

Abstract
The hippocampus is injured in both hypoxia–ischemia (HI) and perinatal iron deficiency that are comorbidities in infants of diabetic mothers and intrauterine growth restricted infants. We hypothesized that preexisting perinatal iron deficiency predisposes the hippocampus to greater injury when exposed to a relatively mild HI injury. Iron-sufficient and iron-deficient rats (hematocrit 40% lower and brain iron concentration 55% lower) were subjected to unilateral HI injury of 15, 30, or 45 mins (n = 12 to 13/HI duration) on postnatal day 14. Sixteen metabolite concentrations were measured from an 11 μL volume on the ipsilateral (HI) and contralateral (control) hippocampi 1 week later using in vivo 1H NMR spectroscopy. The concentrations of creatine, glutamate, myo-inositol, and N-acetylaspartate were lower on the control side in the iron-deficient group (P < 0.02, each). Magnetic resonance imaging showed hippocampal injury in the majority of the iron-deficient rats (58% versus 11%, P < 0.0001) with worsening severity with increasing durations of HI (P = 0.0001). Glucose, glutamate, N-acetylaspartate, and taurine concentrations were decreased and glutamine, lactate and myo-inositol concentrations, and glutamate/glutamine ratio were increased on the HI side in the iron-deficient group (P < 0.01, each), mainly in the 30 and 45 mins HI subgroups (P < 0.02, each). These neurochemical changes likely reflect the histochemically detected neuronal injury and reactive astrocytosis in the iron-deficient group and suggest that perinatal iron deficiency predisposes the hippocampus to greater injury from exposure to a relatively mild HI insult.
Keywords: hippocampus, hypoxia, iron deficiency, ischemia, NMR spectroscopy, rat
Introduction
Perinatal hypoxic–ischemic (HI) injury and brain iron deficiency are common co-morbidities in infants of diabetic mothers and intrauterine growth restricted infants. In comparison to the general population, the incidence of perinatal HI injury is higher in these infants (Kattwinkel, 2006). Approximately 25% of both groups of infants are also at risk for brain iron deficiency with brain iron concentration being 30% to 40% lower in the most severe cases (Georgieff et al, 1995; Petry et al, 1992). Cognitive abnormalities, whose nature suggests the compromise of the developing hippocampus, are common in infants of diabetic mothers and intrauterine growth restricted infants (Ornoy et al, 1999; Siddappa et al, 2004; Silverman et al, 1998). In animal models, both perinatal iron deficiency and HI independently cause hippocampal injury (deUngria et al, 2000; Towfighi et al, 1997; Yager and Thornhill, 1997).
As iron and iron-containing enzymes are essential for normal neurodevelopment, perinatal iron deficiency may predispose the developing hippocampus to greater HI injury. In developing rats, perinatal iron deficiency adversely affects oxidative phosphorylation in the hippocampus, as indexed by decreased cytochrome c oxidase activity selectively in this brain region (deUngria et al, 2000). Furthermore, there is a greater loss of hippocampal cytochrome c oxidase activity 24 h after an acute HI injury in this model (Rao et al, 1999), suggesting poor recoverability of oxidative metabolism in perinatal iron deficiency. Our in vivo 1H NMR spectroscopy study has showed that in addition to altered energy metabolism, the concentrations of excitatory amino acids, glutamate, and aspartate are elevated in the iron-deficient hippocampus (Rao et al, 2003). The potential for excitotoxicity is enhanced under these conditions (Schmidt-Kastner and Freund, 1991; Vexler and Ferriero, 2001). Our previous study has showed that perinatally iron-deficient rats subjected to unilateral HI injury of two and one half hours on postnatal day (P) 7 sustain severe hippocampal injury (Rao et al, 1999).
Whether perinatal iron deficiency increases the vulnerability of the hippocampus to the more prevalent HI injury of mild to moderate severity is not known and is the focus of the present study. We hypothesized that the iron-deficient hippocampus would sustain greater severity of HI injury when compared with the iron-sufficient one. The objective of the study was to assess the effect of increasing durations of unilateral HI injury on the hippocampus in a developing rat model of perinatal iron deficiency using high-field in vivo 1H NMR spectroscopy and histochemistry. 1H NMR spectroscopy is a sensitive, noninvasive method that has been used to evaluate perinatal HI injury in humans and rats (Huppi and Lazeyras, 2001; Malisza et al, 1999). Changes in neurochemical concentrations, typically reported as ratio of each other, have been used to quantify the severity of HI injury (Huppi and Lazeyras, 2001; Robertson et al, 2001). With high magnetic field strengths (e.g. 9.4 T) absolute concentrations of multiple metabolites from a defined brain region, such as the developing hippocampus can be determined (Tkac et al, 2003). In the present study, we elucidated the specific neurochemical and histochemical alterations due to graded, relatively mild HI injury in the iron-deficient hippocampus.
Materials and methods
Animals
The Institutional Animal Care and Use Committee of University of Minnesota approved all experimental protocols (protocol number 0105A99961). Fifty-six iron-deficient (from 9 litters) and 59 iron-sufficient (from 8 litters) male and female Sprague–Dawley rat pups (Harlan Sprague Dawley, Indianapolis, IN, USA) were studied on P14. This postnatal age was chosen due to the neurodevelopmental similarities between the rat brain on P12−13 and the full-term human newborn infant (Romijn et al, 1991). Fetal and neonatal iron deficiency was induced by feeding the pregnant dam a low-iron diet (Formula TD 80396, Harlan-Teklad; Madison, WI, USA; elemental iron concentration: 3 to 6 mg/kg) from gestational day 2 until P7, followed by an iron-supplemented diet (Teklad 4% Mouse/Rat Diet 7001, Harlan-Teklad; elemental iron concentration: 198 mg/kg). This dietary manipulation induces brain iron deficiency of a severity similar to that showed in human infants with perinatal iron deficiency (Petry et al, 1992; Rao et al, 2003) with over 90% reduction in stainable iron in the hippocampus (deUngria et al, 2000). Dams of iron-sufficient pups received the iron-supplemented diet throughout the gestational and post-natal periods. Animals were allowed food and water ad libitum and were maintained on a 12-h day-and-night cycle. Litter size was limited to 8 by random culling soon after birth. Eight pups from one litter in each dietary group were used to measure the body and brain weights, hematocrit, and brain iron concentration. The remaining animals were subjected to unilateral HI injury.
Induction of HI Injury
The immature Levine model was used to induce unilateral HI injury (Vannucci and Vannucci, 2005). Briefly, under inhalation anesthesia (Isoflurane 3% for induction, 1.5% maintenance in 1:1 mixture of O2 and N2O), the right common carotid artery was isolated through a ventral cervical midline incision and was permanently ligated using 4.0 silk. One to two hours after the surgery, pairs of rat pups were exposed to 8% O2 (balance nitrogen) in an airtight glass jar that was partially submerged in a warm water bath (temperature inside the jar: 34±0.5°C). The injury is limited predominantly to the hemisphere ipsilateral to the ligated side (i.e., the side subjected to both ischemia and hypoxia) in this model (Vannucci and Vannucci, 2005). After a randomly assigned predesignated duration of exposure of 15, 30, or 45 mins, the hypoxic gas mixture was rapidly replaced with room air, the jar was opened and the animals were removed. They were reunited with their respective dams after complete recovery.
