

Abstract
The clinically vital antimycotic agent amphotericin B represents the archetypal example of a channel-forming small molecule. The leading model for self-assembly of the amphotericin B channel predicts that C(41) carboxylate and the C(3′) ammonium ions form intermolecular salt bridges/hydrogen bonds that are critical for stability. We herein report a flexible degradative synthesis pathway that enables the removal of either or both of these groups from amphotericin B. We further demonstrate with extensive NMR experiments that deleting these groups does not alter the conformation of the polyene macrolide skeleton. As predicted by the leading model, amphotericin B derivatives lacking the mycosamine sugar that contains the C(3′) ammonium ion are completely inactive against Saccharomyces cerevisiae. However, strikingly – and in strong contradiction with the current model – the amphotericin B derivative lacking the C(41) carboxylate is at least equipotent to the natural product. Collectively, these findings demonstrate that the leading model for the mechanism of action of amphotericin B must be significantly revised – either the C(41) carboxylate is not required for channel formation, or channel formation is not required for antifungal activity.
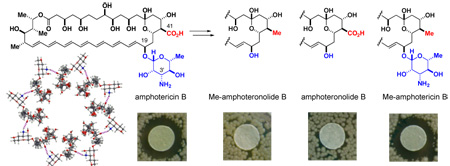
The leading model for the antifungal action of amphotericin B (AmB, 1) involves its self-assembly into a membrane-spanning ion channel.1 This natural product thus represents a potential prototype for small molecules with the capacity to perform ion channel-like functions in living systems. Efforts to harness this potential and/or improve the notoriously poor therapeutic index of this clinically vital antimycotic2 would benefit from a molecular-level understanding of this channel activity.
Although the evidence that AmB can self-assemble in lipid membranes to form discrete ion conducting channels is strong,1,3 the molecular architecture of this channel assemblage and its role in antifungal activity remain poorly understood.4 Despite this, the leading “barrel-stave” model5 is an often cited textbook classic (Fig. 1A).6 Extensive computer modeling studies predict that this complex is stabilized by a ring of salt bridges7a and/or hydrogen bonds7b–c at the channel periphery between oppositely-charged C(41)-carboxylate and C(3′)-ammonium ions. Conspicuously, these two functional groups are installed biosynthetically as post-polyketide synthase (PKS) modifications of the macrolide skeleton, i.e., a P450-mediated oxidation of the C(41)-methyl group and a glycosyl transferase-mediated attachment of mycosamine to the C(19) alcohol (Fig. 1B).8,9
Figure 1.

A. A bird's eye view of the current "barrel stave" model for the AmB channel. Salt bridges and/or hydrogen bonds (dashed lines) between oppositely-charged C(41)-carboxylate and C(3')-ammonium ions are predicted to be critical for channel stabilization. B. These two functional groups are installed as post-PKS modifications of the macrolide skeleton.
Many interesting studies have probed the action of AmB via covalent modification of the C(41)-carboxylic acid and/or C(3′)-amine.4e,10 However, the self-assembly of small molecules can be exquisitely sensitive to steric effects,11 and this phenomenon may complicate this experimental approach. We herein report an alternative strategy that involves synthetically deleting chemical groups appended to the macrolide skeleton and determining the functional consequences.12 This approach has led to the striking observation that, contrary to the current channel model (Fig. 1A), oxidation at C(41) is not required for potent antifungal activity.
