial cases are rare, but substantially more frequent than expected by chance. Indeed, the recurrence risk of autism in sibships is approximately 45 times greater than in the general population.3,5 Furthermore, twin studies have documented a higher concordance rate in monozygotic (60–91%) than in dizygotic twins (0–6%)6,7, indicating that the heritability of autism may be over 90%.7
To localise susceptibility loci in autism, five independent genome scans have been performed and indicated a multiloci aetiology.8–12 Several regions exhibiting elevated maximum LOD scores (MLS) were identified as candidates, on chromosomes 1p, 2q, 6q16-21, 7q31-32, 13q, and 15q. In the Paris Autism Research International Sibpair (PARIS) study, conducted with 51 affected sib pairs (ASP), chromosome 6q21 was the most significant region, with a multipoint MLS of 2.23.9 This region contains the glutamate receptor 6 (GluR6 or GRIK2) gene, an excellent positional and functional candidate for susceptibility to autism.
Glutamate is the principal excitatory neurotransmitter in the brain and acts via two types of glutamate receptors: metabotropic and ionotropic.13 The metabotropic receptors are coupled to various second messenger systems via GTP-binding proteins, whereas the ionotropic glutamate receptors form ligand-gated ion channels and are named according to their protypical agonists: NMDA (N-methyl-D-aspartate) receptors, AMPA (α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate) receptors (GluR1-4), and kainate receptors (GluR6-7, KA1 and KA2). GluR6 is a member of the ionotropic kainate receptors (KARs), expressed during brain development and post-transcriptionally modified by editing.14 Glutamate and its receptors are directly involved in cognitive functions such as memory and learning15 and are candidates for several neurological and psychiatric diseases.16 Based on neuroanatomical studies and similarities between symptoms produced by glutamate antagonists in healthy subjects and those seen in autism, it has been recently proposed that autism could be a hypoglutamatergic disorder.17 Furthermore, animal models of hypoglutamatergia show several similarities with autism, such as defective habituation, restricted behavioural repertoire and inability to change behavioural program.17 Finally, disturbed glutamate concentrations have been reported in autistic patients compared to controls, with high levels in plasma18 and low levels in platelets.19
In the present study, we present the complete genomic structure of the human GluR6 gene, including new isoforms, linkage/association analyses in autistic subjects and the nucleotide variations present in the gene. These studies support the hypothesis that GluR6 may be a susceptibility gene for autism.
Materials and methods
Families
Families with at least one autistic child were recruited by the PARIS study at specialised clinical centres in seven countries (Austria, Belgium, France, Italy, Norway, Sweden, and the United States). Fifty-nine families with at least two children with autism (including one family with three autistic children and one family with two affected siblings and one affected half-sibling) comprise the sample for the linkage analysis and were described previously.9 One hundred and seven families with one autistic child were added for the association study. All the autistic subjects fulfilled the DSM IV criteria for autistic disorder2 and the Autism Diagnostic Interview Revised (ADI-R) algorithm for ICD-10 childhood autism.20 Subjects were included only after thorough clinical, neuropsychological and neurological examination described elsewhere.9 Cases diagnosed with associated organic conditions or other established chromosomal disorders were excluded. The study was approved by the ethical committees of the collaborating institutions. Informed consents were obtained from the parents of each child included in the study.
DNA, RNA analysis and genotyping
Blood samples were collected from both parents and autistic subjects. DNA was extracted and B lymphoblastoid cell lines generated. Most of the microsatellites were amplified and genotyped at the Centre National de Genotypage (Evry, France) using fluorescent primers as described.9 Sequence analysis of the GluR6 gene was performed by direct sequence of the PCR products, using fluorescently labelled Taq DyeDeoxy terminator reaction mix and a 373A automated DNA sequencer (Applied Biosystems). The C/T SNP in intron 14 is located 8 bp from exon 15 and was genotyped with enzyme EcoNI, which digests when C is present. The G/A SNP in exon 15 was genotyped using a mismatch primer, which creates a HindIII site when G is present. These two SNPs are separated by 120 bp. The G/A SNP in exon 16 (M867I) was genotyped with EcoRV, which digests when A (Isoleucine) is present. For the analysis of the GluR6 transcripts, total brain RNAs (Clontech) were reverse transcribed using the Gene Amp RNA PCR kit (Perkin-Elmer Corp., Norwalk, CT) and amplified with forward primers Gluex15, 15b or 15t and reverse primer Gluex16. Primer sequences used in this study are available from our web site (http://www.gs-im3.fr/autism).
Statistical analysis
Nonparametric ASP analysis was performed using ASPEX,21 under an additive model and assuming no dominance variance. ASPEX calculates a nonparametric MLS that is interpreted in the same manner as a LOD score. We also used GENEHUNTER-PLUS22,23 to perform multipoint nonparametric affected-relative-pair analysis. The nonparametric-linkage (NPL) was performed using the “all” statistic of the computer package. GENEHUNTER-PLUS calculates a maximum-likelihood estimate of the NPL statistic converted to a “semiparametric” LOD score: LOD*. For these analyses, LOD* was calculated using the exponential likelihood-based allele-sharing model which is more robust in smaller data sets.23 Like the MLS statistic generated by ASPEX, this LOD* may be interpreted as a traditional LOD score.
Family-based linkage and association analyses were performed using the transmission disequilibrium test (TDT), where preferential allelic transmission from heterozygous parents to affected offspring is tested by applying the (b−c)2/(b+c) statistics and the χ2 (“McNemar test”).24 For association studies, only one affected child per family was included. Haplotypes of the two SNPs located in intron 14 and exon 15 were constructed on the basis of transmission patterns in families in which both parents were genotyped, assuming an absence of recombination between the two loci because of close proximity of the SNPs (120 bp). For each haplotype, a test with 1 df for excess transmission of that haplotype was calculated. Finally, a global test was also performed with H-1 df, where H is the number of haplotypes for which transmission data are available.25 In this study, haplotypes CA and TG, less represented, were pooled. Data presented are nominal P values.