1H NMR Spectroscopy
1H NMR spectroscopy was performed 1 week post-HI injury (i.e., on P21) using a previously described protocol (Tkac et al, 2003). Both or at least one randomly selected pup in each pair that was subjected to HI injury simultaneously (see above) was studied by NMR spectroscopy. Briefly, under inhalation anesthesia (1% to 2% Isoflurane in an equal mixture of O2 and N2O), spontaneously breathing rat pups were placed inside a horizontal 9.4 T/31 cm magnet (Magnex Scientific; Abingdon, UK) equipped with an 11-cm gradient coil (300 mT/m, 500 μs) with strong custom-built shim coil (Magnex Scientific). The magnet was interfaced to a Varian INOVA console (Varian, Inc.; Palo Alto, CA, USA). A quadrature transmit/ receive surface RF coil with two geometrically decoupled single-turn coils with a 14 mm diameter was used (Tkac et al, 2003). All first- and second-order shims were automatically adjusted by FASTMAP (Gruetter, 1993). The water signal was efficiently suppressed using VAPOR water suppression technique (Tkac et al, 1999). Ultra-short echo-time STEAM (TE = 2 ms, TM = 20 ms, TR = 5 secs, NT = 160) combined with outer volume suppression (Tkac et al, 1999) was used to select volumes of interest (VOI) centered on the left (unligated side) and right (ligated side) hippocampus in each rat. The positions of VOI were based on multislice RARE images (echo train length = 8, TE = 60 ms, matrix = 256 × 256, slice thickness = 1 mm) and corresponded to coronal plates 50 to 54 of stereotaxic atlas of the developing rat brain (Sherwood and Timiras, 1970). The VOI was approximately 11 μl (2.5 × 1.5 × 3.0 mm). When there was evidence of tissue loss, the size of the VOI was appropriately adjusted to ensure that only the hippocampus was assessed. The order of NMR spectroscopy from the two sides was chosen at random. The study of a single animal did not exceed 60 mins.
Tissue Preparation
Immediately after 1H NMR spectroscopy, the brains were harvested and processed for histochemical analysis. Animals were deeply anesthetized with sodium pento-barbital (100 mg/kg, i.p.) before in situ transcardial perfusion with 0.9% saline, followed by 5% neutral buffered formaldehyde solution and 5% sucrose in phosphate-buffered saline (PBS), pH 7.4. Brains were removed and postfixed in the same fixative overnight at 4°C, followed by serial overnight passages in increasing sucrose concentrations (20%, 30%, and 40%) in PBS at 4°C for cryoprotection. They were then flash-frozen using dry ice and acetone and were embedded in a tissue-freezing medium (Triangle Biomedical Sciences, Durham, NC, USA) for sectioning. In animals used to assess brain iron concentration, a blood sample was obtained to determine the hematocrit before perfusion with saline and perfusion with formaldehyde was omitted.
Histochemistry
Serial 15 μm coronal frozen sections were obtained at the level of the posteriolateral thalamus using a cryostat (Model CM1900; Leica Instruments GmbH; Nussloch, Germany) at −20°C to −25°C. Sections were mounted on poly-L-lysine coated slides and were stored at −20°C until stained for Nissl substance, degraded myelin basic protein (MBP), and glial fibrillary acidic protein (GFAP).
Rabbit polyclonal antibodies to degraded MBP (Chemicon International; Temecula, CA, USA) and to GFAP (Novus-Biologicals; Littleton, CO, USA) were used to characterize abnormal-appearing oligodendrocyte processes and cell bodies in demyelinated areas (Matsuo et al, 1997) and for characterizing astrocyte morphology (Nedelcu et al, 1999), respectively, in the brain sections adjacent to those used for Nissl histochemistry. Sections were preincubated using avidin-biotin blocking solution (Vector Labs; Burlingame, CA, USA) and were incubated overnight at 4°C with the respective antibodies (dilution 1:1000). After incubation, the slides were rinsed several times with PBS and immunohistochemical staining was performed using the universal VECTASTAIN Elite ABC Kit (Vector Labs; Burlingame, CA, USA). The diaminobenzedine substrate kit (Vector Labs) was used to visualize the bound peroxidase. Sections were treated with nickel chloride solution to obtain a black/gray stain. Neurons in sections used for GFAP staining were counterstained with Gill's hematoxylin. Sections were then dehydrated by a series of ethanol rinses and were mounted using Vecta-Mount (Vector Labs) and coverslipped.
Analyses
Brain iron assay
Brain iron concentration was assayed by atomic absorption spectroscopy and was expressed as micrograms of elemental iron/g wet-tissue weight (Rao et al, 2003).
Magnetic resonance image analysis
Hippocampal injury in the hemisphere ipsilateral to the ligated side (i.e., the HI side) was evaluated in a masked manner and was graded as no discernible injury, injury limited to the hippocampus, or extensive injury involving the entire hippocampus and the adjacent tissues. The number of animals with these injuries in each of the three HI subgroups in the two dietary groups was determined and compared.
Quantification of metabolites
The LCModel (Provencher, 1993) was used to analyze in vivo 1H NMR spectra with the macromolecule signal included in the basis set (Tkac et al, 2003). Unsuppressed water signal was used as an internal reference for quantification. A brain water content of 83% was used in the calculation based on our previous study (Tkac et al, 2003). The following 18 metabolites were quantified from each spectrum: alanine, aspartate, creatine, γ-aminobutyric acid (GABA), glucose, glutamate, glutamine, glutathione, glycerophosphorylcholine (GPC), lactate, myo-inositol, N-acetylaspartate (NAA), N-acetylaspartylglutamate, phosphocreatine, phosphorylcholine (PCho), phosphorylethanolamine, scyllo-inositol, and taurine, and were represented as μmol/g tissue. The glutamate/glutamine and phosphocreatine/creatine ratios were also determined.
Histochemical analysis
The brain sections were visualized through a × 4 objective in a light microscope (Nikon Eclipse E600, Nikon Corporation; Shinagawa-ku; Tokyo, Japan). Digital microscopic images were collected at × 40 magnification using a camera (Nikon Digital Camera DXM1200, Nikon Corporation) and were projected onto the computer monitor using a software program (ACT-1, Nikon Corporation). Hippocampal subareas CA1, CA3ab, CA3c, and the dentate gyrus were identified using the nomenclature of Johansen (1993). Four to six brain sections from six to nine animals per HI subgroup in each dietary group that showed bilateral symmetry, tissue integrity, and uniform background staining were randomly selected for all histochemistry analyses. The presence of injury on the HI side was determined in a masked manner.
For assessing the severity of neuronal injury, the intensity of Nissl staining on the HI and control sides was determined using a computer-assisted program (ImageJ 1.34s; National Institutes of Health, Baltimore, MD, USA) (deUngria et al, 2000; Rao et al, 1999). On the image projected and frozen on the computer screen, a 20 pixel × 20-pixel grid was positioned randomly on five, bilaterally corresponding points within hippocampal subareas on the control and HI sides. As cortical injury is patchy in this model (Vannucci and Vannucci, 2005), staining intensity of 20 lightest points in the parietal cortex on the HI side was compared with that in 20 corresponding locations on the control side. The intensity of Nissl staining was measured in grayscale units (range: 1/white; 254/black), and mean values for each brain region on the HI and control sides were determined and compared.
Reactive astrocytosis in subarea CA1 was determined by counting all GFAP positive cells within a 290 μm × 235 μm grid placed on hippocampal CA1 subarea on [C0]identical positions on the HI and the control sides. Mean numbers of reactive astrocytes on the control and HI side were determined and compared.
Statistical Analysis
Group means of body and brain weights, hematocrit, and brain iron concentration in the two dietary groups were compared using unpaired, two-tailed t-tests. Mortality and presence of injury in the two groups were compared using Pearson χ2 test. The percent Nissl staining intensity losses in the brain regions and the number of GFAP positive astrocytes in the CA1 hippocampal subarea on the HI and control sides were compared using paired t-tests within each HI subgroup. The differences in the GFAP positive astrocytes between the two dietary groups were determined using Wilcoxon signed-rank test.
