

Abstract
Prostate specific membrane antigen (PSMA) is a transmembrane protein highly expressed in advanced and metastatic prostate cancers. The pathological consequence of elevated PSMA expression in not known. Here, we report that PSMA is localized to a membrane compartment in the vicinity of mitotic spindle poles and associates with the anaphase-promoting complex (APC). PSMA expressing cells prematurely degrade cyclin B and exit mitosis due to increased APC activity and incomplete inactivation of APC by the spindle assembly checkpoint. Further, expression of PSMA in a karyotypically stable cell line induces aneuploidy. Thus, these findings provide the first evidence that PSMA has a causal role in the induction of aneuploidy and might play an etiological role in the progression of prostate cancer.
Keywords: Prostate specific membrane antigen, cell cycle, anaphase promoting complex, aneuploidy, prostate cancer
INTRODUCTION
Prostate specific membrane antigen (PSMA) is localized to secretory cells within the prostatic epithelium and is upregulated in advanced prostate carcinoma and metastatic disease (1). PSMA is a type II transmembrane protein with a short, N-terminal cytoplasmic tail, a single transmembrane domain and a large extracellular C-terminal domain and exhibits both N-acetylated alpha-linked acidic peptidase (NAALADase) and folate hydrolase activities (1, 2). While the physiological and pathological functions of PSMA remain unclear, it is of particular interest that PSMA expression is a universal feature of prostate carcinoma. PSMA expression increases with tumor aggressiveness with greatest levels being found in high-grade tumors, metastatic lesions and androgen-independent disease (3). This salient association between PSMA expression levels and tumor stage alludes to a potential etiological role for PSMA in the pathogenesis and progression of prostate cancer.
Progression through mitosis relies on a complex series of precisely coordinated events that is primarily regulated by the periodic rise and fall of mitotic B-type cyclin levels. During the late stages of G2, elevated levels of the mitotic B-type cyclins permit cellular alterations associated with mitosis, such as nuclear envelope breakdown and chromosome condensation. As these cells enter into mitosis, the newly replicated centrosomes migrate to opposing ends of the cell to serve as the mitotic spindle poles. The spindle poles nucleate a dense array of microtubules that attach in a bipolar manner to the kinetochores on the condensed chromosomes (4, 5). As the condensed chromosomes attach to spindle microtubules and become properly aligned at the metaphase plate, a large multi-subunit ubiquitin ligase known as the anaphase promoting complex (APC) becomes activated and degrades cyclin B (5, 6). The degradation of cyclin B allows the cells to exit mitosis and enter a subsequent round of the cell cycle (7).
While the principle function of spindle poles was previously believed to be limited to nucleation of microtubules, more recent evidence alludes to additional roles in signal transduction and cell cycle regulation. It is now evident that centrosomes play a crucial role in the timing of mitotic events, such as the cyclin B degradation in early Drosophila embryos and exit of cytokinesis in animal cells (8, 9). Immunofluorescence analysis has also demonstrated that APC accumulates at the centrosomes and that the spindle poles are where the ubiquitination of cyclin B is initiated (10). Furthermore, several proteins involved in spindle assembly checkpoint (SAC) function and temporal regulation of mitosis have also been localized to the centrosomes and spindle poles, including Mad2, Bub2, and Bfa1 (11-14).
The activity of APC is highly regulated by checkpoint mechanisms that prevent APC activation and entry into anaphase (15, 16). Several mutations and epigenetic defects affecting mitotic checkpoint components, including RASSF1A, Bub1, BubR1, and Mad2, Mad1, have all been identified in various forms of solid and hematological malignancies (17-22). Premature activation of APC would provide less time for proper chromosome attachment and alignment and would thus reduce the fidelity of chromosomal segregation into daughter cells. This type of chromosomal instability (CIN) is by far the most frequent form of genetic instability among solid tumor cells and manifests in gross gains or losses of one or more chromosomes, a condition known as aneuploidy. Aneuploidy is apparent during the earliest stages of malignant transformation, has been implicated as a driving force in the process of tumorigenesis (17-19, 21, 22) and may have a central etiological role in inducing the complex phenotypes associated with cancer by altering the expression and balance of literally thousands of structural and regulatory genes.
In this study, we demonstrate that PSMA, a membrane protein, is localized to a membrane compartment in the vicinity of centrosomes at the spindle poles and associates with APC leading to premature activation of APC and induction of aneuploidy. These studies suggest that PSMA has a causal role in the progression of prostate cancer.
MATERIALS AND METHODS
Cell Lines and Immunofluorescence
PC3 cells expressing PSMA (PC3-PSMA) cells (23, 24), and Madin-Darby Canine Kidney (MDCK) cells expressing PSMA (MDCK-PSMA) or a deletion construct lacking amino acids 103-750 of the extracellular domain of PSMA (MDCK-PSMA-Δ103-750) (25, 26) were described previously. A cytoplasmic tail deletion construct of PSMA (PSMA-ΔCD) (27) was stably expressed in MDCK cells (25). For HCT-116 cells expressing PSMA (HCT-PSMA), PSMA was cloned from LNCaP cells and lentivirus harboring PSMA was transduced. HCT-116 cells expressing GFP (HCT-GFP) cells were generated as control.