Results
Linkage analysis of the GluR6 gene in autistic sib pairs
A complete genomic screen performed by the PARIS study9 revealed the most significant MLS close to D6S283 (single point MLS=1.02). To further study this linkage with autism, a fine mapping of chromosome 6q21 was performed using additional microsatellites flanking D6S283 in 59 ASP (Table 1 and Figure 1). De facto, D6S283 was found to be located in intron 11 of the human GluR6 gene (Figure 2a). To increase the information content for GluR6, two additional intragenic microsatellites, D6S1543 in intron 1 and a polymorphic stretch of TAA in exon 1626 were studied. Allele sharing and identity by descent (IBD) were analysed by ASPEX27 and GENEHUNTER-PLUS22 softwares. ASPEX was used to determine the paternal and the maternal LOD scores. GENEHUNTER-PLUS was used to analyse all families, including one family with two affected siblings and one half-sibling. Results are presented in Table 1 and Figure 1. The most significant LOD scores correspond to the GluR6 markers (ASPEX MLS=3.28; GENEHUNTER-PLUS NPL= 3.28; P= 0.0005; LOD*= 2.44). As shown in Figure 1 and Table 1, maternal sharing was more pronounced than paternal sharing. The region containing the susceptibility gene was bounded by D6S1613 and D6S268, i.e., from 104 to 115 cM (95% confidence limit) and was determined according to the equation of Kruglyak and Lander,28 using the peak IBD score from ASPEX. Due to the large size of the gene (670 kb), three intragenic recombination events were observed. For the region flanking the GluR6 gene, the information content was greater than 80%.
Table 1.
Multi-point LOD scores of 6q21 markers used for the sib-pair analysis
| Markers | Distancea(cM) | ASPEX (58 families)
|
GENEHUNTER (59 families)
|
|||||
|---|---|---|---|---|---|---|---|---|
| pat | mat | mlod | NPL | P value | LOD* | Info | ||
| D6S294 | 79 | 0.00 | 0.00 | 0.00 | −0.32 | 0.63 | 0.037 | 0.79 |
| D6S286 | 90 | 0.24 | −0.03 | 0.21 | 0.36 | 0.35 | 0.035 | 0.88 |
| D6S1644 | 96 | 0.59 | 0.38 | 0.96 | 1.24 | 0.10 | 0.039 | 0.89 |
| D61613 | 97 | 0.65 | 0.44 | 1.09 | 1.46 | 0.071 | 0.50 | 0.95 |
| GluR6 | 108–110 | |||||||
| TAA repeat (exon 16) | 0.85 | 2.42 | 3.28 | 3.26 | 0.00051 | 2.44 | 0.97 | |
| D6S283 (intron 11) | 0.63 | 2.40 | 3.03 | 3.28 | 0.00049 | 2.44 | 0.97 | |
| D6S1543 (intron 1) | 0.99 | 1.99 | 2.98 | 3.10 | 0.00094 | 2.18 | 0.97 | |
| D6S268 | 115 | 0.68 | 0.24 | 0.91 | 2.12 | 0.017 | 1.23 | 0.88 |
| D6S1594 | 117 | 0.68 | −0.00 | 0.67 | 1.82 | 0.035 | 0.87 | 0.89 |
| D6S261 | 120 | 0.56 | −0.07 | 0.49 | 1.74 | 0.040 | 0.78 | 0.90 |
Distance in Kosambi centiMorgans from the Marshfield chromosome 6 sex-averaged linkage map. GluR6 markers are indicated in bold.
Figure 1.
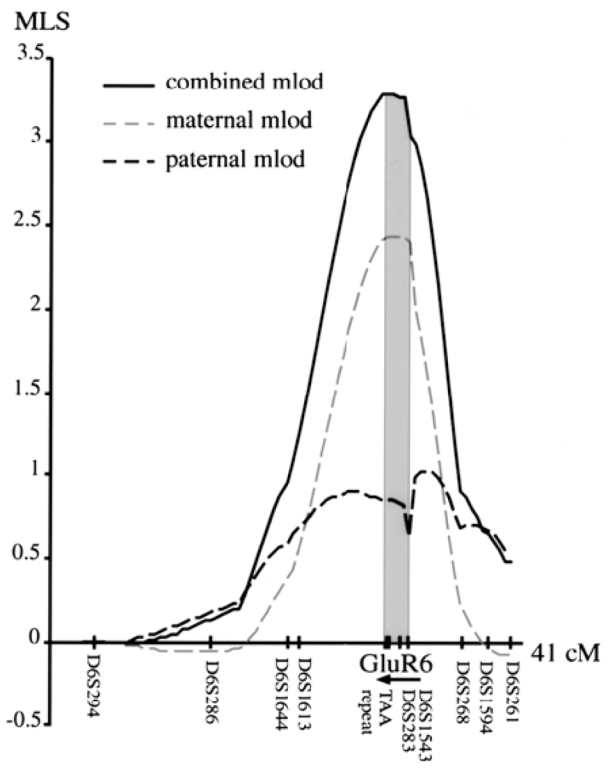
Maternal, paternal and combined LOD scores at each marker along the chromosome 6q21 region calculated by ASPEX.
Figure 2.
