

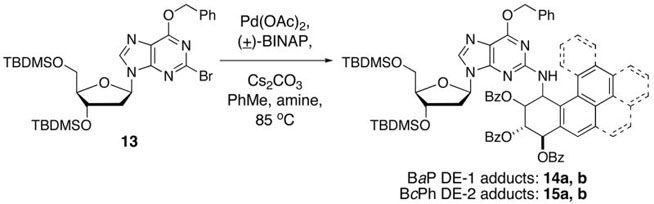
Abstract

Palladium–catalyzed C–N bond formation has been utilized to synthesize covalent 2′–deoxyadenosine (dA) and 2′–deoxyguanosine (dG) adducts of benzo[a]pyrene (BaP) series 1 (syn) and benzo[c]phenanthrene (BcPh) series 2 (anti) diol epoxides. For this (±)-10α-amino-7β,8α,9β-trisbenzoyloxy-7,8,9,10-tetrahydro BaP and (±)-1β-amino-2α,3α,4β-trisbenzoyloxy-1,2,3,4-tetrahydro BcPh were coupled with 6-halo-9-[3,5-bis-O-(tert-butyldimethylsilyl)-β-D-erythro-pentofuranosyl]purine and O6-benzyl-3′,5′-bis-O-(tert-butyldimethylsilyl)-2-bromo-2′-deoxyinosine, using a (±)–BINAP–Pd complex and Cs2CO3. For the synthesis of the dA adducts, both the 6-chloro as well as the 6-bromo purine nucleoside derivatives were analyzed for the C–N coupling reaction with the hydrocarbon amino tribenzoates. With the BaP amino tribenzoate, the 6-chloro nucleoside provided satisfactory results, whereas the 6-bromo analogue proved to be superior with the BcPh amino tribenzoate. Overall lower yields of the dA adducts were obtained with the more hindered fjord region BcPh amino tribenzoate as compared to the bay region BaP amino tribenzoate. In contrast to reactions leading to the dA adducts, C–N reactions of both BaP and BcPh amino tribenzoates with the 2-bromo-2′-deoxyinosine derivative proceeded in comparable yields. This seems to indicate that such Pd-catalyzed adduct forming reactions at the C–6 position may be influenced by steric constraints of the amine component, whereas those at the C–2 are less sensitive. Diastereomeric adduct pairs were separated and characterized by spectral methods and by comparisons to adducts produced by direct displacement reactions as well as those formed from DNA alkylation by diol epoxides.
INTRODUCTION
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous environmental pollutants that result from activities of a modern society. Among the multiple metabolic pathways,1–4 many PAHs that contain a bay or a fjord region undergo metabolic activation to 4 diol epoxides4 that exert their carcinogenic activity by alkylation of cellular DNA.5–7 This reaction with DNA proceeds by C–O bond scission of the oxirane, followed by cis and trans attack by the exocyclic amino groups of the purine bases (Scheme 1).6,7
SCHEME 1.

Cis/trans ring opening of the four isomeric diol epoxides
DNA alkylation upon metabolism of any PAH results in the formation of 16 purine nucleoside adducts.6,7 In order to probe the structural and biological properties of individual diol epoxide–DNA lesions, access to site-specifically modified DNA containing stereochemically defined diol epoxide adducts is critical. Thus, substantial effort has been directed to the development of methods leading to diol epoxide adducted DNA.8,9 Reactions of diol epoxides with nucleosides or nucleotides are not productive avenues for the synthesis of these adducts.8a Therefore, the total synthesis approach requires reversing the nucleophile/eectrophile roles of the nucleoside and the PAH. Reactions of electrophilic purine nucleosides with amino PAH derivatives leads to the covalent diol epoxide–nucleoside adducts that can, after appropriate functionalization, be incorporated into DNA oligomers by solid-phase synthesis methods.9 Typically, only fluorinated purine nucleosides possess sufficient reactivity to undergo SNAr displacement reactions with the relatively unreactive amino derivatives of polycyclic aromatic hydrocarbons.9b,10 However, syntheses of 6-fluoropurine 2′-deoxyriboside11 and 2-fluoro-2′-deoxyinosine12 are non-trivial, multi-step procedures.
Pd catalyzed C–N bond formation is growing in significance for the modification of nucleosides13 and several biologically important nucleoside adducts have been synthesized via its application.14 Our interest in Pd catalyzed synthesis of diol epoxide–nucleoside adducts stemmed from several important considerations, such as cumbersome synthesis of reactive fluorinated nucleosides usually required for displacement reactions with the axially constrained and hindered PAH amines and the fact that some displacement reactions leading to the diol epoxide nucleoside adducts are not particularly efficient. Access to chloro and bromo nucleosides is easier,11,14a,15,16 and Pd catalyzed C–N bond formation presents a potentially facile approach to this important class of biologically relevant, modified nucleosides.
We17 and others18 have independently reported Pd–catalyzed synthesis of nucleoside adducts from the carcinogenic (±)–BaP DE–2. Therefore, the current investigation focused on synthesis of adducts from the stereoisomeric (±)–BaP DE–1 (a bay region derivative) and from (±)–BcPh DE–2 (a fjord region compound, Figure 1), representing two structurally different paradigms. This also provided an opportunity to understand on a comparative basis the efficiency of catalysis reactions on chloro and bromo nucleosides. Pd catalyzed amination of a trissilyl protected 6-chloro purine nucleoside with aliphatic amines was reported to proceed in good yield but reactions with the less nucleophilic aryl amines were low-yielding.19 Prior work has shown direct displacement reactions of hindered PAH amines with 6-chloro purine nucleoside to be inefficient.9b,10 On the other hand, two PAH amines underwent efficient Pd–catalyzed coupling with 6-chloro-9-[3,5-bis-O-(tert-butyldimethylsilyl)-β-D-erythro-pentofuranosyl]purine,17 and both chloro and bromo nucleosides were effective coupling partners in C–N bond formation with azole nitrogens.20 Based on these results we decided to assess the effectiveness of 6-bromo and 6-chloro-9-[3,5-bis-O-(tert-butyldimethylsilyl)-β-D-erythro-pentofuranosyl]purine for Pd catalyzed C–N bond forming reactions with bay and fjord region amino PAH derivatives. In this report we discuss: (a) the efficiencies of 2′-deoxyadenosine adduct syntheses by C–N bond formation with the two C–6 halopurine 2′-deoxyribonucleosides, (b) synthesis of 2′-deoxyguanosine adducts of the 2 PAH diol epoxides, (c) a comparison of the various reactions, and (d) an assessment of the stereochemical integrity in the Pd–catalyzed reactions.
FIGURE 1.

The bay– and fjord–region diol epoxides of BaP and BcPh
RESULTS AND DISCUSSION
Synthesis of BaP amino tribenzoate
Many PAH amino triols and amino triacyl derivatives that are suitable precursors to the nucleoside–diol epoxide adducts can be stereoselectively synthesized by ring opening reactions of the diol epoxides themselves.8,9b,10,21 In the present case, known bromohydrin (±)–1 was synthesized via reaction of BaP dihydrodibenzoate23 with NBS/NaOAc in ~30% H2O–THF,23 and cyclized to the epoxide (±)–223 with NaH in dry THF (Cs2CO3 in THF can be used but the ensuing reactions are not very clean). Ring-opening of 2 to the azidotriol dibenzoate was performed with LiN3 in DMF at room temperature and the resulting crude material was directly converted to the azido tribenzoate (±)–3.21b Finally, reduction of the azide moiety with H2 and Lindlar catalyst yielded the amino tribenzoate (±)–4 (use of excess catalyst was required for efficient conversion, Pd–C could also be used but was less efficient and PtO2 was ineffective).
Synthesis of BcPh amino tribenzoate
Synthesis of the BcPh amino tribenzoate (±)–8 commenced from BcPh DE–2 [(±)–(5)].24 Due to its low solvolytic reactivity, as we had reported earlier,25 ring opening of (±)–5 was conducted in THF–H2O. These conditions eliminate non-benzylic ring opening by the azide, a feature that was observed when DMF was used as solvent.25 Conversion of the azidotriol (±)–6 to the triester (±)–7 was accomplished with PhCOCN/Et3N in DMF.21b Again, catalytic reduction with H2 and excess Lindlar catalyst yielded the amino tribenzoate (±)–8.
Coupling of the BaP and BcPh amino tribenzoates with C–6 halo purine 2′-deoxyribosides
Once the amino tribenzoates (±)–4 and (±)–8 were synthesized, the next step was the analysis of their coupling with each of the halo nucleosides 6-chloro and 6-bromo-9-[3,5-bis-O-(tert-butyldimethylsilyl)-β-D-erythro-pentofuranosyl]purine (9 and 10). For this we selected conditions that we have successfully used previously for the synthesis of BaP DE–2 adducts with 2′-deoxyadenosine.17 The C–N bond formation was conducted with 1.1 molar equiv of amine with the combination of 10 mol% Pd(OAc)2/30 mol% (±)–BINAP/1.4 molar equiv Cs2CO3 in toluene as solvent. The results of these reactions are compiled in Table 1.
TABLE 1.
Coupling of amino tribenzoates (±)–4 and (±)–8 with the two C–6 halo nucleosides 9 and 10a
 | ||||||
|---|---|---|---|---|---|---|
| entry | amine | Mmol | nucleoside | molar [nucleoside] | time (h) | adducts, yieldb |
| 1 |