Immunofluorescence analysis was performed as described (25, 27) using monoclonal antibody (mAb) J591 against the extracellular domain of PSMA (28) and rabbit anti-pericentrin (Covance Research Products, Princeton, NJ). Diamino-2-phenylindole dihydrochloride (DAPI) was used to visualize nuclei and chromosomes. Micrographs were taken with an Axiophot microscope (Zeiss, Jena, Germany) equipped with a triple filter (DAPI/fluorescein isothiocyanate (FITC)/Texas Red).
Cell Synchronization and Cell Cycle Analysis
A G2/M block was induced with 100 nM nocodazole for 10 hours and mitotic cells were harvested by trypsinization and gentle tapping of the culture dish. The mitotic block was released and at each indicated time point, cells were harvested and fixed in 70% ethanol. DNA was stained with 100 μg/ml propidium iodide (PI) and 20 μg/ml ribonuclease A in hypotonic citrate buffer. Samples were analyzed on a FACSCalibur flow cytometer (BD Biosciences,San Jose, CA) as described (29). Analysis of multivariate data was performed with CELLQuest™ software (BD Biosciences) and DNA histograms generated with ModFit LT™ software (Verity Software House).
Immunoblot Analysis and in vitro APC Assay
100 μg of total protein from synchronized and mitotic block-released PC3 and PC3-PSMA cells lysed in 95 mM NaCl, 25 mM Tris, pH 7.4, 0.5 mM EDTA, 2% SDS, 1 mM phenylmethylsulfonyl fluoride, and 5 μg/ml each of antipain, leupeptin, and pepstatin (protease inhibitor cocktail) were used for immunoblot analysis using anti-cyclin B1 (Santa Cruz Biotechnology Inc, Santa Cruz, CA). Proteins were detected by enhanced chemiluminescence (ECL) and quantified by densitometry. Immunoblot analysis of non-synchronized and G2/M PC3 and PC3-PSMA cells were performed using anti-Mad2, BubR1, Cdc20 (30) and Cdc27 (BD Transduction Laboratories, San Jose, CA) antibodies.
In vitro APC assays were performed using an in vitro transcribed and translated N-terminal fragment of cyclin B1 (cyclin B1-N1-102) as substrate. Amino acids 1-102 of cyclin B1 were amplified by PCR and subcloned into pCDNA3. 35S-methionine labeled cyclin B1-N1-102 was obtained using the TNT quick-coupled Transcription/Translation system (Promega, Madison, WI). Cell pellets of synchronized PC3 and PC3-PSMA cells were snap frozen in liquid nitrogen and cell lysates were prepared by incubating for 30 minutes in an ice-cold hypotonic buffer (20 mM Hepes pH 7.6, 20 mM NaF, 1.5 mM MgCl2, 1 mM DTT, 5 mM KCl, 20 mM β-glycerophosphate, 250 μM NaVO3, 1 mM PMSF, and EDTA-free protease inhibitors) followed by brief homogenization. 30 μg of total protein from cell lysate supernatants (1 hour centrifugation at 13,000 rpm at 4°C in a micro centrifuge) were added to reaction buffer containing 20 mM Tris pH 7.5, 20 mM NaCl, 5 mM MgCl2, 5 mM ATP-γ-S, 20 μg/ml MG-132, 0.5 μg Ubc10, 20 μM ubiquitin, 1 μm ubiquitin aldehyde, protease inhibitors, and 2 μl of in vitro translated 35S-cyclin B1-N1-102. Reactions were incubated at 37 °C for 60 minutes. Samples were separated by SDS-PAGE (4-15% gradient gel), enhanced with salicylate, and subjected to autoradiography.
Co-Immunoprecipitations and in vitro Binding Assays
Cell lysates of non-synchronized cells or from PSMA-positive prostate tumor tissue lysates were prepared in 20 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 % Triton-X-100, 1 mM β-glycerolphosphate, 1 mM NaVO3, 2.5 mM Na-pyrophosphate, 50 mM NaF, and protease inhibitor cocktail. Cdc 27 was immunoprecipitated using goat anti-Cdc 27 (Santa Cruz Biotechnology Inc.) and PSMA with mAb J591 for 16 hours at 4°C. Co-immunoprecipitating proteins were detected by immunoblotting. In vitro pull-down assays from PC3 cells were performed using a purified GST-PSMA fusion protein as described (24). A monoclonal anti-Cdc27 antibody (BD Transduction Laboratories) was used to detect Cdc27 in both co-immunoprecipitates and GST pull-downs.
Cytogenetics and Fluorescence in situ Hybridization (FISH)
Early passages from HCT cells were analyzed by standard cytogenetics. About 25 metaphases were analyzed and karyotyped to determine chromosome number and structure consistency. FISH was used to determine the aneuploidy in cells obtained from a series of cell-passages (2, 15, 34, and 45) of HCT-PSMA and HCT-GFP cells. In order to establish a generalized mechanism of chromosome number variation, FISH analyses with DNA-probes specific for the centromeres of chromosome 3, 7, 17, and 9p21 region, respectively were used (Abbott-Vysis, Abbott Park, IL). These probes are labeled variously as a cocktail for Multicolor FISH analysis (Urovysion probe panel). Cells that easily detached were fixed and hybridized with the Urovysion probe panel following the manufacturer’s recommendation. Interphase cells with multicolor probe signals were examined under a fluorescent microscope (Axiophot, Zeiss) equipped with appropriate filters. Approximately, 1000 nuclei were analyzed for each passage and cell-type.