The synthetic manipulation of AmB is made challenging by its sensitivity to light, oxygen, and acid as well as its minimal solubility in most organic solvents and water. Nevertheless, we ultimately developed a flexible degradative pathway that enables the synthetic reversal of either13 or both of the two post-PKS modifications predicted to be critical for self-assembly of the AmB channel (Scheme 1). Synthesis of the novel MeAmdeB 2 (Fig. 1B) commenced with the conversion of AmB into the suitably protected and more soluble nonasilylated N-Fmoc methyl ketal 7.13b The C(41)-carboxylic acid was then selectively reduced to the corresponding primary alcohol via the intermediacy of a 2-pyridinethiol ester.14 Subsequent iodination with PPh3/I2 15 yielded the desired C(41)-iodomethyl derivative 8. Oxidative deglycosylation of this intermediate using DDQ16 in the presence of CaCO3 smoothly generated enone 9. Reduction of the C(19)-ketone with NaBH4/MeOH resulted in a ~2:1 mixture of diastereomers, while use of the (S)-CBS oxazaborolidine catalyst17 provided the desired 19-R isomer in a synthetically useful 6:1 d.r. (see SI). A subsequent reductive cleavage of the C(41)-iodide was achieved with NaBH418 in DMPU to yield advanced intermediate 10. Global desilylation with HF/pyridine, hydrolysis of the methyl ketal, and preparative high performance liquid chromatography (HPLC) yielded diastereomerically pure MeAmdeB 2. The funneling of intermediates 7 and 8 into modified versions of this flexible pathway enabled the preparation of the remaining two targeted derivatives AmdeB 313a and MeAmB 413b,c (Scheme 1, Fig. 1B).
Scheme 1a.

a Reagents and conditions (a) (i) Fmoc-succinimide, pyridine, DMF:MeOH 2:1, 23 °C, 12 h; (ii) CSA, THF:MeOH 1:1, 0 °C, 1 h, 90% (two steps); (b) TESOTf, 2,6 lutidine, hexanes, 0 °C, 3 h, 96%; (c) 2-thiopyridyl chloroformate, Et3N, Et2O, 0 °C, 30 min, 91%; (d) LiBH4, Et2O, 23 °C, 2 h, 88%; (e) I2, PPh3, imidazole, THF, 0 °C, 1 h, 78%; (f) DDQ, CaCO3, THF, 23 °C, 10 min, 67%; (g) (S)-CBS oxazaborolidine, Me2S·BH3, CH2Cl2, −10 °C, 30 min, 6:1 d.r., 79%; (h) NaBH4, DMPU, 23 °C, 6 h, 78%; (i) (i) HF/pyridine, THF:pyridine 3:2, 0 °C, 6 h; (ii) AcOH:H2O:THF 1:1:2, 23 °C, 30 min; HPLC, 38% (two steps); (j) allyl bromide, i-Pr2NEt, DMF, 23 °C, 8 h, 86%; (k) DDQ, CaCO3, THF, 23 °C, 20 min, 65%; (l) NaBH4, THF:MeOH 3:1, 0 °C, 30 min, >20:1 d.r., 77%; (m) HF/pyridine, THF:pyridine 5:3, 0 → 23 °C, 6 h, 56%; (n) CSA, THF:H2O 2:1, 23 °C, 5h; HPLC, 81%; (o) Pd(PPh3)4, thiosalicylic acid, THF, 23 °C, 13 h; HPLC, 50%; (p) HF/pyridine, THF:pyridine 5:3, 0 → 23 °C, 6.5 h, 73%; (q) NaBH4, DMSO, 23 °C, 8 h, 58%; (r) (i) CSA, THF:H2O 2:1, 23 °C, 30 min; (ii) piperidine, DMSO:MeOH 4:1, 23 °C, 3 h; HPLC, 56% (two steps).
Because the AmB framework is known to be quite rigid,19 we postulated that the ground-state conformation would be unchanged by these appendage deletions, thereby further facilitating the interpretation of structure/function data generated with this approach. To confirm this hypothesis, we determined the ground-state conformation of compounds 1–4 (or more soluble analogs, see SI for details) using Monte Carlo methods constrained by extensive NOESY and phase-sensitive COSY NMR data processed using amplitude-constrained multiplet evaluation.20 As shown in Fig. 2, the conformation of the macrolide skeleton was unaltered by these appendage deletions (root mean square deviation for all four compounds = 0.081 Å).
