

Abstract
In an effort to probe the reaction mechanism of VanX, the D-ala-D-ala dipeptidase required for high-level vancomycin resistance in bacteria, stopped-flow kinetic and rapid-freeze quench EPR studies were conducted on the Co(II)-substituted enzyme when reacted with D-ala-D-ala. The intensity of the Co(II) ligand field band at 550 nm decreased (ε550 = 140 to 18 M−1cm−1) when VanX was reacted with substrate, suggesting that the coordination number of the metal increases from 5 to 6 upon substrate binding. The stopped-flow trace was fitted to a kinetic mechanism that suggests the presence of an intermediate whose breakdown is rate-limiting. Rapid-freeze quench EPR studies verified the presence of a reaction intermediate that exhibits an unusually low hyperfine constant (33 G), which suggests a bidentate coordination of the intermediate to the metal center. The EPR studies also identified a distinct enzyme product complex. The results were used to offer a detailed reaction mechanism for VanX that can be used to guide future inhibitor design efforts.
VanX is a Zn(II)-containing metalloenzyme that is required for high-level vancomycin resistance in bacteria.1 The intracellular role of VanX is to hydrolyze vancomycin-binding D-ala-D-ala dipeptides that are used to synthesize the normal bacterial peptidoglycan layer. Many vancomycin-resistant bacteria produce D-ala-D-lactate depsipeptides that can be inserted into the peptidoglycan layer; however, these depsipeptides bind vancomycin 103 weaker than their dipeptide counterparts.2, 3 Reynolds et al. reported that Enterococcus faecium strains containing functional VanX produced vancomycin-susceptible D-ala-D-ala containing precursors to vancomycin-resistant D-ala-D-lactate containing precursors in a ratio of 1:49.3 The removal of VanX activity changed this ratio to 1:1. Clearly, VanX is an excellent target for the generation of inhibitors. Towards this effort, several inhibitors have been reported for VanX;4–9 however, none of these inhibitors are clinically-useful. Rational design or redesign of these inhibitors requires structural and mechanistic information about the enzyme.10 A crystal structure of VanX has been reported, and this structure showed a single Zn(II) ion coordinated by 2 histidines and 1 aspartate.11 More recent spectroscopic studies have demonstrated that the Zn(II) in VanX is 5-coordinate,12 with the other two ligands probably being solvent molecules. The crystal structure demonstrated that the active site of VanX is contained in a very narrow cavity,11 which explains the stringent substrate specificity exhibited by the enzyme.3, 5 To date, there have been very few mechanistic studies reported on VanX,5, 13 presumably due to the lack of a substrate that can be used in a continuous assay. To circumvent this problem, we utilized stopped-flow optical and rapid-freeze quench EPR studies to probe the metal ion in Co(II)-substituted VanX during catalysis.
Zn(II)-containing and Co(II)-substituted VanX-maltose binding protein fusions were prepared and characterized as previously described.12 The lack of a chromophoric substrate prompted the evaluation alternative methods of investigating the catalytic reaction of VanX. No time-resolved changes in tryptophan fluorescence were observed upon reaction of Zn-VanX with D-ala-D-ala, but changes in the ligand field electronic absorption at 550 nm of Co-VanX were observed upon reaction at 2 °C (Figure 1). The time-dependence of the absorption was characterized by an initial rapid decrease (0 – 10 ms), followed by first a slow (15 – 35 ms) and then a more rapid reappearance of absorption at over the next 120 milliseconds. The molar absortivity of the Co(II) ligand field band at 550 nm was 140 M−1cm−1 for the enzyme in both the initial and final states, suggesting a coordination number of 5. The molar absorptivity of the enzyme after the first 15 milliseconds was 22 M−1cm−1, indicative of a catalytic intermediate with distorted six-fold coordination of Co(II).
Figure 1.

Stopped-flow Vis trace (data points) of the reaction of 356 µM Co(II)-substituted VanX with 356 µM D-ala-D-ala at 2 °C in 20 mM Tris, pH 7.6, containing 200 mM NaCl. The line was generated by using the Applied Photophysics ProK software and the mechanism and rate constants shown in Scheme I.
The simplest kinetic mechanism that allows for reversible binding of substrate and product, the presence of a reaction intermediate, and one substrate/two products is given in Scheme I.
Scheme 1.

