

Abstract
A protein-DNA dimerizer constructed from a DNA-binding pyrrole-imidazole polyamide and the peptide FYPWMK facilitates binding of the natural transcription factor Exd to an adjacent DNA site. Previous dimerizers have been constructed with the peptide attached to an internal pyrrole monomer in an overall branched oligomer. Linear oligomers constructed by attaching the peptide to the polyamide C-terminus expand the range of protein-DNA dimerization to six additional DNA sites. Replacing the FYPWMK hexapeptide with a WM dipeptide, which was previously functional in branched compounds, does not lead to a functional linear dimerizer. Instead, inserting an additional lysine generates a minimal, linear WMK tripeptide conjugate that maintains the activity of the larger FYPWMK dimerizers in a single DNA-binding site orientation. These studies provide insight into the importance of linker length and composition, binding site spacing and orientation, and the protein-binding domain content that are important for the optimization of protein DNA-dimerizers suitable for biological experiments.
Introduction
Transcription in living cells is controlled by precise spatial and temporal recognition of DNA by protein transcription factors.1 These proteins often bind to DNA as homo- and heterodimers or larger complexes which increases their overall DNA-binding site size, specificity, and affinity.2 Transcription factors are usually modular, whereby the DNA-binding and dimerization, or protein-binding domains, are functionally and structurally independent.3 Small molecules called artificial transcription factors have been reported to mimic the modular components of these endogenous proteins.4 Protein-DNA dimerizers are a type of artificial transcription factor which facilitate binding of proteins to an adjacent DNA site, mimicking natural multiprotein-DNA complexes.5 Our long-term goal is to ask whether protein-DNA dimerizers can serve as artificial regulators of gene expression in a living cell. From a design point of view, we are exploring what is the minimum size and shape (branched or linear) that will be optimal for cell uptake with adequate functional potency in the transcriptional apparatus. It has been previously shown that branched dimerizers facilitate protein-binding at three different binding sites (Figure 1a).5c Here, we describe linear structural motifs which expand the number of recognition sites targetable by protein-DNA dimerizers. The optimal DNA site spacings and necessary protein-binding elements have also been determined, which culminated in the discovery of a minimal, three amino acid protein-binding domain that yields strong dimerization at DNA-binding sites reaching 6 base pairs apart.
Figure 1.

Design of linear protein-DNA dimerizers. (a) Comparison of branched polyamide-peptide conjugates to linear conjugates, both used as protein-DNA dimerizers for Exd. (open circles = N-methylpyrrole, dark circles = N-methylimidazole, half circle = γ-aminobutyric acid, diamond = β-alanine, half-circle with plus = N,N-dimethylaminopropylamine, free-hand line = linker domain, and black triangle = protein-binding domain). (b) Diagram of a linear conjugate in the proximal orientation with the protein-binding domain directed toward Exd’s DNA-binding site. (c) Diagram of a linear conjugate in the distal orientation with the protein-binding domain directed away from Exd’s DNA-binding site. (d) An illustrative model of the linear protein-DNA dimerizer 1 in complex with Exd and DNA as shown schematically in panel b.
Protein-DNA dimerizers can mimic homeobox (HOX) transcription factors by binding cooperatively to DNA with the protein extradenticle (Exd).5 The conserved class of HOX transcription factors is known to bind relatively short DNA sites by themselves, but in complex with co-factor proteins, they bind larger DNA sequences.6 The increase in their composite DNA recognition site is thought to be important for their function in developmental pathways.6 For instance in D. melanogaster, the HOX protein ultrabithorax (Ubx) binds the site 5′-TTAT-3′ alone, but in complex with Exd the composite site increases by four base pairs to 5′-TGATTTAT-3′.7a A crystal structure of the ternary Ubx/Exd/DNA complex revealed how both Ubx and Exd’s major groove binding homeodomains interact via Ubx’s N-terminal YPWM peptide motif.7a The conserved YPWM motif spans the DNA minor groove between the two proteins and inserts a hydrophobic tryptophan into the complementary Exd binding pocket. Additional crystal structures of HoxB1/Pbx/DNA and HoxA9/Pbx/DNA which contain human homologues of Ubx and Exd have also been reported in which the conserved tryptophan of the YPWM motif mediates the majority of the protein-protein contacts.7b–c Protein-DNA dimerizers have been constructed by replacing the natural DNA-binding homeodomain of Ubx with a synthetic pyrrole-imidazole polyamide which binds the DNA minor groove.5 Polyamides are ideal small-molecule DNA binding domains since they can be programmed to bind to a broad repertoire of sequences using an aromatic amino acid pairing code.8a–b Hairpin polyamides by themselves have shown the ability to inhibit protein-DNA complex formation in biological contexts.8c–d Protein-DNA dimerizers have the potential to be more specific regarding cellular targets relative to parent hairpin DNA binders by virtue of their ability to bind larger cognate DNA sites with proteins and through incorporation of a unique protein-binding recognition interface.
In this study we explore the range of DNA sites in which Exd-DNA dimerization is observed in the presence of linear polyamide-peptide conjugates using a series of DNA duplexes containing incremental A,T base pair spacings between the polyamide and Exd binding sites. Linear oligomers were generated by attaching an FYPWMK peptide to the polyamide C-terminus incorporating linkers of different lengths and composition, i.e. 1 and 2 (Figure 2). The synthesis of linear conjugates is simpler than branched ones since the peptide can be coupled directly to a standard pyrrole-imidazole polyamide without the need for the synthesis of advanced building blocks. Once the scope of recognition sequences with these larger constructs was defined, efforts were made to discover a minimal protein-binding domain for dimerizers in this linear configuration. It is shown that WM dipeptides (with both L and D stereochemistry of the W) which previously enhanced Exd-DNA binding in the branched oligomer5d failed to yield stable Exd-DNA complexes. Exd dimerization activity was recovered by inserting a lysine back into the peptide to generate minimal WMK tripeptide conjugates. The presence of a positively charged ammonium ion in the linker and lysine seem to be favorable for complex formation possibly due to interactions with the negatively charged DNA backbone.
Figure 2.

Structures of compounds 1–14.
Results and Discussion
Dimerization in Proximal and Distal Orientations
Exd is expected to bind 5′-TGAT-3′ as found in the Ubx/Exd/DNA crystal structure7a and the hairpin polyamide ImImPyPy-γ-PyPyPyPy-β-codes for the sequence 5′-WGGWWW-3′ (where W = A or T).8a–b In the proximal orientation, the polyamide binding site is oriented such that the protein-binding YPWM peptide is projected adjacent to Exd (Figure 1b). In the distal orientation, the polyamide binding site was inverted such that the peptide is projected away from Exd (Figure 1c). For dimerization of Exd at a given DNA site to occur, the protein-binding peptide must project over the DNA phosphate backbone and interact favorably with the binding pocket of the major groove binding Exd protein (Figure 1d). Dimerization was expected to be dependent on both the length and composition of the linker between the polyamide and the YPWM-containing peptide as well as the size of the spacer region between the polyamide’s and protein’s binding sites.