|
0.032 | 9 | 0.028 | 22 | 11a, b: 32% |
| 2 | 0.044 | 9 | 0.04 | 8 | 11a, b: 43% | |
| 3 | 0.028 | 9 | 0.1 | 4 | 11a, b: 59% | |
| 4 | 0.024 | 10 | 0.1 | 4 | 11a, b: 57% | |
| 5 | 0.222 | 9 | 0.1 | 5 | 11a, b: 73% | |
|
| ||||||
| 6 |

|
0.028 | 9 | 0.025 | 6 | 12a, b: 37% |
| 7 | 0.029 | 9 | 0.1 | 4 | 12a, b: 36% | |
| 8 | 0.026 | 10 | 0.1 | 4 | 12a, b: 50% | |
| 9 | 0.265 | 10 | 0.1 | 4 | 12a, b: 45% | |
Reactions were conducted in PhMe at 85 °C.
Yield is of isolated, purified products and represents the combined yield of diastereomeric adduct pairs.
The results in Table 1 show some interesting features and also provide meaningful comparisons between the reactions of the two amines. In the case of the BaP amino tribenzoate (±)–4 the reactions appeared to be dependent upon the concentration of the nucleoside (limiting reagent) and coupling yield increased with concentration (entries 1–3). When comparing yields of 11a, b from the reactions of the two halo nucleosides, as we had demonstrated in an earlier communication,18 chloro nucleoside 9 underwent C–N bond formation in comparable yield to the bromo analogue 10 (entry 3 versus 4). In contrast to these results, reactions with BcPh amino tribenzoate (±)–8 appeared to be different. Nucleoside concentration had a smaller influence on product yield and more notably, reactions with bromo nucleoside 10 provided superior results. Yields of adducts 12a, b from the more hindered fjord region BcPh amino tribenzoate were lower than those from the bay region BaP derivative.
Coupling of the BaP and BcPh amino tribenzoates with O6-benzyl-2-bromo-2′-deoxyinosine
Since we have faced difficulties in the preparation of 3′,5′-bis-O-(tert-butyldimethylsilyl)-2-chloro-2′-deoxyinosine,26 the readily available 2-bromo analogue 13 was used to synthesize the 2′-deoxyguanosine adducts.14a Using the same catalyst composition described above for the synthesis of the 2′-deoxyadenosine adducts, C–N bond formation between the PAH amino tribenzoates (±)–4, (±)–8 and 13 was conducted at a nucleoside concentration of 0.1 M. The results from these experiments are shown in Table 2.
TABLE 2.
Coupling of amino tribenzoates (±)–4 and (±)–8 with 3′,5′-bis-O-(tert-butyldimethylsilyl)-2-bromo-2′-deoxyinosine 13a
 | ||||
|---|---|---|---|---|
| Entry | Amine | mmol | time (h) | adducts, yieldb |
| 1 |

|
0.023 | 18 | 14a, b: 68% |
| 2 | 0.222 | 16 | 14a, b: 73% | |
| 3 |

|
0.029 | 18 | 15a, b: 68% |
| 4 | 0.265 | 16 | 15a, b: 70% | |
Reactions were conducted in PhMe at 85 °C at a 0.1 M concentration of the nucleoside.
Yield is of isolated, purified products and represents the combined yield of diastereomeric adduct pairs.
The most noteworthy feature in Table 2 is the observation that comparably good product yields were obtained from the two amino benzoates. This contrasts with the results in Table 1 where the yield of the 2′-deoxyadenosine adducts from the more hindered BcPh amino tribenzoate was lower than that from the BaP amino tribenzoate. This result seems to imply that some subtle influences, possibly steric features associated with the PAH amines, come into play in C–N bond formation at the C-6 position of purine nucleosides but such effects do not seem to influence C–N bond formation at the C-2 position. Most impressively, good yields of both the BaP and BcPh adducts are realized via this method. Although these compounds can be directly used for DNA assembly by methods we have previously described,8d for reasons indicated later unequivocal structure confirmation was undertaken.
Structure confirmation of the synthetic PAH diol epoxide–nucleoside adducts
In products 11a, b and 12a, b the benzoates in each pair of diastereomeric adducts were cleaved with NH3/MeOH and the resulting trihydroxy compounds were acetylated (Scheme 4). In the case of 14a, b and 15a, b the previously described acyl group interchange was followed by removal of the O6 benzyl group by hydrogenolysis (Scheme 4). Small amounts of the diastereomeric adducts were separated by preparative TLC after these manipulations, and as described in the next section the disilyl triacetates 16a–19b were analyzed in detail.
SCHEME 4.

Preparation of the adduct triacetates for characterization
Evaluation of stereochemical integrity in the adduct-forming reactions
The presence of a hydrogen atom α to the amino group raises the possibility of loss of stereochemical integrity, and loss of chirality in Pd–mediated C–N bond formation via β–hydride migration has been documented in the literature.28 The use of a bis phosphine ligand provides amelioration of this problem, possibly by closing open coordination sites at the metal center.28 In the present cases stereochemical integrity is of utmost importance since loss of chirality at the amine bearing carbon would alter the structure and conformation of the biologically important adducts that are to be used for structure–biological studies.
In our preliminary communication where we had initially disclosed studies on Pd–mediated C–N bond formation as a route to carcinogen–nucleoside conjugates,17 we had addressed this potential problem by comparison of the products to those that were synthesized by displacement of fluoride from a fluoro nucleoside. In the present cases, chirality attrition would result in a cis relative stereochemical arrangement of the nucleoside and the adjacent acetoxyl group (Figure 2). Chiroptical methods alone would not be adequate to distinguish between the trans and cis products due to close similarities in their CD spectra.28
FIGURE 2.
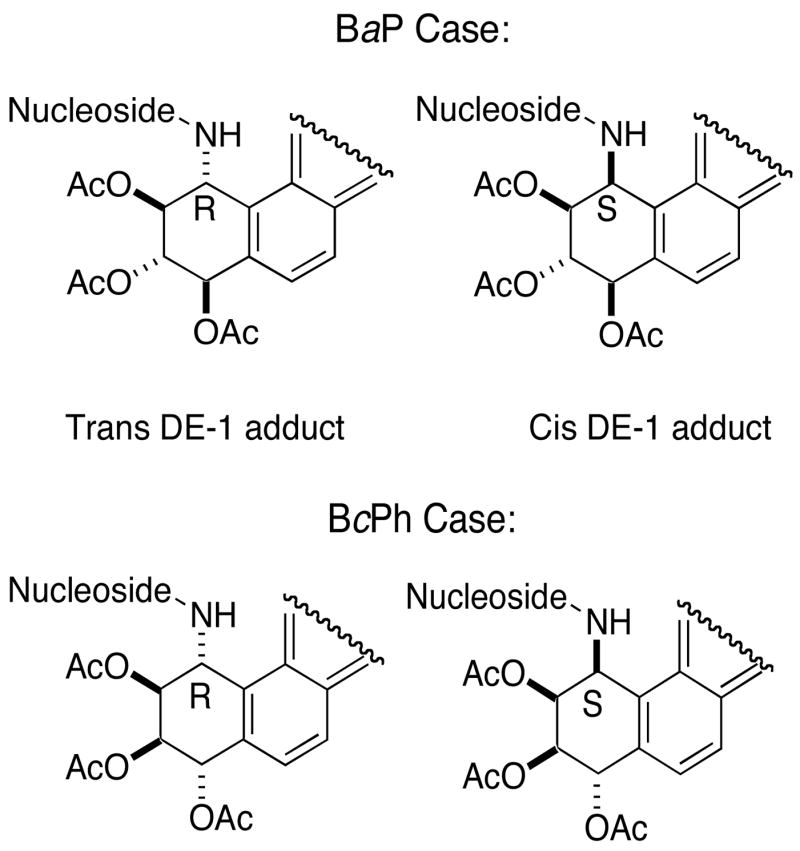
Loss of stereochemical integrity would result in the cis rather than the trans adducts.
Therefore, the coupling constants of the tetrahydro ring protons in the adducts obtained by the Pd-catalyzed method (Table 3) were compared to cis and trans adducts that are known in the literature and/or have been unequivocally synthesized (see the Supporting Information for this data). In the BaP case, the trans ring-opened 2′-deoxyadenosine adducts (16a, b) synthesized in this study showed J7,8 = 5.8–6.0, J8,9 = 6.6–6.9 and J9,10 = 3.9–4.2 Hz and these are consistent with those for adducts derived from 6-fluoro-9-[3,5-bis-O-(tert-butyldimethylsilyl)-β-D-erythro-pentofuranosyl]purine (J7,8 = 6.2, J8,9 = 7.0 and J9,10 = 4.0 Hz). The latter products are derived via a SNAr mechanism where chirality attrition is not a question. Further, the J values of the trans adducts are quite different from those of the isomeric cis compounds where they are J7,8 = 7.9–8.0, J8,9 = 11.5 and J9,10 = 4.5 Hz.9e The coupling constants for the trans ring-opened 2′-deoxyguanosine adducts (18a, b) obtained in the present study were J7,8 = 6.5–6.9, J8,9 = 6.5–7.0 and J9,10 = 4.0–4.4 Hz. Not only are these values very similar to those reported for the adducts obtained from 3′,5′-bis-O-(tert-butyldimethylsilyl)-2-fluoro-2′-deoxyinosine by fluoride displacement (J7,8 = 6.1–6.9, J8,9 = 6.6–6.8 and J9,10 = 3.9–4.4 Hz),29 but again these are also markedly different from those of the isomeric cis ring-opened adducts (J7,8 = 8.2, J8,9 = 11.8 and J9,10 = 2.2 Hz).30
TABLE 3.
Coupling constants for the tetrahydro ring protons in the adductsa
| benzo[a]pyrene series | ||
|---|---|---|
| 2′-deoxyadenosine adducts | 2′-deoxyguanosine adductsd | |
|
|
||