Immuno-Electron Microscopy
Cells fixed in ice-cold methanol were incubated with mAb J591 as described for immunofluorescence, washed, and then incubated with 10 nm gold-conjugated anti-mouse secondary antibody. After washing, cells were fixed in 2.5% glutaraldehyde in cacodylate buffer, scraped off the plate and cell pellets were processed by conventional electron microscope procedures.
RESULTS
To elucidate whether expression of PSMA itself has a role in prostate cancer progression, full-length protein was expressed in PC3 cells (PC3-PSMA), a prostate cancer cell line devoid of endogenous PSMA (23). In addition to its plasma membrane localization (24), PSMA was distinctly evident in the vicinity of mitotic spindle poles around centrosomes (Fig. 1). Localization to the mitotic spindle pole regions was also observed in stable clones of MDCK cells expressing PSMA (MDCK-PSMA-1) (25) (Fig.1). Strikingly, cells with multiple centrosomes showed PSMA localized around each additional centrosome in both PC3-PSMA and MDCK-PSMA cells (Fig. 1, inserts). Localization to the spindle pole region was contingent upon the cytoplasmic tail of PSMA, as removal of this domain (PSMA-ΔCD) resulted in the loss of localization around the centrosomes with expression primarily limited to the plasma membrane (27) and trans Golgi network (Fig. 1). Furthermore, a PSMA deletion mutant lacking most of its extracellular domain (MDCK-PSMA-Δ103-750) (26) showed distinct localization in the vicinity of mitotic spindle poles (Fig. 1) indicating that the glutamate carboxy peptidase activity of PSMA which is localized to the extracellular domain (1) is not necessary for its spindle pole localization. Immunogold electron microscopy further confirmed localization of PSMA around the centrosomes at the spindle pole region. At higher magnification a distinct labeling proximal to membranes in the centrosomal region was visible (Fig. 2).
Figure 1.

Localization of PSMA at the mitotic spindle poles. Note distinct colocalization of PSMA (green) with centrosomes (red) at the mitotic spindle poles in PC3-PSMA, MDCK-PSMA-1, and MDCK-PSMA-Δ103-750 cells but not in MDCK-PSMA-ΔCD cells. Scale bars, 8 μm. PSMA, prostate-specific membrane antigen.
Figure 2.

Immunoelectron microscopy showing PSMA localization around the centrosomes. A, Shows a cartoon of the electron micrograph shown in B. A, B, The arrows indicate centriolar cylinders and the asterisks indicate the gold particles (10 nm) decorating PSMA. One of the spindle poles is shown in this image. The condensed chromatin from metaphase chromosomes is indicated by an arrowhead in this mitotic cell. Insets in B are magnified images of regions corresponding to arrows in B. In C the magnification facilitates visualization of gold particles on membrane like structures indicated by arrows. Bars in A-C, 0.5 μm. PSMA, prostate-specific membrane antigen.
Given the function of spindle poles in cell division, we hypothesized that localization of PSMA in the vicinity of spindle poles is associated with a role in mitosis. Cells were synchronized in mitosis by nocodazole and cell cycle progression after its removal was monitored by flow cytometry (Table 1 and Fig. 3). Nocodazole treatment blocked over 90% of cells in G2/M with a DNA content of 4N, for both PC3 and MDCK cells, irrespective of PSMA expression, suggesting efficient synchronization of cells in G2/M (Fig. 3). PC3-PSMA cells and two independent clones of MDCK-PSMA cells (MDCK-PSMA-1 and MDCK-PSMA-2) exited mitosis in an accelerated manner compared to control cells that do not express PSMA (Fig. 3, PC3 and MDCK-pCDNA3, respectively). Strikingly, MDCK-PSMA-ΔCD cells entered G1 slower than either MDCK-PSMA clone, but with a rate closer to that shown by MDCK-pCDNA3 (empty vector) cells (Fig. 3). The MDCK cells exhibited slower recovery from nocodazole release, necessitating the observation of later time points. Furthermore, MDCK-PSMA-Δ103-750 cells entered G1 faster than MDCK-pCDNA3 cells and similar to MDCK-PSMA cells (Fig. 3). Taken together, these results demonstrated that the cytoplasmic tail is essential for PSMA localization to the mitotic spindle pole region and accelerated exit from mitosis.
Table 1.
Cell cycle progression in nocodazole blocked cells
| Cell lines | % of cells in G1 ± SEM after release of nocodazole block | ||
|---|---|---|---|
| PC3 PC3-PSMA |
Time (minutes) | ||
| 60 | 90 | 120 | |
| 3 ± 1 25 ± 2 |
43 ± 3 52 ± 1 |
69 ± 3 79 ± 1 |
|
| MDCK-pCDNA3 MDCK-PSMA-1 MDCK-PSMA-2 MDCK-PSMA-ΔCD MDCK-PSMA-Δ103-750 |
Time (hours) | ||
| 2 | 4 | 8 | |
| 25 ± 1 43 ± 1 57 ± 2 25 ± 1 22 ± 1 |
29 ± 1 53 ± 3 64 ± 2 39 ± 1 43 ± 0 |
41 ± 2 64 ± 1 81 ± 3 52 ± 1 68 ± 3 |
|
| HCT-GFP HCT-PSMA |
Time (hours) | ||
| 1 | 2 | 4 | |
| 6 ± 1 11 ± 1 |
22 ± 0 43 ± 1 |
28 ± 2 47 ± 3 |
|
Data represent the means ± SEM of two (PC3, HCT) and three (MDCK) independent experiments.