Figure 2.

Superposition of the ground state conformation of the macrolactone skeletons of compounds 1–4 (or their more soluble analogs, see SI for details).
The impact of deleting these functional groups on antifungal activity against Saccharomyces cerevisiae was qualitatively evaluated using a disc diffusion assay.21 As shown in Fig. 3A, derivatives 2 and 3, both of which lack the mycosamine appendage, were completely inactive.22 In stark contrast, and counter to the current channel model, MeAmB 4 was found to be roughly equipotent to the natural product. This striking result was confirmed quantitatively in a broth dilution assay23 (MIC: AmB = 2 µM, MeAmB = 1 µM) (Fig. 3B). Similar results were observed in both assays with clinically-relevant Candida albicans (Fig. 3). Clearly, post-PKS oxidation of the AmB macrolide at C(41) is not required for potent antifungal activity.
Figure 3.

A. Disc diffusion assay with S. cerevesiae (40 µg of compound per disc). Similar results were achieved with C. albicans (SI). B. Broth dilution assays; values represent the average of three experiments.
These findings stand in strong contradiction with the current model for the mechanism of action of AmB (Fig. 1A). There are at least two possible explanations: oxidation at C(41) may not be required for channel formation and/or channel formation may not be required for antifungal activity.4 An extensive series of biophysical studies with compounds 1–4 are planned to distinguish between these possibilities. In preliminary studies using pyranine-impregnated liposomes,24 MeAmB demonstrates membrane-permeabilizing activity similar to that of AmB (SI).
These results also demonstrate that the deletion of appended functional groups represents a powerful approach for probing the still poorly understood activity of AmB. The general application of this strategy to systematically dissect the structure/function relationships that underlie this potentially prototypical channel-forming small molecule is currently underway in our laboratories.
Supplementary Material
Detailed synthesis, NMR, and assay procedures, spectral data, and spectra; full citations for refs 4e, 7b, 8a–b, 10a–b, 10e, 13a–d, 15, 18, 19a, and 24. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
We gratefully acknowledge Bristol-Myers Squibb Company for a gift of AmB, and the NIH (GM080436), Dreyfus Foundation, and UIUC for funding. We also thank C. Rienstra, B. Wylie, V. Mainz, P. Molitor, F. Lin, F. Delaglio, and J. Baudry for assistance with NMR studies, NMRPipe, MOE, and Fig. 1A, P. Orlean, J. Grimme and R. Hellman for assistance with yeast assays, and J. Morrissey, S. Smith, K. Suslick, G. Fujii, and S.M. Chiang for assistance with liposome studies.
REFERENCES
- 1.Reviews: Bolard J. Biochim. Biophys. Acta. 1986;864:257–304. doi: 10.1016/0304-4157(86)90002-x. Hartsel SD, Hatch C, Ayenew W. J. Liposome Res. 1993;3:377–408. Cereghetti DM, Carreira EM. Synthesis. 2006;6:914–942. Hartsel S, Bolard J. Trends Pharm. Sci. 1996;17:445–449. doi: 10.1016/s0165-6147(96)01012-7.