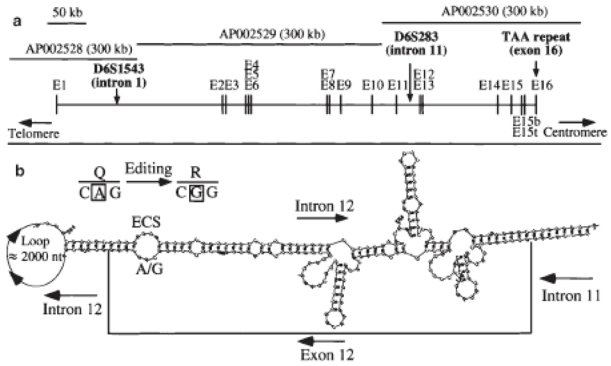
Structure of the human GluR6 gene. a. Genomic organisation of the human GluR6 gene. Exon sequences were identified by BLAST analysis, using the human GluR6 cDNA sequence (NM_021956) and three genomic sequences (AP002528, AP002529, AP002530); b. Predicted exon-intron dsRNA around the Q/R editing site (intron 12 containing the ECS) of GluR6 pre-mRNA by the RNA mfold software.59 The nucleotides of intron 12 omitted from presentation are indicated in the loop.
Genomic organisation of the human GluR6 gene
The sequence of the complete GluR6 gene is now available in the Genbank database (accession numbers: AP002528, AP002529 and AP002530). The gene spans 670 kb and includes 16 exons in the open reading frame (ORF) (Figure 2a). Orientation of the GluR6 gene (from telomere to centromere) was deduced from the three intragenic recombination events. In intron 12, we identified a sequence similar to exon 12 that contains the editing site complementary sequence (ECS) required for double-strand RNA (dsRNA) formation and deamination of adenosine by the enzyme DRADA.29 This modification at the nucleic acid level (A/G) leads to an amino acid change (Q621R) at the protein level. The predicted exon-intron dsRNA around the Q/R editing site of GluR6 pre-mRNA is depicted in Figure 2b. This structure, requiring a distant intronic sequence, is homologous to the mouse gene.29 Two other sites are edited in exon 11 of the GluR6 gene, leading to the amino acid changes 1567 V and Y571C. For these edited sites, the dsRNA structure is unknown. Finally, two alternatively spliced exons are located in intron 15. Insertion of exon 15bis or 15ter in the mRNA, between exons 15 and 16, leads to a different COOH terminal sequence with a premature ending of the protein (see below).
Sequence analysis of the GluR6 gene in autistic patients
The complete ORF, ECS, and the two alternatively spliced exons 15bis and 15ter were sequenced in 33 patients selected from ASP with IBD2 (23 patients) or IBD1 (10 patients). The location of all SNPs identified is shown in Figure 3a. Only one nucleotide variation, in exon 16, leads to a modification of the protein sequence at amino acid M867I. The Methionine 867 is highly conserved during evolution from Xenopus laevi to human and is close to a protein kinase C putative phosphorylation site (Figure 3b). By RT-PCR performed on human control brain RNA, we found three different isoforms of the GluR6 mRNA leading to three different C-terminal sequences at the protein level, generated by alternative splicing of exon 15bis or 15ter (Figures 3c and 3d). In mouse, the transcript corresponding to exon 15bis was already reported.30 Exons 15bis and 15ter are spliced-out in the major transcript GluR6a. By contrast, in the presence of exon 15bis or 15ter, a premature stop codon leads to different and shorter C-terminal ends (Figure 3d). The amino acid change M867I takes place in the C-terminal domain of the protein specific to the GluR6a isoform and is absent in GluR6b and GluR6c. These isoforms are conserved between mouse and human but show no homology with known domains (not shown).
Figure 3.
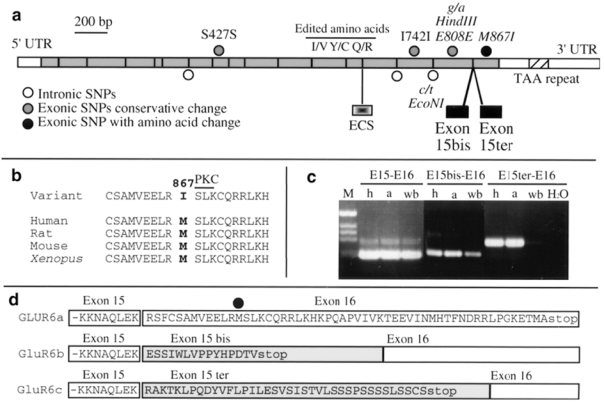
Sequence analysis and isoforms of the human GluR6 transcript, a. Location of all SNPs identified in this study, b. Conservation of the Methionine 867 in different species and location of the protein kinase C (PKC) phosphorylation consensus site; c. RT-PCR performed on human hippocampus (h), amygdala (a) and whole brain (wb) RNA; forward primers were chosen in exon 15, 15bis or 15ter; the reverse primer is the same for all PCRs and is located in exon 16. d. C-terminal protein sequences of the different GluR6 isoforms.
Linkage disequilibrium and association analyses of the GluR6 gene in autistic subjects
TDT was performed in 59 ASP and 107 trios using three SNPs, located in intron 14 (EcoNI), exon 15 (HindIII) and exon 16 (M867I) (Figure 3a). , The frequency of the C allele in intron 14 was 67% in parents and 68% in autistic subjects. The G allele in exon 15 was present in 59% of parents and 60% of autistic subjects. The Ile allele in exon 16 was found in 7.7% of autistic subjects (males 8.2%; females 6.4%), in 4.7% of parents (fathers 3.6%; mothers 5.8%), and in 3.9% of controls (N=200). There was no significant difference between samples for any of the SNPs and no deviation from Hardy-Weinberg equilibrium.