Figure 3.

Analysis of the cell cycle progression in PSMA expressing cells after release of the mitotic block. Cells were blocked in mitosis as described in Materials and Methods. The mitotic block was released for 60, 90, and 120 minutes for PC3 clones (A, B) and 2, 4, and 8 hours for MDCK clones (C-G) and analyzed for cell cycle progression. Profiles shown are representative of the data shown in Table 1.
Entry and exit from mitosis are regulated primarily through control of (cyclin dependent kinase) CDK1 activity via ubiquitin-mediated proteolysis of cyclin B (7, 31). As the chromosomes attach to spindle microtubules and become properly aligned at the metaphase plate, APC becomes activated and targets key substrates, including cyclin B, for degradation (32, 33). The mitotic spindle checkpoint halts the action of APC and acts to restrain cells from entering anaphase (16). We hypothesized that accelerated exit from mitosis of PSMA expressing cells is due to impaired spindle checkpoint with increased APC activity leading to premature degradation of cyclin B. In PC3-PSMA cells, during nocodazole arrest and following mitotic release, cyclin B1 levels were consistently lower than in control PC3 cells (Fig. 4A), suggesting increased activity and/or incomplete inactivation of the APC in PSMA expressing cells. To test this possibility, we developed an in vitro assay to monitor APC’s ubiquitin ligase activity on cyclin B. Since APC is a large protein complex consisting of >10 subunits and the active enzyme has not been successfully purified in vitro (34, 35), we used total cell lysates as the source of APC. Since UbcH10 is the specific ubiquitin-conjugating enzyme (E2) for APC substrates and no other activity has been ascribed to this enzyme, our APC assay specifically detects APC activity. Furthermore, in mitotic HeLa cell lysates ubiquitination of cyclin B1 was greatly amplified by the addition of increasing amounts of UbcH10 and inhibited by dominant negative UbcH10 (our unpublished data). Relative to PC3 cells, the APC ubiquitin ligase activity for cyclin B was substantially higher at 0, 60, and 120 minutes after release from nocodazole block in PC3-PSMA cells (Fig. 4B). Therefore, increased APC activity in nocodazole blocked PC3-PSMA cells indicated that APC is incompletely inactivated by the mitotic spindle checkpoint, and confirmed our hypothesis that PSMA expression leads to a weakened SAC function in prostate cancer cells.
Figure 4.

Cyclin B1 levels and APC activation and association with PSMA. A, Immunoblot analysis of cyclin B1 in PC3 and PC3-PSMA cells after release of the mitotic block. Data represent the means ± SD of two independent experiments. B, In vitro APC ubiquitin ligase activity assay in PC3 and PC3-PSMA cells after 60 and 120 min release from nocodazole block.
The APC function is monitored by a complex pattern of regulation (32).Mad2 and BubR1 are well-characterized negative regulators of APC (6), while Cdc20 activates APC function (36). However, immunoblot analysis revealed that asynchronous populations of PC3 and PC3-PSMA cells expressed similar levels of the APC regulators Mad2, BubR1, and Cdc20, and of the core subunit of the APC, Cdc27 (Fig. 5A). The levels of BubR1, Cdc20, and Cdc27 were all increased to a similar extent in mitotically active PC3 cells independent of PSMA expression. Due to phosphorylation, Cdc27 and BubR1 showed multiple bands with the banding pattern and intensity of these bands being comparable in PC3 and PC3-PSMA cells (Fig. 5A). Co-immunoprecipitation analysis using anti-Cdc27 antibody revealed similar levels of Mad2, BubR1, and Cdc20 associated with APC in both PC3 and PC3-PSMA cells (Fig. 5B) and in addition, Mad2, BubR1 or Cdc20 were not detected in PSMA immunoprecipitates (data not shown), making it unlikely that PSMA activates APC by altering association of these regulators with APC.
Figure 5.

Association of PSMA with APC. A, Immunoblot analysis of Mad2, BubR1, Cdc20, and Cdc27 in interphase (I) and Mitotic (M) cells. B, Co-immunoprecipitation of Mad2, BubR1, and Cdc20 with Cdc27 in PC3 and PC3-PSMA cells. Equal amounts of Cdc 27 used in the immunoprecipitation (IP) was confirmed by immunoblotting. C, Co-immunoprecipitation of PSMA with Cdc27 (left top) and Cdc27 co-immunoprecipitating PSMA (left bottom) in PC3-PSMA and MDCK-PSMA cells. Note Cdc27 does not co-immunoprecipitate with PSMA-ΔCD. Affinity precipitation of Cdc27 by GST-PSMA from PC3 cell lysate (right). D, Co-immunoprecipitation of Cdc27 with PSMA in prostate cancer tissues (top). PSMA levels in same tissues are shown (bottom).
We then tested whether PSMA associates with the core APC complex. We observed that in both PC3-PSMA and MDCK-PSMA cells Cdc27 co-immunoprecipitated with PSMA (Fig. 5C, left panel) but not with the cytoplasmic tail deletion mutant of PSMA in MDCK-PSMA-ΔCD cells confirming that the cytoplasmic tail mediates the association of PSMA with APC. These results were further verified by in vitro pull-down assays from PC3-PSMA cell lysates using a GST-fusion protein containing the cytoplasmic tail of PSMA (GST-PSMA-CD) (Fig. 5C, right panel). We were also able to co-immunoprecipitate PSMA and Cdc27 in lysates made from six PSMA-positive tumor tissue samples (Fig. 5D). Taken together, these results demonstrated that the cytoplasmic tail of PSMA associates with Cdc27 in both cultured cells and in prostate tumor tissues and this association results in an increased APC activity in PSMA expressing cells.