Neurochemical concentrations were analyzed as follows: first, to assess whether acute hypoxia alone had an effect on the unligated side, the concentrations of metabolites on the left side in the three HI subgroups were compared within each dietary group. Then the difference in the concentration of each neurochemical between the HI side and the control side was determined for each animal. Distributions of these differences were examined for approximation to normality using histograms and box plots. The effect of iron deficiency on the mean concentration difference of each neurochemical was determined using univariate ANOVAs with dietary group and HI duration as fixed factors. For neurochemicals demonstrating a main effect of diet, the effect at a specific HI duration was established by post hoc analysis using Bonferroni adjusted t-tests.
The effect of iron deficiency on the mean Nissl staining intensity loss in brain areas was determined using univariate ANOVA, and the effect at a specific HI duration was established by post hoc analysis using Bonferroni adjusted t-tests. A statistical software package (SPSS, Version 13.0, SPSS Inc., Chicago, IL, USA) was used for all analysis. Data are presented as mean±s.e.m. Significance was set at P < 0.05, except for the post hoc analysis, where P < 0.02 was considered significant.
Results
When compared with the iron-sufficient group, iron-deficient rats had lower mean body weight (32.2±0.8 versus 29.1±0.3 g), hematocrit (40.0±0.6 versus 24.2±0.7%), and brain iron concentration (8.23±0.40 versus 4.07±0.17 μg/g) (P < 0.01, each) on P14. The mean brain weight was similar (0.86± 0.00 versus 0.88±0.01 g) in the two groups.
There was no mortality during surgery in either group. Survival rates after HI injury were similar in the two groups (88% in the iron-deficient and 85% in the iron-sufficient). All deaths occurred during exposure to hypoxia and there was no delayed mortality in either group. In the iron-sufficient group, all deaths occurred in the 45 mins HI subgroup, whereas in the iron-deficient group, death occurred in the 30 mins, and 45 mins HI subgroups. Of the survivors, 90% in the iron-deficient and 82% in the iron-sufficient group were studied by 1H NMR spectroscopy.
The MRI showed either no discernable injury, injury limited to the hippocampus or extensive injury involving the hippocampus and the adjacent cerebral cortex on the HI side (Figure 1). The number of rats with these injuries in the two dietary groups is showed in Table 1. More iron-deficient animals had hippocampal injury on MRI than did iron-sufficient animals (58% versus 11%, P < 0.0001). Furthermore, there was dependency between duration of hypoxia and the extent of hippocampal injury in the iron-deficient group (P = 0.0001), but not in the iron-sufficient group (P = 0.08). Standardized residuals showed a duration-dependent effect of HI injury on the extent of hippocampal injury in the iron-deficient group, such that higher duration of HI was associated with more severe injury (Table 1).
Figure 1.
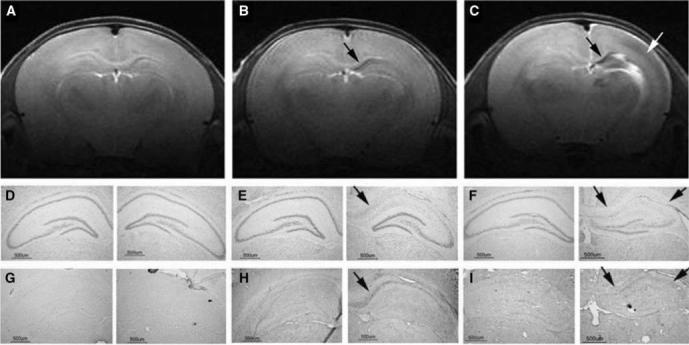
Post-hypoxic–ischemic (HI) hippocampal injury. Magnetic resonance imaging and brain sections from three iron-deficient rats obtained 1 week after a unilateral HI injury of 15, 30, or 45 mins on postnatal day 14 showed no hippocampal injury (A, D, and G), neuronal injury and demyelination limited to CA1 hippocampal subarea (arrows in B, E, and H) and injury involving the whole hippocampus (black arrows in C, F, and I) and the adjacent parietal cortex (white arrow in C) on the HI side, respectively. The distribution of rats with these injuries in the two dietary groups is given in Table 1. (15 μm coronal brain sections, Nissl histochemistry, and immunohistochemistry for degraded myelin basic protein on adjacent brain sections. See text for details of RARE image acquisition).
Table 1.
Post-HI hippocampal injury
| Dietary groups and HI subgroups |
Injury on MRI analysis |
Injury on histochemical analysis |
|||
|---|---|---|---|---|---|
| None | Localized to hippocampus | Hippocampus and adjacent cerebral cortex | Localized to hippocampus | Hippocampus and adjacent cerebral cortex | |
| Iron-sufficient (min) | |||||
| 15 | 12 | 0 | 0 | 0 | 0 |
| 30 | 11 | 0 | 1 | 0 | 1 |
| 45 | 9 | 3 | 0 | 0 | 2 |
| Iron-deficient (min)a,b,c | |||||
| 15 | 11 | 1 | 0 | 1 | 1 |
| 30 | 1 | 7 | 5 | 1 | 5 |
| 45 | 4 | 2 | 7 | 1 | 5 |
Values = number of rats with hippocampal injury on the HI hemisphere detected by MRI or histochemistry 1 week post-HI injury on postnatal day 14. For MRI analysis, N = 12 in each HI subgroup in the iron-sufficient group and N = 12, 13, and 13 in 15, 30, and 45 min HI subgroups, respectively, in the iron-deficient group. For histochemical analysis, N = 6 in each HI subgroup in the iron-sufficient group and N = 6, 9, and 8 in 15, 30, and 45 min HI subgroups, respectively, in the iron-deficient group. All analyses are by Pearson χ2.
Hippocampal injury significantly more common in the iron-deficient group on MRI (P < 0.0001) and histochemistry (P = 0.02) when compared with the iron-sufficient group.
Dependency between duration of hypoxia and the extent of hippocampal injury on MRI was present only in the iron-deficient group (P = 0.0001).
Injury in the parietal cortex significantly more common in the iron-deficient group (P < 0.0001).
The representative 1H NMR spectra from the control and the HI sides are shown in Figure 2. All metabolites except scyllo-inositol could be reliably quantified from each spectrum. Cramer Rao lower bounds, which are estimates of accuracy of the fitted concentrations, were < 25% for all metabolites, except alanine and N-acetylaspartylglutamate, and were ≤4% for creatine + phosphocreatine, myo-inositol, NAA, and taurine in each of the 148 measured spectra. This corresponded to an estimated error of calculated concentration of ≤0.3 μmol/g. Because of the close spectral similarity, reliable distinction between GPC and PCho was not possible. Therefore, their sum (GPC + PCho) was used in the analysis. Thus, the neurochemical profile consisted of 16 neurochemicals and two neurochemical ratios. Additional peaks, possibly representing mobile lipids were also present on the HI side in some animals.
Figure 2.
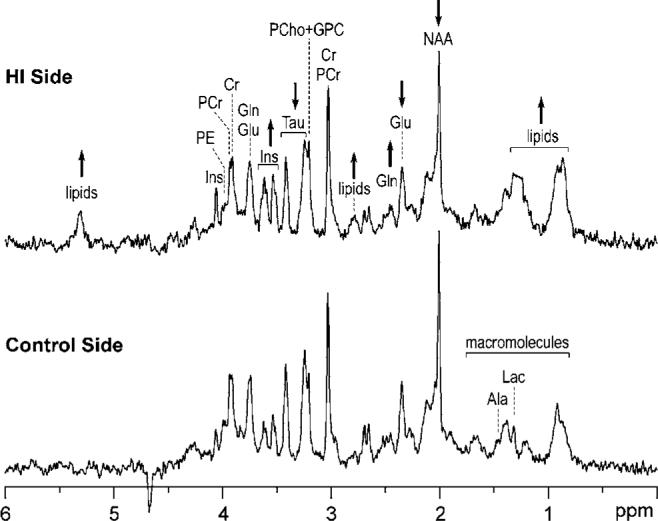
1H NMR spectra of the rat hippocampus post-HI injury. 1H NMR spectra from the hippocampi on the HI (top) and control (bottom) hemispheres from an iron-deficient rat were obtained one week after an acute HI injury of 30 mins on postnatal day 14 (whose MRI is shown in Figure 1b). Arrows indicate the direction of changes in certain neurochemicals on the HI side when compared with the control side. Additional peaks due to lipid signals are also present on the HI side. See text for NMR spectroscopy details. Abbreviations: Ala, alanine; Cr, creatine; Glu, glutamate; Gln, glutamine; GPC, glycerophosphorylcholine; Lac, lactate; Ins, myo-inositol; NAA, N-acetylaspartate; PCr, phosphocreatine; PCho, phosphorylcho-line; PE, phosphorylethanolamine; and Tau, taurine.