- 2.Kontoyiannis DP, Lewis RE. Lancet. 2002;359:1135–1144. doi: 10.1016/S0140-6736(02)08162-X. [DOI] [PubMed] [Google Scholar]
- 3.(a) Andreoli TE, Monahan M. J. Gen. Physiol. 1968;52:300–325. doi: 10.1085/jgp.52.2.300. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cass A, Finkelstein A, Krespi V. J. Gen. Physiol. 1970;56:100–124. doi: 10.1085/jgp.56.1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ermishkin LN, Kasumov KM, Potzeluyev VM. Nature. 1976;262:698–699. doi: 10.1038/262698a0. [DOI] [PubMed] [Google Scholar]; (d) Borisova MP, Ermishkin LN, Silberstein AYA. Biochim. Biophys. Acta. 1979;553:450–459. doi: 10.1016/0005-2736(79)90300-6. [DOI] [PubMed] [Google Scholar]; (e) Mickus DE, Levitt DG, Rychnovsky SD. J. Am. Chem. Soc. 1992;114:359–360. [Google Scholar]; (f) Sykora J, Yilma S, Neely WC, Vodyanoy V. Langmuir. 2003;19:858–864. [Google Scholar]
- 4.(a) HsuChen CC, Feingold DS. Nature. 1974;251:656–659. doi: 10.1038/251656a0. [DOI] [PubMed] [Google Scholar]; (b) Chen WC, Chou D-L, Feingold DS. Antimicrob. Agents Chemother. 1978;13:914–917. doi: 10.1128/aac.13.6.914. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brajtburg J, Powderly WG, Kobayashi GS, Medoff G. Antimicrob. Agents Chemother. 1990;34:183–188. doi: 10.1128/aac.34.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Beggs WH. Antimicrob. Agents Chemother. 1994;38:363–364. doi: 10.1128/aac.38.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zumbuehl A, et al. Angew. Chem. Int. Ed. 2004;43:5181–5185. doi: 10.1002/anie.200460489. [DOI] [PubMed] [Google Scholar]
- 5.(a) Finkelstein A, Holz R. In: Membranes vol. 2. Lipid Bilayers and Antibiotics. Eisenman G, editor. New York: Marcel Dekker, Inc.; 1973. pp. 377–408. [Google Scholar]; (b) De Kruijff B, Demel RA. Biochim. Biophys. Acta. 1974;339:57–70. doi: 10.1016/0005-2736(74)90332-0. [DOI] [PubMed] [Google Scholar]; (c) Andreoli TE. Ann. N.Y. Acad. Sci. 1974;235:448–468. doi: 10.1111/j.1749-6632.1974.tb43283.x. [DOI] [PubMed] [Google Scholar]
- 6.Petty HR. Molecular Biology of Membranes, Structure and Function. New York: Plenum Press; 1993. [Google Scholar]
- 7.(a) Khutorsky VE. Biochim. Biophys. Acta. 1992;1108:123–127. doi: 10.1016/0005-2736(92)90015-e. [DOI] [PubMed] [Google Scholar]; (b) Baginski M, et al. Mol. Pharmacol. 1997;52:560–570. doi: 10.1124/mol.52.4.560. [DOI] [PubMed] [Google Scholar]; (c) Baginski M, Resat H, Borowski E. Biochim. Biophys. Acta. 2002;1567:63–78. doi: 10.1016/s0005-2736(02)00581-3. [DOI] [PubMed] [Google Scholar]
- 8.(a) McNamara CM, et al. J. Chem. Soc. Perkin Trans. 1998;1:83–87. [Google Scholar]; (b) Caffrey P, et al. Chem. Biol. 2001;8:713–723. doi: 10.1016/s1074-5521(01)00046-1. [DOI] [PubMed] [Google Scholar]
- 9.To the best of our knowledge, these two post-PKS modifications are unique to the polyene macrolides and they always co-appear.