The elevated APC activity in PSMA expressing cells resulting in premature degradation of cyclin B1 would provide less time for chromosome segregation, increasing the likelihood of aneuploidy. In order to assess the significance of PSMA expression on genomic stability, we expressed PSMA in HCT-116 cells (HCT-PSMA). Derived from colorectal carcinoma, the HCT-116 cell line possesses a nearly diploid karyotype and is an established model for studying chromosomal instability (37). The diploid karyotype of this cell line was confirmed by standard karyotype analysis (our unpublished data). Moreover, since aneuploidy is an irreversible event and since prostate cancer cells expressing PSMA such as LNCaP are aneuploid, RNAi mediated knockdown of PSMA in these cells is less likely to provide information as to whether PSMA expression is associated with aneuploidy. Therefore, HCT-116 cells were used as a model to test whether PSMA expression induces aneuploidy. PSMA co-localized with centrosomes in HCT-PSMA cells, and these cells exited mitosis faster than control HCT-GFP cells (Table 1). Fluorescence in situ hybridization (FISH) was done using probes specific for chromosomes 3, 7, 17 centromeres, and 9p21 region, respectively. No chromosomal abnormalities were observed between HCT-PSMA and HCT-GFP cells at passage 2 (Fig. 6B). However, at higher passage numbers of 15, 34, and 45, all these chromosomes showed various degrees of aneuploidy in HCT-PSMA cells (Fig. 6C-G). The frequency of aneuploidy increased with passage number, and by passage 45 there was significant increase in the number of aneuploidy in PSMA expressing cells (P<0.001, chi square = 13.25; n=1000). In addition, clear evidence of micronuclei formation was observed (Fig. 6E, arrow, and F) which provided further evidence for chromosomal instability (38).
Figure 6.
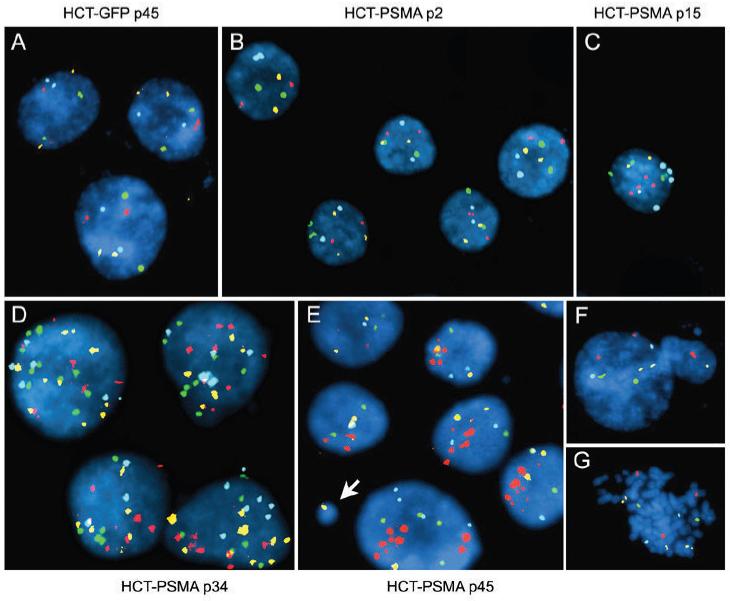
PSMA expressing human cells are genetically unstable. HCT-PSMA and HCT-GFP cells were analyzed by multicolor FISH with Chromosome 3 labeled with Spectrum Red (CEP3), Chromosome 7 with Spectrum Green (CEP7), Chromosome 17 with Spectrum Aqua (CEP 17), and 9p21 region (p16 gene) with Spectrum Gold. Control HCT-GFP cells at passage 45 (A), and HCT-PSMA cells at passage 2 (B), are diploid whereas HCT-PSMA cells at passage 15 (C), 34 (D), and 45 (E) show aneuploidy. Micronucleus formation (E, arrow and F) and abnormal metaphase, (G) in HCT-PSMA cells at passage 45 are shown.
DISCUSSION
In this study, we report that PSMA is localized to a membrane compartment in the vicinity of the mitotic spindle poles. The J591 monoclonal antibody against PSMA used in this study has been extensively used for immunofluoresecence, immunogold labeling, immunoblot and immunoprecipitation analyses cited in numerous publications (24, 26-28, 39). This antibody detects a single band in immunoblot and immunoprecipitation analysis (25, 28) and cells lacking PSMA expression do not reveal any background staining (27, 39). Therefore, the staining observed around the centrosomes is specific to the localization of PSMA at this site.
