

Abstract
The coupling of electronic and biological functionality through self-assembly is an interesting target in supramolecular chemistry. We report here on a set of diacetylene-derivatized peptide amphiphiles (PAs) that react to form conjugated polydiacetylene backbones following self-assembly into cylindrical nanofibers. The polymerization reaction yields highly conjugated backbones when the peptidic segment of the PAs has a linear, as opposed to a branched, architecture. Given the topotactic nature of the polymerization, these results suggest that a high degree of internal order exists in the supramolecular nanofibers formed by the linear PA. On the basis of microscopy, the formation of a polydiacetylene backbone to covalently connect the β-sheets that help form the fibers does not disrupt the fiber shape. Interestingly, we observe the appearance of a polydiacetylene (PDA) circular dichroism band at 547 nm in linear PA nanofibers suggesting the conjugated backbone in the core of the nanostructures is twisted. We believe this CD signal is due to chiral induction by the β-sheets, which are normally twisted in helical fashion. Heating and cooling shows simultaneous changes in β-sheet and conjugated backbone structure, indicating they are both correlated. At the same time, poor polymerization in nanofibers formed by branched PAs indicates that less internal order exists in these nanostructures and, as expected, then a circular dichroism signal is not observed for the conjugated backbone. The general variety of materials investigated here has the obvious potential to couple electronic properties and in vitro bioactivity. Furthermore, the polymerization of monomers in peptide amphiphile assemblies by a rigid conjugated backbone also leads to mechanical robustness and insolubility, two properties that may be important for the patterning of these materials at the cellular scale.
Introduction
Over the past two decades, there has been a great interest in self-assembly of peptide-containing amphiphilic molecules into various supramolecular architectures. Depending on molecular design, different architectures have been constructed including spheres,1–3 tubes,3–5 cylinders,6–10 and twisted cylinders,11 among others.12–14 In principle, peptide-based amphiphiles can be designed to be biodegradable and biocompatible, and therefore, they have great potential as self-assembling materials for in vivo imaging,15 drug delivery,12,16 and as bioactive scaffolds for cell signaling.17–20
Peptide amphiphiles, by definition, have components that contain hydrophobic and hydrophilic segments, which can lead to ordered aggregation of these molecules to minimize interfacial energies with solvent or incompatible molecular segments. Interest in self-assembling peptide-based amphiphiles has extended from peptides to derivatized peptides and peptidic block copolymers.21 A few examples of peptide-only amphiphiles include cyclic peptides,4,16,22 short cationic and anionic surfactant-like peptides,7,23 and β-sheet forming peptides.24 Another family of peptide amphiphiles includes those with lipid alkyl tails. Yamada et al. designed a system using a tripeptide unit with double alkyl tails to form cylindrical micelles in organic solvent.8,25 These molecules were recently redesigned by our laboratory to form helical nanofibers with controllable pitch by varying the size of substituents.11 Single- and double-tailed alkyl units have also been synthesized to form protein-like architectures further modified to contain epitope-binding peptide sequences for bioactivity and sensing studies.17,26 Amphiphilic peptide polymers include the work of Deming27 and Conticello,9 whose use of polypeptides and copolymers have led to interesting materials for drug delivery.
Our group was the first to demonstrate that peptide amphiphiles containing fatty acid hydrophobic segments could be designed to form high-aspect-ratio cylindrical nanofibers that could also incorporate a large diversity of bioactive epitopes. This is now well understood to be the result of β-sheet formation between amino acids situated next to the hydrophobic segments. At appropriate concentrations (0.1–2 wt %), these one-dimensional nanofibers also form self-supporting gels in aqueous solution through changes in pH which reduce or eliminate net charge in the molecules, temperature, or addition of screening salts.6a,28 Work by Jiang et al.29 and Paramonov et al.30 have shown that the β-sheets tend to align parallel to the z-axis of the fiber. Thus, the general design of our PA molecules is highly tolerant to changes in the peptide sequence without compromising the formation of cylindrical nanofibers and self-supporting gels. This tolerance has enabled the synthesis of many PAs containing different epitopes and chromophores, as well as changes in molecular architecture of the peptide from linear to branched31 structures. Thus, even PA molecules with highly tapered or non-tapered shapes that in ordinary surfactants would yield spheres and lamellar aggregates, respectively, end up assembling as nanofibers. This is the result of β-sheet formation, and we therefore refer to these PAs as “βPAs”. The structural characterization of these supramolecular fibers consisting of β-sheets and hydrophobic collapse is not trivial since they are not crystals but do contain internal order.29 The mechanical integrity of the PA gels that are formed is also limited due to the absence of any internal covalent bonds connecting the PA monomers. In our original work, we showed that cysteine residues could be used to internally crosslink the nanofibers. In the present work, we have used a different approach to create internal covalent bonds among PA monomers that transforms the components of nanofibers to polymers with a conjugated backbone. This has the additional advantage that the backbone could be used to design functions based on its electronically conjugated nature.
An efficient polymerization of diacetylenes yielding highly conjugated backbones requires positioning of atoms in a crystalline lattice with little displacement before and after the reaction also known as a topochemical reaction.32–34 The reaction proceeds via covalent bonding between the C1 and C4 carbons of the diacetylenes, resulting in an alternating ene-yne conjugated backbone (Scheme 1). In aqueous environments, these materials often appear red or blue colored, with the blue color indicating greater order in the conjugated π-system.35 As the conjugated π-system becomes less ordered, the observed color shifts from blue to red. Charych36 and Jelinek37 utilized polydiacetylenes as chromatic sensors in lipid-like vesicles, and Tirrell and co-workers incorporated diacetylenes into the alkyl tails of peptide amphiphiles to create polymerized vesicles38 and Langmuir—Blodgett films39 as sensitive biosensors in solution. In those studies, cell/protein attachment disrupts the diacetylene film or vesicle, inducing an observable color change from blue to red. Peptide amphiphiles containing diacetylenes that self-assemble into nanoribbons have also been used in magnetic alignment studies to demonstrate macroscopic ordering of peptidic structures.40 Diacetylene-derivatized alkyl tails with variable hydrophilic head groups41 or oligopeptidic polymers42 also form interesting twisted nanostructures.