|
16a (10S) | 18a (10S) |
| J9,10 = 4.2b (2.9)c | J9,10 = 4.4 | |
| J8,9 = 6.9b (3.3)c | J8,9 = 7.0 | |
| J7,8 = 6.0b (3.1)c | J7,8 = 6.9 | |

|
16b (10R) | 18b (10R) |
| J9,10 = 3.9b (2.9)c | J9,10 = 4.0 | |
| J8,9 = 6.6b (3.6)c | J8,9 = 6.5 | |
| J7,8 = 5.8b (3.1)c | J7,8 = 6.5 | |
|
| ||
| benzo[c]phenanthrene series | ||
|
| ||
| 2′-deoxyadenosine adducts | 2′-deoxyguanosine adductsd | |
|
|
||

|
17a (1S) | 19a (1S) |
| J1,2 = 4.5b (3.3)c | J1,2 = 4.4 | |
| J2,3 = 2.9b (2.9)c | J2,3 = 2.7 | |
| J3,4 = 8.3b (7.3)c | J3,4 = 8.5 | |

|
17b (1R) | 19b (1R) |
| J1,2 = 4.1b (3.7)c | J1,2 = 4.3 | |
| J2,3 = 2.4b (2.9)c | J2,3 = 2.6 | |
| J3,4 = 8.3b (7.4)c | J3,4 = 8.9 | |
Data obtained at 500 MHz under ambient conditions.
Obtained in acetone–d6
Obtained in deacidified CDCl3.
Obtained in DMSO–d6.
In the BcPh case, the differences in the J values for the isomeric cis and trans ring opened adducts are smaller. Thus, data for the 2′-deoxyadenosine adducts (17a, b) were compared to those reported for the products arising from fluoride displacement chemistry25 as well as adduct peracetates obtained from alkylation of DNA by (±)–BcPh DE–2,31 and a very close match was observed (see the Supporting Information for a comprehensive tabulation). Best comparisons for the 2′-deoxyguanosine adducts (19a, b) were data for the peracetates of adducts derived from DNA alkylation by (±)–BcPh DE–2.31 The observed coupling constants for 19a, b are J3,4 = 8.5–8.9, J2,3 = 2.6–2.7 and J1,2 = 4.3–4.4 Hz and these match well with J3,4 = 8.4–8.5, J2,3 = 1.6–2.6 and J1,2 = 4.1–4.5 Hz reported for the corresponding adduct peracetates (see the Supporting Information).
These comparisons led us to the conclusion that chirality scrambling was not a problem within the detection limits of the analyses used. An interesting observation that stemmed from these studies is that the BaP adducts 16a, b showed a solvent-dependent conformational change that is reflected in the coupling constants of the tetrahydro ring protons. The J7,8 = 5.8–6.0, J8,9 = 6.6–6.9 and J9,10 = 3.9–4.2 Hz observed in acetone-d6 dramatically changed to J7,8 = 3.1, J8,9 = 3.3–3.6 and J9,10 = 2.9 Hz in CDCl3. Since the coupling constants provide an understanding of the orientation of the substituents in the tetrahydrobenzo ring, a decrease in the values is an indication of substantially more axially disposed substituents in CDCl3 as compared to acetone–d6.
In evaluating the conformational equilibria of the BaP DE–1 adducts 16a, 16b the NMR coupling constants were compared to those of products obtained by a trans ring opening of BaP DE–1 with various nucleophiles.23 There are 4 principal conformers that can be considered for 16a, 16b, and those for the 10S diastereomer (16a) are shown in Figure 3. Among these, conformer I was generated by imposing large dihedral angles between vicinal protons and the structure was optimized. For conformers II–IV possible dihedral angle constraints were imposed based upon the observed J values and the structures were optimized followed by further minimization using semi-empirical AM1. Conformer I that has all equatorial substituents can be readily eliminated since this should display large coupling constants between all the axial protons and would require a quasi-equatorial orientation of the bay-region purine substituent as well (for this reason no further AM1 minimization was considered). This analysis is comparable to that reported for the ring opening products of BaP DE–1.23 Conformer II appears to be preferred in CDCl3, a solvent of lower polarity, and dihedral angles (absolute values) after AM1 minimization are H7–C–C–H8 = 55.3, H8–C–C–H9 = 48.4 and H9–C–C–H10 = 54.6. A possible stabilizing feature in such a conformer could be an intramolecular hydrogen bond between the NH proton and the axial acyl group on the same face. In analogy to the conformation of 16a, a C-10 anilino adduct arising by a trans ring opening of BaP DE–1 has also been proposed to exist in a conformation with all axial substituents in CDCl3, possibly stabilized by an intramolecular hydrogen bond.23 In the more polar acetone– d6 there are two possible conformers that could account for the observed J values. These are III, where the absolute values of the dihedral angles in the minimized structure are H7–C–C–H8 = 147.4, H8–C–C–H9 = 145.6 and H9–C–C–H10 = 48.4, and IV with dihedral angles H7–C–C–H8 = 35.4, H8–C–C–H9 = 144.7 and H9–C–C–H10 = 136.7. Both III and IV adopt a boat or a flattened half chair conformation, and this analysis is again comparable to that reported for the trans ring opening products of BaP DE–1.23 In conformer III the C-7, C-8 substituents are diequatorially disposed whereas in conformer IV these are diaxial. Between III and IV, the latter also has a quasi-equatorial purine substituent in the sterically congested bay region (quasi-axial in III). These reasons render III more preferable to IV. In the case of the BcPh adducts 17a, b although small differences in the coupling constants are observed in CDCl3 and acetone–d6, these are not as dramatic. Such an analysis was not conducted with the 2′-deoxyguanosine adducts as good line shape for these is typically only observed in DMSO– d6 and not in CDCl3.
Figure 3.