Although PSMA is localized to a compartment around the centrosomes, it is important to note that PSMA is not a core centrosomal component. Treatment of cells with nocodazole completely abrogated PSMA localization around the centrosome (our unpublished data), unlike core centrosomal components that still associate with centrosomes following nocodazole treatment. We suggest that PSMA is localized to a membrane compartment around the centrosomes at the vicinity of spindle poles. Recent studies have indicated that important regulators of endocytic traffic, such as clathrin (40, 41), myosin Vb and rab11 (42), as well as cargo molecules, such as the polymeric IgA receptor (42) are localized to the spindle poles. PSMA is internalized via clathrin coated pits (28) and expression of K44A dynamin which inhibits clathrin-dependent endocytosis prevents internalization of PSMA (27). These results are consistent with the idea of the presence of a membrane compartment in the vicinity of the spindle poles that is involved in clathrin-mediated endocytosis. At this point it is not known whether the PSMA localized to the spindle pole is internalized from the plasma membrane or delivered to this site via a biosynthetic route. The origin and nature of this compartment and how PSMA is targeted to this membrane compartment will be addressed in future studies. Interestingly, LNCaP cells that express high levels of endogenous PSMA and which have been widely used as a model in prostate cancer research did not reveal PSMA localization around centrosomes. Further analysis in this cell line revealed that this cell line has defects in the organization of key membrane compartments such as Golgi apparatus and endosomes (our unpublished observations). Studies are in progress to address the difference in the localization of PSMA in LNCaP cells.
We have demonstrated that PSMA expressing cells exit mitosis prematurely. While the effects on cell cycle are small, they are reproducible and significant and have been confirmed in multiple cell lines. Cells expressing the cytoplasmic tail deletion mutant of PSMA progress through the cell cycle similar to control cells without PSMA expression, and PSMA-ΔCD does not associate with APC establishing further specificity to our experiments. It is important to note that PSMA association with APC probably does not inhibit the spindle checkpoint per se, but rather partially uncouples APC activity either directly or indirectly from spindle checkpoint control. Several reports have described spindle checkpoint proteins such as early mitotic inhibitor (Emi1) and tumor suppressor RASSF1A to inhibit APC by interacting with the activator Cdc20 (22, 36, 43). Recently, the oncoprotein Tax of the human T lymphotropic virus type 1 (HTLV-1) has been shown to activate APC by directly binding to Cdc20 and leaving the spindle checkpoint intact (44). A protein called Xnf7 is unusual in that it inhibits the APC by interaction with Cdc27 (45). Here we presented evidence for a protein that mediates APC activation by interaction with Cdc27 and impairing the mitotic spindle checkpoint. This could be achieved by maintaining high concentrations of the dynamic active APC complex at the spindle poles, a site for initiation of cyclin B1 ubiquitination during mitosis (46). A mechanism for inactivating SAC has recently been ascribed to the non-degrading ubiquitination of the mitotic activator subunit Cdc20 by the APC in conjunction with the E2 UbcH10, causing disassociation of checkpoint proteins from Cdc20 (47). Perhaps the increased APC activity observed in the PSMA expressing cells is sufficient to impair SAC activity. Conversely, the deubiquitinating enzyme USP44 deubiquitinates Cdc20 and drives the assembly of the inhibitory SAC complexes (48). PSMA could potentially act at any of these steps to increase APC activity. Consequently, the spindle checkpoint would be impaired in performing its function to keep APC inactive. Treatment of cells with strong inhibitors of the spindle checkpoint such as Hesperadin or ZM447439 (Aurora kinase inhibitor) results in these cells entering endoreduplication cycles and exhibiting polyploidy within 24-48 hours. In contrast, in PSMA expressing cells aneuploidy is not observed until the 15th passage. The effect is subtle and we believe more relevant to the biological evolution of aneuploidy in a tumor cell.
PSMA is not expressed at high levels in normal prostatic epithelial cells. However, expression is significantly elevated during the progression of cancer, with the greatest levels observed in high-grade tumors, metastatic lesions, and androgen independent disease. Thus, increased expression of PSMA directly correlates with the aggressiveness of the disease. Low expression of PSMA per se might not constitute a factor leading to aneuploidy, since low levels of PSMA are found in benign prostate tumors and some normal non-prostatic tissues (1, 2). Our data strongly suggest that upon increased expression of PSMA, the ability of its cytoplasmic tail to associate with the APC complex dysregulates APC function leading to aneuploidy in cancer cells. Thus, PSMA function might not be required for normal cell cycle progression but its elevated expression and mislocalization at the spindle pole region and association with APC is an accidental and pathological consequence in cancer cells leading to aneuploidy. An understanding of the putative oncogenic role of PSMA could have major implications for the management and therapy of prostate cancer. Targeted therapies against PSMA expressing prostate cancer cells are already being evaluated for their potential to treat prostate cancer (49). Since PSMA expression is closely associated with aneuploidy, such anti-PSMA therapeutic strategies might have the advantage of specifically targeting the most aggressive and aneuploid cells.
ACKNOWLEDGEMENTS
We thank Drs. Warren Heston (Cleveland Clinic Foundation, OH) for PSMA cDNA, Michel Sadelain (Memorial Sloan Kettering Cancer Center, NY) for PC3-PSMA and Bert Vogelstein (Johns Hopkins University, MD) for HCT-116 cells, Neil Bander (Weill Medical School Cornell University, NY) for mAb J591, Wei Dai (University of Oklahoma, OK) for anti-BubR1 antibody, and Kuan-Teh Jeang (National Institute of Health, MD) for anti-Mad2 antibody. We thank Dr. Gregory Payne for critical reading of the manuscript.
Financial support: This study was supported by grants from DOD W81XWH-04-1-0113 and NIH DK56216.