Scheme 1.
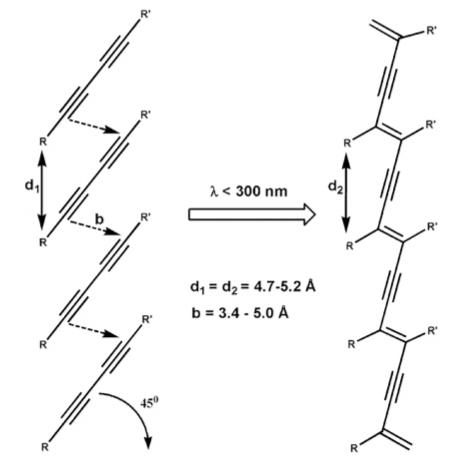
Polymerization Reaction of Diacetylenes When UV Irradiated
We report here on the polymerization of diacetylenes in the alkyl tails of our PA structures known to form cylindrical nanofibers driven by hydrophobic collapse of β-sheets. The PAs utilized in the study have either linear or branched architecture in their peptide segments. Previous work in our laboratory involved probing the internal structure of PA nanofibers using fluorescence techniques.43 These studies revealed that the interior of nanofibers is well solvated by water, but more densely packed and geometrically constrained near the core compared to the periphery. This would be consistent with an internal structure consisting of β-sheets that aggregate as a result of hydrophobic interactions. Hartgerink et al. has also supported this finding, reporting that bulky amino acids reduce hydrogen-bonding and hinder self-assembly of PAs as they are placed further into the nanofiber interior.30 More recently, our group has also determined that there is order in the inner hydrophobic core of the nanofibers containing alkyl chains.29 In the present work, we have placed the diacetylene groups in the hydrophobic alkyl segments and studied the polymerization both as a probe for the nanofiber’s internal structure as well as a strategy to obtain novel properties.
Results and Discussion
We synthesized the two PAs shown in Scheme 2 containing a diacetylene moiety in the alkyl tail. The peptide sequence of linear PA 1 has the sequence Lys-Lys-Leu-Leu-Ala-Lys-(OC25H40) whereas PA 2 contains an additional lysine residue to create its branched architecture Lys-Lys(Lys)-Leu-Leu-Ala-Lys(OC25H40).
Scheme 2.

Diacetylene PAs: Linear PA (1); Branched PA (2)
Both linear 1 and branched 2 PAs were found to form gels at concentrations of 2.0 wt % in water when exposed to ammonium hydroxide vapor in a sealed chamber for five to 10 min. The presence of base reduces and screens the net charges on PA molecules thus driving self-assembly into nanofibers through hydrogen bonding and hydrophobic collapse. Samples were irradiated at 256 nm either before or after gelation, both resulting in the well-known colorimetric change associated with diacetylene polymerization, from colorless to an intense blue color (see Figure 1). Irradiated samples that had not been exposed to ammonium hydroxide vapor also polymerize after exposure to UV light, indicating that the PA aggregates exist in solution even without the base-induced gelation procedure. The irradiation of PA solutions changes color to dark purple (3 in Figure 1), whereas irradiation of the gel results in an intense blue color (4 in Figure 1). The purple color suggests the presence of both conformationally disordered (red) and ordered (blue) states in the PDA backbones formed. Irradiation of the branched PAs under the same conditions resulted in similar color changes. After polymerization, the PA gels also appeared more mechanically robust relative to nonirradiated gels. This change in mechanical properties is clearly due to the formation of polydiacetylene backbones within the nanofibers. We are currently investigating the complex mechanical behavior of these materials and hope to report the results in a follow-up paper. We concluded from these initial observations that PA nanofibers consisting of either linear or branched molecules have sufficient internal order to support the topotactic polymerization of diacetylenes in their hydrophobic core.
Figure 1.

Linear PA samples from left to right: (1) PA solution without irradiation, (2) gelled PA without irradiation, (3) PA solution with irradiation, and (4) gelled PA with irradiation. Samples were irradiated for 2 min in this image. Nonirradiated samples occasionally appeared tinted blue due to polymerization either from stray UV light exposure or thermal excitation.
A blue color following diacetylene polymerization in peptide amphiphiles was observed in the low curvature planar assemblies of large vesicles (70–150 nm) by Tirrell et al.38 Since diacetylene polymerization is a topotactic reaction, the color change due to backbone conjugation can be correlated to the degree of molecular ordering in the reactive assemblies. This ordering should be compromised by high curvature in the assemblies and in fact the blue color indicative of long, effective conjugation length was not observed in spherical micelles by Huo et al.35 and Okada et al.36d It is therefore interesting that in our nanoscale cylindrical assemblies with high curvature (5–8 nm) we do observe an intense blue color. This suggests a high degree of internal order in the cylindrical nanostructures formed by the βPAs studied here.
Spectroscopic Analysis of Nanofibers
As mentioned previously, polydiacetylenes show colorimetric changes switching between blue to red when their conjugated backbone is perturbed from ordered to disordered states, respectively. We utilized UV—vis spectroscopy and circular dichroism (CD) to monitor diacetylene absorption bands after irradiation.
Using PA solutions diluted to concentrations of 50, 100, and 200 μM, UV—vis absorption was monitored for 45 s as shown in Figure 2. For the spectra of both linear and branched PA samples, two major absorption peaks are observed near 530 nm (red state) and 630 nm (blue state), which we attribute to two different backbone conformational states. The band observed at 550 nm for PA 1 spectra appears to be a superposition of the 530 nm band and the band at 585 nm of the blue state.30 The presence of ordered (blue) and disordered (red) states in both the linear and branched PAs spectra is clearly revealed in the spectra obtained. After several seconds of continued irradiation, the intensity of the blue state band decreases and that of the red state band increases. Ultimately, even as samples continued to be irradiated for a total of 2 min, the rapid decrease of the band at 630 tends to be coupled with the growing band at 550 nm. The increase of the red state at 550 nm suggests that continued polymerization of the polydiacetylenes results in an increased level of disorder in the polymer backbones formed. This has been also observed in monolayers by Evans et al.44 demonstrating that extended UV exposure time decreases the effective conjugation length of polydiacetylene backbones. It should be noted that similar absorption changes with irradiation were observed for PA gels that were diluted to the concentrations stated above. This UV—vis observation also indicates the PA molecules studied here under nongelating conditions exist in assembled states. Compared to the linear PA, the UV spectra of the branched PAs show significantly lower absorption. Because both are known to self-assemble into fibers, this suggests that diacetylene alkyl tails in the branched PA nanofibers are less efficiently ordered to support diacetylene polymerization. At lower absorption intensities, a band at 320 nm can be observed in the branched PA, which we assign as an exciton absorption peak as characterized by Weiser and co-workers.45 We infer that the reduced efficiency of diacetylene reaction for polymerization is the result of less efficient packing of the branched peptide segments within the nanostructure.