Iron deficiency had an effect on the hippocampal concentrations of creatine, glutamate, myo-inositol, and N-acetylaspartate on the control side (P < 0.02, each; Figure 3). Post hoc analysis showed that concentration of NAA in all three HI subgroups (P < 0.01, each), and that of myo-inositol in the 15 mins HI subgroup (P = 0.02) on the control side were lower in the iron-deficient group when compared with the iron-sufficient group. The duration of HI injury did not have an effect on any of the neurochemical concentrations and neurochemical ratios on the unligated side in either dietary group (Figure 3).
Figure 3.

Post-HI neurochemical profile of the rat hippocampus on the control side. Values = mean±s.e.m. neurochemical concentration or ratio on the unligated left side one week after a unilateral HI injury of 15, 30, or 45 mins on postnatal day 14 in the iron-sufficient (A) and the iron-deficient (B) groups, N as in Table 1. *Significant differences between the iron-sufficient and iron-deficient groups (P≤0.02, each; ANOVA). §Significant differences between the dietary groups in these HI subgroups (P < 0.02, each; Bonferroni-adjusted t tests). None of the neurochemicals or ratios differed among HI subgroups within each dietary group. Abbreviations: Ala, alanine; Asp, aspartate; Cr, creatine; GABA, γ-aminobutyric acid; Glc, glucose; Glu, glutamate; Gln, glutamine; GSH, glutathione; GPC, glycerophosphorylcholine; Lac, lactate; Ins, myo-inositol; NAA, N-acetylaspartate; NAAG, N-acetylaspartylglutamate; PCr, phosphocreatine; PCho, phosphorylcholine; PE, phosphorylethanolamine; and Tau, taurine.
There was a main effect of iron deficiency on the following neurochemicals on the HI side: glucose, glutamate, glutamine, lactate, myo-inositol, NAA, and taurine concentrations and glutamate/glutamine ratio (P < 0.01 each; Figure 4). The effect of iron deficiency on most of the neurochemicals was present in the 30 and 45 mins HI subgroups (P < 0.02, each). Iron deficiency did not have an effect on alanine, aspartate, creatine, GABA, glutathione, GPC + PCho, N-acetylaspartylglutamate, phosphocreatine, phosphorylethanolamine concentrations, and phosphocreatine/creatine ratio.
Figure 4.
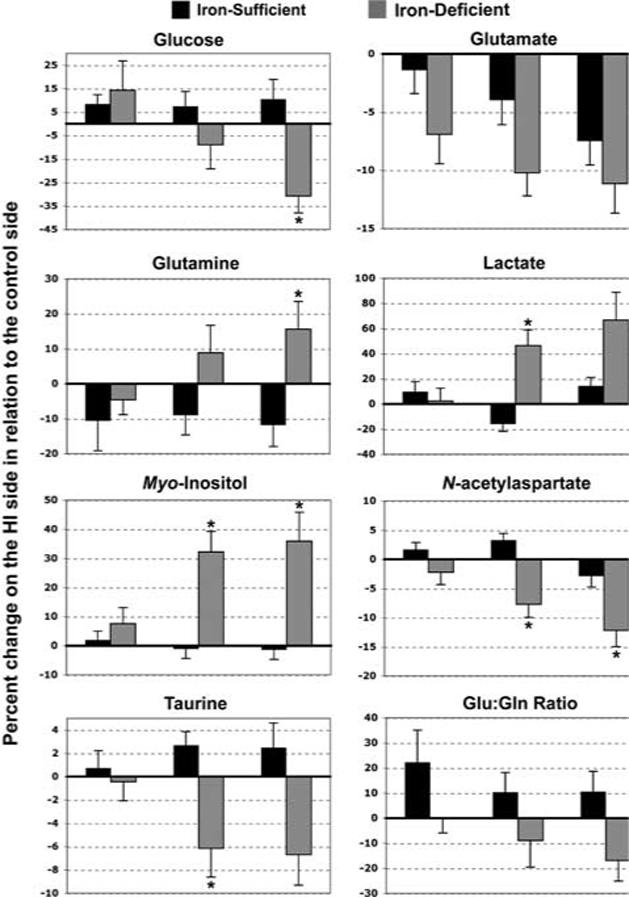
Post HI neurochemical changes in the rat hippocampus on the HI side. Values = mean±s.e.m. changes in neurochemical concentrations and glutamate/glutamine ratio on the HI side as a percentage of those on the control side 1 week after a unilateral HI injury of 15, 30, or 45 mins on postnatal day 14, N as in Table 1. There was a main effect of iron deficiency for each neurochemical and glutamate/gluta-mine ratio (P < 0.01, each; ANOVA). *Significant differences between iron-sufficient and iron-deficient groups in these HI subgroups (P < 0.02, each; Bonferroni-adjusted Student's t-tests). Abbreviation: Glu:Gln ratio, glutamate/glutamine ratio.
Histochemical changes in the hippocampus and adjacent parietal cortex mirrored the anatomic MRI changes (Figure 1). More rats in the iron-deficient group had neuronal injury (53% versus 13%, P = 0.02; Table 1) and myelin breakdown in the hippocampus than those in the iron-sufficient group. The loss of Nissl staining intensity varied among the dietary groups as well as among the hippocampal subareas (P < 0.01, each; Table 2). Post hoc analysis showed that Nissl staining intensity was significantly decreased in subarea CA1 on the HI side in the 30 mins HI subgroup in the iron-deficient group (P < 0.01; Table 2). The intensity of Nissl staining was not affected in other hippocampal subareas in the iron-deficient group and in any hippocampal subarea in the iron-sufficient group.
Table 2.
Regional loss of Nissl staining intensity post HI injury
| Dietary groups and HI subgroups |
Hippocampal subareaa |
||||
|---|---|---|---|---|---|
| CA1 | CA3ab | CA3c | Dentate gyrus | Parietal cortex | |
| Iron-sufficient | |||||
| 15 min (n = 6) | 3.3 ± 4.2 | 0.0 ± 2.3 | 0.0 ± 2.3 | 0.3 ± 1.1 | 0.9 ± 0.2 |
| 30 min (n = 6) | 0.6 ± 6.2 | 0.0 ± 2.6 | 4.8 ± 5.4 | 0.0 ± 4.3 | 0.6 ± 0.2 |
| 45 min (n = 6) | 6.8 ± 7.1 | 0.0 ± 2.1 | 0.1 ± 3.0 | 1.0 ± 1.9 | 0.4 ± 0.3 |
| Iron-deficientb | |||||
| 15 min (n = 6) | 3.7 ± 4.9 | 0.0 ± 1.2 | 0.0 ± 3.3 | 7.8 ± 4.6 | 0.2 ± 0.1 |
| 30 min (n = 9) | 18.8 ± 3.0c | 6.8 ± 6.9 | 2.5 ± 3.7 | 2.6 ± 2.3 | 0.3 ± 0.2 |
| 45 min (n = 8) | 17.6 ± 6.1 | 2.1 ± 3.9 | 0.0 ± 2.9 | 1.8 ± 2.3 | 2.8 ± 1.7 |
Values = mean ± s.e.m. % loss of Nissl staining intensity on the HI hemisphere when compared with the corresponding brain region on the control hemisphere.