Kinetic mechanism of VanX. Mechanism was used to fit stopped-flow data in Figure 1.
The stopped-flow trace (Figure 1) was fitted using Applied Photophysics ProK software with the following assumptions: (1) binding of substrate and products occurs at the diffusion controlled limit (108 M−1s−1), 14 (2) chemistry steps are essentially irreversible (k−2 and k−3 set to <1), (3) dissociation constants for both D-ala molecules are in the millimolar range, (4) E, EP1, and EP1P2 contain a five-coordinate metal center while ES and EI contain a six-coordinate center. In analyses with ProK, values for k−1, k2, and k3 were allowed to float for χ2 minimizations, while the other constants were fixed. The best fit, when using the rate constants in Scheme I, of the data is shown in Figure 1. Fittings with values of k−2 and k−3 >> 1 s−1 returned unreasonable values for kcat and Km. The errors in k−1, k2, and k3 from the fit are less than 5%. The theoretical values for kcat (k2k3k4k5 / [k4k5(k2+k−2+k3)+k−3k5(k2+k−2)+k2k3(k4+k5)]) and Km (k−1k−2(k−3k5+k4k5)+k4k5(k2k3+k−1k3) / [k1(k4k5(k2+k−2+k3)+k−3k5(k2+k−2)+ k2k3(k4+k5)]) for the mechanism in Scheme I are 24 s−1 and 126 µM, respectively. These theoretical values compare favorably with experimentally-determined kcat and Km values of 40 s−1 and 100 µM, respectively, at 2 °C. The data presented in Figure 1 represent the optimum conditions for the experiment, where (1) each of the reaction intermediates can be observed from the resting state onwards and (2) the Co(II) d-d bands are sufficiently intense as to be discriminating in terms of fit parameters. Numerous other data were collected at different concentrations. Due to the kinetics of the initial step and the inherently low intensity of Co(II) d-d transitions, the data were characterized by either a lack of a contribution of the early fast phase of the reaction or else by poor signal-to-noise in the data, at high and low enzyme concentrations, respectively. Nevertheless, fits to these data using the parameters presented in Scheme I show that the proposed mechanism is consistent with these other data.
To further probe the structure of Co(II) during the reaction, EPR spectra were carried out on Co-VanX before, during, and after reaction with D-ala-D-ala (Figure 2). The resting state is characterized by two EPR signals (Figure 2A,B), each consistent with 5-coordinate Co(II).12 The EPR signal from a rapid-freeze-quenched sample corresponding to 10.5 ms reaction time (Figure 2B,C) was characterized by a sharp feature at 1010 G (geff. = 6.8) and broad underlying absorption from 1800 – 4000G. Precise spin Hamiltonian parameters could not be obtained for the broad region, but simulation indicated that the spectrum is consistent with an essentially axial S = 3/2, MS = |±1/2〉 species with greal (x,y) ~ 2.65, and thus, consistent with 5- or 6-coordinate Co(II).15 Closer examination of the 200 – 1200 G region revealed resolved hyperfine structure, indicative of a very highly-constrained geometry, just as expected for an intermediate that binds similarly as the phosphinate analog of D-ala-D-ala.11 Even more striking evidence for bound intermediate, however, was the magnitude of the hyperfine coupling, 33 G (Figure 2G). This compares to a typical value of ~ 90 G for protein-bound Co(II) and 40 – 50 G for Co(II) when a constituent of a four- or five-atom substrate-derived ring system.15 The hyperfine coupling of 33 G from the Co-VanX intermediate indicates substantial paramagnetic electron density delocalization from the Co(II) ion, likely due to bidentate coordination of intermediate to Co(II) with concomittant loss of one of the solvent molecules, perhaps as the nucleophile. Interestingly, upon exhaustion of substrate the signal observed from Co-VanX (Figure 2E,F) appears to be distinct from the signals observed in the resting enzyme (Figure 2B).
Figure 2.