- 10.(a) Chéron M, et al. Biochem. Pharmacol. 1988;37:827–836. doi: 10.1016/0006-2952(88)90168-2. [DOI] [PubMed] [Google Scholar]; (b) Matsumori N, et al. J. Am. Chem. Soc. 2002;124:4180–4181. doi: 10.1021/ja012026b. [DOI] [PubMed] [Google Scholar]; (c) Matsumori N, Sawada Y, Murata M. J. Am. Chem. Soc. 2005;127:10667–10675. doi: 10.1021/ja051597r. [DOI] [PubMed] [Google Scholar]; (d) Matsumori N, Sawada Y, Murata M. J. Am. Chem. Soc. 2006;128:11977–11984. doi: 10.1021/ja063433w. [DOI] [PubMed] [Google Scholar]; (e) Umegawa Y, et al. Tetrahedron Lett. 2007;48:3393–3396. [Google Scholar]
- 11.(a) Mathias JP, Simanek EE, Whitesides GM. J. Am. Chem. Soc. 1994;116:4326–4340. [Google Scholar]; (b) Görbitz CH, Etter MC. J. Am. Chem. Soc. 1992;114:627–631. [Google Scholar]
- 12.(a) Lemieux RU. Acc. Chem. Res. 1996;29:373–380. [Google Scholar]; (b) Tor Y. ChemBioChem. 2003;4:998–1007. doi: 10.1002/cbic.200300680. [DOI] [PubMed] [Google Scholar]; (c) Morgan AJ, Wang YK, Roberts MF, Miller SJ. J. Am. Chem. Soc. 2004;126:15370–15371. doi: 10.1021/ja047360x. [DOI] [PubMed] [Google Scholar]; (d) Nam J, Shin D, Rew Y, Boger DL. J. Am. Chem. Soc. 2007;129:8747–8755. doi: 10.1021/ja068573k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) In pioneering studies, Nicolaou and coworkers reported an alternative degradative synthesis of 3: Nicolaou KC, et al. J. Am. Chem. Soc. 1988;110:4660–4672. (b) A degradative synthesis of 4 is described in the patent literature: Driver MJ, et al. # 5,204,330. U.S. Patent. 1993 MacPherson DT, et al. Recent Advances in the Chemistry of Antiinfective Agents. 1993:205–222. (d) An impure sample of 4 was prepared via genetic engineering of the producer: Carmody M, et al. J. Biol. Chem. 2005;280:34420–34426. doi: 10.1074/jbc.M506689200.
- 14.Corey EJ, Clark DA. Tetrahedron Lett. 1979;20:2875–2878. [Google Scholar]
- 15.Garegg PJ, et al. J. Chem. Soc., Perkin Trans. 1980;I:2866–2869. [Google Scholar]
- 16.Kennedy RM, Abiko A, Masamune S. Tet. Lett. 1988;29:447–450. [Google Scholar]
- 17.Corey EJ, Bakshi RK, Shibata S, Chen CP, Singh VK. J. Am. Chem. Soc. 1987;109:7925–7926. [Google Scholar]
- 18.Hutchins RO, et al. J. Org. Chem. 1978;43:2259–2267. [Google Scholar]
- 19.(a) Sowinski P, et al. Magn. Res. Chem. 1992;30:275–279. [Google Scholar]; (b) Matsumori N, Houdai T, Murata M. J. Org. Chem. 2007;72:700–706. doi: 10.1021/jo061309p. [DOI] [PubMed] [Google Scholar]
- 20.Delaglio F, Zhengrong W, Bax A. J. Magn. Res. 2001;149:276–281. doi: 10.1006/jmre.2001.2297. [DOI] [PubMed] [Google Scholar]
- 21.NCCLS Performance Standards for Antimicrobial Disk Susceptibility Tests, M2-A8 Approved Standard-8th Ed. Vol. 23, Number 1, 2003.
- 22.To the best of our knowledge, this is the first demonstration that the sugar moiety of AmB is required for biological activity. The relative importance of the three polar functional groups at C(2′), C(3′), and C(4′) remains unclear.
- 23.NCCLS Reference Method for Broth Dilution Antifungal Susceptibility Testing, M27-A2, Approved standard-2nd Ed. Vol. 22, Number 15, 2002.
- 24.Fujii G, et al. Biochemistry. 1997;36:4959–4968. doi: 10.1021/bi962894z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed synthesis, NMR, and assay procedures, spectral data, and spectra; full citations for refs 4e, 7b, 8a–b, 10a–b, 10e, 13a–d, 15, 18, 19a, and 24. This material is available free of charge via the Internet at http://pubs.acs.org.