Nomenclature based on Johansen (1993).
Main effect of iron-deficiency on Nissl staining intensity loss was seen for subarea CA1 (P = 0.03, univariate ANOVA).
Significantly different between iron-sufficient and iron-deficient groups (P < 0.01, Bonferroni-adjusted t-test).
There were more reactive astrocytes in the CA1 subarea on the HI side in each of the three HI subgroups only in the iron-deficient group (P < 0.01, each; Figure 5). As a whole, iron-deficient group had more reactive astrocytes on the HI side than the iron-sufficient group (P < 0.001; Figure 5). In some iron-deficient animals with extensive hippocampal injury on the HI side, neuronal injury, and reactive astrocytosis were present also in the CA1 hippo-campal subarea on the control side (Figure 5c).
Figure 5.
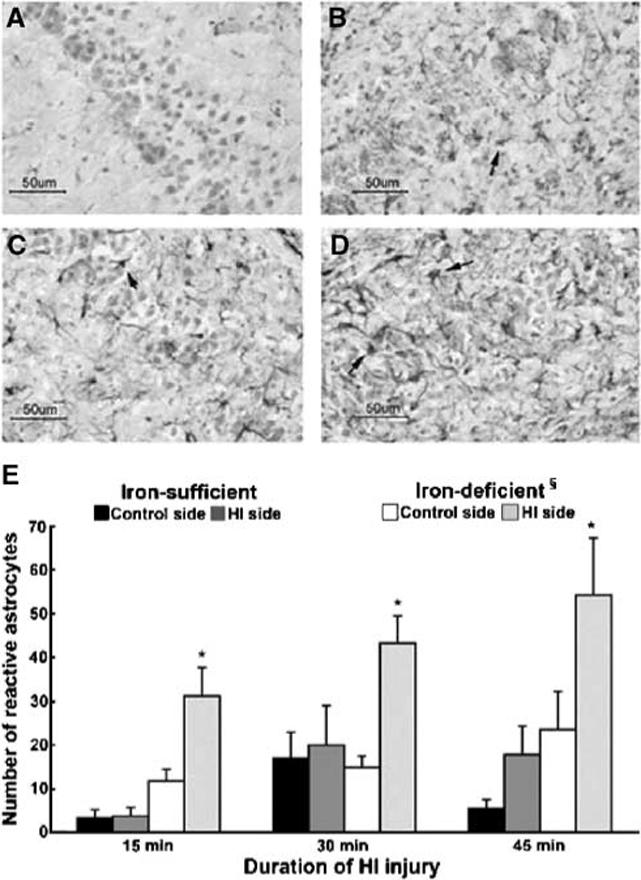
Post-HI reactive astrocytosis in the hippocampal CA1 subarea. Brain sections on the control side (A and C) and HI side (B and D) are from the same iron-deficient rats shown in Figure 1, and were obtained 1 week after an acute HI injury of 30 mins (A and B) or 45 mins (C and D). Arrows point to reactive astrocytes amidst degenerating neurons in CA1 subarea of the hippocampus on the HI side (B and D) and extending to the control side in the animal with more extensive injury (C). (E) The number of reactive astrocytes on the control and HI sides in the two dietary groups. §Significant difference between the iron-sufficient and iron-deficient groups (P < 0.001; Wilcoxon Signed-Rank test). *Significant differences between the control and HI sides in the iron-deficient group (P < 0.01 each; paired t-tests). (A–D: 15 μm coronal brain sections, GFAP immunohistochemistry for astrocytes and Gill's hematoxylin histochemistry for neurons. Values in (E) = mean±s.e.m. GFAP positive cells within a 290 μm × 235 μm grid placed on identical positions on CA1 hippocampal subarea bilaterally, N as in Table 2).
Injury in the parietal cortex adjacent to the hippocampus also was more common in the iron-deficient group (58% versus 18%, P < 0.01; Table 1). However, unlike the well-demarcated hippocampal injury, cortical injury was patchy and was restricted to the middle cortical layers. Neither iron-deficiency nor duration of HI injury had an effect on Nissl staining intensity loss in the parietal cortex (Table 2).
Discussion
The findings of the study suggest that the vulnerability of the developing hippocampus to a relatively mild HI injury is increased when there is coexisting perinatal brain iron deficiency. Magnetic resonance imaging and histochemistry showed hippocampal injury in all animals that sustained brain injury in both dietary groups. In animals with more extensive injury involving the cerebral cortex, hippocampal injury was invariably present. 1H NMR spectroscopy reliably quantified the severity of hippocampal injury. Multiple neurochemicals were altered on the HI side in the iron-deficient group. Specific neurochemicals were affected and others, such as creatine, were spared. Moreover, neuro-chemical changes were not uniform; while the concentrations of some were increased, those of others were decreased. These findings suggest that neurochemical alterations likely represent changes in specific cellular and metabolic processes in the iron-deficient hippocampus subjected to acute HI injury.
There was loss of NAA on the HI side in the 30 and 45 mins HI subgroups in the iron-deficient group. In adult gerbils subjected to global ischemia, decreased NAA determined by in vivo 1H NMR spectroscopy correlates with neuronal loss in the CA1 subarea of the hippocampus (Sager et al, 2001). In the present study also, histochemistry showed decreased Nissl staining intensity in the CA1 subarea, suggesting that decreased NAA likely represents neuroaxonal injury in the iron-deficient hippocampus. However, unlike the adult brain, NAA is also present in immature oligodendrocytes in the developing brain (Bhakoo and Pearce, 2000; Urenjak et al, 1992). Iron, as well as energy demand of oligodendrocytes in the developing brain is high (Connor and Menzies, 1996), and these cells are highly vulnerable to HI injury (Skoff et al, 2001). Therefore, decreased NAA in the present study may represent tissue injury involving both neurons and oligodendrocytes (Huppi and Lazeyras, 2001).
Neuronal injury was accompanied by demyelination and reactive astrocytosis in CA1 subarea of the hippocampus in the iron-deficient group. The corresponding increases in myo-inositol and gluta-mine concentrations are the likely in vivo markers of these processes. In human infants and animal models, elevated myo-inositol is considered a marker of disrupted osmotic regulation and/or reactive astrocytosis in the injured brain (Bitsch et al, 1999; Robertson et al, 2001; Schuhmann et al, 2003). Glial fibrillary acidic protein expression and inositol phosphate accumulation increase concomitantly after an ischemic brain injury in rats and is maximum 7 days later (Fortuna et al, 1997).
There was demyelination at the site of injury in the iron-deficient group. In adult animals, demyeli-nation commences within 1 to 3 days after focal cerebral ischemia (Irving et al, 2001). Failure of Na +-K + ATPase in the axons, oligodendrocyte injury, and wallerian degeneration secondary to gray matter injury are postulated to be responsible for this demyelination (Schabitz et al, 2000). There is limited information on post-HI demyelination in the developing hippocampus. In rats, myelinated fibers in the hippocampus are first seen between P15 and P17 and progressively increase until P25, when the adult pattern is achieved (Meier et al, 2004; Savaskan et al, 1999). As acute HI in our study predated the onset of hippocampal myelination, either an ongoing injury, such as the secondary energy failure that occurs 24 to 48 h later (Vannucci and Vannucci, 2005), or wallerian degeneration of the nascent myelin due to hippocampal neuronal injury may be responsible for the myelin breakdown observed in the iron-deficient group. Altered myelin production and maintenance associated with iron deficiency (Beard et al, 2003) also may have been responsible for the enhanced demyelination. Whether perinatal HI injury results in demyelination in the human hippocampus, where myelinated fibers are first seen at 39 weeks but peak myelination occurs after birth (Arnold and Trojanowski, 1996), is not known.