Possible conformations of the 10S BaP DE–1 deoxyadenosine adduct 16a (generated via MacSpartan Pro V 1.0.3). For simplicity the saccharide of the nucleoside has been replaced with a hydrogen atom.
CONCLUSIONS
In summary, our studies show that Pd–mediated amination chemistry is an effective method leading to nucleoside adducts of PAH diol epoxides. This method appears to be broadly applicable to the synthesis of bay- and fjord-region diol epoxide–nucleoside adducts, and in the present case has led to the synthesis of nucleoside adducts from BaP DE–1 and BcPh DE–2. There are some interesting differences in these reactions. The adduct forming reactions of the less hindered amino tribenzoate derived from BaP DE–1 provide good yields of the N6-2′-deoxyadenosine adducts, whereas corresponding reactions with the more hindered amino tribenzoate derived from BcPh DE–2 are lower yielding. This suggests that subtle factors, possibly steric congestion, associated with the PAH amino tribenzoates are at play in these reactions. Also, reactions with the BaP amino tribenzoate proceed well with the C–6 chloro nucleoside, whereas those with the BcPh amino tribenzoate proceed better with the C–6 bromo nucleoside. In each case, the pure adduct diastereomers were separated after replacement of the PAH benzoyl groups with acetyl esters. Quite in contrast to reactions at the C–6 position, C–N bond–forming reactions of both BaP and BcPh amino tribenzoates at the C–2 position of silyl protected O6-benzyl-2-bromo-2′-deoxyinosine proceed in comparably good yields. This seems to suggest that structural factors associated with the PAH component are not quite as critical in reactions at the C–2. Here again, the benzoyl groups on the PAH were replaced with acetyls and facile debenzylation delivered the PAH diol epoxide 2′-deoxyguanosine adducts quite efficiently. The adduct diastereomers were readily separated after the O6-deprotection. The pure adduct diastereomers from each reaction were compared with known compounds. On the basis of these comparisons it was rationalized that loss of chirality at the amine-bearing carbon does not compete with product formation. This factor is important from the standpoint that these compounds are to be used for structural and biochemical experimentation after incorporation into DNA oligomers. Chemistry leading to site-specific DNA modification by these diol epoxide–nucleoside adducts is underway and results are forthcoming.
EXPERIMENTAL
(±)-7β,8α-Bisbenzoyloxy-9α-bromo-10β-hydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene (1).23
To a solution of (±)-7β,8α-bisbenzoyloxy-7,8-dihydrobenzo[a]pyrene22 (1.4 g, 2.8 mmol) in THF–H2O (350 mL:140 mL) were added freshly recrystallized N-bromosuccinimide (654 mg, 3.6 mmol) and NaOAc (695 mg, 8.4 mmol). The mixture was stirred under N2 gas at room temperature under subdued light. Tlc showed the reaction to be complete after 18 h and the solvents were evaporated. The residue was dissolved in ethyl acetate and washed twice with water. The organic layer was separated, dried over Na2SO4 and evaporated. The resulting product was chromatographed on silica gel using CH2Cl2 and product obtained was then washed twice with a 1:1 ether–hexane to afford 1 g (60%) of (±)–1 as a white powder.
(±)-7β,8α-Bisbenzoyloxy-9β,10β-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene (2).23
A stirring suspension of NaH (24 mg, 1 mmol) in THF (5 mL) was flushed with N2 gas and cooled in an ice bath. Compound 1 (300 mg, 0.507 mmol) was added and the mixture turned brownish. After stirring at 0 °C for 1 h, the mixture was allowed to warm to room temperature over 30 minutes. Tlc indicated the reaction to be complete. The mixture was diluted with 1:1 Et2O–EtOAc and washed twice with water. The organic layer was dried over Na2SO4 and evaporated to leave a solid. This solid was washed twice with 1:1 Et2O–hexane with sonication to yield 217 mg (84%) of (±)–2 as a pinkish powder.
(±)-10α-Azido-7β,8α,9β-trisbenzoyloxy-7,8,9,10-tetrahydrobenzo[a]pyrene (3).21b
The diol epoxide dibenzoate (±)–2 (657 mg, 1.28 mmol) and LiN3 (629.6 mg, 12.86 mmol) were allowed to stir in DMF (15 mL) at room temperature overnight. The mixture was diluted with EtOAc and washed with water. The organic layer was dried over Na2SO4 and evaporated. This crude material was directly benzoylated by addition of dry DMF (30 mL), dry Et3N (1.5 mL, 10.7 mmol) followed by benzoyl cyanide (0.839 g, 6.4 mmol). The mixture was allowed to stir at room temperature for 30 minutes, quenched with methanol and diluted with EtOAc. The mixture was washed twice with brine, the organic layer was dried over Na2SO4 and evaporated. The resulting yellow oil was chromatographed on a silica gel column using CH2Cl2. The product was washed twice with 1:1 Et2O–hexane to yield 513 mg (61%) of (±)–3 as a white powder.
(±)-10α-Amino-7β,8α,9β-trisbenzoyloxy-7,8,9,10-tetrahydrobenzo[a]pyrene (4).21b
To a suspension of Lindlar catalyst (875 mg) in 1:1 THF-EtOH (70 mL) was added the azidotribenzoate (±)–3 (175 mg, 0.27 mmol). The suspension was stirred under a hydrogen balloon for 2 h and 20 min, at which time tlc indicated that all starting material had been consumed. The mixture was filtered through Celite and the solvents were evaporated. The resulting pinkish oil was chromatographed on a silica gel column using 1:100 MeOH–CH2Cl2 and the resulting product was washed twice with 1:1 Et2O–hexane to yield 121 mg (72%) of (±)–4 as a white powder.
(±)-1β-Azido-2α,3α,4β-trihydroxy-1,2,3,4-tetrahydrobenzo[c]phenanthrene (6).25
BcPh diol epoxide (±)–5 (244 mg, 0.876 mmol) and sodium azide (569.9 mg, 8.76 mmol) were stirred in 1:1 THF–H2O (100 mL) at 40 °C. Tlc showed disappearance of the starting material after 40 h. The mixture was extracted with EtOAc. The aqueous layer was extracted one more time with EtOAc and the combined organic layers were washed with water, dried over Na2SO4 and evaporated. The product was taken up in the minimum volume of Et2O and added to hexane under sonication. The precipitated product was isolated and treated in the same manner one more time. The resulting white powder was dried to yield 225 mg (80%) of (±)–6.
(±)-1β-Azido-2α,3α,4β-trisbenzoyloxy-1,2,3,4-tetrahydrobenzo[c]phenanthrene (7)
The azido triol (±)–6 (39.5 mg, 0.123 mmol) was dissolved in dry DMF (1.5 mL). Dry Et3N (171.5 μL, 1.23 mmol) and benzoyl cyanide (161.3 mg, 1.23 mmol) were added and the reaction mixture was allowed to stir for 45 min at room temperature. The reaction was quenched with sat aq NaHCO3 and the mixture was extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4 and evaporated. The product was chromatographed on silica gel using 2:1 toluene–CH2Cl2. Finally, some minor impurities were removed by washing the product twice with 1:1 Et2O–hexane. This process yielded 47 mg (60%) of (±)–7 as a white powder. Rf (SiO2/2:1 toluene–CH2Cl2) = 0.53. 1H NMR (600 MHz, CDCl3): δ 9.00 (d, 1H, Ar–H, J = 8.8), 8.14 (d, 2H, Ar–H, J = 8.3), 8.01 (d, 1H, Ar–H, J = 8.3), 7.97 (d, 2H, Ar–H, J = 8.3), 7.82 (d, 1H, Ar–H, J = 8.8), 7.79–7.68 (m, 7H), 7.58 (t, 1H, Ar–H, J = 7.7), 7.53 (t, 1H, Ar–H, J = 7.4), 7.48 (t, 1H, Ar–H, J = 7.4), 7.47 (d, 1H, Ar–H, J = 7.8), 7.46 (d, 1H, Ar–H, J = 7.8), 7.36 (t, 2H, Ar–H, J = 7.8), 7.31 (t, 2H, Ar–H, J = 7.4), 7.14 (d, 1H, H4, J = 7.3), 6.35 (dd, 1H, H3, J = 7.3, 2.6), 6.18 (dd, 1H, H2, J = 4.9, 2.6), 5.97 (d, 1H, H1, J = 4.9). HRMS calculated for C39H31N4O6 (M+ + NH4): 651.2238, found: 651.2239.