Accordingly, two polyamide-peptide conjugates were constructed that incorporated either a tertiary amine (1) or a PEG linker (2). The distance between the terminal amine of the linker to the carbonyl carbon of the methionine of the peptide was determined computationally in the gas-phase for MM2-energy minimized linkers to be approximately 23 and 30 Å for 1 and 2, respectively. A series of DNA duplexes which contained an incremental A,T spacer region were screened by electrophoretic mobility shift (EMSA), or gel shift assays, to determine the range of protein recruitment to the DNA at room temperature (20 °C). A total of nine different duplexes (47–55 bps) in the proximal orientation with spacer regions between −1 and 7 bps were used, covering distances between the polyamide and expected protein-binding domain position from approximately 11 to 30 Å. Negative spacer regions indicate an overlap in the expected binding sites.
It might be expected that since 2 contains a longer linker than 1, it would facilitate Exd binding at sites further away from the polyamide binding site. Instead, it was observed that 1 dimerizes Exd effectively on composite sites containing spacer regions between 2 to 6 base pairs, whereas 2 only dimerizes Exd on sites with spacer regions of 4 to 5 base pairs (Figure 3 and Table 1). Thus, compound 2 has a narrower recognition profile, in contrast to 1 which is more promiscuous. The greater tolerance for multiple binding sites by compound 1 may be due to its chemical composition. It is possible that the presence of the positively charged ammonium ion in 1 may interact more favorably with the phosphate backbone than the PEG linker of 2. Additionally, the dihedral angles along the -OCH2CH2O- axes of PEG are known to favor gauche conformations which is expected to form a partially helical linker spanning approximately 24 Å in the context of 2.9 There is probably an energetic barrier along the transition from a helical, all-gauche conformation to the fully-extended, all-trans form that explains why no dimerization is observed at distances greater than 24 Å. Control experiments at all of these sites show that Exd by itself, or in the presence of parent polyamide ImImPyPy-γ-PyPyPyPy-β-Dp 15, or the double alanine mutants 3 and 4, does not lead to stable complex formation (Supplemental Figure 1). Thus, the polyamide domain by itself does not appear to contribute to the binding of Exd through allosteric interactions as observed in previously reported systems.5d Furthermore, these control experiments show that dimerization of Exd is dependent on the central tryptophan and methionine residues in this context.
Figure 3.
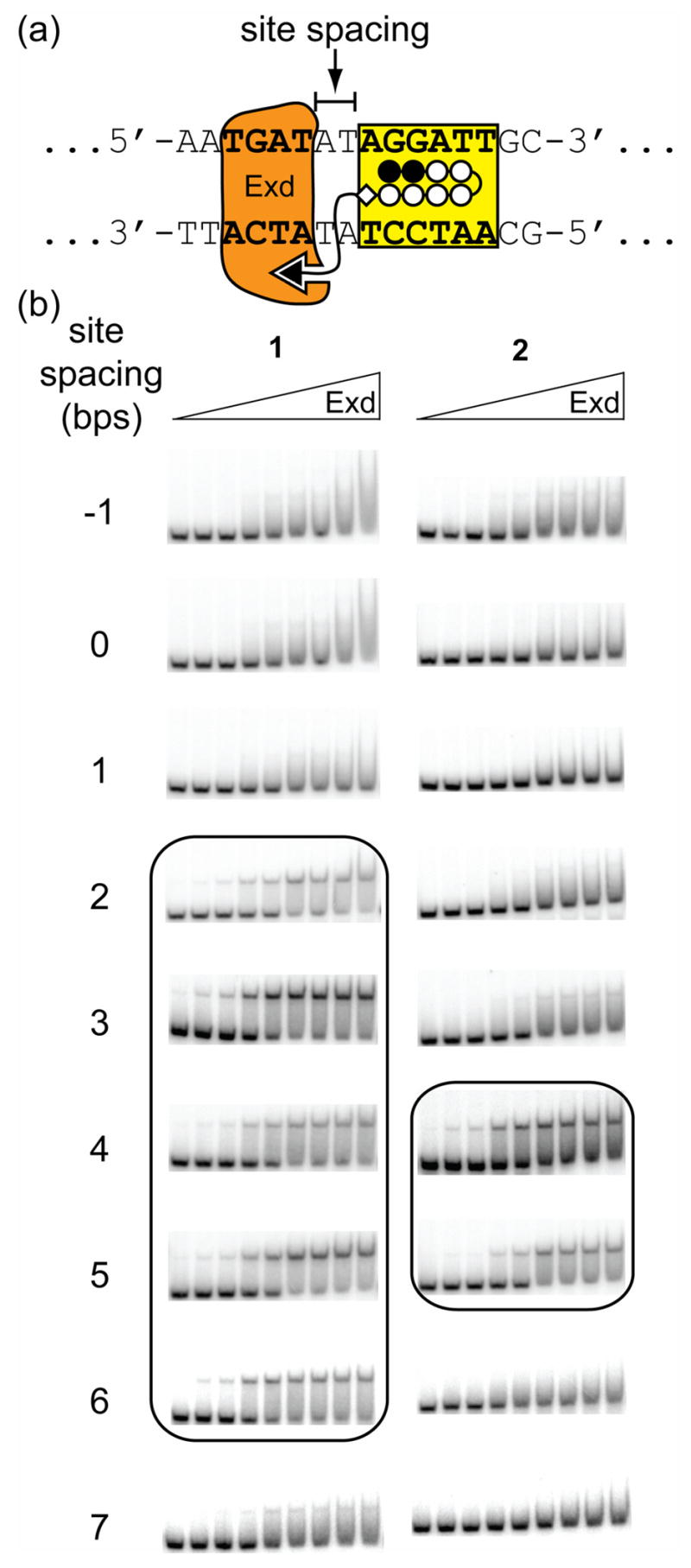
Representative gel shift results for Exd in the presence of linear protein-DNA dimerizers with differently sized linkers on two proximally-oriented DNA sites. Conjugate 1 has an amine-containing linker (~23 Å long) and conjugate 2 has a PEG-type linker (~30 Å long). (a) Schematic of the positions of the Exd and polyamide DNA-binding sites showing the A,T spacer region which was varied. (b) Gel shift experiments show that 1 forms a stable complex with Exd and DNA with site spacings between 2–6 bps, whereas 2 only forms stable complexes from 4–5 bps. A concentration of 50 nM of the indicated compound was added to all lanes and the concentration of Exd from left to right is 10 pM, 50 pM, 100 pM, 500 pM, 1 nM, 5 nM, 10 nM, 50 nM, and 100 nM.