Lactate levels were elevated on the HI side in the iron-deficient group. During and immediately after the HI injury, lactate levels are elevated in both cerebral hemispheres, probably due to anaerobic glycolysis secondary to decreased oxygen delivery (Malisza et al, 1999; Payen et al, 1996). In the present study, lactate levels continued to be elevated one week post-HI injury, when perfusion and oxygenation presumably have been restored at the site of injury (Phillis and O'Regan, 2003). Such persistent elevation in lactate is postulated to be due to retention in damaged cells, as well as due to metabolic activity in phagocytic cells at the site of injury (Malisza et al, 1999; Schuhmann et al, 2003). In the present study, increased lactate levels were associated with decreased glucose concentrations, potentially suggesting persistent alterations in substrate utilization in the iron-deficient hippocampus subjected to HI injury.
Glutamate concentrations were decreased and glutamine concentrations were increased in the iron-deficient group, likely reflecting the tissue composition (loss of glutamate-containing neurons and infiltration of glutamine-containing astrocytes) at the site of HI injury. However, they may also represent a disrupted glutamine–glutamate equilibrium between neurons and glia (Wallin et al, 2000). Typically, glutamate released into the extracellular space is taken up by nearby astrocytes, converted to glutamine through the action of the enzyme gluta-mine synthase and trafficked back to the neurons for reconversion to glutamate, thereby completing the so-called glutamate–glutamine cycle (Daikhin and Yudkoff, 2000; Magistretti et al 1999). The decreased glutamate and increased glutamine concentrations in the present study implies that after acute HI injury, the neuronal–glial glutamate–glutamine equilibrium may be shifted in favor of increased glutamine synthesis in the iron-deficient hippocampus (Aas et al, 1993; Dao et al, 1991; Wallin et al, 2000; Phillis and O'Regan, 2003). Such metabolic shift has been showed in in vivo rat models of glutamate receptor overstimulation (Tkac et al, 2001).
In contrast to the findings in our previous study that assessed the effects of iron deficiency without superimposed hypoxia or HI injury on the hippo-campus (Rao et al, 2003), the concentrations of creatine, glutamate, myo-inositol, and N-acetylaspartate were lower in the hippocampus on the unligated side in the iron-deficient group. Similar neurochemical changes have been showed in the hippocampus exposed to chronic hypoxia that was also iron-deficient (Raman et al, 2005). Taken together, these neurochemical changes suggest that hypoxia without ischemia may alter neuronal and glial integrities and energy metabolism in an iron-deficient hippocampus. A threshold effect may be present since a duration-dependent effect of hypoxia on the neurochemicals was not present. Additional studies are necessary to determine the effect of acute hypoxia on the iron-deficient hippocampus.
We used a well-established model of unilateral HI injury (Vannucci and Vannucci, 2005) in our study. The survival rate and the pattern of injury were similar to those reported at this postnatal age (Towfighi et al, 1997; Yager and Thornhill, 1997). Injury was not selective to the hippocampus even though this structure was the focus of the present study. Both MRI and histochemistry showed injury in the adjacent parietal cortex in animals with extensive hippocampal injury in both dietary groups. This is consistent with the reported distribution of injury in this model at this postnatal age (Towfighi et al, 1997; Yager and Thornhill, 1997). We did not evaluate the neurochemical alterations in the cortex or in any other brain regions, and the histochemical assessment beyond the hippocampus was limited to the adjacent parietal cortex. The lack of evidence for injury by densitometric analysis in this area may be due to the limitation of this method to accurately quantify the cortical injury secondary to its patchy nature (Nelson and Silverstein, 1994). Additional studies are necessary to establish the neurochemical and histologic changes in the cerebral cortex and other brain regions due to HI injury in perinatal iron deficiency.
In addition to brain iron deficiency, anemia that is inevitable in our dietary model may have played a significant role in brain injury. In human infants, severe iron deficiency anemia has been associated with hemiplegia, presumably due to cerebral hypoxia, altered viscosity, and thrombosis (Yager and Hartfield, 2002). As we did not measure cerebral blood flow changes and blood viscosity in our study, it is not possible to differentiate the relative roles of brain iron deficiency and anemia for our results. Future studies controlling for anemia are necessary to establish the role of brain iron deficiency in acute HI injury. Similarly, the brain iron deficiency in our model was slightly more severe than that showed in infants of diabetic mothers and intrauterine growth restricted infants with fetal and neonatal iron deficiency (Petry et al, 1992; Georgieff et al, 1995). Moreover, these infants typically do not have anemia. Therefore, the results of the study are more applicable to perinatal iron deficiency with anemia secondary to maternal dietary iron deficiency that is commonly seen in disadvantaged populations (Rao and Georgieff, 2001). To our knowledge, the brain iron concentration of the offspring of iron-deficient mothers has not been assessed. Therefore, caution is necessary while extrapolating the study results to human clinical conditions.
Nevertheless, the study shows the importance of optimal iron homeostasis during brain development. Through its role in the production of free radicals, iron is thought to play a significant role in perinatal HI injury (Palmer et al, 1999; Vexler and Ferriero, 2001). High iron content, combined with immature antioxidant mechanisms, is postulated to predis-pose the developing brain to HI injury (Vexler and Ferriero, 2001). In neonatal rats subjected to acute HI injury, iron is histochemically detectable within 2−4 h in brain regions that subsequently undergo injury (Palmer et al, 1999). Administration of the iron chelator desferoxamine before or soon after HI injury reduces injury in neonatal rats (Palmer et al, 1994). Conversely, iron deficiency also appears to increase the risk of HI injury in the developing brain (Yager and Hartfield, 2002).
In conclusion, high field 1H NMR spectroscopy allowed us to determine the dose–response effect of graded durations of mild HI injury in the iron-deficient hippocampus. The ability to detect and quantify the neurochemical changes from specific brain regions attests to the sensitivity and usefulness of this method for studies of this nature. Because it is noninvasive, the method can be extended to human infants to detect and monitor the progression of perinatal HI injury. Increased vulnerability of the hippocampus and other brain regions to mild HI injury may be responsible for the long-term cognitive deficits in human infants with perinatal iron deficiency.
Acknowledgements
The assistance of Jeff Long, PhD, with statistical analysis, Jane Wobken with histochemical studies and Ann Fandrey with manuscript preparation are gratefully acknowledged.
This paper was presented in part at the annual meetings of Pediatric Academic Societies: (1) Rao R, Tkac I, Townsend E, Gruetter R, Nelson C, Georgieff M (2002) Evaluation of acute and long-term effects of graded hypoxia ischemia on hippocampal meta-bolites in iron deficient vs. iron sufficient developing rats by magnetic resonance spectroscopy at 9.4 T. Pediatr Res 51:442A and (2) Ennis K, Townsend E, Wobken J, Georgieff M, Rao R (2004) Perinatal iron deficiency increases the vulnerability of the developing hippocampus to mild hypoxic–ischemic injury. Pediatr Res 55:179A.
This work was supported in part by Grants HD29421 and HD33692 from the National Institutes of Health. Dr Rao was a scholar of the Child Health Research Center and University Pediatrics Foundation at University of Minnesota during this research. The Center for Magnetic Resonance Research is in part supported by a center grant from the National Center for Regional Resources (RR08079) and the MIND Institute. The 9.4 Tesla magnet is funded in part by a gift from the WM Keck Foundation.