Abbreviations List
- APC
anaphase-promoting complex
- CDK
cyclin-dependent kinase
- CIN
chromosomal instability
- DAPI
diamino-2-phenylindole dihydrochloride
- ECL
enhanced chemiluminescence
- FISH
Fluorescence in situ hybridization
- FITC
fluorescein isothiocyanate
- MDCK
Madin Darby Canine Kidney
- MDCK-PSMA
Madin Darby canine kidney cells expressing PSMA
- NAALADase
N-acetylated alpha-linked acidic peptidase
- PC3-PSMA
PC3 cells expressing prostate specific membrane antigen
- PI
propidium iodide
- PSMA
Prostate specific membrane antigen
- SAC
spindle assembly checkpoint
REFERENCES
- 1.Rajasekaran AK, Anilkumar G, Christiansen JJ. Is prostate-specific membrane antigen a multifunctional protein? Am J Physiol Cell Physiol. 2005;288:C975–81. doi: 10.1152/ajpcell.00506.2004. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh A, Heston WD. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J Cell Biochem. 2004;91:528–39. doi: 10.1002/jcb.10661. [DOI] [PubMed] [Google Scholar]
- 3.Wright GL, Jr., Grob BM, Haley C, et al. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology. 1996;48:326–34. doi: 10.1016/s0090-4295(96)00184-7. [DOI] [PubMed] [Google Scholar]
- 4.Cleveland DW, Mao Y, Sullivan KF. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell. 2003;112:407–21. doi: 10.1016/s0092-8674(03)00115-6. [DOI] [PubMed] [Google Scholar]
- 5.Georgi AB, Stukenberg PT, Kirschner MW. Timing of events in mitosis. Curr Biol. 2002;12:105–14. doi: 10.1016/s0960-9822(01)00662-5. [DOI] [PubMed] [Google Scholar]
- 6.Fang G. Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol Biol Cell. 2002;13:755–66. doi: 10.1091/mbc.01-09-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Potapova TA, Daum JR, Pittman BD, et al. The reversibility of mitotic exit in vertebrate cells. Nature. 2006;440:954–8. doi: 10.1038/nature04652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piel M, Nordberg J, Euteneuer U, Bornens M. Centrosome-dependent exit of cytokinesis in animal cells. Science. 2001;291:1550–3. doi: 10.1126/science.1057330. [DOI] [PubMed] [Google Scholar]
- 9.Wakefield JG, Huang JY, Raff JW. Centrosomes have a role in regulating the destruction of cyclin B in early Drosophila embryos. Curr Biol. 2000;10:1367–70. doi: 10.1016/s0960-9822(00)00776-4. [DOI] [PubMed] [Google Scholar]
- 10.Kraft C, Herzog F, Gieffers C, et al. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. Embo J. 2003;22:6598–609. doi: 10.1093/emboj/cdg627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fraschini R, Formenti E, Lucchini G, Piatti S. Budding yeast Bub2 is localized at spindle pole bodies and activates the mitotic checkpoint via a different pathway from Mad2. J Cell Biol. 1999;145:979–91. doi: 10.1083/jcb.145.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howell BJ, Hoffman DB, Fang G, Murray AW, Salmon ED. Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J Cell Biol. 2000;150:1233–50. doi: 10.1083/jcb.150.6.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin DY, Spencer F, Jeang KT. Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell. 1998;93:81–91. doi: 10.1016/s0092-8674(00)81148-4. [DOI] [PubMed] [Google Scholar]
- 14.Pereira G, Manson C, Grindlay J, Schiebel E. Regulation of the Bfa1p-Bub2p complex at spindle pole bodies by the cell cycle phosphatase Cdc14p. J Cell Biol. 2002;157:367–79. doi: 10.1083/jcb.200112085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bharadwaj R, Yu H. The spindle checkpoint, aneuploidy, and cancer. Oncogene. 2004;23:2016–27. doi: 10.1038/sj.onc.1207374. [DOI] [PubMed] [Google Scholar]
- 16.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–93. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 17.Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–3. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 18.Grady WM. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004;23:11–27. doi: 10.1023/a:1025861527711. [DOI] [PubMed] [Google Scholar]
- 19.Hernando E, Orlow I, Liberal V, Nohales G, Benezra R, Cordon-Cardo C. Molecular analyses of the mitotic checkpoint components hsMAD2, hBUB1 and hBUB3 in human cancer. Int J Cancer. 2001;95:223–7. doi: 10.1002/1097-0215(20010720)95:4<223::aid-ijc1038>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 20.Iwanaga Y, Chi YH, Miyazato A, et al. Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res. 2007;67:160–6. doi: 10.1158/0008-5472.CAN-06-3326. [DOI] [PubMed] [Google Scholar]
- 21.Michel L, Diaz-Rodriguez E, Narayan G, Hernando E, Murty VV, Benezra R. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc Natl Acad Sci U S A. 2004;101:4459–64. doi: 10.1073/pnas.0306069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song MS, Song SJ, Ayad NG, et al. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat Cell Biol. 2004;6:129–37. doi: 10.1038/ncb1091. [DOI] [PubMed] [Google Scholar]
- 23.Chang SS, Reuter VE, Heston WD, Bander NH, Grauer LS, Gaudin PB. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res. 1999;59:3192–8. [PubMed] [Google Scholar]