Table 1.
Summary of select proximal orientation gel shiftsa
| Base pairs between binding sites | Estimated proximal distance (Å)b | Exd-DNA binding with 1 | Exd-DNA binding with 2 |
|---|---|---|---|
| −1 | 23.8 | − | − |
| 0 | 20.0 | − | − |
| 1 | 15.0 | − | − |
| 2 | 11.2 | + | − |
| 3 | 11.2 | + | w |
| 4 | 14.2 | + | + |
| 5 | 19.4 | + | + |
| 6 | 25.4 | + | − |
| 7 | 29.8 | w | − |
A ‘+’, ‘w’, and ‘−’ indicate a strong, weak, or no complex, respectively. Binding constants (Ka’s) for ‘+’ entries varied between 0.8 ± 0.3 to 2.7 ± 1.0 × 109 M−1 and the amount of complex formed was ≥ 0.4 (Supplemental Figure 5).
measured from the Met C to the linker N experiments show that dimerization of Exd is dependent on the central tryptophan and methionine residues in this context.
In the distal orientation, 5 different spacer regions (5 duplexes, 46–50 bps) were investigated from −2 to 2 bps covering a theoretical distance of approximately 15 to 30 Å between the polyamide and expected protein-binding domain position. In this arrangement, Exd dimerization was observed only with compound 1 at the −2 spacer site (Figure 4 and Table 2). Control experiments at the −2 site show that Exd by itself, in the presence of parent polyamide ImImPyPy-γ-PyPyPyPy-β-Dp 15, or double alanine mutant 3, does not lead to stable complex formation (Supplemental Figure 2).
Figure 4.
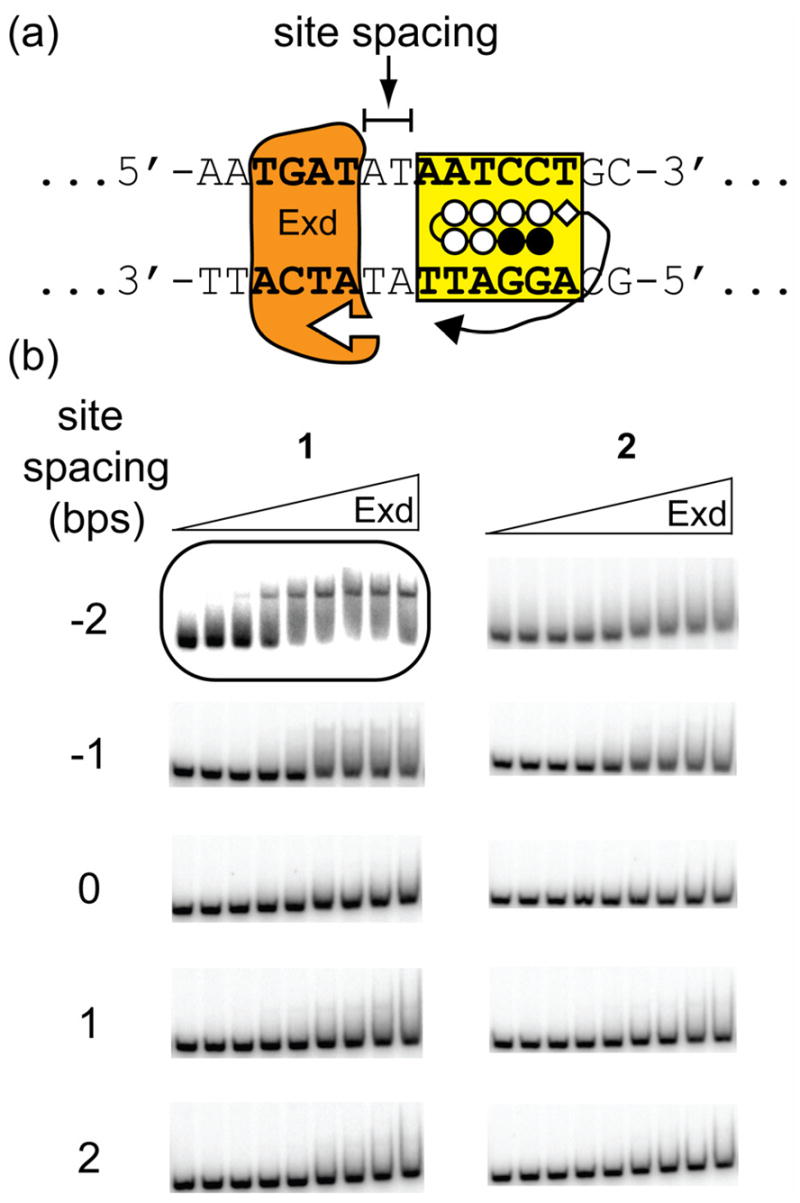
Representative gel shift results for Exd in the presence of linear protein-DNA dimerizers with differently sized linkers on two distally-oriented DNA sites. (a) Schematic of the positions of the Exd and polyamide DNA-binding sites showing the A,T spacer region which was varied. (b) Gel shift experiments show that 1 forms a stable complex with Exd and DNA when there is a 2 bp overlap, i.e. a site spacing of −2, between the two sites, whereas 2 does not form a complex with Exd at any of these sites. Concentrations of the compounds and Exd are the same as described in Figure 3.
Table 2.
Summary of select distal orientation gel shiftsa
| Base pairs between binding sites | Estimated distal distance (Å)b | Exd-DNA binding with 1 | Exd-DNA binding with 2 |
|---|---|---|---|
| −2 | 15.1 | + | − |
| −1 | 19.5 | − | − |
| 0 | 23.8 | − | − |
| 1 | 28.1 | − | − |
| 2 | 31.5 | − | − |
A ‘+’ and ‘−’ indicate the presence or absence of a complex, respectively. The binding constant (Ka) for the ‘+’ entry is 2.5 ± 1.8 × 109 M−1 and the amount of complex formed was 0.5 ± 0.1.
measured from the Met C to the linker N
Minimization of the Protein-binding Domain
As mentioned above, we have previously reported that in the context of a branched polyamide-peptide conjugate, the protein-binding domain could be reduced to a WM dipeptide.5d Thus, compounds 5–8 were synthesized to see if an analogous linear dipeptide conjugate would also dimerize Exd. Gel shift experiments with 5 and 6 using DNA duplexes with spacings of 0 to 6 in the proximal orientation, and −2 in the distal orientation did not lead to any significant gel shift (Supplemental Figure 3). A weak complex was observed with compound 6 at the 3 bp proximal spacing (Θapp = 0.3 ± 0.1 at 100 nM Exd and 50 nM 6), but was not significant enough to reliably determine the Exd-DNA binding affinity. Similarly, the WM conjugates bearing the PEG linker (7 and 8) failed to yield any discernible gel shift with DNA duplexes containing proximal spacings of 1 to 6 bps or at the −2 bp distal spacing (Supplemental Figure 3).