The TDT was first calculated as a test for linkage using all probands (Table 2). This test looks at all affected offspring of heterozygous parents for a given allele. For each allele, the frequency of transmission to the affected children is compared with the frequency of non-transmission.24 When male autistic subjects were considered, a significant linkage disequilibrium (LD) was found for the maternal transmission of allele C of intron 14 (TDTall χ2 = 9.6; df = 1; P = 0.002) and for allele G of exon 15 (TDTall χ2 = 12.5; df = 1; P = 0.0004). LD was also present when the population of ASP and trios were analysed separately. The sample size of affected females with heterozygous parents was too small to perform statistical analyses. When all subjects from both sexes were analysed, LD was less pronounced but still significant (allele C intron 14: TDTall χ2 = 6.7; df = 1; P = 0.009 and allele G exon 15: TDTall χ2 = 7.2; df = 1; P = 0.007). TDT performed on 36 unaffected sibs revealed no segregation distortion (allele C intron 14: 6 T versus 6 NT; and allele G exon 15: 4 T versus 4 NT). Although the M867I change was found in only a small number of parents, the Isoleucine allele seemed to be transmitted more than expected (TDTall χ2 = 7.4; df = 1; P = 0.007). In contrast with the maternal transmission, the paternal transmission of all GluR6 alleles was as expected in the absence of linkage.
Table 2.
TDT analyses for GluR6 alleles in autistic subjects
| Autistic subjects | Maternal transmission
|
Paternal transmission
|
||||
|---|---|---|---|---|---|---|
| Intron 14 allele C | Exon 15 allele G | Exon 16 allele Ile | Intron 14 allele C | Exon 15 allele G | Exon 16 allele Ile | |
| Autistic males (ASP; trios) | ||||||
| All affected | ||||||
| T | 39 (24; 15) | 35 (19; 16) | 10 (8; 2) | 24 | 19 | 3 |
| NT | 16 (10; 6) | 11 (8; 3) | 1 (0; 1) | 24 | 29 | 4 |
| χ2 | 9.6 (5.8; 3.9) | 12.5 (4.5; 8.9) | 7.4 | 0 | 2.1 | - |
| P value | 0.002 (0.015; 0.05) | 0.0004 (0.03; 0.003) | 0.007 | ns | ns | - |
| Only one affected | ||||||
| T | 29 (14; 15) | 26 (10; 16) | 8 (6; 2) | 20 | 20 | 2 |
| NT | 12 (6; 6) | 10 (7; 3) | 1 (0; 1) | 19 | 23 | 3 |
| χ2 | 7 (3.2; 3.9) | 7.1 (0.5; 8.9) | 5.4 | 0.03 | 0.2 | - |
| P value | 0.008 (ns; 0.05) | 0.008 (ns; 0.003) | 0.02 | ns | ns | - |
| Autistic females (ASP; trios) | ||||||
| All affected | ||||||
| T | 8 (3; 5) | 6 (3; 3) | 2 (1; 1) | 7 | 11 | 1 |
| NT | 9 (5; 4) | 9 (5; 4) | 2 (2; 0) | 13 | 9 | 0 |
| χ2 | 0.1 | 0.6 | - | 1.31 | 0.47 | - |
| P value | ns | ns | - | ns | ns | - |
| All autistic subjects (ASP; trios) | ||||||
| All affected | ||||||
| T | 47 (27; 20) | 41 (22; 19) | 12 (9; 3) | 31 | 30 | 4 |
| NT | 25 (15; 10) | 20 (13; 7) | 3 (2; 1) | 37 | 38 | 4 |
| χ2 | 6.7 (3 .4; 3. 3) | 7.2 (2.3; 5.5) | 5.4 (4.45; -) | 0.5 | 0.9 | - |
| P value | 0.009 (ns; ns) | 0.007 (ns; 0.018) | 0.02 (0.035; -) | ns | ns | - |
T: transmitted; NT: not transmitted.
Results with significance <0.05 are indicated in bold.
TDT was also calculated using only one proband per family (single affected) in order to have independent data. In this situation, TDT is a valid test for association.24 When maternal transmission to male autistic subjects was tested, there was a significant association for allele C of intron 14 (TDT association χ2 = 7; df = 1; P = 0.008) and allele G of exon 15 (TDT association χ2 = 7.1; df = 1; P = 0.008). No significant association was observed when female autistic subjects or paternal transmission were analysed.
Finally, to detect LD between specific GluR6 haplotypes and autism, we reconstructed paternal and maternal haplotypes transmitted to autistic subjects (Table 3). There are two major haplotypes, CG and TA; the other haplotypes, CA and TG, are less represented. The ancestral haplotype is probably CG, based on the sequence of the chimpanzee DNA (not shown). Thus, the most represented haplotypes are separated by two mutational events leading to TA. The Isoleucine allele is always associated with a CA haplotype. There was a significant difference in the maternal transmission of the different haplotypes in male probands (global χ2 = 12.7; df = 2; P = 0.002), with haplotype CG being more transmitted (TDT χ2 = 11.5; df = 1; P = 0.0007) and haplotype TA less transmitted (TDT χ2 = 4.8; df = 1; P = 0.03). When subjects from both sexes were tested, the difference in maternal transmission was still significant (global χ2 = 9.9; df = 2; P = 0.007), with haplotype CG being more transmitted (TDT χ2 = 9; df = 1; P = 0.003).
Table 3.