Figure 2.
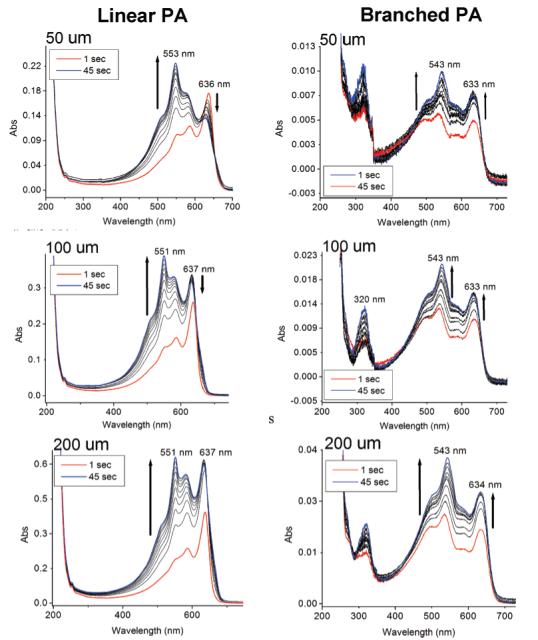
Time-dependent UV spectroscopy of linear (left) and branched (right) PAs at varying concentrations. Samples were monitored in 5 s time-intervals for 45 s. The spectra also show data of 1 s after irradiation.
CD experiments were initially performed to ensure that the β-sheet structures, which help drive PA nanofiber formation, were still present after UV irradiation. The β-sheet band at 220 nm was observed both before and after irradiation, implying that polymerization does not completely disrupt the hydrogen-bonded structures (Figures 3 and 5). Even at 80 °C, the 220 nm band is still present, although markedly reduced in intensity. Random-coil bands can also be observed at 200 nm possibly due to limited PA aggregation at the low concentration of the experiment (100 μM).
Figure 3.
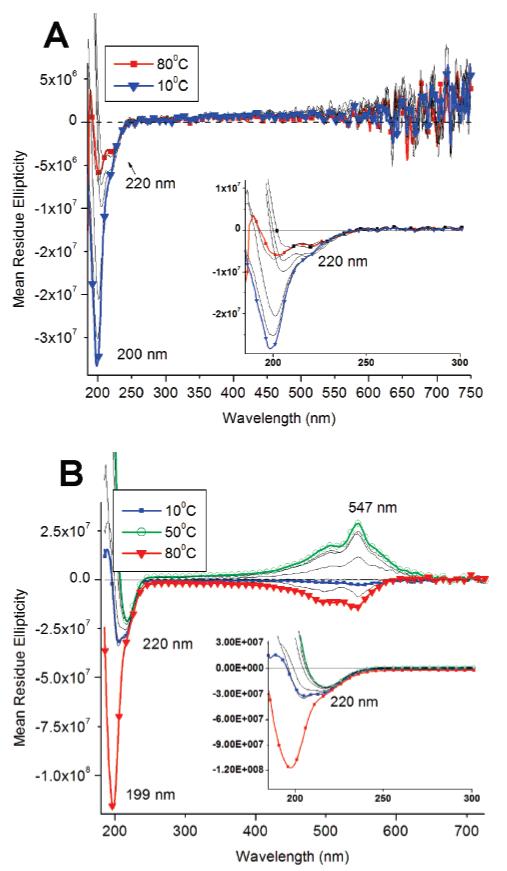
Temperature-dependent CD spectroscopy of linear PAs before (A) and after (B) irradiation. Samples were monitored from 80 °C – 10 °C and irradiated for 2 min.
Figure 5.
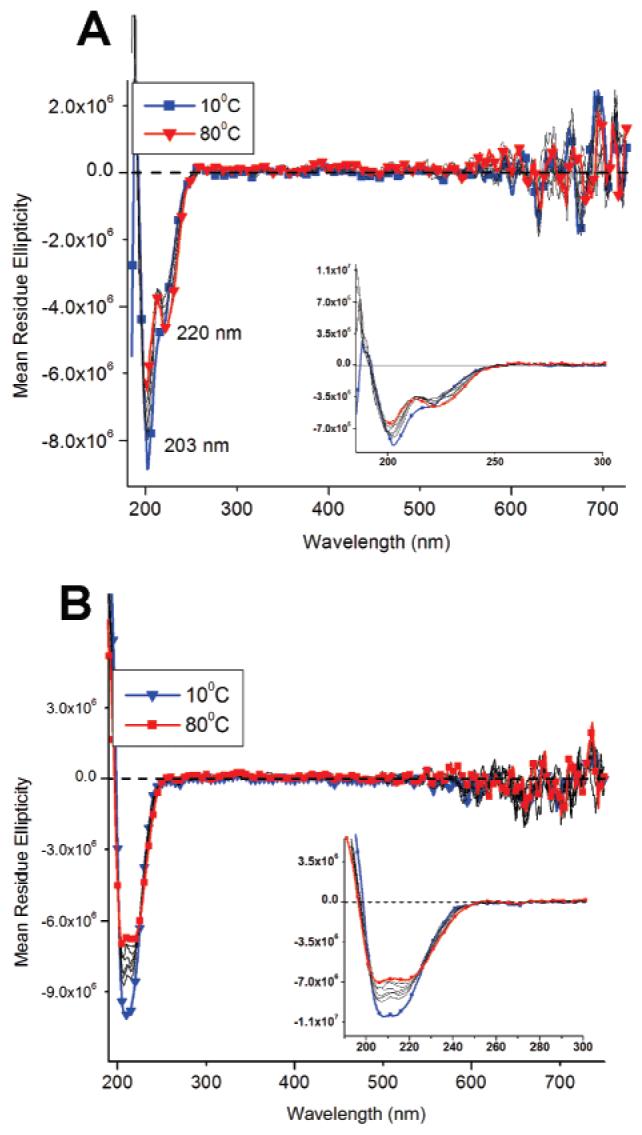
Temperature-dependent CD spectroscopy of branched PAs before (A) and after (B) irradiation. Samples were monitored from 80–10 °C and irradiated for 2 min.