(±)-1β-Amino-2α,3α,4β-trisbenzoyloxy-1,2,3,4-tetrahydrobenzo[c]phenanthrene (8)
The azido tribenzoate (±)–7 (239 mg, 0.38 mmol) and Lindlar catalyst (1.195 g) were stirred in 1:1 THF–EtOH (95 mL) under a hydrogen balloon for 2 h and 20 min while protected from light. Tlc showed disappearance of the starting material and formation of a new spot. The mixture was filtered through Celite and the residue was washed twice with EtOAc. The filtrate was dried over Na2SO4 and the solvents were evaporated. The compound was purified by chromatography on a silica gel column using 95:5 toluene–EtOAc. Finally, some minor contaminants were removed by washing the product with 1:1 Et2O–hexane to provide 199 mg (87%) of (±)–8 as a white powder. Rf (SiO2/95:5 toluene–EtOAc) = 0.26. 1H NMR (600 MHz, CDCl3): δ 9.51 (d, 1H, Ar–H, J = 8.6), 8.13 (d, 2H, Ar–H, J = 7.8), 7.97 (d, 2H, Ar–H, J = 7.8), 7.93 (t, 2H, Ar–H, J = 7.6), 7.77 (d, 1H, Ar–H, J = 8.6), 7.75 (d, 2H, Ar–H, J = 7.7), 7.72 (t, 2H, Ar–H, J = 8.3), 7.66 (d, 1H, Ar–H, J = 8.1), 7.65(t, 1H, Ar–H, J = 7.3), 7.57 (t, 1H, Ar–H, J = 7.6), 7.50 (t, 1H, Ar–H, J = 7.6), 7.47 (t, 1H, Ar–H, J = 7.7), 7.46 (d, 1H, Ar–H, J = 7.7), 7.44 (d, 1H, Ar–H, J = 7.6), 7.35 (t, 2H, Ar–H, J = 7.6), 7.30 (t, 2H, Ar–H, J = 7.7), 7.06 (d, 1H, H4, J = 7.8), 6.65 (dd, 1H, H3, J = 7.8, 2.5), 6.04 (app. t, 1H, H2, J = 3.5), 5.55 (d, 1H, H1, J = 4.4). HRMS calculated for C39H30NO6 (M+ + H): 608.2068, found: 608.2070.
N6-[10-(7,8,9-trisbenzoyloxy-7,8,9,10-tetrahydrobenzo[a]pyrenyl)]-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyadenosine (11a, b)
(a) Pd–mediated coupling of 9 and (±)–4
Into an oven-dried, screw-cap vial equipped with a stirring bar, were placed Pd(OAc)2 (4.6 mg, 20.5μmol) and (±)–BINAP (37.7 mg, 60.5 μmol). Toluene (2 mL) was added and the mixture was stirred at room temperature for 5 minutes. Cs2CO3 (92.0 mg, 0.282 mmol) followed by (±)–4 (140.0 mg, 0.222 mmol) and then chloro nucleoside 9 (100.6 mg, 0.202 mmol) were added. The vial was flushed with N2 gas, sealed with a Teflon-lined cap and heated in a sand bath that was maintained at 85 °C. The reaction was monitored by tlc and judged to be complete after 5 hours at which time the mixture was cooled, diluted with Et2O and washed twice with brine. The organic layer was dried over Na2SO4and the solvent was evaporated. Chromatography of the crude mixture on a silica gel column using 4:40:56 acetone–hexane–CH2Cl2 yielded 161 mg (73%) of the diastereomeric mixture of adducts 11a, b as a white powder. Rf 11a, b (SiO2/4:40:56 acetone–hexane–CH2Cl2) = 0.61.
(b) Pd–mediated coupling of 10 and (±)–4
A similar C–N reaction between bromo nucleoside 10 (11.7 mg, 21.5 μmol) and (±)–4 (15.0 mg, 23.7 μmol) was conducted using Pd(OAc)2 (0.5 mg, 2.2 μmol), (±)–BINAP (4.0 mg, 6.4 μmol) and Cs2CO3 (9.9 mg, 30.4 μmol) in toluene (0.21 mL). This reaction was completed within 4 h and yielded, after purification, 13.4 mg (57%) of the adduct diastereomers as a white powder.
Synthesis and characterization of N6-[10-(7,8,9-trisacetoxy-7,8,9,10-tetrahydrobenzo[a]pyrenyl)]-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyadenosine (16a, b)
The diastereomeric mixture of adducts 11a, b (10 mg) was dissolved in MeOH saturated with NH3 (2.5mL). The reaction was allowed to proceed for 15 hours at 55 °C at which time tlc showed the reaction to be complete. The mixture was cooled to room temperature and carefully evaporated. The product was dried under vacuum to give 8 mg of crude material. This crude material was taken in pyridine (100 μL), acetic anhydride (100 μL) and a few crystals of DMAP were added, and the mixture was allowed to stir at room temperature overnight. The reaction mixture was diluted with Et2O and washed with 1 M aq HCl, sat aq NaHCO3 and twice with water. The organic layer was dried over Na2SO4 and mixture was evaporated to yield 9 mg of a crude mixture of diastereoisomers 16a, b that were separated using preparative tlc (SiO2, 1 mm, 20 × 20 cm and elution with 92:8 CH2Cl2-EtOAc) to yield 3.2 mg of the less–polar adduct and 2.8 mg of the more–polar adduct (combined yield of 72%).
The less–polar diastereomer was the 10S isomer (16a) as determined by the presence of a positive CD band at 282 nm in its CD spectrum. 1H NMR (500 MHz, CDCl3): δ 8.63 (s, 1H, purine–H2), 8.29 (d, 1H, Ar–H11, J = 9.4), 8.21 (d, 1H, Ar–H1 or H3, J = 7.8), 8.18 (d, 1H, Ar–H3 or H1, J = 7.8), 8.19 (s, 1H, Ar–H6), 8.09 (d, 1H, Ar–H5, J = 9.2), 8.08 (d, 1H, Ar–H4, J = 9.2), 8.06 (s, 1H, purine–H8), 8.05 (d, 1H, Ar–H12, J = 9.4), 8.02 (t, 1H, Ar–H2, J = 7.8), 6.77 (dd, 1H, H10, J = 9.1, 2.9), 6.67 (d, 1H, H7, J = 3.1), 6.46 (t, 1H, H1′, J = 6.3), 6.23 (d, 1H, NH, J = 9.1), 5.69 (t, 1H, H8, J = 3.3), 5.64 (t, 1H, H9, J = 2.9), 4.59 (app. q, 1H, H3′, J = 4.9), 4.00 (app. q, 1H, H4′, J = 3.7), 3.87 (dd, 1H, H5′, J = 11.4, 4.2), 3.75 (dd, 1H, H5″, J = 11.4, 3.1), 2.64 (app. quint, 1H, H2′, J = 13.1, 5.4), 2.46 (ddd, 1H, H2″, J = 13.1, 5.8, 4.5), 2.18, 2.11, 2.05 (3s, 9H, OCOCH3), 0.92, 0.86 (2s, 18H, tert-Bu), 0.11, 0.10, 0.05, 0.03 (4s, 12H, SiCH3). HRMS calcd for C48H62N5O9Si2 (M+ + H) 908.4081, found 908.4090.
The more–polar diastereomer was the 10R isomer (16b) as determined by the presence of a negative CD band at 282 nm in its CD spectrum. 1H NMR (500 MHz, CDCl3): δ 8.62 (s, 1H, purine–H2), 8.35 (d, 1H, Ar–H11, J = 9.4), 8.21 (d, 1H, Ar–H1 or H3, J = 7.6), 8.17 (d, 1H, Ar–H3 or H1, J = 7.6), 8.20 (s, 1H, Ar–H6), 8.10 (d, 1H, Ar–H5, J = 9.2), 8.06 (s, 1H, purine–H8), 8.08 (d, 1H, Ar–H4, J = 9.2), 8.03 (d, 1H, Ar–H12, J = 9.4), 8.02 (t, 1H, Ar–H2, J = 7.6), 6.76 (br, 1H, H10), 6.67 (d, 1H, H7, J = 3.1), 6.46 (t, 1H, H1′, J = 6.4), 6.22 (br d, 1H, NH, J = 7.9), 5.69 (t, 1H, H8, J = 3.6), 5.64 (t, 1H, H9, J = 2.9), 4.62 (app. q, 1H, H3′, J = 4.3), 4.00 (app. q, 1H, H4′, J = 3.7), 3.88 (dd, 1H, H5′, J = 11.4, 4.3), 3.77 (dd, 1H, H5″, J = 11.4, 3.0), 2.63 (app. quint, 1H, H2′, J = 13.1, 6.2), 2.43 (ddd, 1H, H2″, J = 13.1, 6.0, 4.6), 2.18, 2.09, 2.04 (3s, 9H, OCOCH3), 0.91, 0.86 (2s, 18H, tert-Bu), 0.094, 0.092, 0.05, 0.04 (4s, 12H, SiCH3). HRMS calcd for C48H62N5O9Si2 (M+ + H) 908.4081, found 908.4084.
N6-[1-(2,3,4-trisbenzoyloxy-1,2,3,4-tetrahydrobenzo[c]phenanthrenyl)]-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyadenosine (12a, b)
(a) Pd–mediated coupling of 10 and (±)–8