TDT analyses for GluR6 haplotypes in autistic subjects
| Maternal transmission
|
Paternal transmission
|
|||||||
|---|---|---|---|---|---|---|---|---|
| CG | TA | CA+TG | χ2 sum P value | CG | TA | CA+TG | χ2 sum P value | |
| Autistic males | ||||||||
| T | 37 | 16 | 8+2 | χ2=12.7 | 24 | 21 | 8+0 | χ2=0.06 |
| NT | 13 | 31 | 14+5 | df=2 | 23 | 21 | 7+2 | df=2 |
| χ2 | 11.5 | 4.8 | 2.8 | P = 0.002 | 0.02 | 0 | 0.07 | ns |
| P value | 0.0007 | 0.03 | ns | ns | ns | ns | ||
| Autistic females | ||||||||
| T | 7 | 8 | 3+0 | 8 | 10 | 1+1 | ||
| NT | 7 | 8 | 3+0 | 8 | 8 | 4+0 | ||
| χ2 | 0 | 0 | - | 0 | 0.2 | - | ||
| P value | ns | ns | - | ns | ns | - | ||
| All autistic subjects | ||||||||
| T | 44 | 24 | 11+2 | χ2 = 9.9 | 32 | 31 | 9+1 | χ2=0.3 |
| NT | 20 | 39 | 17+5 | df=2 | 31 | 29 | 11+2 | df=2 |
| χ2 | 9 | 3.6 | 2.3 | P = 0.007 | 0.02 | 0.07 | 0.4 | ns |
| P value | 0.003 | ns | ns | ns | ns | ns | ||
T: transmitted; NT: not transmitted. Haplotypes were constructed with the C/T SNP in intron 14 and the G/A SNP in exon 15.
Discussion
Linkage and association of the GluR6 gene with autism
In this study, using a relatively high number of autistic subjects (227 children: 59 ASP and 107 trios) and two different strategies (sib-pair analysis and TDT), we found a significant linkage between the GluR6 gene and autism. The sib-pair analysis showed an excess of allele sharing in ASP, which was more pronounced when maternal alleles were considered. TDT analysis, performed with all affected subjects, also showed a significant maternal transmission disequilibrium. The MLS generated in this study by the ASP method represents one of the highest identified for autism but still fails to meet the threshold for definite significance.31 The linkage region extends over 11 cM, from 104 to 115 cM (95% confidence intervals), with GluR6 located at 108–110 cM. These findings suggest that either GluR6 or a gene nearby is in linkage with autism. When test of linkage is carried out with the sib-pair analysis, there is no requirement for association with GluR6, since linkage is detected as an excess of sharing independently of the allele type. In contrast, TDT, performed with specific GluR6 SNPs, can detect linkage only in the presence of association.25 In our study, LD and association were observed in the trio sample, independently from the ASP sample used to detect linkage on 6q21. On the basis of these genetic results, we suggest that GluR6 or a nearby gene may be implicated in the predisposition to autism.
An excess of maternal allele sharing was observed using the sib-pair analysis, and consistent with this observation, LD and association studies were significant only when maternal transmission was tested. These findings can be the consequence of meiotic drive or imprinting. Meiotic drive results in non-Mendelian segregation of alleles to offspring.32 TDT performed on unaffected sibs revealed no segregation distortion, arguing against this possibility, but the sample size is too small to rule out this hypothesis. Genomic imprinting consists of a specific allelic expression depending on its parental origin. Interestingly, other candidate regions for predisposition to autism, such as 7q and s15q11-13,33–34 have been reported to contain several imprinted genes. In addition, an imprinted locus on the X chromosome has been hypothesised to be responsible for male susceptibility to the syndrome.35 An imprinted region has been described near chromosome 6q21, at 6q23–24, in association with neonatal diabetes.36 Furthermore, it has been shown recently that GluR7, a very close paralogue of GluR6, shows unequal allelic expression in the human brain, a characteristic of genes subject to genomic imprinting.37 Studies to determine the imprinting status of GluR6 are underway.
The present findings also showed that LD and association were significant when male autistic subjects were considered and less when female were included in the analysis. This observation is interesting considering the well-known gender difference in the genetic predisposition to autism. Interestingly, a recent study reported that a subgroup of individuals with idiopathic autism, defined on the basis of both a normal physical examination and a structurally normal brain by MRI, exhibit a male to female sex ratio of 23:1, whereas in the subgroup with morphological anomalies, the sex ratio is reduced to 1.7:1.4 Thus, differences in the genetic predisposition to autism may underlie these differences in the spectrum of the phenotype between the two sexes.
In this study, LD and association analyses were performed with informative but apparently non-functional SNPs. One nucleotide variation changing the protein (M867I) may be functionally relevant. This amino acid change takes place in the intracytoplasmic C-terminal region, in a highly conserved domain of the protein, which is subject to specific regulation by alternative splicing and plays an important role in the organisation and electrophysiological properties of GluR6. In particular, recombinant GluR6 proteins deleted in the C-terminal region are not associated with the synapse-associated protein PSD95/SAP90 and therefore may not be targeted to the synapse.38 Furthermore, interaction with PSD95/SAP90 alters GluR6 function by reducing desensitisation. In our families, the isoleucine allele was somewhat more frequent in autistic subjects (8%) compared to controls (4%) and exhibited more maternal transmission. However, replication studies are needed in other families from different populations and the consequence of the M867I change at the functional level needs to be elucidated.
GluR6 as a genetic factor in autism?
Dysfunction of GluR6 may participate in the impairments in communication and learning in autistic subjects. KARs have two major activities: they contribute to the excitatory postsynaptic current/potential in response to glutamate and modulate the release of neurotransmitters (γ-aminobutyric acid [GABA] and glutamate) through presynaptic action.39–41 Moreover, KARs are subjected to developmental and activity-dependant regulation42 and GluR6 function is modulated by protein phosphorylation.43 GluR6 is abundantly expressed in brain regions involved in learning and memory (such as the hippocampus) as well as in motor and motivational aspects of behaviour (such as basal ganglia and the cerebellum). Mutations in GluR6 could thus have important consequences in the integration of excitatory synaptic signals controlling these aspects of behaviour. Although the behaviour of GluR6-deficient mice has not been fully investigated,44 these mice appear to have difficulties in sensorimotor coordination and motivational aspects of locomotor activity (C Mulle, personal communication).