Interestingly, we also observed a polydiacetylene CD band at 547 nm in irradiated samples of linear PA 1 (Figure 3b), which was not expected since this portion of the molecule is not intrinsically chiral. Hanks and co-workers have also shown that hydrogen-bonded, achiral assemblies containing the diacetylene polymer do not display CD signal, but substitution of chiral side groups result in an induced polydiacetylene CD signal.46 Figure 3 shows temperature-dependent CD spectra of non-irradiated and irradiated PA solutions at 100 μM concentrations. The signal at 547 nm was not detected in the nonirradiated sample, so it is definitely connected with formation of the conjugated backbone in the core of the nanofibers. Further analysis of the CD spectrum (Figure 3b) shows the PDA band changes sign at 80 °C from its maximum negative intensity to a maximum positive intensity at 50 °C, then decreasing to negative intensity at 10 °C. While the interpretation of the unusual CD signals in the polydiacetylene region is still unclear, we suggest they may be attributed to the spectral combination of exciton coupling of the diacetylene polymer47 and the induced CD signal from adjacent β-sheets. β-Sheets are known to twist naturally in helical fashion48 and this can result in an induced chiral environment to the polydiacetylene backbone. We expect the chirality of the polydiacetylene region to be dependent on the chemical sequence of the β-sheet more so than the overall peptide length, assuming a longer sequence does not disrupt the self-assembled β-sheets within the nanofiber. Coincidentally, the preferred distance between diacetylene monomers is similar to the distance between hydrogen bonds between parallel β-sheets (∼5 Å). Mori et al. have studied polymerization of diacetylene monomers using a β-sheet template and showed that the polymerization occurs parallel to the hydrogen bonds.49 The strong CD band at 547 nm from the irradiated linear PA not only shows that the PDA is in a chiral environment but suggests that β-sheets are twisted within the nanofibers. Also, the induced chiral structure of conjugated backbone is not in immediate proximity to the β-sheets, which also supports the importance of the internal order in the nanofibers.
At the same time, there is no observable CD signal for the PDA backbone in the branched PA 2. This observation, together with the low UV—vis signal intensity from the branched PA, shows a clear structural difference between the interior core of the branched and linear PA assemblies. The packing geometry of the branched PA inhibits the polymerization reaction. Figure 4 shows an idealized model of a polydiacetylene (red) backbone following the twisting of the β-sheet (blue).
Figure 4.
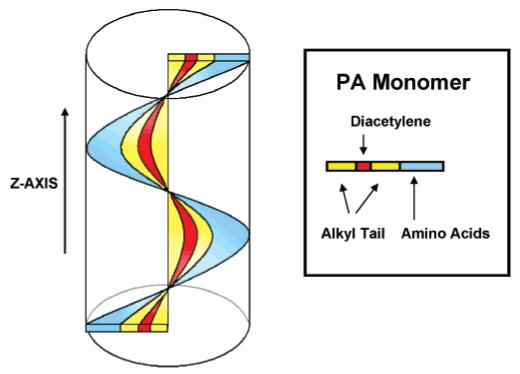
Proposed three-dimensional model of the PA nanofiber. Polymerization of the diacetylene (red) occurs along the z-axis of the fiber following the directionality of the beta-sheet (blue).
The CD signal changes observed at 547 nm for the polymerized PA nanofibers suggests a structural relationship between the PA β-sheets and the polydiacetylene backbone. At 80 °C, when the β-sheet is destabilized and partly melted, the PDA conformation appears to change (Figure 3b) as evidenced by a change from a positive CD signal at room temperature to a negative CD signal, implying structural reorganization within the PA assemblies, possibly even involving the disappearance of nanofibers. Unlike the spectrum of the nonirradiated PA spectum, which shows a diminished random-coil signal, the irradiated PA spectrum shows the random-coil signature from peptidic segments to be dominant at high temperatures. The continued presence of the random coil at high temperatures for the irradiated samples is possibly due to the diacetylene polymer preventing the reorganized PA structures from completely disassembling. As the temperature decreases, the hydrogen bonds of β-sheets re-form and the PDA conformation adjusts in order to accommodate the β-sheet structure. As samples are cooled the sign of the CD signal at 547 nm also changes. The reason for this sign reversal is not clear, but one may speculate that the details of β-sheet structure (e.g., the nature of twisting) after heating the PDA-containing assemblies to 80 °C and then cooling to room temperature may be different. The annealed nanofibers containing conjugated backbones may be equilibrating to a new internal structure in which β-sheets and PDA backbones establish a different chiral environment. The dominance of β-sheet structure in the nanofiber assemblies at room temperature is clear as their negative CD signal signature becomes more prominent at lower temperatures. Interestingly, temperature-dependent CD experiments of the branched PAs showed no significant difference before and after irradiation (Figure 5) and, as mentioned previously, a PDA signal was not observed at 547 nm. Both β-sheet and random-coil bands at 220 and 200 nm, respectively, were present at high temperatures for both irradiated and nonirradiated branched PA spectra.