It was noted that the substitution of FYPWMK (1 and 2) with WM (5–8) eliminated the lysine between the presumed linker domain and the core WM dipeptide of the YPWM motif. Thus, WMK tripeptide conjugates 9–12 were synthesized to ensure identical linker length and composition between the polyamide and the WM moiety. These compounds were examined by gel shift using duplexes with proximal spacings between 3 and 6 bps shown above to enable dimerization of Exd with the larger conjugates 1 and 2. Both tertiary amine containing WMK conjugates (9 and 10) yielded a complex with Exd only at the 6 bp proximal site, but the PEG containing WMK conjugates (11 and 12) did not lead to an observable shift in any of the orientations (Figure 5 and Supplemental Figure 4b). Both 9 and 10 at the proximal 6 bp spacing site yielded similar binding affinities (Ka = 9.2 ± 0.6 × 108 M−1 and 7 ± 2 × 108 M−1, respectively) and amounts of complex formed (0.50 ± 0.01 and 0.53 ± 0.05, respectively) indicating no significant difference in Exd binding by either the L- or D-tryptophan peptides in this context. The binding affinities of Exd in the presence of 9 and 10 are within 4-fold of those observed for the larger conjugates 1 and 2 (Tables 1 and 2). In comparing 9 and 10 to 1, however, it appears that the use of smaller and presumably lower-affinity protein-binding domains leads to a narrower site spacing tolerance. In general, larger higher-affinity protein-binding domains may tolerate suboptimal linker distances and may generate more promiscuous protein-DNA dimerizers.
Figure 5.
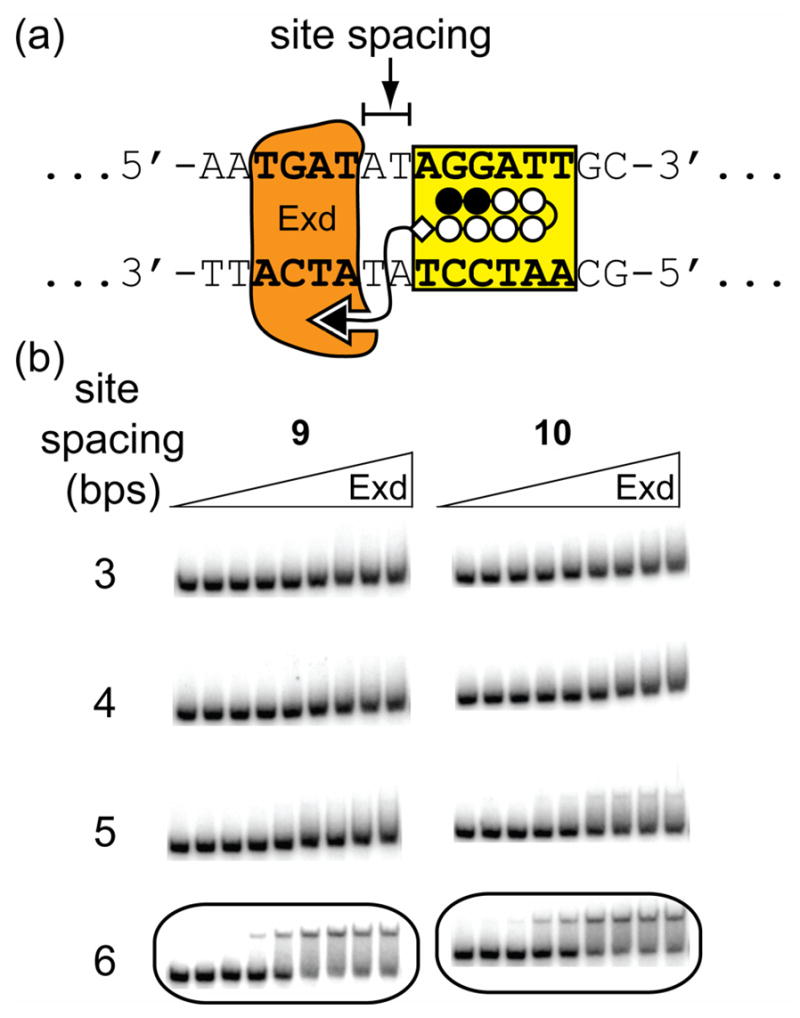
Representative gel shift results for Exd in the presence of linear WMK tripeptide conjugates on select proximally-oriented DNA sites. (a) Schematic of the positions of the Exd and polyamide DNA-binding sites showing the A,T spacer region which was varied. (b) Gel shift experiments show that both 9 and 10 form stable complexes with Exd and DNA only when there is a 6 bp site spacing. Concentrations of the compounds and Exd are the same as described in Figure 3.
Finally, to determine the principal role of the lysine in the WMK tripeptide conjugates 9 and 10, their lysine side-chains were acetylated to create conjugates 13 and 14, respectively. These compounds retain the same linker length as 9 and 10 between the polyamide and the core WM dipeptide, but should eliminate the positive charge on the lysine while maintaining comparable steric bulk. Both 13 and 14 were not able to form a complex in the presence of Exd (Supplemental Figure 4c). This suggests that the lysine side-chain makes a favorable electrostatic interaction to stabilize the Exd-DNA-dimerizer complex. Although a direct interaction with the Exd protein cannot be ruled out, the presumed requirement for the linker domain to traverse the DNA backbone suggests that the primary role of this lysine group may be to interact favorably with the negatively charged phosphate groups of DNA. In comparing to natural systems it was observed that Ubx contains an alanine at this position,7a but lysine and arginine are often found immediately adjacent to the C-terminal end of the YPWM motif in many HOX proteins.10
Calculation of Distances
The superposition of a polyamide crystal structure11 (PDB code 365D) over specific DNA sites in the Ubx/Exd/DNA crystal structure7a (PDB code 1B8I) readily allows the measurement of theoretical distances between the polyamide tail and the YPWM peptide that the linker domain must traverse (Figure 6 and Tables 1 and 2). In the proximal orientation the shortest distance occurs somewhere between the 2 and 3 bp spacings (~11 Å) and increases in both directions in a non-linear fashion due to the helical nature of the DNA grooves. In the distal orientation, the polyamide positions the linker past the minimum distance of approximately 11 Å observed in the proximal orientation. Thus, even at a 2 bp overlap in the distal orientation the distance is approximately 15 Å, which is longer than the shortest distance in the proximal orientation.
Figure 6.