In addition, abnormal regulation of GluR6 expression could play a role in the high occurrence of epileptic seizures in autistic subjects.45 Administration of kainate in the rat brain produces seizures and is considered a model for epilepsy.46 GluR6-deficient mice are less susceptible to systemic administration of kainate44 and over-expression of GluR6 in rat hippocampus produces spontaneous seizures.47
Finally, a dysfunction in GluR6 mRNA editing process may also be implicated in the autistic phenotype. Editing is a complex post-transcriptional regulation leading to a modification of the mRNA sequence compared to the related genomic sequence.48 Mutations in genes involved in this process leads to altered behavioural phenotypes.49 Editing of GluR6 at the Q/R site modifies the calcium permeability of the receptor channel.50 The extent of editing is developmentally regulated in the rat brain and highest levels of edited forms are reached after birth.51 In this study, we have identified the sequence of the human ECS in intron 12 required for the editing at the Q/R site,29 but sequence analysis of the autistic subjects did not reveal differences compared to controls. However, it has been shown that slight modification of dsRNA structure dramatically alters the editing process.29 Since GluR6 pre-mRNA is long (670 kb), we can not exclude that sequence variation may affect the editing process. Moreover, GluR6 can also undergo editing at two other sites (I/V and Y/C), but sequences involved in this editing are unknown. Further work should directly ascertain the proportion of edited GluR6 forms in brain samples from autistic subjects. Interestingly, a marked decrease (9%) of unedited fraction of GluR6 (Q) subunit has been observed in surgically excised hippocampus from patients with refractory epilepsy.52
GluR6 may also be candidate for other psychiatric and neurological diseases. In the 6q21 region, three independent studies have reported an elevated linkage with schizophrenia.53–55 Two markers, D6S424 (102 cM) and D6S423 (125 cM), flanking the GluR6 gene show the most significant linkage (P < 0.0003). Since hypoglutamatergia is a well-established hypothesis for schizophrenia,56 GluR6 is a strong candidate. In support of this hypothesis, partial loss of hippocampal GluR6 mRNAs was observed in schizophrenic patients.57 Finally, two independent studies have suggested that the age of onset of Huntington disease is associated with GluR6 genotype variation.58
In conclusion, we observed an excess of allele sharing, LD, and association between GluR6 and autism. These results apparently contrast with other studies using whole genome scans, which have not found significant MLS for chromosome 6q16-23. Although genetic heterogeneity between study populations cannot be excluded, the linkage to 6q observed in the original PARIS study may have been a consequence of a higher information content using the GluR6 intragenic microsatellite (D6S283). Moreover, the data presented here indicate that LD between GluR6 and autism is seen mainly through maternal transmission. Therefore, we suggest that chromosome 6q21 and GluR6 should be reconsidered and tested for linkage and association in other samples, and a meta-analysis should be done to determine the relative strength of the present findings. Finally, these analyses are exclusively based on genetic data, so further functional studies should be performed to determine the potential role of GluR6 in the predisposition to autism.
Acknowledgments
We thank the patients and their families who made this research possible, Dr. Ken McElreavey and Dr. Christophe Mulle for helpful discussions and comments during the writing of the manuscript, and Johann Tassin for technical assistance. Genotyping facilities were provided by the Centre National de Genotypage (CNG), Evry, France. This work was supported by the Délégation de la Recherche Clinique de l’Assistance Publique-Hôpitaux de Paris, the French Research Ministry (Actions Concertées Incitatives), France Télécom, and the Swedish Medical Research Council. C.B. was supported by a Cure Autism Now Foundation Award.
References
- 1.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. American Psychiatric Press; Washington DC: 1994. [Google Scholar]
- 2.Kanner L. Autistic disturbances of affective contact. Nerv Child. 1943;2:217–250. [PubMed] [Google Scholar]
- 3.Gillberg C, Wing L. Autism: not an extremely rare disorder. Acta Psychiatr Scand. 1999;99:399–406. doi: 10.1111/j.1600-0447.1999.tb00984.x. [DOI] [PubMed] [Google Scholar]
- 4.Miles JH, Hillman RE. Value of a clinical morphology examination in autism. Am J Med Genet. 2000;91:245–253. [PubMed] [Google Scholar]
- 5.Ritvo ER, Freeman BJ, Pingree C, Mason-Brothers A, Jorde L, Jenson WR, et al. The UCLA-University of Utah epidemiologic survey of autism: prevalence. Am J Psychiatry. 1989;146:194–199. doi: 10.1176/ajp.146.2.194. [DOI] [PubMed] [Google Scholar]
- 6.Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 7.Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, Jakobsson G, Bohman M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry. 1989;30:405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- 8.International Molecular Genetic Study of Autism Consortium. A full genome screen for autism with evidence for linkage to a region on chromosome 7q. Hum Mol Genet. 1998;7:571–578. doi: 10.1093/hmg/7.3.571. [DOI] [PubMed] [Google Scholar]