Into an oven-dried, screw-cap vial equipped with a stirring bar, were placed Pd(OAc)2 (5.4 mg, 24.1 μmol) and (±)–BINAP (45.0 mg, 72.3 μmol). Toluene (2.4 mL) was added and the mixture was stirred at room temperature for 5 minutes. Cs2CO3 (109.9 mg, 0.337 mmol) followed by (±)–8 (161.0 mg, 0.265 mmol) and then bromo nucleoside 10 (131.0 mg, 0.241 mmol) were added. The vial was flushed with N2 gas, sealed with a Teflon-lined cap and heated in a sand bath that was maintained at 85 °C. The reaction was monitored by tlc and judged to be complete after 4 hours at which time the mixture was cooled, diluted with Et2O and washed twice with brine. The organic layer was dried over Na2SO4 and the solvent was evaporated. Chromatography of the crude mixture on a silica gel column using 99:1 CH2Cl2–EtOAc yielded 116 mg (45%, 75% based upon recovered (±)–8) of the diastereomeric mixture of adducts 12a, b as a white powder. Rf 12a, b (SiO2/99:1 CH2Cl2–EtOAc) = 0.37.
(b) Pd–mediated coupling of 9 and (±)–8
A similar C–N reaction between chloro nucleoside 9 (13.4 mg, 26.8 μmol) and (±)–8 (17.9 mg, 29.5 μmol) was conducted using Pd(OAc)2 (0.6 mg, 2.7 μmol), (±)–BINAP (5.0 mg, 8.0 μmol) and Cs2CO3 (12.2 mg, 37.4 μmol) in toluene (0.26 mL). This reaction was completed within 4 h and yielded, after purification, 10.5 mg (36%, 39% based upon recovered (±)–8) of the adduct diastereomers as a white powder.
Synthesis and characterization of N6-[1-(2,3,4-trisacetoxy-1,2,3,4-tetrahydrobenzo[c]phenanthrenyl)]-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyadenosine (17a, b).25
The diastereomeric mixture of adducts 12a, b (10 mg) was dissolved in MeOH saturated with NH3 (2.5mL). The reaction was allowed to proceed for 15 hours at 55 °C at which time tlc showed the reaction to be complete. The mixture was cooled to room temperature and carefully evaporated. The product was dried under vacuum to give 8.4 mg of crude material. This crude material was taken in pyridine (100 μL), acetic anhydride (100 μL) and a few crystals of DMAP were added, and the mixture was allowed to stir at room temperature overnight. The reaction mixture was diluted with Et2O and washed with 1 M aq HCl, sat aq NaHCO3 and twice with water. The organic layer was dried over Na2SO4 and mixture was evaporated to yield 8 mg of the mixture of diastereoisomers 17a, b that were separated using preparative tlc (SiO2, 1 mm, 20 × 20 cm and elution with 90:10 CH2Cl2-EtOAc) to yield 2.8 mg of the less–polar adduct and 2.9 mg of the more–polar adduct (combined yield of 69%).
The less–polar diastereomer was the 1S isomer (17a) as determined by the presence of a positive CD band at 248 nm in its CD spectrum. 1H NMR (500 MHz, CDCl3): δ 8.66 (s, 1H, purine–H2), 8.52 (d, 1H, Ar–H12, J = 8.6), 8.09 (s, 1H, purine–H8), 7.98 (d, 1H, Ar–H6, J = 8.3), 7.87 (dd, 1H, Ar–H9, J = 8.1, 1.3), 7.78 (d, 1H, Ar–H8, J = 8.7), 7.74 (d, 1H, Ar–H7, J = 8.7), 7.54 (d, 1H, Ar–H5, J = 8.3), 7.50 (t, 1H, Ar–H10, J = 7.6), 7.23 (dd, 1H, Ar–H11, J = 8.6, 7.8), 6.54 (br s, 1H, NH), 6.51 (t, 1H, H1′, J = 6.3), 6.47 (d, 1H, H1, J = 3.3), 6.39 (d, 1H, H4, J = 7.3), 6.30 (t, 1H, H2, J = 2.9), 5.95 (dd, 1H, H3, J = 7.3, 2.9), 4.61 (app. quint, 1H, H3′, J = 4.3), 4.01 (app. q, 1H, H4′, J = 3.5), 3.86 (dd, 1H, H5′, J = 11.3, 4.1), 3.77 (dd, 1H, H5″, J = 11.3, 2.9), 2.62 (app. quint, 1H, H2′, J = 13.3. 6.2), 2.48 (ddd, 1H, H2″, J = 13.3, 6.1, 4.3), 2.24, 2.02, 1.89 (3s, 9H, OCOCH3), 0.92, 0.89 (2s, 18H, tert-Bu), 0.12, 0.10, 0.07, 0.06 (4s, 12H, SiCH3).
The more–polar diastereomer was the 1R isomer (17b) as determined by the presence of a negative CD band at 248 nm in its CD spectrum. 1H NMR (500 MHz, CDCl3): δ 8.65 (s, 1H, purine–H2), 8.56 (d, 1H, Ar–H12, J = 8.6), 8.03 (s, 1H, purine–H8), 7.98 (d, 1H, Ar–H6, J = 8.3), 7.87 (dd, 1H, Ar–H9, J = 8.0, 1.4), 7.78 (d, 1H, Ar–H8, J = 8.7), 7.73 (d, 1H, Ar–H7, J = 8.7), 7.55 (d, 1H, Ar–H5, J = 8.3), 7.50 (t, 1H, Ar–H10, J = 7.5), 7.25 (dd, 1H, Ar–H11, J = 8.5, 7.8), 6.54 (br s, 1H, NH), 6.47 (t, 1H, H1′, J = 6.6), 6.40 (d, 1H, H1, J = 3.7), 6.39 (d, 1H, H4, J = 7.4), 6.29 (t, 1H, H2, J = 3.7), 5.94 (dd, 1H, H3, J = 7.4, 2.9), 4.63 (app. quint, 1H, H3′, J = 3.8), 4.03 (app. q, 1H, H4′, J = 3.7), 3.89 (dd, 1H, H5′, J = 11.2, 4.4), 3.79 (dd, 1H, H5″, J = 11.2, 3.4), 2.69 (app. quint, 1H, H2′, J = 13.2. 6.2), 2.44 (ddd, 1H, H2″, J = 13.2, 6.0, 3.7), 2.23, 2.02, 1.89 (3s, 9H, OCOCH3), 0.92, 0.89 (2s, 18H, tert-Bu), 0.12, 0.11, 0.07, 0.05 (4s, 12H, SiCH3).
N2-[10-(7,8,9-trisbenzoyloxy-7,8,9,10-tetrahydrobenzo[a]pyrenyl)]-O6-benzyl-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (14a, b)
Into an oven-dried, screw-cap vial equipped with a stirring bar, were placed Pd(OAc)2 (4.5 mg, 20.0μmol) and (±)–BINAP (37.7 mg, 60.6 μmol). Toluene (2.0 mL) was added and the mixture was stirred at room temperature for 5 minutes. Cs2CO3 (92.0 mg, 0.282 mmol) followed by (±)–4 (140.0 mg, 0.222 mmol) and then bromo nucleoside 13 (131.0 mg, 0.202 mmol) were added. The vial was flushed with N2 gas, sealed with a Teflon-lined cap and heated in a sand bath that was maintained at 85 °C. The reaction was monitored by tlc and judged to be complete after 16 hours at which time the mixture was cooled, diluted with Et2O and washed twice with brine. The organic layer was dried over Na2SO4 and the solvent was evaporated. Chromatography of the crude mixture on a silica gel column using 4:40:56 acetone–hexane–CH2Cl2 yielded 176.2 mg (73%) of the diastereomeric mixture of adducts 14a, b as a white powder. Rf 14a, b (SiO2/4:40:56 acetone–hexane–CH2Cl2) = 0.54.
Synthesis and characterization of N2-[10-(7,8,9-trisacetoxy-7,8,9,10-tetrahydrobenzo[a]pyrenyl)]-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (18a, b).29
(a) Step 1: synthesis of the adduct triacetate
The diastereomeric mixture of adducts 14a, b (18.9 mg) was dissolved in MeOH saturated with NH3 (2.5 mL). The reaction was allowed to proceed for 15 hours at 55 °C at which time tlc showed the reaction to be complete. The mixture was cooled to room temperature, carefully evaporated and the product was dried under vacuum. This crude material was taken in pyridine (300 μL), acetic anhydride (300 μL) and a few crystals of DMAP were added, and the mixture was allowed to stir at room temperature overnight. The reaction mixture was diluted with Et2O and washed with 1 M aq HCl, sat aq NaHCO3 and twice with water. The organic layer was dried over Na2SO4 and the mixture was evaporated to yield 11.5 mg of the mixture of the triacetyl adduct diastereoisomers that was purified by preparative tlc (SiO2, 1 mm, 20 × 20 cm and elution with 95:5 CH2Cl2–EtOAc). This adduct mixture was used for the second step.
(a) Step 2: debenzylation
To a solution of the adduct mixture (11.2 mg) in 1:1 THF–MeOH (0.12 mL) was added 5% Pd–C on (5mg). The reaction mixture was stirred under 1 atm H2pressure (balloon) for 5 hours at which time tlc indicated the reaction to be complete. The mixture was filtered through Celite and evaporated to dryness. The two diastereoisomers were separated using preparative tlc (SiO2, 1 mm, 20 × 20 cm and elution with 35:40:25 acetone–hexane–CH2Cl2) to yield 3.1 mg of the less–polar adduct and 3.2 mg of the more–polar adduct (combined yield of 43 % over the two steps).