Microscopic Imaging
Transmission electron microscopy (TEM) was performed on both branched and linear PAs before and after irradiation to observe morphological changes. The micrographs are shown in Figures 6 and 7. Both samples are clearly composed of long nanofibers with diameters in the range of 5 to 8 nm. There was no detectable change in their diameters before and after irradiation, indicating that the topochemical reaction does not alter fiber morphology. Although both formed fibers, the branched PA fibers seemed more bundled and aggregated which could be explained by enhanced inter-fiber interactions as a result of less efficient packing among PA molecules within individual nanofibers. Additional micrographs were taken on polymerized linear PAs at pH 2, 4, 7, 10, and 12. Polymerization of 1 wt % samples at all these pH values resulted in blue colored materials, regardless of pH. TEM images did not show clear morphological differences among the samples, with diameters in the range of 5 to 8 nm (see Supporting Information).
Figure 6.

TEM images of linear PA: nonirradiated (A) and irradiated (B). Samples were stained with phosphotungstic acid for approximately 1–3 min. Irradiated samples were exposed (256 nm) for 2 min.
Figure 7.
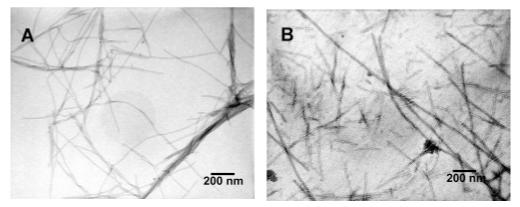
TEM images of branched PA: nonirradiated (A) and irradiated (B). Samples were stained with phosphotungstic acid for approximately 1 – 3 min. Irradiated samples were exposed (256 nm) for 2 min.
Atomic force microscopy (AFM) was also used to investigate the structure of samples. The AFM of linear PA samples, both irradiated and non-irradiated, revealed nanofibers with heights of approximately 5 nm and microns in length (Figure 8). Slight undulations along the fiber were sometimes observed in the irradiated samples, possibly due to structural defects that became trapped during the polymerization reaction giving the fiber a “bumpy” morphology. In the samples of the branched PA samples (Figure 9), AFM images also revealed nanofibers before and after irradiation. The fibers in this case appeared shorter with more morphology defects compared to the linear PA nanofibers.
Figure 8.
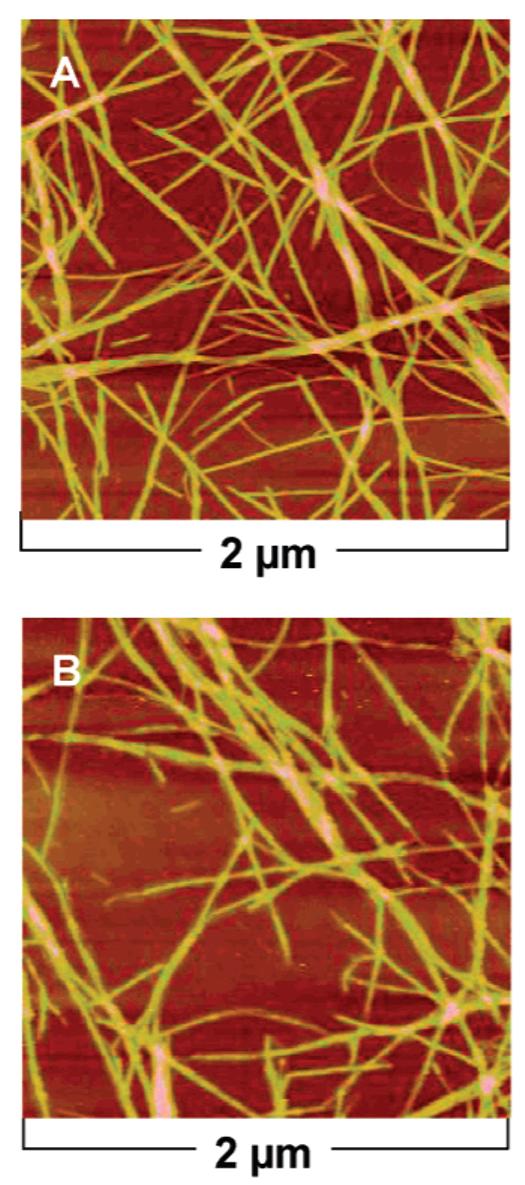
AFM height profiles of linear PA nonirradiated (A) and irradiated (B). Samples were drop-casted and imaged on silicon substrates after 2 min of irradiation.
Figure 9.
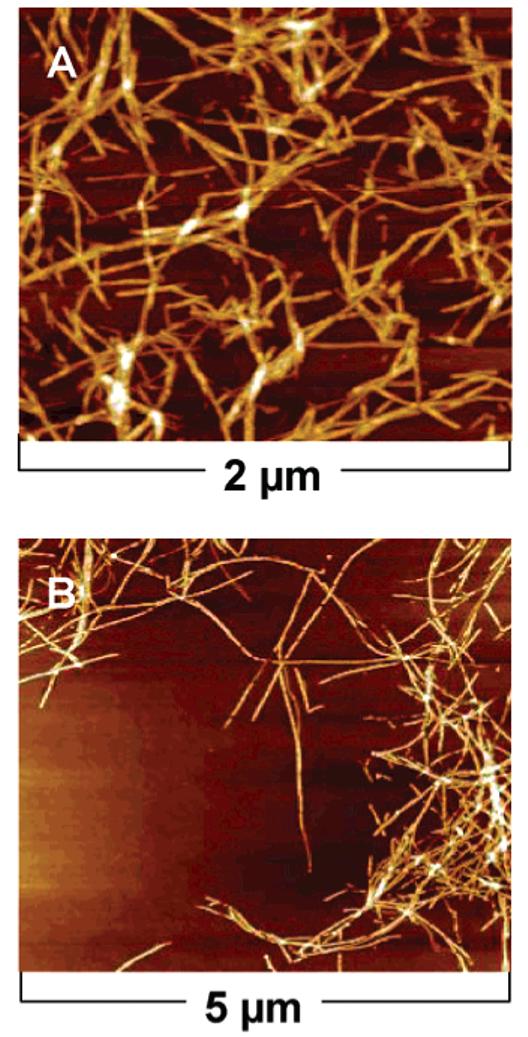
AFM height profiles of branched PA nonirradiated (A) and irradiated (B). Samples were drop-casted and imaged on silicon substrates after 2 min of irradiation.