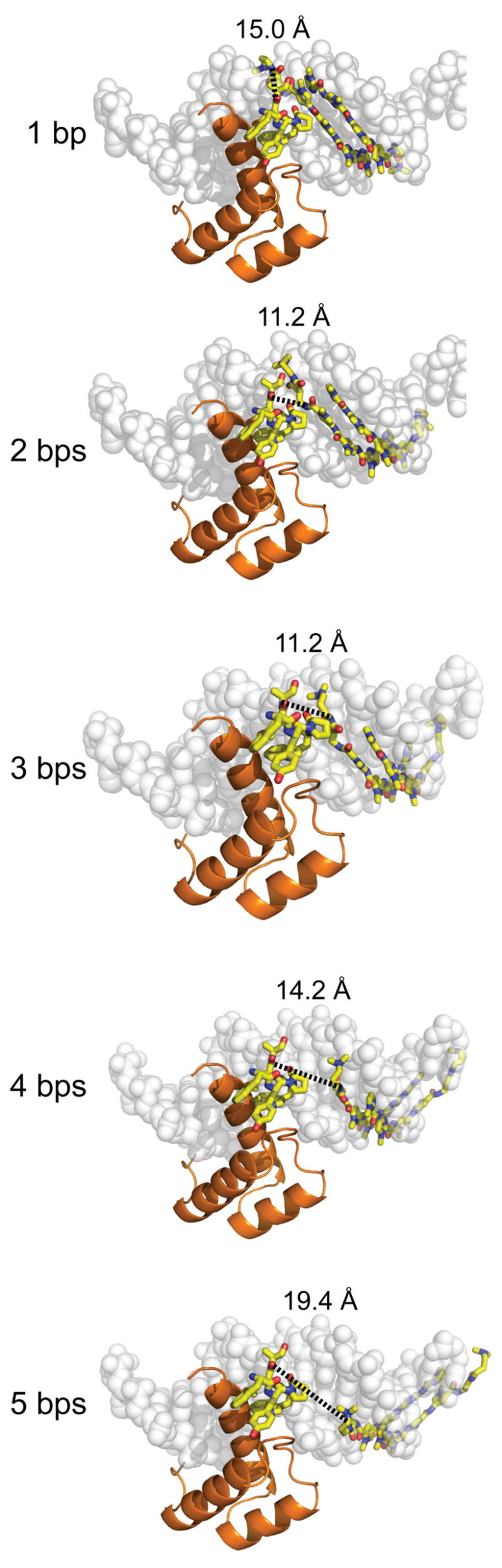
A series of estimated distance measurements are shown between the polyamide tail and the methionine carbonyl of the YPWM protein-binding domain which each dimerizer linker domain must traverse. The models of the polyamides binding to the DNA were generated by superimposing the DNA-binding site from a representative polyamide-DNA crystal structure (PDB code = 365D) over the Ubx/Exd/DNA crystal structure (PDB code 1B8I) and deleting the Ubx protein except for its YPWM protein-binding domain. In the proximal orientation, there exists a minimum distance of ~11 Å between the site spacings of 2–3 bps which increases non-linearly in each direction as the polyamide slides along the helical DNA minor groove.
The results cannot be completely rationalized using distance measurements, however. For instance, comparing proximal sites with spacers of similar distances, such as 1 and 4 bps (14–15 Å), or 0 and 5 bps (19–20 Å) reveal that sites with similar distance constraints do not necessarily lead to similar abilities to enhance Exd binding to DNA. This apparent discrepancy could be explained by the fact that the measured distances do not account for the interference by the phosphate backbone which the linker domain must cross to place the protein-binding domain in the necessary location. Additionally, the linker domain must bend back approximately 180° in the 1 and 0 bp sites to bring the protein-binding domain into the correct binding orientation whereas the 4 and 5 bp sites project the protein-binding domain directly toward its Exd binding pocket. Such bending of the linker domain may be energetically unfavorable.
Similarly, in spite of the overlapping range of possible distances for the distal binding orientations (~11–30 Å) when compared to the proximal sites (~15–30 Å), only the −2 bp distal site led to a stable complex with Exd. Unfortunately, greater overlap sites in the distal orientation cannot be addressed without changing the structure of the polyamide which was held constant for the purposes of this study. Movement of the polyamide binding site to the −3 bp site may still lead to a functional binding orientation, but the overlapped −4 bp site will probably not be tolerated since Arg2 of the Exd homeodomain contacts the thymine base at this location in the minor groove.7a
Interestingly, a complex was observed at the 6 bp (strong complex) and 7 bp (weak complex) proximal arrangements (≥ 25 Å spacing) with compound 1 even though the fully extended linker (~23 Å) was predicted not to reach in either case. This suggests there may have been some structural adaptation by the complex to enable the YPWM-Exd interaction. It is noted that TATA DNA sequences are known to be bent by TATA-binding protein12 and it is thought that such sequences are inherently flexible.13 Thus, the use of a variable poly-AT spacer may have enabled such DNA distortions. Either the Exd structure or the placement of the YPWM peptide in the Exd binding pocket may also allow structural deviations.
Conclusions
A set of linear polyamide-peptide conjugates have been created which expand the recognition scope of protein-DNA dimerizers targeted towards Exd. In particular, conjugate 1 was shown to enhance Exd’s binding to DNA when the protein and polyamide binding sites were −2, 2, 3, 4, 5, and 6 bps apart. We have previously reported branched dimerizers that facilitate Exd binding to −1,5c 0,5 and 1 bp5c orientations. Thus, a set of solutions from −2 to 6 bps has been completed using these linear conjugates. Furthermore, the protein-binding domain was minimized to a WMK tripeptide (9 and 10) that functioned nearly as well as the larger conjugates (1 and 2). Remarkably, the range of tolerated orientations appears to be tunable not only by the length and chemical nature of the linker (compare 1 to 2), but also by the content of the protein-binding domain (compare 1 to 9).