The less–polar diastereomer was the 10S isomer (18a) as determined by the presence of a positive CD band at 251 nm in its CD spectrum. 1H NMR (500 MHz, DMSO–d6): δ 0.68 (s, 1H, guanine ring NH), 8.35 (d, 1H, Ar–H3 or H1, J = 7.8), 8.31 (d, 1H, Ar–H1 or H3, J = 7.8), 8.29 (d, 1H, Ar–H11, J = 9.3), 8.24 (s, 2H, Ar–H4 and H5), 8.23 (s, 2H, Ar–H6), 8.22 (d, 1H, Ar–H12, J = 9.3), 8.11 (t, 1H, Ar–H2, J = 7.8), 7.95 (s, 1H, purine–H8), 7.30 (br s, 1H, exocyclic NH), 6.63 (d, 1H, H7, J = 6.9), 6.24 (t, 1H, H1′, J = 7.1), 6.18 (dd, 1H, H10, J = 7.5, 4.4), 5.85 (dd, 1H, H9, J = 7.0, 4.4), 5.42 (t, 1H, H8, J = 6.9), 4.50 (dt, 1H, H3′, J = 5.1, 4.6), 3.88 (dt, 1H, H4′, J = 5.4, 4.3), 3.79 (dd, 1H, H5′, J = 11.5, 5.3), 3.73 (dd, 1H, H5″, J = 11.5, 4.9), 2.55 (app. quint, 1H, H2′, J = 12.9, 6.4), 2.29 (ddd, 1H, H2″, J = 12.9, 6.6, 5.6), 2.27, 2.07, 1.98 (3s, 9H, OCOCH3), 0.87, 0.85 (2s, 18H, tert-Bu), 0.06, 0.05, 0.44, 0.04 (4s, 12H, SiCH3).
The more–polar diastereomer was the 10R isomer (18b) as determined by the presence of a negative CD band at 250 nm in its CD spectrum. 1H NMR (500 MHz, DMSO–d6): δ 10.65 (s, 1H, guanine ring NH), 8.35 (d, 1H, Ar–H3 or H1, J = 7.8), 8.31 (d, 1H, Ar–H1 or H3, J = 7.8), 8.29 (d, 1H, Ar–H11, J = 9.3), 8.24 (s, 2H, Ar–H4 and H5), 8.23 (s, 1H, Ar–H6), 8.15 (d, 1H, Ar–H12, J = 9.3), 8.11 (t, 1H, Ar–H2, J = 7.8), 7.96 (s, 1H, purine–H8), 7.24 (br s, 1H, exocyclic NH), 6.63 (d, 1H, H7, J = 6.5), 6.23 (t, 1H, H1′, J = 6.7), 6.18 (dd, 1H, H10, J = 7.8, 4.0), 5.80 (dd, 1H, H9, J = 6.5, 4.0), 5.43 (t, 1H, H8, J = 6.5), 4.45 (m 1H, H3′), 3.83 (m, 1H, H4′), 3.75 (dd, 1H, H5′, J = 10.8, 7.3), 3.71 (dd, 1H, H5″, J = 10.8, 4.4), 3.08 (app. quint, 1H, H2′, J = 13.9, 6.6), 2.28 (ddd, 1H, H2″, J = 12.9, 7.2, 5.2), 2.25, 2.07, 1.99 (3s, 9H, OCOCH3), 0.87, 0.64 (2s, 18H, tert-Bu), 0.10, 0.08, −0.02, −0.31 (4s, 12H, SiCH3).
N2-[1-(2,3,4-trisbenzoyloxy-1,2,3,4-tetrahydrobenzo[c]phenanthrenyl)]-O6-benzyl-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (15a, b)
Into an oven-dried, screw-cap vial equipped with a stirring bar, were placed Pd(OAc)2 (5.4 mg, 24.1μmol) and (±)–BINAP (45.0 mg, 72.3 μmol). Toluene (2.4 mL) was added and the mixture was allowed to stir at room temperature for 5 minutes. Cs2CO3 (109.9 mg, 0.337 mmol) followed by (±)–8 (161.0 mg, 0.265 mmol) and then bromo nucleoside 13 (156.0 mg, 0.24 mmol) were added. The vial was flushed with N2 gas, sealed with a Teflon-lined cap and heated in a sand bath that was maintained at 85 °C. The reaction was monitored by tlc and judged to be complete after 16 hours at which time the mixture was cooled, diluted with Et2O and washed twice with brine. The organic layer was dried over Na2SO4 and the solvent was evaporated. Chromatography of the crude mixture on a silica gel column using 99:1 CH2Cl2–EtOAc yielded 197 mg (70%) of the diastereomeric mixture of adducts 15a, b as a white powder. Rf 15a, b (SiO2/99:1 CH2Cl2–EtOAc) = 0.49.
Synthesis and characterization of N2-[1-(2,3,4-trisacetoxy-1,2,3,4-tetrahydrobenzo[c]phenanthrenyl)]-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (19a, b).32
(a) Step 1: synthesis of the adduct triacetate
The diastereomeric mixture of adducts 15a, b (23.1 mg) was dissolved in MeOH saturated with NH3 (2.5 mL). The reaction was allowed to proceed for 15 hours at 55 °C at which time tlc showed the reaction to be complete. The mixture was cooled to room temperature, carefully evaporated and the product was dried under vacuum. This crude material was taken in pyridine (300 μL), acetic anhydride (300 μL) and a few crystals of DMAP were added, and the mixture was allowed to stir at room temperature overnight. The reaction mixture was diluted with Et2O and washed with 1 M aq HCl, sat aq NaHCO3 and twice with water. The organic layer was dried over Na2SO4 and mixture was evaporated to yield 16.0 mg of the mixture of the triacetyl adduct diastereoisomers that was purified by preparative tlc (SiO2, 1 mm, 20 × 20 cm and elution with 95:5 CH2Cl2–EtOAc). This adduct mixture was used for the second step.
(a) Step 2: debenzylation
To a solution of the adduct mixture (16.0 mg) in 1:1 THF–MeOH (0.16 mL) was added 5% Pd–C on (5mg). The reaction mixture was stirred under 1 atm H2 pressure (balloon) for 5 hours at which time tlc indicated the reaction to be complete. The mixture was filtered through Celite and evaporated to dryness. The two diastereoisomers were separated using preparative tlc (SiO2, 1 mm, 20 × 20 cm and elution with 35:35:30 acetone–hexane–CH2Cl2) to yield 4.4 mg of the less–polar adduct and 7.0 mg of the more–polar adduct (combined yield of 64% over the two steps).
The less–polar diastereomer was the 1S isomer (19a) as determined by the presence of a positive CD band at 257 nm in its CD spectrum. 1H NMR (500 MHz, DMSO–d6): δ 10.16 (s, 1H, guanine ring NH), 8.52 (d, 1H, Ar–H12, J = 8.9), 8.18 (d, 1H, Ar–H9, J = 8.4), 8.05 (d, 1H, Ar–H6, J = 8.2), 8.04 (s, 1H, purine–H8), 7.95 (d, 1H, Ar–H7, J = 8.8), 7.92 (d, 1H, Ar–H8, J = 8.8), 7.64 (br d, 1H, exocyclic NH, J = 6.5), 7.60 (t, 1H, Ar–H10, J = 7.4), 7.58 (d, 1H, Ar–H5, J = 8.5), 7.34 (t, 1H, Ar–H11, J = 8.2), 6.43 (d, 1H, H4, J = 8.5), 6.32 (t, 1H, H1′, J = 6.6), 6.06 (dd, 1H, H2, J = 4.4, 2.7), 6.04 (dd, 1H, H1, J = 6.5, 4.4), 5.74 (dd, 1H, H3, J = 8.5, 2.7), 4.50 (dt, 1H, H3′, J = 6.0, 3.7), 3.86 (app. q, 1H, H4′, J = 4.5), 3.77 (dd, 1H, H5′, J = 11.3, 5.2), 3.72 (dd, 1H, H5″, J = 11.3, 4.5), 2.68 (app. quint, 1H, H2′, J = 13.4, 7.7), 2.35 (ddd, 1H, H2″, J = 13.4, 6.4, 4.0), 2.23, 1.99, 1.90 (3s, 9H, OCOCH3), 0.89, 0.83 (2s, 18H, tert-Bu), 0.07, 0.06, 0.058, 0.054 (4s, 12H, SiCH3).
The more–polar diastereomer was the 1R isomer (19b) as determined by the presence of a negative CD band at 257 nm in its CD spectrum. 1H NMR (500 MHz, DMSO– d6): δ 10.24 (s, 1H, guanine ring NH), 8.44 (d, 1H, Ar–H12, J = 8.9), 8.19 (d, 1H, Ar–H9, J = 8.5), 8.06 (d, 1H, Ar–H6, J = 8.1), 8.03 (s, 1H, purine–H8), 7.97 (d, 1H, Ar–H7, J = 8.9), 7.93 (d, 1H, Ar–H8, J = 8.9), 7.87 (br s, 1H, exocyclic NH), 7.61 (t, 1H, Ar–H10, J = 7.4), 7.57 (d, 1H, Ar–H5, J = 8.5), 7.25 (t, 1H, Ar–H11, J = 8.3), 6.44 (d, 1H, H4, J = 8.9), 6.23 (dd, 1H, H1′, J = 9.2, 5.4), 6.10 (t, 1H, H2, J = 3.5), 5.95 (dd, 1H, H1, J = 6.4, 4.3), 5.75 (dd, 1H, H3, J = 8.9, 2.6), 4.38 (d, 1H, H3′′, J = 5.1), 3.55 (t, 1H, H4′, J = 10.3), 3.76 (dd, 1H, H5′, J = 10.3, 4.4), 3.46 (ddd, 1H, H5″, J = 10.3, 4.1), 3.23 (ddd, 1H, H2′, J = 13.4, 9.2, 4.9), 2.12 (dd, 1H, H2″, J = 13.4, 5.4), 2.24, 2.00, 1.95 (3s, 9H, OCOCH3), 0.86, 0.52 (2s, 18H, tert-Bu), 0.08, 0.07, −0.54, −0.66 (4s, 12H, SiCH3).
Supplementary Material
General experimental methods, tabulation of the coupling constant data for the tetrahydro ring protons in the BaP and BcPh adducts, CD spectra of adduct pairs 16a/16b, 17a/17b, 18a/18b, 19a/19b, 1H NMR spectra of (±)–7, (±)–8, 16a, 16b, 17a, 17b, 18a, 18b, 19a and 19b. This information is available free of cost via the Internet at www.pubs.acs.org.
SCHEME 2.