References
- Aas JE, Berg-Johnsen J, Hegstad E, Laake JH, Langmoen IA, Ottersen OP. Redistribution of glutamate and glutamine in slices of human neocortex exposed to combined hypoxia and glucose deprivation in vitro. J Cereb Blood Flow Metab. 1993;13:503–15. doi: 10.1038/jcbfm.1993.65. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Trojanowski JQ. Human fetal hippocampal development: I. Cytoarchitecture, myeloarchitecture, and neuronal morphologic features. J Comp Neurol. 1996;367:274–92. doi: 10.1002/(SICI)1096-9861(19960401)367:2<274::AID-CNE9>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Beard JL, Wiesinger JA, Connor JR. Pre- and postweaning iron deficiency alters myelination in Sprague–Dawley rats. Dev Neurosci. 2003;25:308–15. doi: 10.1159/000073507. [DOI] [PubMed] [Google Scholar]
- Bhakoo KK, Pearce D. In vitro expression of N-acetyl aspartate by oligodendrocytes: implications for proton magnetic resonance spectroscopy signal in vivo. J Neurochem. 2000;74:254–62. doi: 10.1046/j.1471-4159.2000.0740254.x. [DOI] [PubMed] [Google Scholar]
- Bitsch A, Bruhn H, Vougioukas V, Stringaris A, Lassmann H, Frahm J, et al. Inflammatory CNS demyelination: histopathologic correlation with in vivo quantitative proton MR spectroscopy. Am J Neuroradiol. 1999;20:1619–27. [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. 1996;17:83–93. doi: 10.1002/(SICI)1098-1136(199606)17:2<83::AID-GLIA1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Daikhin Y, Yudkoff M. Compartmentation of brain glutamate metabolism in neurons and glia. J Nutr. 2000;130:1026S–31S. doi: 10.1093/jn/130.4.1026S. [DOI] [PubMed] [Google Scholar]
- Dao DN, Ahdab-Barmada M, Schor NF. Cerebellar glutamine synthetase in children after hypoxia or ischemia. Stroke. 1991;22:1312–6. doi: 10.1161/01.str.22.10.1312. [DOI] [PubMed] [Google Scholar]
- deUngria M, Rao R, Wobken JD, Luciana M, Nelson CA, Georgieff MK. Perinatal iron deficiency decreases cytochrome c oxidase (CytOx) activity in selected regions of neonatal rat brain. Pediatr Res. 2000;48:169–76. doi: 10.1203/00006450-200008000-00009. [DOI] [PubMed] [Google Scholar]
- Fortuna S, Pestalozza S, Lorenzini P, Bisso GM, Morelli L, Michalak H. Transient global brain hypoxia–ischemia in adult rats: neuronal damage, glial proliferation, and alterations in inositol phospholipid hydrolysis. Neurochem Int. 1997;31:563–9. doi: 10.1016/s0197-0186(97)00005-3. [DOI] [PubMed] [Google Scholar]
- Georgieff MK, Mills MM, Gordon K, Wobken JD. Reduced neonatal liver iron concentrations after uteroplacental insufficiency. J Pediatr. 1995;127:308–14. doi: 10.1016/s0022-3476(95)70317-9. [DOI] [PubMed] [Google Scholar]
- Gruetter R. Automatic, localized in vivo adjustment of all first- and second-order shim coils. Magn Reson Med. 1993;29:804–11. doi: 10.1002/mrm.1910290613. [DOI] [PubMed] [Google Scholar]
- Huppi PS, Lazeyras F. Proton magnetic resonance spectroscopy ((1)H-MRS) in neonatal brain injury. Pediatr Res. 2001;49:317–20. doi: 10.1203/00006450-200103000-00003. [DOI] [PubMed] [Google Scholar]
- Irving EA, Bentley DL, Parsons AA. Assessment of white matter injury following prolonged focal cerebral ischaemia in the rat. Acta Neuropathol (Berl) 2001;102:627–35. doi: 10.1007/s004010100416. [DOI] [PubMed] [Google Scholar]
- Johansen FF. Interneurones in rat hippocampus after cerebral ischemia: morphometric, functional, and therapeutic investigations. Acta Neurol Scand. 1993;150:1–32. [PubMed] [Google Scholar]
- Kattwinkel J. Overview and principles of resuscitation. In: Kattwinkel J, editor. Textbook of neonatal resuscitation. 5th ed. American Academy of Pediatrics and American Heart Association; Chapel Hill, NC: 2006. pp. 1–28. [Google Scholar]
- Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy on demand. Science. 1999;283:496–7. doi: 10.1126/science.283.5401.496. [DOI] [PubMed] [Google Scholar]
- Malisza KL, Kozlowski P, Ning G, Bascaramurty S, Tuor UI. Metabolite changes in neonatal rat brain during and after cerebral hypoxia–ischemia: a magnetic resonance spectroscopic imaging study. NMR Biomed. 1999;12:31–8. doi: 10.1002/(sici)1099-1492(199902)12:1<31::aid-nbm544>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Matsuo A, Lee GC, Terai K, Takami K, Hickey WF, McGeer EG, et al. Unmasking of an unusual myelin basic protein epitope during the process of myelin degeneration in humans: a potential mechanism for the generation of autoantigens. Am J Pathol. 1997;150:1253–66. [PMC free article] [PubMed] [Google Scholar]
- Meier S, Brauer AU, Heimrich B, Nitsch R, Savaskan NE. Myelination in the hippocampus during development and following lesion. Cell Mol Life Sci. 2004;61:1082–94. doi: 10.1007/s00018-004-3469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelcu J, Klein MA, Aguzzi A, Boesiger P, Martin E. Biphasic edema after hypoxic–ischemic brain injury in neonatal rats reflects early neuronal and late glial damage. Pediatr Res. 1999;46:297–304. doi: 10.1203/00006450-199909000-00008. [DOI] [PubMed] [Google Scholar]
- Nelson C, Silverstein FS. Acute disruption of cytochrome oxidase activity in brain in a perinatal rat stroke model. Pediatr Res. 1994;36:12–9. doi: 10.1203/00006450-199407001-00003. [DOI] [PubMed] [Google Scholar]
- Ornoy A, Wolf A, Ratzon N, Greenbaum C, Dulitzky M. Neurodevelopmental outcome at early school age of children born to mothers with gestational diabetes. Arch Dis Child. 1999;81:F10–4. doi: 10.1136/fn.81.1.f10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C, Roberts RL, Bero C. Deferoxamine posttreatment reduces ischemic brain injury in neonatal rats. Stroke. 1994;25:1039–45. doi: 10.1161/01.str.25.5.1039. [DOI] [PubMed] [Google Scholar]
- Palmer C, Menzies SL, Roberts RL, Pavlick G, Connor JR. Changes in iron histochemistry after hypoxic–ischemic brain injury in the neonatal rat. J Neurosci Res. 1999;56:60–71. doi: 10.1002/(SICI)1097-4547(19990401)56:1<60::AID-JNR8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Payen JF, LeBars E, Wuyam B, Tropini B, Pepin JL, Levy P, et al. Lactate accumulation during moderate hypoxic hypoxia in neocortical rat brain. J Cereb Blood Flow Metab. 1996;16:1345–52. doi: 10.1097/00004647-199611000-00032. [DOI] [PubMed] [Google Scholar]
- Petry CD, Eaton MA, Wobken JD, Mills MM, Johnson DE, Georgieff MK. Iron deficiency of liver, heart, and brain in newborn infants of diabetic mothers. J Pediatr. 1992;121:109–14. doi: 10.1016/s0022-3476(05)82554-5. [DOI] [PubMed] [Google Scholar]