Experimental Section
Peptide Amphiphile Synthesis
All amino acids and Rink MBHA resin were purchased from Novabiochem Corporation (San Diego, CA). 10,12-Pentacosadiynoic acid was purchased from Fluka and Alfa Aesar. n-Pentacosanoic acid was supplied by TCI America, Inc. All other reagents and solvents for peptide synthesis were purchased from Aldrich or Mallinckrodt and used as provided. Peptide amphiphiles were manually synthesized by Fmoc solid-phase peptide synthesis using orthogonal protecting strategies. Coupling of the first amino acid (Fmoc-Lys(Mtt)-OH) on to the resin was followed by deprotection of the Mtt group (5% TFA cleaving cocktail which consisted of TFA, TIPS, and water (95:2.5:2.5) in DCM) to couple the 10,12-pentacosadiynoic acid (4 equiv acid: 4 equiv HBTU: 6 equiv DIEA). All subsequent couplings were done using HBTU (4 equiv), DIEA (6 equiv), and the appropriate Fmoc-protected amino acid (4 equiv). Fmoc deprotection occurred using 20% piperidine solution in DMF. The PAs were cleaved from the resin using the TFA cleaving cocktail. Compound purity was analyzed by an analytical reverse-phase high-performance liquid chromatography (RP—HPLC) on an Agilent HP 1050 system equipped with a Waters Atlantis C18 column (5 μm particle size, 150 × 4.6 mm or 250 × 4.6 mm). Purification by RP—HPLC was done using a preparative Varian HPLC system equipped with a Waters Atlantis C18 preparative column (5 μm particle size, 250 × 30.0 mm). Confirmation of mass and purity included ESI (LCQ Advantage) and MALDI-TOF—MS (Voyager DE Pro).
Polymerization
Polymerizations of self-assembled diacetylene PAs were performed by irradiating at 254 nm using a compact handheld 4-W UV lamp (UVP Model UVGL-25). Samples were prepared in aqueous solution and irradiated at approximately 3 cm from the source for 2 min in a cuvette, unless otherwise noted.
Spectroscopic Characterization
UV—vis spectroscopy was performed using a Cary 500 Spectrometer for concentration and time-dependent irradiation experiments at room temperature. A model J-715 Jasco Circular Dichroism Spectrometer was used for concentration and temperature-dependent CD experiments. Before data acquisition, all samples were allowed to equilibrate at the set temperature for 5 min and averaged over of four runs.
Microscopy
AFM was performed on a DI SPM instrument using tapping mode on silicon substrates that were pre-washed using piranha cleaning solution, water, and isopropyl alcohol. Standard silicon AFM tips were purchased from Asylum Research (Santa Barbara, CA). Samples were prepared by drop casting 1 μL of a 0.1 wt % solution onto the substrate and allowing the solvent to evaporate in air. Samples were initially prepared as 2 wt % solutions and gelled, then irradiated and diluted as necessary.
TEM was performed on a Hitachi H-8100 TEM using 200 keV accelerating voltage. Samples were prepared similarly to those described in the AFM procedure by drop casting 1 μL of 0.1 wt % PA solution in aqueous solution on to a carbon-coated copper grid (SPI Supplies; West Chester, PA). The samples on the grid were stained for 1–3 min in phosphotungstic acid, gently rinsed in water and blotted dry.
Conclusions
We successfully polymerized supramolecular PA nanofibers with the polydiacetylene backbone while retaining the shape of the nanostructure. The efficiency of polymerization is decreased in branched architectures due by poor molecular packing within the fiber. The β-sheet structure that is characteristic of these nanofibers is able to induce a chiral structure in the conjugated backbone, reflecting a high degree of internal order in these structures. Polymerization of these systems with conjugated backbones may offer new possibilities for mechanical and optical properties, particularly in the context of biological applications.
Supplementary Material
Acknowledgment
This work was funded by the National Institutes of Health (NIH) NIH-NIDCR Grant No. 1R01DE015-920-01A1, and by the National Science Foundation (NSF) through a fellowship for L.H. from the MRSEC at Northwestern University Grant No. NSF DMR-0520513. We are also grateful for the use of instruments in the Analytical Services Center, the Keck Biophysics Facility, the Electron Probe Instrumentation Center, the Nanoscale Integrated Fabrication, Testing and Instrumentation Center, and the Institute for BioNanotechnology in Medicine at Northwestern University. We are also thankful for valuable discussions with L. C. Palmer, L. S. Li, D. A. Stone, H. Jiang, and S. Soukasene, all from the authors’ laboratory.