- 24.Anilkumar G, Rajasekaran SA, Wang S, Hankinson O, Bander NH, Rajasekaran AK. Prostate-specific membrane antigen association with filamin A modulates its internalization and NAALADase activity. Cancer Res. 2003;63:2645–8. [PubMed] [Google Scholar]
- 25.Christiansen JJ, Rajasekaran SA, Moy P, et al. Polarity of prostate specific membrane antigen, prostate stem cell antigen, and prostate specific antigen in prostate tissue and in a cultured epithelial cell line. Prostate. 2003;55:9–19. doi: 10.1002/pros.10203. [DOI] [PubMed] [Google Scholar]
- 26.Christiansen JJ, Rajasekaran SA, Inge L, et al. N-glycosylation and microtubule integrity are involved in apical targeting of prostate-specific membrane antigen: implications for immunotherapy. Mol Cancer Ther. 2005;4:704–14. doi: 10.1158/1535-7163.MCT-04-0171. [DOI] [PubMed] [Google Scholar]
- 27.Rajasekaran SA, Anilkumar G, Oshima E, et al. A novel cytoplasmic tail MXXXL motif mediates the internalization of prostate-specific membrane antigen. Mol Biol Cell. 2003;14:4835–45. doi: 10.1091/mbc.E02-11-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, Moy P, Kim S, et al. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997;57:3629–34. [PubMed] [Google Scholar]
- 29.Krishan A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol. 1975;66:188–93. doi: 10.1083/jcb.66.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu H, Lan Z, Li W, et al. p55CDC/hCDC20 is associated with BUBR1 and may be a downstream target of the spindle checkpoint kinase. Oncogene. 2000;19:4557–62. doi: 10.1038/sj.onc.1203803. [DOI] [PubMed] [Google Scholar]
- 31.King RW, Deshaies RJ, Peters JM, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–9. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- 32.Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–56. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- 33.Sudakin V, Ganoth D, Dahan A, et al. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–97. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herzog F, Peters J. Large[hyphen (true graphic)]Scale Purification of the Vertebrate Anaphase[hyphen (true graphic)]Promoting Complex/Cyclosome. In: Deshaies RJ, editor. Methods in Enzymology. Academic Press; 2005. pp. 175–95. [DOI] [PubMed] [Google Scholar]
- 35.Tang Z, Yu H. Functional analysis of the spindle-checkpoint proteins using an in vitro ubiquitination assay. In: Schoenthal AH, editor. Checkpoint Controls and Cancer. Humana Press; Totowa, New Jersey: 2004. pp. 227–42. [DOI] [PubMed] [Google Scholar]
- 36.Yu H. Regulation of APC-Cdc20 by the spindle checkpoint. Curr Opin Cell Biol. 2002;14:706–14. doi: 10.1016/s0955-0674(02)00382-4. [DOI] [PubMed] [Google Scholar]
- 37.Fodde R, Kuipers J, Rosenberg C, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–8. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- 38.Rajagopalan H, Jallepalli PV, Rago C, et al. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- 39.Liu H, Rajasekaran AK, Moy P, et al. Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Cancer Res. 1998;58:4055–60. [PubMed] [Google Scholar]
- 40.Okamoto CT, McKinney J, Jeng YY. Clathrin in mitotic spindles. Am J Physiol Cell Physiol. 2000;279:C369–74. doi: 10.1152/ajpcell.2000.279.2.C369. [DOI] [PubMed] [Google Scholar]
- 41.Royle SJ, Bright NA, Lagnado L. Clathrin is required for the function of the mitotic spindle. Nature. 2005;434:1152–7. doi: 10.1038/nature03502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hobdy-Henderson KC, Hales CM, Lapierre LA, Cheney RE, Goldenring JR. Dynamics of the apical plasma membrane recycling system during cell division. Traffic. 2003;4:681–93. doi: 10.1034/j.1600-0854.2003.00124.x. [DOI] [PubMed] [Google Scholar]
- 43.Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell. 2001;105:645–55. doi: 10.1016/s0092-8674(01)00361-0. [DOI] [PubMed] [Google Scholar]
- 44.Liu B, Hong S, Tang Z, Yu H, Giam CZ. HTLV-I Tax directly binds the Cdc20-associated anaphase-promoting complex and activates it ahead of schedule. Proc Natl Acad Sci U S A. 2005;102:63–8. doi: 10.1073/pnas.0406424101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Casaletto JB, Nutt LK, Wu Q, et al. Inhibition of the anaphase-promoting complex by the Xnf7 ubiquitin ligase. J Cell Biol. 2005;169:61–71. doi: 10.1083/jcb.200411056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82–7. doi: 10.1038/10049. [DOI] [PubMed] [Google Scholar]
- 47.Reddy SK, Rape M, Margansky WA, Kirschner MW. Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature. 2007;446:921–5. doi: 10.1038/nature05734. [DOI] [PubMed] [Google Scholar]
- 48.Stegmeier F, Rape M, Draviam VM, et al. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature. 2007;446:876–81. doi: 10.1038/nature05694. [DOI] [PubMed] [Google Scholar]
- 49.Bander NH, Nanus DM, Milowsky MI, Kostakoglu L, Vallabahajosula S, Goldsmith SJ. Targeted systemic therapy of prostate cancer with a monoclonal antibody to prostate-specific membrane antigen. Semin Oncol. 2003;30:667–76. doi: 10.1016/s0093-7754(03)00358-0. [DOI] [PubMed] [Google Scholar]