In combination with the previous results for branched dimerizers,5 the work presented here provides design principles for dimerizers suitable for use in cell culture and fruit fly experiments. Developmental processes in fruit flies have been perturbed using polyamides.14 The question arises whether a linear or branched protein-DNA dimerizer can interfere specifically with the normal function of Exd and lead to distinct fruit fly phenotypes. All compounds in this study function at temperatures suitable for experiments in fruit flies (20 °C) and the unnatural D-Trp WMK conjugate (10) is expected to be resistant to endogenous proteases. 15
The unique availability of multiple high-resolution structural models of HOX-DNA complexes provided a notable advantage for the design and refinement of these dimerizers. The growing number of high-resolution structures of multiprotein-DNA complexes should facilitate the design of additional protein-DNA dimerizers that function in other systems. Thus, there is a need for further structural studies on protein-DNA complexes that should aid the development of these molecules. This structure-based design approach contrasts with the screening efforts used for the development of small activation domains for other artificial transcription factors due to the paucity of such high-resolution models.4h–i,17
Experimental Section
General
Fmoc protected amino acids and 2-chlorotritylchloride resin was purchased from Novabiochem. Super Acid Sensitive Resin (SASRIN™) was purchased from Bachem. Boc-β-Ala-PAM resin was purchased from Peptides International. Trifluoroacetic acid (TFA) was purchased from Halocarbon. Molecular biology grade ethylenediaminetetraactic acid (EDTA), potassium glutamate, and acetylated BSA were purchased from Sigma. DTT was purchased from ICN. Other chemicals were purchased from Aldrich or EM Sciences and used without further purification. All other solvents were purchased from EM Sciences and were reagent grade. Water (18 MΩ) was purified using a Millipore MilliQ water purification system. Biological experiments were performed using Ultrapure Distilled Water (DNase/RNase free) purchased from Gibco. The pH of buffers was adjusted using a Beckman 340 pH/Temp Meter. All buffers were sterilized by filtration through either a Nalgene 0.2 μm cellulose nitrate filtration device or a Pall Acrodisc® Syringe Filter HT Tuffryn® membrane (0.2 μm). DNA oligonucleotides were obtained from Integrated DNA Technologies. [γ-32P]-adenosine-5′-triphosphate (≥ 6000 Ci/mmol) was obtained from Perkin Elmer. Polynucleotide kinase (PNK) was obtained from Roche. UV spectra were recorded in water using an Agilent 8453 UV-Visible spectrophotometer. All polyamide concentrations were determined using an extinction coefficient ε = 69,500 M−1·cm−1 at a λmax near 310 nm. Matrix-assisted, LASER desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) was performed using a Applied Biosystems Voyager DE Pro Spectrometer. Electrospray ionization (ESI) mass spectrometry was performed using a Finnigan LCQ ion trap mass spectrometer. Analytical High-Pressure Liquid Chromatography (HPLC) was performed with a Beckman Gold system equipped with a diode array 168 Detector and a 125 Solvent module using a Phenomenex Gemini C18 column (5 μm particle size, 250 × 4.60 mm). Preparative HPLC was performed with a Beckman Gold system equipped with a 127P Solvent Module and a single wavelength 166P Detector monitoring 310 nm using a Waters DeltaPak C18 reverse phase column (100 μm particle size, 25 × 100mm) or a Phenomenex Gemini C18 reverse phase column (5 μm particles, 110 Å pore, 250 × 21.2 mm). For both analytical and preparative HPLC, solvent A was 0.1% (v/v) aqueous TFA and solvent B was acetonitrile. In general, compound purity was assessed using an HPLC analytical method employing a gradient of ~4.12% B/min starting from 10% B with a flow rate of 1.5 mL/min. Final preparative HPLC purification was performed by ramping from 10–55% B over 75 min using the Gemini column. If the purity of the compound was less than 95%, a second HPLC purification was performed employing a slow gradient through the previous % B elution of the compound.
Synthesis
Polyamide monomers were prepared as described previously.18 Polyamides were synthesized according to standard synthetic procedures,18 and cleaved from the resin with either 3,3′-diamino-N-methyl-dipropylamine, 4,7,10-trioxa-1,13-tridecanediamine, or 3-(dimethylamino)-propylamine. Before conjugation to peptides, polyamides were HPLC purified and characterized by MALDI-TOF MS, UV-Vis spectroscopy, and analytical HPLC. Protected peptides were synthesized using SASRIN or 2-chlorotrityl resin using standard Fmoc protocols19 and characterized by ESI MS and analytical HPLC. Polyamide-peptide conjugates were prepared as described previously using 0.1 M DIEA in DMF using excess PyBOP (~5 equiv) followed by deprotection with 80% TFA, 5% TIS, 5% EDT, and 10% DCM.5a Conjugates were then purified by preparative HPLC and characterized by MALDI-TOF or ESI MS, UV-Vis spectroscopy, and analytical HPLC. 1: (MALDI-TOF) [M+H] calcd for C113H146N31O19S 2274.1, observed 2274.0; 2: (MALDI-TOF) [M+H] calcd for C116H151N30O22S 2349.1, observed 2349.0; 3: (MALDI-TOF) [M+H] calcd for C103H137N30O19 2099.1, observed 2098.8; 4: (MALDI-TOF) [M+H] calcd for C106H142N29O22 2174.1, observed 2174.0; 5: (ESI) [M+H] calcd for C84H109N26O14S 1737.8, observed 1737.6; 6: (MALDI-TOF) [M+H] calcd for C84H109N26O14S 1737.8, observed 1737.4; 7: (ESI) [M+H] calcd for C87H114N25O17S 1812.9, observed 1812.5; 8: (MALDI-TOF) [M+Na] calcd for C87H113N25O17SNa 1834.8, observed 1834.6; 9: (MALDI-TOF) [M+H] calcd for C90H121N28O15S 1865.9, observed 1865.5; 10: (MALDI-TOF) [M+H] calcd for C90H121N28O15S 1865.9, observed 1865.8; 11: (MALDI-TOF) [M+H] calcd for C93H126N27O18S 1941.0, observed 1941.0; 12: (MALDI-TOF) [M+H] calcd for C93H126N27O18S 1941.0, observed 1940.9. Acetylated compounds 13 and 14 were synthesized by treating 200–300 nmol of 9 and 10, respectively, with 8–9 equivalents of acetic anhydride in 0.1 M DIEA in DMF and purified by preparative HPLC. 13: (MALDI-TOF) [M+H] calcd for C92H123N28O16S 1907.9, observed 1908.2; 14: (MALDI-TOF) [M+H] calcd for C92H123N28O16S 1907.9, observed 1908.0; 15: (MALDI-TOF) [M+H] calcd for C58H72N21O10 1222.6, observed 1222.7.
Preparation of 32P-labeled DNA duplexes
DNA oligos were first annealed by heating to 95 °C for 5 min and allowing the solution to cool slowly to room temperature for ~2–3 hours. The dsDNA duplexes were then 5′-labeled using polynucleotide kinase using the manufacturer’s protocols. Labeled duplexes were purified by gel electrophoresis and band extraction from a 6% nondenaturing polyacrylamide gel, diluted to 10 kpcm/μL, stored at −78 °C, and used within 2 weeks.