EPR spectra of 1 mM Co-VanX in the resting state (A, B), after 10 ms reaction with 5 mM D-ala-D-ala (C, D), and after complete reaction with D-ala-D-ala (E, F). The traces G show the hyperfine region of spectrum C with a simulation overlaid indicating AI = 7/2(59Co) = 3.5 × 10−3 cm−1 (33 G). Spectra A, C, E and G were recorded at 9.63 GHz, 11 K, 2 mW; B, D and F were recorded at 5 K, 200 mW.
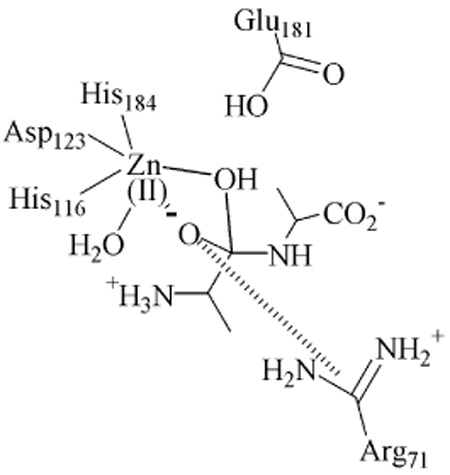
Thus, the combined data from the present study indicate that (i) there is a metal-ion-bound intermediate, EI, of VanX formed during the reaction with D-ala-D-ala, (ii) EI is bound in a bidentate fashion to the metal ion, and (iii) a distinct product complex exists. With these data, a reaction mechanism can be offered (Figure 3). Substrate binds to the metal site with the assistance of protonated Arg71 (not shown for clarity), 13 generating a 6-coordinate metal ion. Attack by hydroxide, which is generated by Zn(II) and Glu181, 13 results in an intermediate in which the hitherto sp2-hybridized carbon has become tetrahedral and is bidentately bound to 6-coordinate Zn(II). The source of the proton on the C-terminus of alanine is unknown; however, this proton could be donated from Glu181, the nucleophile, or another active site residue. Rate-limiting breakdown of this intermediate and loss of the C-terminal D-ala yields an enzyme-product complex that is five-coordinate. These findings represent the first mechanistic information on VanX and provide useful information for the rational design or redesign of clinically useful inhibitors of the enzyme.
Figure 3.

Proposed reaction mechanism of VanX.
Acknowledgement
The authors would like to thank the National Institutes of Health (GM40052 to M.W.C.), (AI056231 to B.B.), and (EB001980 to the Medical College of Wisconsin) for funding this work. We would also like to thank Professor Ken Johnson for helpful discussions.
References
- 1.Lessard IAD, Walsh CT. Proc. Nat. Acad. Sci. 1999;96:11028–11032. doi: 10.1073/pnas.96.20.11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McComas CC, Crowley BM, Boger DL. J. Am. Chem. Soc. 2003;125:9314–9315. doi: 10.1021/ja035901x. [DOI] [PubMed] [Google Scholar]
- 3.Reynolds PE, Depardieu F, Dutka-Malen S, Arthur M, Courvalin P. Mol. Microbiol. 1994;13(6):1065–1070. doi: 10.1111/j.1365-2958.1994.tb00497.x. [DOI] [PubMed] [Google Scholar]
- 4.Wu Z, Walsh CT. J. Am. Chem. Soc. 1996;118:1785–1786. [Google Scholar]
- 5.Wu Z, Wright GD, Walsh CT. Biochemistry. 1995;34:2455–2463. doi: 10.1021/bi00008a008. [DOI] [PubMed] [Google Scholar]
- 6.Wu Z, Walsh CT. Proc. Nat. Acad. Sci. 1995;92:11603–11607. doi: 10.1073/pnas.92.25.11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang KW, Brandt JJ, Chatwood LL, Crowder MW. Bioorg. Med. Chem. Lett. 2000;10(10):1087–1089. doi: 10.1016/s0960-894x(00)00186-4. [DOI] [PubMed] [Google Scholar]
- 8.Yaouancq L, Anissimova M, Badet-Denisot MA, Badet B. Eur. J. Org. Chem. 2002;(21):3573–3579. [Google Scholar]
- 9.Araoz R, Anhalt E, Rene L, Badet-Denisot MA, Courvalin P, Badet B. Biochemistry. 2000;39:15971–15979. doi: 10.1021/bi001408b. [DOI] [PubMed] [Google Scholar]
- 10.Crowder MW. Curr. Drug Targets. 2006;6(2):147–158. doi: 10.2174/187152606784112146. [DOI] [PubMed] [Google Scholar]
- 11.Bussiere DE, Pratt SD, Katz L, Severin JM, Holzman T, Park CH. Molecular Cell. 1998;2:75–84. doi: 10.1016/s1097-2765(00)80115-x. [DOI] [PubMed] [Google Scholar]
- 12.Breece RM, Costello A, Bennett B, Sigdel TK, Matthews ML, Tierney DL, Crowder MW. J. Biol. Chem. 2005;280(12):11074–11081. doi: 10.1074/jbc.M412582200. [DOI] [PubMed] [Google Scholar]
- 13.McCafferty DG, Lessard IAD, Walsh CT. Biochemistry. 1997;36:10498–10505. doi: 10.1021/bi970543u. [DOI] [PubMed] [Google Scholar]
- 14.Fierke CA, Johnson KA, Benkovic SJ. Biochemistry. 1987;26(13):4085–4082. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- 15.Bennett B. Curr. Topics Biophys. 2002;26(1):49–57. [Google Scholar]