- Phillis JW, O'Regan MH. Characterization of modes of release of amino acids in the ischemic/reperfused rat cerebral cortex. Neurochem Int. 2003;43:461–7. doi: 10.1016/s0197-0186(03)00035-4. [DOI] [PubMed] [Google Scholar]
- Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med. 1993;30:672–9. doi: 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- Raman L, Tkac I, Ennis K, Georgieff MK, Gruetter R, Rao R. In vivo effect of chronichypoxia on the neurochemical profile of the developing rat hippocampus. Brain Res Dev Brain Res. 2005;156:202–9. doi: 10.1016/j.devbrainres.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Rao R, de Ungria M, Sullivan D, Wu P, Wobken JD, Nelson CA, et al. Perinatal brain iron deficiency increases the vulnerability of rat hippocampus to hypoxic ischemic insult. J Nutr. 1999;129:199–206. doi: 10.1093/jn/129.1.199. [DOI] [PubMed] [Google Scholar]
- Rao R, Georgieff MK. Neonatal iron nutrition. Semin Neonatol. 2001;6:425–35. doi: 10.1053/siny.2001.0063. [DOI] [PubMed] [Google Scholar]
- Rao R, Tkac I, Townsend EL, Gruetter R, Georgieff MK. Perinatal iron deficiency alters the neuro-chemical profile of the developing rat hippocampus. J Nutr. 2003;133:3215–21. doi: 10.1093/jn/133.10.3215. [DOI] [PubMed] [Google Scholar]
- Robertson NJ, Lewis RH, Cowan FM, Allsop JM, Counsell SJ, Edwards AD, et al. Early increases in brain myo-inositol measured by proton magnetic resonance spectroscopy in term infants with neonatal encephalopathy. Pediatr Res. 2001;50:692–700. doi: 10.1203/00006450-200112000-00011. [DOI] [PubMed] [Google Scholar]
- Romijn HJ, Hofman MA, Gramsbergen A. At what age is the developing cerebral cortex of the rat comparable to that of the full-term newborn human baby? Early Hum Dev. 1991;26:61–7. doi: 10.1016/0378-3782(91)90044-4. [DOI] [PubMed] [Google Scholar]
- Sager TN, Topp S, Torup L, Hanson LG, Egestad B, Moller A. Evaluation of CA1 damage using single-voxel 1H-MRS and un-biased stereology: can non-invasive measures of N-acetyl-asparate following global ischemia be used as a reliable measure of neuronal damage? Brain Res. 2001;892:166–75. doi: 10.1016/s0006-8993(00)03274-1. [DOI] [PubMed] [Google Scholar]
- Savaskan NE, Plaschke M, Ninnemann O, Spillman AA, Schwab ME, Nitsch R, et al. Myelin does not influence the choice behaviour of entorhinal axons but strongly inhibits their outgrowth length in vitro. Eur J Neurosci. 1999;11:316–26. doi: 10.1046/j.1460-9568.1999.00430.x. [DOI] [PubMed] [Google Scholar]
- Schabitz WR, Li F, Fisher M. The N-methyl-D-aspartate antagonist CNS 1102 protects cerebral gray and white matter from ischemic injury following temporary focal ischemia in rats. Stroke. 2000;31:1709–14. doi: 10.1161/01.str.31.7.1709. [DOI] [PubMed] [Google Scholar]
- Schuhmann MU, Stiller D, Skardelly M, Bernarding J, Klinge PM, Samii A, et al. Metabolic changes in the vicinity of brain contusions: a proton magnetic resonance spectroscopy and histology study. J Neurotrauma. 2003;20:725–43. doi: 10.1089/089771503767869962. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Freund TF. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience. 1991;40:599–636. doi: 10.1016/0306-4522(91)90001-5. [DOI] [PubMed] [Google Scholar]
- Sherwood NM, Timiras PS. A stereotaxic atlas of the developing rat brain. University of California Press; Berkeley: 1970. pp. 14–203. [Google Scholar]
- Siddappa AM, Georgieff MK, Wewerka S, Worwa C, Nelson CA, DeRegnier RA. Iron deficiency alters auditory recognition memory in newborn infants of diabetic mothers. Pediatr Res. 2004;55:1034–41. doi: 10.1203/01.pdr.0000127021.38207.62. [DOI] [PubMed] [Google Scholar]
- Silverman BL, Rizzo TA, Cho NH, Metzger BE. effects of the intrauterine environment. The Northwestern University Diabetes in Pregnancy Center. Diabetes Care. 1998;21(Suppl 2):B142–9. [PubMed] [Google Scholar]
- Skoff RP, Bessert DA, Barks JD, Song D, Cerghet M, Silverstein FS. Hypoxic–ischemic injury results in acute disruption of myelin gene expression and death of oligodendroglial precursors in neonatal mice. Int J Dev Neurosci. 2001;19:197–208. doi: 10.1016/s0736-5748(00)00075-7. [DOI] [PubMed] [Google Scholar]
- Tkac I, Keene CD, Pfeuffer J, Low WC, Gruetter R. Metabolic changes in quinolinic acid-lesioned rat striatum detected non-invasively by in vivo (1)H NMR spectroscopy. J Neurosci Res. 2001;66:891–8. doi: 10.1002/jnr.10112. [DOI] [PubMed] [Google Scholar]
- Tkac I, Rao R, Georgieff MK, Gruetter R. Developmental and regional changes in the neurochemical profile of the rat brain determined by in vivo 1H NMR spectroscopy. Magn Reson Med. 2003;50:24–32. doi: 10.1002/mrm.10497. [DOI] [PubMed] [Google Scholar]
- Tkac I, Starcuk Z, Choi IY, Gruetter R. In vivo 1H NMR spectroscopy of rat brain at 1 ms echo time. Magn Reson Med. 1999;41:649–56. doi: 10.1002/(sici)1522-2594(199904)41:4<649::aid-mrm2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Towfighi J, Mauger D, Vannucci RC, Vannucci SJ. Influence of age on the cerebral lesions in an immature rat model of cerebral hypoxia–ischemia: a light microscopic study. Brain Res Dev Brain Res. 1997;100:149–60. doi: 10.1016/s0165-3806(97)00036-9. [DOI] [PubMed] [Google Scholar]
- Urenjak J, Williams SR, Gadian DG, Noble M. Specific expression of N-acetylaspartate in neurons, oligodendrocyte-type-2 astrocyte progenitors, and immature oligodendrocytes in vitro. J Neurochem. 1992;59:55–61. doi: 10.1111/j.1471-4159.1992.tb08875.x. [DOI] [PubMed] [Google Scholar]
- Vannucci RC, Vannucci SJ. Perinatal hypoxic–ischemic brain damage: evolution of an animal model. Dev Neurosci. 2005;27:81–6. doi: 10.1159/000085978. [DOI] [PubMed] [Google Scholar]
- Vexler ZS, Ferriero DM. Molecular and biochemical mechanisms of perinatal brain injury. Semin Neonatol. 2001;6:99–108. doi: 10.1053/siny.2001.0041. [DOI] [PubMed] [Google Scholar]
- Wallin C, Puka-Sundvall M, Hagberg H, Weber SG, Sandberg M. Alterations in glutathione and amino acid concentrations after hypoxia–ischemia in the immature rat brain. Brain Res Dev Brain Res. 2000;125:51–60. doi: 10.1016/s0165-3806(00)00112-7. [DOI] [PubMed] [Google Scholar]
- Yager JY, Hartfield DS. Neurologic manifestations of iron deficiency in childhood. Pediatr Neurol. 2002;27:85–92. doi: 10.1016/s0887-8994(02)00417-4. [DOI] [PubMed] [Google Scholar]
- Yager JY, Thornhill JA. The effect of age on susceptibility to hypoxic-ischemic brain damage. Neurosci Biobehav Rev. 1997;21:167–74. doi: 10.1016/s0149-7634(96)00006-1. [DOI] [PubMed] [Google Scholar]