- 9.Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Rastam M, Sponheim E, et al. Genome-wide scan for autism susceptibility genes. Paris Autism Research International Sibpair Study. Hum Mol Genet. 1999;8:805–812. doi: 10.1093/hmg/8.5.805. [DOI] [PubMed] [Google Scholar]
- 10.Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, et al. A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet. 1999;65:493–507. doi: 10.1086/302497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrett S, Beck JC, Bernier R, Bisson E, Braun TA, Casavant TL, et al. An autosomal genomic screen for autism. Collaborative Linkage Study of Autism. Am J Med Genet. 1999;88:609–615. doi: 10.1002/(sici)1096-8628(19991215)88:6<609::aid-ajmg7>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- 12.Buxbaum JD, Silverman JM, Smith CJ, Kilifarski M, Reichert J, Hollander E, et al. Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. Am J Hum Genet. 2001;68:1514–1520. doi: 10.1086/320588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 14.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 15.Shimizu E, Tang YP, Rampon C, Tsien JZ. NMDA receptor-dependent synaptic reinforcement as a crucial process for memory consolidation. Science. 2000;290:1170–1174. doi: 10.1126/science.290.5494.1170. [DOI] [PubMed] [Google Scholar]
- 16.Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130:1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- 17.Carlsson ML. Hypothesis: is infantile autism a hypoglutamatergic disorder? Relevance of glutamate - serotonin interactions for pharmacotherapy. J Neural Transm. 1998;105:525–535. doi: 10.1007/s007020050076. [DOI] [PubMed] [Google Scholar]
- 18.Moreno-Fuenmayor H, Borjas L, Arrieta A, Valera V, Socorro-Candanoza L. Plasma excitatory amino acids in autism. Invest Clin. 1996;37:113–128. [PubMed] [Google Scholar]
- 19.Rolf LH, Haarmann FY, Grotemeyer KH, Kehrer H. Serotonin and amino acid content in platelets of autistic children. Acta Psychiatr Scand. 1993;87:312–316. doi: 10.1111/j.1600-0447.1993.tb03378.x. [DOI] [PubMed] [Google Scholar]
- 20.Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 21.Hinds D, Risch N. The ASPEX package: affected sib-pair mapping. 1996 ftp://lahmed.stanford.edu/pub/aspex.
- 22.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 23.Kong A, Cox NJ. Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM) Am J Hum Genet. 1993;52:506–516. [PMC free article] [PubMed] [Google Scholar]
- 25.Spielman RS, Ewens WJ. The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet. 1996;59:983–989. [PMC free article] [PubMed] [Google Scholar]
- 26.Paschen W, Blackstone CD, Huganir RL, Ross CA. Human GluR6 kainate receptor (GRIK2): molecular cloning, expression, polymorphism, and chromosomal assignment. Genomics. 1994;20:435–440. doi: 10.1006/geno.1994.1198. [DOI] [PubMed] [Google Scholar]
- 27.Hauser ER, Boehnke M, Guo SW, Risch N. Affected-sib-pair interval mapping and exclusion for complex genetic traits: sampling considerations. Genet Epidemiol. 1996;13:117–137. doi: 10.1002/(SICI)1098-2272(1996)13:2<117::AID-GEPI1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 28.Kruglyak L, Lander ES. High-resolution genetic mapping of complex traits. Am J Hum Genet. 1995;56:1212–1223. [PMC free article] [PubMed] [Google Scholar]
- 29.Herb A, Higuchi M, Sprengel R, Seeburg PH. Q/R site editing in kainate receptor GluR5 and GluR6 pre-mRNAs requires distant intronic sequences. Proc Natl Acad Sci USA. 1996;93:1875–1880. doi: 10.1073/pnas.93.5.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregor P, O’Hara BF, Yang X, Uhl GR. Expression and novel subunit isoforms of glutamate receptor genes GluR5 and GluR6. Neuroreport. 1993;4:1343–1346. doi: 10.1097/00001756-199309150-00014. [DOI] [PubMed] [Google Scholar]
- 31.Lander E, Kruglyak L. Genetic dissection of complex traits - guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 32.Lyttle TW. Cheaters sometimes prosper: distortion of mendelian segregation by meiotic drive. Trends Genet. 1993;9:205–210. doi: 10.1016/0168-9525(93)90120-7. [DOI] [PubMed] [Google Scholar]
- 33.Ashley-Koch A, Wolpert CM, Menold MM, Zaeem L, Basu S, Donnelly SL, et al. Genetic studies of autistic disorder and chromosome 7. Genomics. 1999;61:227–236. doi: 10.1006/geno.1999.5968. [DOI] [PubMed] [Google Scholar]
- 34.Wolpert CM, Menold MM, Bass MP, Qumsiyeh MB, Donnelly SL, Ravan SA, et al. Three probands with autistic disorder and isodicentric chromosome 15. Am J Med Genet. 2000;96:365–372. doi: 10.1002/1096-8628(20000612)96:3<365::aid-ajmg25>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 35.Skuse DH. Imprinting, the X-chromosome, and the male brain: explaining sex differences in the liability to autism. Pediatr Res. 2000;47:9–16. doi: 10.1203/00006450-200001000-00006. [DOI] [PubMed] [Google Scholar]
- 36.Gardner RJ, Mackay DJ, Mungall AJ, Polychronakos C, Siebert R, Shield JP, et al. An imprinted locus associated with transient neonatal diabetes mellitus. Hum Mol Genet. 2000;9:589–596. doi: 10.1093/hmg/9.4.589. [DOI] [PubMed] [Google Scholar]