Footnotes
Supporting Information Available: HPLC, MALDI, and ESI of the PAs 1-2, a photograph of the PA 1 polymerized gel, and pH-dependent TEM micrographs of PA 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Neumann R, Ringsdorf H. J. Am. Chem. Soc. 1986;108:487–490. doi: 10.1021/ja00263a021. [DOI] [PubMed] [Google Scholar]
- (2).Murakami Y, Nakano A, Yoshimatsu A, Uchitomi K, Matsuda Y. J. Am. Chem. Soc. 1984;106:3613–3623. [Google Scholar]
- (3).Vauthey S, Santoso S, Gong HY, Watson N, Zhang SG. Proc. Natl. Acad. Sci. U.S.A. 2002;99:5355–5360. doi: 10.1073/pnas.072089599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ghadiri MR, Granja JR, Milligan RA, McRee DE, Khazanovich N. Nature. 1993;366:324–327. doi: 10.1038/366324a0. [DOI] [PubMed] [Google Scholar]
- (5).Shimizu T, Masuda M, Minamikawa H. Chem. ReV. 2005;105:1401–1443. doi: 10.1021/cr030072j. [DOI] [PubMed] [Google Scholar]
- (6) (a).Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]; (b) Hartgerink JD, Beniash E, Stupp SI. Proc. Natl. Acad. Sci. U.S.A. 2002;99:5133–5138. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang SG. Nat. Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- (8).Yamada N, Ariga K, Naito M, Matsubara K, Koyama E. J. Am. Chem. Soc. 1998;120:12192–12199. [Google Scholar]
- (9).Zimenkov Y, Dublin SN, Ni R, Tu RS, Breedveld V, Apkarian RP, Conticello VP. J. Am. Chem. Soc. 2006;128:6770–6771. doi: 10.1021/ja0605974. [DOI] [PubMed] [Google Scholar]
- (10).Wang K, Keasling JD, Muller SJ. Int. J. Biol. Macromol. 2005;36:232–240. doi: 10.1016/j.ijbiomac.2005.06.006. [DOI] [PubMed] [Google Scholar]
- (11).Li L-S, Jiang H, Messmore BW, Bull SR, Stupp SI. Angew. Chem., Int. Ed. 2007;46:5873–5876. doi: 10.1002/anie.200701328. [DOI] [PubMed] [Google Scholar]
- (12).Lear JD, Wasserman ZR, Degrado WF. Science. 1988;240:1177–1181. doi: 10.1126/science.2453923. [DOI] [PubMed] [Google Scholar]
- (13).Lu K, Jacob J, Thiyagarajan P, Conticello VP, Lynn DG. J. Am. Chem. Soc. 2003;125:6391–6393. doi: 10.1021/ja0341642. [DOI] [PubMed] [Google Scholar]
- (14).Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, Deming TJ. Nature. 2002;417:424–428. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- (15) (a).Bull SR, Guler MO, Bras RE, Venkatasubramanian PN, Stupp SI, Meade TJ. Bioconjugate Chem. 2005;16:1343–1348. doi: 10.1021/bc050153h. [DOI] [PubMed] [Google Scholar]; (b) Bull SR, Guler MO, Bras RE, Meade TJ, Stupp SI. Nano Lett. 2005;5:1–4. doi: 10.1021/nl0484898. [DOI] [PubMed] [Google Scholar]
- (16).Ghadiri MR, Granja JR, Buehler LK. Nature. 1994;369:301–304. doi: 10.1038/369301a0. [DOI] [PubMed] [Google Scholar]
- (17) (a).Fields GB, Lauer JL, Dori Y, Forns P, Yu YC, Tirrell M. Biopolymers. 1998;47:143–151. doi: 10.1002/(SICI)1097-0282(1998)47:2<143::AID-BIP3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; (b) Pakalns T, Haverstick KL, Fields GB, McCarthy JB, Mooradian DL, Tirrell M. Biomaterials. 1999;20:2265–2279. doi: 10.1016/s0142-9612(99)00157-x. [DOI] [PubMed] [Google Scholar]
- (18).Kaiser ET, Kezdy FJ. Science. 1984;223:249–255. doi: 10.1126/science.6322295. [DOI] [PubMed] [Google Scholar]
- (19).Holmes TC, de Lacalle S, Su X, Liu GS, Rich A, Zhang SG. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6728–6733. doi: 10.1073/pnas.97.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20) (a).Harrington DA, Cheng EY, Guler MO, Lee LK, Donovan JL, Claussen RC, Stupp SI. J. Biomed. Mater. Res. A. 2006;78A:157–167. doi: 10.1002/jbm.a.30718. [DOI] [PubMed] [Google Scholar]; (b) Rajangam K, Behanna HA, Hui MJ, Han XQ, Hulvat JF, Lomasney JW, Stupp SI. Nano Lett. 2006;6:2086–2090. doi: 10.1021/nl0613555. [DOI] [PubMed] [Google Scholar]; (c) Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, Stupp SI. Science. 2004;303:1352–1355. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- (21).Lowik D, van Hest JCM. Chem. Soc. ReV. 2004;33:234–245. doi: 10.1039/b212638a. [DOI] [PubMed] [Google Scholar]
- (22) (a).Ghadiri MR, Soares C, Choi C. J. Am. Chem. Soc. 1992;114:825–831. [Google Scholar]; (b) Ghadiri MR, Kobayashi K, Granja JR, Chadha RK, McRee DE. Angew. Chem., Int. Ed. 1995;34:93–95. [Google Scholar]; (c) Hartgerink JD, Granja JR, Milligan RA, Ghadiri MR. J. Am. Chem. Soc. 1996;118:43–50. [Google Scholar]; (d) Rapaport H, Kim HS, Kjaer K, Howes PB, Cohen S, Als-Nielsen J, Ghadiri MR, Leiserowitz L, Lahav M. J. Am. Chem. Soc. 1999;121:1186–1191. [Google Scholar]
- (23) (a).Zhang SG, Zhao XJ. J. Mater. Chem. 2004;14:2082–2086. [Google Scholar]; (b) Zhang SG. Biotechnol. AdV. 2002;20:321–339. doi: 10.1016/s0734-9750(02)00026-5. [DOI] [PubMed] [Google Scholar]; (c) Zhang SG, Marini DM, Hwang W, Santoso S. Curr. Opin. Chem. Biol. 2002;6:865–871. doi: 10.1016/s1367-5931(02)00391-5. [DOI] [PubMed] [Google Scholar]; (d) von Maltzahn G, Vauthey S, Santoso S, Zhang SU. Langmuir. 2003;19:4332–4337. [Google Scholar]