Synthesis of the BaP amino tribenzoate
SCHEME 3.

Synthesis of the BcPh amino tribenzoate
Acknowledgments
This work was supported in part by NIH Grant S06 GM008168–24S1, 2 S06 GM008168–27 (NIGMS) and NSF Grant CHE–0314326 to M.K.L. P.P. acknowledges support via a PSC–CUNY 37 award. Acquisition of a mass spectrometer was funded by NSF Grant CHE–0520963 (M.K.L.). Infrastructural support at CCNY via NIH RCMI Grant G12 RR03060 is gratefully acknowledged. Wavefunction, Inc. is thanked for their technical support.
References
- 1.Earlier studies on PAHs and carcinogenesis has been reviewed in: Yang SK, Silverman BD, editors. Polycyclic Aromatic Hydrocarbon Carcinogenesis: Structure-Activity Relationships. I and II CRC Press; Boca Raton, FL: 1988. Harvey RG, editor. Polycyclic Aromatic Hydrocarbons: Chemistry and Carcinogenicity. Cambridge University Press; Cambridge: 1991. Harvey RG, editor. Polycyclic Hydrocarbons and Carcinogenesis. ACS Symposium Series 283; Washington, DC: 1985. For a detailed survey of the chemistry of polycyclic aromatic hydrocarbons, please see: Harvey RG, editor. Polycyclic Aromatic Hydrocarbons. Wiley-VCH; NY: 1997.
- 2.(a) Cavalieri EL, Rogan EG. Xenobiotica. 1995;25:677–688. doi: 10.3109/00498259509061885. [DOI] [PubMed] [Google Scholar]; (b) Cavalieri EL, Rogan EG. In: The Handbook of Environmental Chemistry. Neilson AH, editor. 3J. Springer-Verlag; Heidelberg: 1998. pp. 81–117. [Google Scholar]
- 3.(a) Penning TM, Burczynski ME, Hung CF, McCoull KD, Palackal NT, Tsuruda LS. Chem Res Toxicol. 1999;12:1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]; (b) Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Chem Res Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 4.Jerina DM, Sayer JM, Agarwal SK, Yagi H, Levin W, Wood AW, Conney AH, Pruess-Schwartz D, Baird WM, Pigott MA, Dipple A. In: Biological Reactive Intermediates III. Kocsis JJ, Jollow DJ, Witmer CM, Nelson JO, Snyder R, editors. Plenum Press; New York: 1986. pp. 11–30. [DOI] [PubMed] [Google Scholar]
- 5.Harvey RG, Geacintov NE. Acc Chem Res. 1988;21:66–73. [Google Scholar]
- 6.Jerina DM, Chadha A, Cheh AM, Schurdak ME, Wood AW, Sayer JM. In: Biological Reactive Intermediates IV. Witmer CM, Snyder R, Jollow DJ, Kalf GF, Kocsis JJ, Sipes IG, editors. Plenum Press; New York: 1991. pp. 533–553. [Google Scholar]
- 7.Dipple A. In: DNA Adducts: Identification and Biological Significance. Hemminki K, Dipple A, Shuker DEG, Kadlubar FF, Sagerbäck D, Bartsch H, editors. Scientific Publication No. 125, International Agency for Research on Cancer; Lyon: 1994. pp. 107–129. [Google Scholar]
- 8.(a) Lakshman MK, Sayer JM, Jerina DM. J Am Chem Soc. 1991;113:6589–6594. [Google Scholar]; (b) Lakshman MK, Sayer JM, Jerina DM. J Org Chem. 1992;57:3438–3443. [Google Scholar]; (c) Lakshman MK, Sayer JM, Yagi H, Jerina DM. J Org Chem. 1992;57:4585–4590. [Google Scholar]; (d) Chaturvedi S, Lakshman MK. Carcinogenesis. 1996;17:2747–2752. doi: 10.1093/carcin/17.12.2747. [DOI] [PubMed] [Google Scholar]
- 9.(a) Cosman M, Ibanez V, Geacintov NE, Harvey RG. Carcinogenesis. 1990;11:1667–1672. doi: 10.1093/carcin/11.9.1667. [DOI] [PubMed] [Google Scholar]; (b) Kim SJ, Harris CM, Jung KY, Koreeda M, Harris TM. Tetrahedron Lett. 1991;32:6073–6076. [Google Scholar]; (c) Steinbrecher T, Becker A, Stezowski JJ, Oesch F, Seidel A. Tetrahedron Lett. 1993;34:1773–1774. [Google Scholar]; (d) DeCorte BL, Tsarouhtsis D, Kuchimanchi S, Cooper MD, Horton P, Harris CM, Harris TM. Chem Res Toxicol. 1996;9:630–637. doi: 10.1021/tx9501795. [DOI] [PubMed] [Google Scholar]; (e) Pilcher AS, Yagi H, Jerina DM. J Am Chem Soc. 1998;120:3520–3521. [Google Scholar]; (f) Cooper MD, Hodge RP, Tamura PJ, Wilkinson AS, Harris CM, Harris TM. Tetrahedron Lett. 2000;41:3555–3558. [Google Scholar]
- 10.Lakshman M, Lehr RE. Tetrahedron Lett. 1990;31:1547–1550. [Google Scholar]
- 11.Robins MJ, Basom GL. Can J Chem. 1973;51:3161–3169. [Google Scholar]
- 12.Zajc B, Lakshman MK, Sayer JM, Jerina DM. Tetrahedron Lett. 1992;33:3409–3412. [Google Scholar]
- 13.(a) Lakshman MK. J Organomet Chem. 2002;653:234–251. [Google Scholar]; (b) Lakshman MK. Curr Org Synth. 2005;2:83–112. [Google Scholar]
- 14.For some examples of Pd-catalyzed C–N bond formation leading to biologically important nucleoside derivative, please see: Harwood EA, Sigurdsson ST, Edfeldt NBF, Reid BR, Hopkins PB. J Am Chem Soc. 1999;121:5081–5082.De Riccardis F, Bonala RR, Johnson F. J Am Chem Soc. 1999;121:10453–10460.Wang Z, Rizzo CJ. Org Lett. 2001;3:565–568. doi: 10.1021/ol006968h.Schoffers E, Olsen PD, Means JC. Org Lett. 2001;3:4221–4223. doi: 10.1021/ol016900h.Gillet LCJ, Shärer OD. Org Lett. 2002;4:4205–4208. doi: 10.1021/ol026474f.Lakshman MK, Ngassa FN, Bae S, Buchanan DG, Hahn HG, Mah H. J Org Chem. 2003;68:6020–6030. doi: 10.1021/jo030113b.Chakraborti D, Colis L, Schneider R, Basu AK. Org Lett. 2003;5:2861–2864. doi: 10.1021/ol034904b.Elmquist CE, Stover JS, Wang Z, Rizzo CJ. J Am Chem Soc. 2004;126:11189–11201. doi: 10.1021/ja0487022.Stover JS, Rizzo CJ. Org Lett. 2004;6:4985–4988. doi: 10.1021/ol047851m.Dai Q, Ran C, Harvey RG. Org Lett. 2005;7:999–1002. doi: 10.1021/ol0475358.
- 15.Maruenda H, Chenna A, Liem LK, Singer B. J Org Chem. 1998;63:4385–4389. [Google Scholar]
- 16.Lakshman MK, Keeler JC, Hilmer JH, Martin JQ. J Am Chem Soc. 1999;121:6090–6091. [Google Scholar]
- 17.Lakshman MK, Gunda P. Org Lett. 2003;5:39–42. doi: 10.1021/ol027084w. [DOI] [PubMed] [Google Scholar]
- 18.Johnson F, Bonala R, Tawde D, Torres MC, Iden CR. Chem Res Toxicol. 2002;15:1489–1494. doi: 10.1021/tx0256174. [DOI] [PubMed] [Google Scholar]
- 19.Barends J, van der Linden JB, van Delft FL, Koomen GJ. Nucleosides Nucleotides. 1999;18:2121–2126. [Google Scholar]
- 20.Lagisetty P, Russon LM, Lakshman MK. Angew Chem, Int Ed. 2006;45:3660–3663. doi: 10.1002/anie.200504565. [DOI] [PubMed] [Google Scholar]
- 21.(a) Lakshman M, Nadkarni DV, Lehr RE. J Org Chem. 1990;55:4892–4897. [Google Scholar]; (b) Lakshman MK, Chaturvedi S, Lehr RE. Synth Commun. 1994;24:2983–2988. [Google Scholar]
- 22.McCaustland DJ, Engel JF. Tetrahedron Lett. 16;1975:2549–2552. [Google Scholar]
- 23.Yagi H, Thakker DR, Hernandez O, Koreeda M, Jerina DM. J Am Chem Soc. 1977;99:1604–1611. doi: 10.1021/ja00447a053. [DOI] [PubMed] [Google Scholar]
- 24.Sayer JM, Yagi H, Croisy-Delcey M, Jerina DM. J Am Chem Soc. 1981;103:4970–4972. [Google Scholar]
- 25.Lakshman MK, Yeh HJC, Yagi H, Jerina DM. Tetrahedron Lett. 1992;33:7121–7124. [Google Scholar]
- 26.Pottabathini N, Bae S, Pradhan P, Hahn HG, Mah H, Lakshman MK. J Org Chem. 2005;70:7188–7195. doi: 10.1021/jo050847j. [DOI] [PubMed] [Google Scholar]
- 27.Wagaw S, Rennels RA, Buchwald SL. J Am Chem Soc. 1997;119:8451–8458. [Google Scholar]
- 28.Cheng SC, Hilton BD, Roman JM, Dipple A. Chem Res Toxicol. 1989;2:334–340. doi: 10.1021/tx00011a011. [DOI] [PubMed] [Google Scholar]
- 29.See the supporting information for: Custer L, Zajc B, Sayer JM, Cullinane C, Phillips DR, Cheh AM, Jerina DM, Bohr VA, Mazur SJ. Biochemistry. 1999;38:569–581. doi: 10.1021/bi9813330.
- 30.Kroth H, Yagi H, Seidel A, Jerina DM. J Org Chem. 2000;65:5558–5564. doi: 10.1021/jo000522x. [DOI] [PubMed] [Google Scholar]
- 31.Agarwal SK, Sayer JM, Yeh HJC, Pannell LK, Hilton BD, Pigott MA, Dipple A, Yagi H, Jerina DM. J Am Chem Soc. 1987;109:2497–2504. [Google Scholar]
- 32.Kroth H, Yagi H, Sayer JM, Kumar S, Jerina DM. Chem Res Toxicol. 2001;14:708–719. doi: 10.1021/tx0002637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General experimental methods, tabulation of the coupling constant data for the tetrahydro ring protons in the BaP and BcPh adducts, CD spectra of adduct pairs 16a/16b, 17a/17b, 18a/18b, 19a/19b, 1H NMR spectra of (±)–7, (±)–8, 16a, 16b, 17a, 17b, 18a, 18b, 19a and 19b. This information is available free of cost via the Internet at www.pubs.acs.org.