- 37.Schiffer HH, Swanson GT, Masliah E, Heinemann SF. Unequal expression of allelic kainate receptor GluR7 mRNAs in human brains. J Neurosci. 2000;20:9025–9033. doi: 10.1523/JNEUROSCI.20-24-09025.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia EP, Mehta S, Blair LA, Wells DG, Shang J, Fukushima T, et al. SAP90 binds and clusters kainate receptors causing incomplete desensitization. Neuron. 1998;21:727–739. doi: 10.1016/s0896-6273(00)80590-5. [DOI] [PubMed] [Google Scholar]
- 39.Chittajallu R, Braithwaite SP, Clarke VR, Henley JM. Kainate receptors: subunits, synaptic localization and function. Trends Pharmacol Sci. 1999;20:26–35. doi: 10.1016/s0165-6147(98)01286-3. [DOI] [PubMed] [Google Scholar]
- 40.Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron. 2001;29:209–216. doi: 10.1016/s0896-6273(01)00191-x. [DOI] [PubMed] [Google Scholar]
- 41.Mulle C, Sailer A, Swanson GT, Brana C, O’Gorman S, Bettler B, et al. Subunit composition of kainate receptors in hippocampal interneurons. Neuron. 2000;28:475–484. doi: 10.1016/s0896-6273(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 42.Kidd FL, Isaac JT. Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature. 1999;400:569–573. doi: 10.1038/23040. [DOI] [PubMed] [Google Scholar]
- 43.Wang LY, Taverna FA, Huang XP, MacDonald JF, Hampson DR. Phosphorylation and modulation of a kainate receptor (GluR6) by cAMP-dependent protein kinase. Science. 1993;259:1173–1175. doi: 10.1126/science.8382377. [DOI] [PubMed] [Google Scholar]
- 44.Mulle C, Sailer A, Perez-Otano I, Dickinson-Anson H, Castillo PE, Bureau I, et al. Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature. 1998;392:601–605. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- 45.Rapin I, Katzman R. Neurobiology of autism. Ann Neurol. 1998;43:7–14. doi: 10.1002/ana.410430106. [DOI] [PubMed] [Google Scholar]
- 46.Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- 47.Telfeian AE, Federoff HJ, Leone P, During MJ, Williamson A. Overexpression of GluR6 in rat hippocampus produces seizures and spontaneous nonsynaptic bursting in vitro. Neurobiol Dis. 2000;7:362–374. doi: 10.1006/nbdi.2000.0294. [DOI] [PubMed] [Google Scholar]
- 48.Niswender CM. Recent advances in mammalian RNA editing. Cell Mol Life Sci. 1998;54:946–964. doi: 10.1007/s000180050225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reenan RA. The RNA world meets behavior: A-->I pre-mRNA editing in animals. Trends Genet. 2001;17:53–56. doi: 10.1016/s0168-9525(00)02169-7. [DOI] [PubMed] [Google Scholar]
- 50.Egebjerg J, Heinemann SF. Ca2+ permeability of unedited and edited versions of the kainate selective glutamate receptor GluR6. Proc Natl Acad Sci USA. 1993;90:755–759. doi: 10.1073/pnas.90.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmitt J, Dux E, Gissel C, Paschen W. Regional analysis of developmental changes in the extent of GluR6 mRNA editing in rat brain. Dev Brain Res. 1996;91:153–157. doi: 10.1016/0165-3806(95)00175-1. [DOI] [PubMed] [Google Scholar]
- 52.Grigorenko EV, Bell WL, Glazier S, Pons T, Deadwyler S. Editing status at the Q/R site of the GluR2 and GluR6 glutamate receptor subunits in the surgically excised hippocampus of patients with refractory epilepsy. Neuroreport. 1998;9:2219–2224. doi: 10.1097/00001756-199807130-00013. [DOI] [PubMed] [Google Scholar]
- 53.Cao Q, Martinez M, Zhang J, Sanders AR, Badner JA, Cravchik A, et al. Suggestive evidence for a schizophrenia susceptibility locus on chromosome 6q and a confirmation in an independent series of pedigrees. Genomics. 1997;43:1–8. doi: 10.1006/geno.1997.4815. [DOI] [PubMed] [Google Scholar]
- 54.Levinson DF, Holmans P, Straub RE, Owen MJ, Wildenauer DB, Gejman PV, et al. Multicenter linkage study of schizophrenia candidate regions on chromosomes 5q, 6q, 10p, and 13q: schizophrenia linkage collaborative group III. Am J Hum Genet. 2000;67:652–663. doi: 10.1086/303041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinez M, Goldin LR, Cao Q, Zhang J, Sanders AR, Nancarrow DJ, et al. Follow-up study on a susceptibility locus for schizophrenia on chromosome 6q. Am J Med Genet. 1999;88:337–343. [PubMed] [Google Scholar]
- 56.Meador-Woodruff JH, Healy DJ. Glutamate receptor expression in schizophrenic brain. Brain Res Rev. 2000;31:288–294. doi: 10.1016/s0165-0173(99)00044-2. [DOI] [PubMed] [Google Scholar]
- 57.Porter RH, Eastwood SL, Harrison PJ. Distribution of kainate receptor subunit mRNAs in human hippocampus, neocortex and cerebellum, and bilateral reduction of hippocampal GluR6 and KA2 transcripts in schizophrenia. Brain Res. 1997;751:217–231. doi: 10.1016/s0006-8993(96)01404-7. [DOI] [PubMed] [Google Scholar]
- 58.Rubinsztein DC, Leggo J, Chiano M, Dodge A, Norbury G, Rosser E, et al. Genotypes at the GluR6 kainate receptor locus are associated with variation in the age of onset of Huntington disease. Proc Natl Acad Sci USA. 1997;94:3872–3876. doi: 10.1073/pnas.94.8.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walter AE, Turner DH, Kim J, Lyttle MH, Muller P, Mathews DH, et al. Coaxial stacking of helixes enhances binding of oligoribonucleotides and improves predictions of RNA folding. Proc Natl Acad Sci USA. 1994;91:9218–9222. doi: 10.1073/pnas.91.20.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]