Electrophoretic Mobility Shift Assays (EMSAs)
EMSA, or gel shift, experiments were performed at 20 °C as described previously.5d Polyamide conjugates were pre-incubated with ~5 kcpm/reaction of 32P-labeled duplexes (46–55 bps, Supplemental Figures 6 and 7) at room temperature (20 °C) in buffer for 1 h. Exd protein was then added to the reaction solution and the solution was allowed to equilibrate for an additional hour. The final concentrations of the components were 50 nM of the indicated compound, 30 mM potassium glutamate, 10 mM HEPES, 0.2 mM DTT, 10% glycerol, and 0.1 mg/mL acetylated BSA. The final concentrations for Exd were the following: 10 pM, 50 pM, 100 pM, 500 pM, 1 nM, 5 nM, 10 nM, 50 nM, and 100 nM. A volume of 10 μL of the solution was loaded onto a, pre-run (15 min) non-denaturing 9% polyacrylamide (29:1 acrylamide:bisacrylamide/3% glycerol, 1X TBE) 0.8 mm thick gel and developed at 180–190V and 13–15 mA for 1.5 h at room temperature. Gels were then dried and exposed to a phosphor screen overnight, which were scanned by a Molecular Dynamics 400S phosphorimager. Recombinant Exd (residues 238–320) was expressed5 and characterized5d as described previously. The Exd concentration was determined by measuring the UV absorption at 280 nm using a calculated20 extinction coefficient of 12,600 M−1·cm−1. The amount of Exd bound to the DNA template was determined as described previously5d and fit to the Hill equation21 with n = 1. All binding affinities were measured in triplicate.
Theoretical distance measurements
The illustrative models shown in Figure 6 were created by superimposing the DNA backbone of the atomic coordinates from a representative polyamide-DNA crystal structure11 (PDB code 365D) over the desired site on the DNA backbone from the Ubx/Exd/DNA crystal structure7a (PDB code 1B8I) using a fit algorithm from SwissPDB Viewer (ver 3.7). The DNA from the polyamide crystal structure and the Ubx homeodomain were then deleted to generate a model from which the distance measurements were be made.
The illustrative models shown in Figure 7 were made by first superimposing a polyamide-DNA crystal structure as described above. The linker between the polyamide and the FYPWMA peptide from Ubx, the linker, and the γ-aminobutyric acid of the polyamide hairpin, the lysine side-chain, and the acetyl moiety were constructed and minimized using an augmented MM3 force field and short molecular dynamics simulations (<100 ps) with all other coordinates locked using CAChe Workspace (ver. 6.1.12.33). The DNA shown in the figure is the same as the DNA from the Ubx/Exd/DNA structure.
Figure 7.

Models of linear-protein DNA dimerizers. (a) A proximally-oriented binding site with a 5 bp spacer with dimerizer 1 is shown. (b) A distally-oriented binding orientation with a 2 bp overlap (i.e. −2 bp spacer) between the polyamide and Exd DNA-binding sites is shown for dimerizer 1.
Supplementary Material
Supplemental Figures 1–7 including gel shifts for compounds 3–8 and 11–15, graphs showing the relationship between site spacing, binding affinity and the fraction of DNA shifted, and the full DNA sequences used for gel shifts. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the National Institute of Health (NIH GM51747 to P.B.D.). A fellowship from the Rose Hills Foundation (to R.L.S.) is gratefully acknowledged. We thank Drs. P. Snow and G. Hathaway (Beckman Institute, California Institute of Technology) for technical assistance with protein expression, purification, and identification.
References
- 1.(a) Mitchell PJ, Tjian R. Science. 1989;245:371–378. doi: 10.1126/science.2667136. [DOI] [PubMed] [Google Scholar]; (b) Lee TI, Young RA. Annu Rev Genet. 2000;34:77–137. doi: 10.1146/annurev.genet.34.1.77. [DOI] [PubMed] [Google Scholar]; (c) Kadonaga JT. Cell. 2004;116:247–257. doi: 10.1016/s0092-8674(03)01078-x. [DOI] [PubMed] [Google Scholar]; (d) Levine M, Tjian R. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 2.(a) Klemm JD, Schreiber SL, Crabtree GR. Annu Rev Immunol. 1998;16:569–592. doi: 10.1146/annurev.immunol.16.1.569. [DOI] [PubMed] [Google Scholar]; (b) Wolberger C. Annu Rev Biophys Biomol Struct. 1999;28:29–56. doi: 10.1146/annurev.biophys.28.1.29. [DOI] [PubMed] [Google Scholar]; (c) Ogata K, Sato K, Tahirov T. Curr Opin Struct Biol. 2003;13:40–48. doi: 10.1016/s0959-440x(03)00012-5. [DOI] [PubMed] [Google Scholar]
- 3.Ptashne M, Gann A. Genes & Signals. Cold Spring Harbor Laboratory Press; Plainview, NY: 2002. [Google Scholar]
- 4.Kuznetsova S, Ait-Si-Ait S, Nagibneva I, Troalen F, Le Villain J, Harel-Bellan A, Svinarchuk F. Nucleic Acids Res. 1999;27:3995–4000. doi: 10.1093/nar/27.20.3995.Mapp AK, Ansari AZ, Ptashne M, Dervan PB. Proc Natl Acad Sci USA. 2000;97:3930–3935. doi: 10.1073/pnas.97.8.3930.Ansari AZ, Mapp AK, Nguyen DH, Dervan PB, Ptashne M. Chem Biol. 2001;8:583–592. doi: 10.1016/s1074-5521(01)00037-0.Liu B, Han Y, Corey DR, Kodadek T. J Am Chem Soc. 2002;124:1838–1839. doi: 10.1021/ja0164226.Stanojevic D, Young RA. Biochemistry. 2002;41:7209–7216. doi: 10.1021/bi015906b.Arora PS, Ansari AZ, Best TP, Ptashne M, Dervan PB. J Am Chem Soc. 2002;124:13067–13071. doi: 10.1021/ja0208355.Liu B, Han Y, Ferdus A, Corey DR, Kodadek T. Chem Biol. 2003;10:909–916. doi: 10.1016/j.chembiol.2003.09.008.Kwon Y, Arndt HD, Mao Q, Choi Y, Kawazoe Y, Dervan PB, Uesugi M. J Am Chem Soc. 2004;126:15940–15941. doi: 10.1021/ja0445140.Xiao X, Yu P, Lim HS, Sikder D, Kodadek T. Angew Chem, Int Ed. 2007;46:2865–2868. doi: 10.1002/anie.200604485.For reviews, see: Ansari AZ, Mapp AK. Curr Opin Chem Biol. 2002;6:765–772. doi: 10.1016/s1367-5931(02)00377-0.Arndt HD. Angew Chem, Int Ed. 2006;45:4552–4560. doi: 10.1002/anie.200600285.