- (24).Schneider JP, Pochan DJ, Ozbas B, Rajagopal LP, Kretsinger J. J. Am. Chem. Soc. 2002;124:15030–15037. doi: 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- (25) (a).Yamada N, Imai T, Koyama E. Langmuir. 2001;17:961–963. [Google Scholar]; (b) Yamada N, Koyama E, Imai T, Matsubara K, Ishida S. Chem. Commun. 1996:2297–2298. [Google Scholar]
- (26) (a).Berndt P, Fields GB, Tirrell M. J. Am. Chem. Soc. 1995;117:9515–9522. [Google Scholar]; (b) Yu YC, Berndt P, Tirrell M, Fields GB. J. Am. Chem. Soc. 1996;118:12515–12520. [Google Scholar]; (c) Yu YC, Tirrell M, Fields GB. J. Am. Chem. Soc. 1998;120:9979–9987. [Google Scholar]
- (27) (a).Minich EA, Nowak AP, Deming TJ, Pochan DJ. Polymer. 2004;45:1951–1957. [Google Scholar]; (b) Pochan DJ, Schneider JP, Kretsinger J, Ozbas B, Rajagopal K, Haines L. J. Am. Chem. Soc. 2003;125:11802–11803. doi: 10.1021/ja0353154. [DOI] [PubMed] [Google Scholar]
- (28).Niece KL, Hartgerink JD, Donners J, Stupp SI. J. Am. Chem. Soc. 2003;125:7146–7147. doi: 10.1021/ja028215r. [DOI] [PubMed] [Google Scholar]
- (29).Jiang HZ, Guler MO, Stupp SI. Soft Matter. 2007;3:454–462. doi: 10.1039/b614426h. [DOI] [PubMed] [Google Scholar]
- (30).Paramonov SE, Jun HW, Hartgerink JD. J. Am. Chem. Soc. 2006;128:7291–7298. doi: 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- (31).Guler MO, Soukasene S, Hulvat JF, Stupp SI. Nano Lett. 2005;5:249–252. doi: 10.1021/nl048238z. [DOI] [PubMed] [Google Scholar]
- (32).Cantow H-J. Polydiacetylenes. Vol. 63. Springer-Verlag; Berlin: 1984. [Google Scholar]
- (33).Fouassier JP, Tieke B, Wegner G. Isr. J. Chem. 1979;18:227–232. [Google Scholar]
- (34).Tieke B, Lieser G, Wegner G. J. Polym. Sci., Part A: Polym. Chem. 1979;17:1631–1644. [Google Scholar]
- (35).Huo Q, Wang SP, Pisseloup A, Verma D, Leblanc RM. Chem. Commun. 1999:1601–1602. [Google Scholar]
- (36) (a).Charych DH, Nagy JO, Spevak W, Bednarski MD. Science. 1993;261:585–588. doi: 10.1126/science.8342021. [DOI] [PubMed] [Google Scholar]; (b) Reichert A, Nagy JO, Spevak W, Charych D. J. Am. Chem. Soc. 1995;117:829–830. [Google Scholar]; (c) Spevak W, Nagy JO, Charych DH. AdV. Mater. 1995;7:85–89. [Google Scholar]; (d) Okada S, Peng S, Spevak W, Charych D. Acc. Chem. Res. 1998;31:229–239. [Google Scholar]
- (37) (a).Jelinek R. Drug DeV. Res. 2000;50:497–501. [Google Scholar]; (b) Kolusheva S, Boyer L, Jelinek R. Nat. Biotechnol. 2000;18:225–227. doi: 10.1038/72697. [DOI] [PubMed] [Google Scholar]; (c) Kolusheva S, Kafri R, Katz M, Jelinek R. J. Am. Chem. Soc. 2001;123:417–422. doi: 10.1021/ja0034139. [DOI] [PubMed] [Google Scholar]; (d) Kolusheva S, Shahal T, Jelinek R. J. Am. Chem. Soc. 2000;122:776–780. [Google Scholar]; (e) Orynbayeva Z, Kolusheva S, Livneh E, Lichtenshtein A, Nathan I, Jelinek R. Angew. Chem., Int. Ed. 2005;44:1092–1096. doi: 10.1002/anie.200462393. [DOI] [PubMed] [Google Scholar]
- (38).Biesalski M, Tu R, Tirrell MV. Langmuir. 2005;21:5663–5666. doi: 10.1021/la0504558. [DOI] [PubMed] [Google Scholar]
- (39).Biesalski MA, Knaebel A, Tu R, Tirrell M. Biomaterials. 2006;27:1259–1269. doi: 10.1016/j.biomaterials.2005.08.002. [DOI] [PubMed] [Google Scholar]
- (40).Lowik DWPM, Shklyarevskiy IO, Ruizendaal L, Christianen PCM, Maan JC, van Hest JCM. AdV. Mater. 2007;19:1191–1195. [Google Scholar]
- (41) (a).Cheng Q, Stevens RC. Langmuir. 1998;14:1974–1976. [Google Scholar]; (b) Cheng Q, Yamamoto M, Stevens RC. Langmuir. 2000;16:5333–5342. [Google Scholar]; (c) Ma GY, Cheng Q. Langmuir. 2005;21:6123–6126. doi: 10.1021/la050376w. [DOI] [PubMed] [Google Scholar]; (d) Huang X, Liu M. Chem. Commun. 2002;15:1590–1591. doi: 10.1039/b204554k. [DOI] [PubMed] [Google Scholar]
- (42).Jahnke E, Lieberwirth I, Severin N, Rabe JP, Frauenrath H. Angew. Chem., Int. Ed. 2006;45:5383–5386. doi: 10.1002/anie.200600610. [DOI] [PubMed] [Google Scholar]
- (43) (a).Guler MO, Hsu L, Soukasene S, Harrington DA, Hulvat JF, Stupp SI. Biomacromolecules. 2006;7:1855–1863. doi: 10.1021/bm060161g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tovar JD, Claussen RC, Stupp SI. J. Am. Chem. Soc. 2005;127:7337–7345. doi: 10.1021/ja043764d. [DOI] [PubMed] [Google Scholar]; (c) Guler MO, Claussen RC, Stupp SI. J. Mater. Chem. 2005;15:4507–4512. [Google Scholar]
- (44).Cai M, Mowery MD, Menzel H, Evans CE. Langmuir. 1999;15:1215–1222. [Google Scholar]
- (45) (a).Berréhar J, Lapersonne-Meyer C, Schott M, Weiser G. Chem. Phys. 2004;303:129–136. [Google Scholar]; (b) Horvath A, Weiser G, Lapersonne-Meyer C, Schott M. Phys. ReV. B. 1996;53:13507–13514. doi: 10.1103/physrevb.53.13507. [DOI] [PubMed] [Google Scholar]
- (46).Deb P, Yuan Z, Ramsey L, Hanks TW. Macromolecules. 2007;40:3533–3537. [Google Scholar]
- (47).Wilson EG. J. Phys. C: Solid State Phys. 1975;8:727–742. [Google Scholar]
- (48).Chothia C. J. Mol. Biol. 1973;75:295–302. doi: 10.1016/0022-2836(73)90022-3. [DOI] [PubMed] [Google Scholar]
- (49).Mori T, Shimoyama K, Fukawa Y, Minagawa K, Tanaka M. Chem. Lett. 2005;34:116–117. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.