- 5.(a) Arndt HD, Hauschild KE, Sullivan DP, Lake K, Dervan PB, Ansari AZ. J Am Chem Soc. 2003;125:13322–13323. doi: 10.1021/ja0371395. [DOI] [PubMed] [Google Scholar]; (b) Hauschild KE, Metzler RE, Arndt HD, Moretti R, Raffaelle R, Dervan PB, Ansari AZ. Proc Natl Acad Sci USA. 2005;102:5008–5013. doi: 10.1073/pnas.0501289102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Warren CL, Kratochvil NCS, Hauschild KE, Foister S, Brezinski ML, Dervan PB, Phillips GN, Jr, Ansari AZ. Proc Natl Acad Sci USA. 2006;103:867–872. doi: 10.1073/pnas.0509843102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Stafford RL, Arndt HD, Brezinski ML, Ansari AZ, Dervan PB. J Am Chem Soc. 2007;129:2660–2668. doi: 10.1021/ja067971k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Peifer M, Wieschaus E. Genes Dev. 1990;4:1209–1223. doi: 10.1101/gad.4.7.1209. [DOI] [PubMed] [Google Scholar]; (b) Chan SK, Jaffe L, Capovilla M, Botas J, Mann RS. Cell. 1994;78:603–615. doi: 10.1016/0092-8674(94)90525-8. [DOI] [PubMed] [Google Scholar]; (c) van Dijk MA, Murre C. Cell. 1994;78:617–624. doi: 10.1016/0092-8674(94)90526-6. [DOI] [PubMed] [Google Scholar]; (d) Mann RS, Chan SK. Trends Genet. 1996;12:258–262. doi: 10.1016/0168-9525(96)10026-3. [DOI] [PubMed] [Google Scholar]
- 7.(a) Passner JM, Ryoo HD, Shen LY, Mann RS, Aggarwal AK. Nature. 1999;397:714–719. doi: 10.1038/17833. [DOI] [PubMed] [Google Scholar]; (b) Piper DE, Batchelor AH, Chang CP, Clearly ML, Wolberger C. Cell. 1999;96:587–597. doi: 10.1016/s0092-8674(00)80662-5. [DOI] [PubMed] [Google Scholar]; (c) LaRonde-LeBlanc NA, Wolberger C. Genes Dev. 2003;17:2060–2072. doi: 10.1101/gad.1103303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Dervan PB. Bioorg Med Chem. 2001;9:2215–2235. doi: 10.1016/s0968-0896(01)00262-0. [DOI] [PubMed] [Google Scholar]; (b) Dervan PB, Edelson BS. Curr Opin Struct Biol. 2003;13:284–299. doi: 10.1016/s0959-440x(03)00081-2. [DOI] [PubMed] [Google Scholar]; (c) Olenyuk BZ, Zhang GJ, Klco JM, Nickols NG, Kaelin WG, Dervan PB. Proc Natl Acad Sci USA. 2004;101:16768–16773. doi: 10.1073/pnas.0407617101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nickols NG, Dervan PB. Proc Natl Acad Sci USA. 2007;104:10418–10423. doi: 10.1073/pnas.0704217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kreuzer HJ, Wang RLC, Grunze M. New J Phys. 1999;1:21.1–21.16. [Google Scholar]
- 10.Duboule D, editor. Guidebook to the Homeobox Genes. Oxford University Press; Oxford: 1994. [Google Scholar]
- 11.Kielkopf CL, Baird EE, Dervan PB, Rees DC. Nat Struct Biol. 1998;5:104–109. doi: 10.1038/nsb0298-104. [DOI] [PubMed] [Google Scholar]
- 12.(a) Kim Y, Geiger JH, Hahn S, Sigler PB. Nature. 1993;365:512–520. doi: 10.1038/365512a0. [DOI] [PubMed] [Google Scholar]; (b) Kim JL, Nikolov DB, Burley SK. Nature. 1993;365:520–527. doi: 10.1038/365520a0. [DOI] [PubMed] [Google Scholar]; (c) Kim JL, Burley SK. Nat Struct Biol. 1994;1:638–653. doi: 10.1038/nsb0994-638. [DOI] [PubMed] [Google Scholar]; (d) Juo ZS, Chiu TK, Leiberman PM, Baikalov I, Berk AJ, Dickerson RE. J Mol Biol. 1996;261:239–254. doi: 10.1006/jmbi.1996.0456. [DOI] [PubMed] [Google Scholar]
- 13.Davis NA, Majee SS, Kahn JD. J Mol Biol. 1999;291:249–265. doi: 10.1006/jmbi.1999.2947. [DOI] [PubMed] [Google Scholar]
- 14.(a) Janssen S, Durussel T, Laemmli UK. Mol Cell. 2000;6:999–1011. doi: 10.1016/s1097-2765(00)00099-x. [DOI] [PubMed] [Google Scholar]; (b) Janssen S, Cuvier O, Muller M, Laemmli UK. Mol Cell. 2000;6:1013–1024. doi: 10.1016/s1097-2765(00)00100-3. [DOI] [PubMed] [Google Scholar]
- 15.Direct observation of the cell uptake of these compounds is not possible using confocal microscopy without the conjugation of a fluorescent dye (see reference 16), so we must infer cell uptake indirectly for compounds 1–15 from the biological data. Experiments with polyamide-peptide-fluorophore conjugates to assess cell uptake are underway.
- 16.(a) Belistky JM, Leslie SJ, Arora PS, Beerman TA, Dervan PB. Bioorg Med Chem. 2002;10:3313–3318. doi: 10.1016/s0968-0896(02)00204-3. [DOI] [PubMed] [Google Scholar]; (b) Best TP, Edelson BS, Nickols NG, Dervan PB. Proc Natl Acad Sci USA. 2003;100:12063–12068. doi: 10.1073/pnas.2035074100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Edelson BS, Best TP, Olenyuk B, Nickols NG, Doss RM, Foister S, Heckel A, Dervan PB. Nucleic Acids Res. 2004;32:2802–2818. doi: 10.1093/nar/gkh609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minter AR, Brennan BB, Mapp AK. J Am Chem Soc. 2004;126:10504–10505. doi: 10.1021/ja0473889. [DOI] [PubMed] [Google Scholar]
- 18.Baird EE, Dervan PB. J Am Chem Soc. 1996;118:6141–6146. [Google Scholar]
- 19.Wellings DA, Atherton E. Methods Enzymol. 1997;289:44–67. doi: 10.1016/s0076-6879(97)89043-x. [DOI] [PubMed] [Google Scholar]
- 20.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 21.Cantor CR, Schimmel PR. Biophysical Chemistry of Macromolecules, Part 3: The Behavior of Biological Macromolecules. W. H. Freeman and Company; New York: 1980. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figures 1–7 including gel shifts for compounds 3–8 and 11–15, graphs showing the relationship between site spacing, binding affinity and the fraction of DNA shifted, and the full DNA sequences used for gel shifts. This material is available free of charge via the Internet at http://pubs.acs.org.