

Abstract

Treatment of aryl iodides with indium metal in the presence of lithium chloride leads to the formation of an organoindium reagent capable of participating in cross-coupling reactions under transition-metal catalysis. Combination with aryl halides in the presence of 5 mol% Cl2Pd(dppf) furnishes biaryl compounds in good yields; similarly, reaction with acyl halides or allylic acetates/carbonates in the presence of 5–10 mol % palladium catalyst leads to arylketones and allylic substitution products, respectively, in moderate yields. The reactions are tolerant of the presence of protic solvents, and ~85% of the indium metal employed can be recovered by reduction of the residual indium salts with zinc(0).
Organoindium compounds have been demonstrated to participate in a wide range of transition-metal-mediated processes for carbon–carbon bond formation.1 These environmentally benign reagents are air- and moisture-stable,2 and can undergo cross-coupling reactions in an atom-efficient manner. One current limitation in the use of organoindiums is the necessity of preparing the reagents by the combination of organomagnesium or organolithium reagents with indium trichloride. As a result of the highly reactive nature of lithium or Grignard organometallics,3 the scope of indium reagents that can be prepared by this method may be limited.4 Furthermore, indium(III) salts such as InCl3 are currently under investigation as possible mutagens.5 A direct method of preparing organoindium reagents for transition-metal-mediated reactions is thus highly desirable.
Knochel has shown that highly functionalized organozinc and organomagnesium reagents can be formed efficiently from the corresponding organic halides and zinc or magnesium metal in the presence of lithium chloride.6 These organometallics readily engage in subsequent transition-metal-mediated carbon–carbon bond-forming reactions. A recent patent from the same group7 has indicated that this method may also be extended to the preparation of organometals containing Cu, Sn, Mn, and In, although Zn was preferable to all others tested. Despite the utility of zinc-mediated processes, an involved procedure for activation of the zinc metal (including use of TMSCl and iodine) is necessary, and the separation of the organozinc reagent solution from any remaining finely dispersed zinc dust is often problematic (zinc metal impurities can interfere with the subsequent cross-coupling reaction).8 Furthermore, the zinc organometallics so formed are both air- and moisture-sensitive. In this Note we wish to describe our studies on the utility, scope, and limitations of the direct reaction of aryl halides with indium metal in the preparation organoindium reagents.
Because of the ease with which allylic indium reagents are formed upon combination of indium metal and allyl halides in polar solvents such as water,9 we reasoned that minimal activation of the indium metal would be necessary to obtain successful insertion reactions with organic halides. Indeed, when methyl 4-iodobenzoate was treated with a slight excess (1.5 equiv) of In•2LiCl (prepared by heating 1 equiv of indium metal with 2 equiv of dry LiCl at 110 °C in vacuo for 20 min) in DMF at 80 °C for 12 h, only trace amounts of the starting iodide were evident by TLC, and GC–MS analysis of a reaction aliquot revealed >95% consumption of stating material. A key to the success of this method was the use of indium metal containing 0.1% magnesium10 as an anticaking reagent; when the magnesium was omitted, the indium metal clumped together on the walls of the reaction flask, and thus a significant excess of indium metal was needed in order to achieve optimal conversion. With In/0.1% Mg, after 12 h at 80 °C it was noticed that the remaining indium metal had also clumped together in the bottom of the reaction flask, thus facilitating the separation of the reagent solution for the subsequent cross-coupling. Addition of the DMF solution of the indium reagent to 4-iodotoluene (0.67 equiv) and 5 mol % Cl2Pd(dppf) and heating at 80 °C for 2h led to the clean formation of biaryl 3 in 85% yield (Table 1, entry 1). DMF was the most suitable solvent for the two-step process, since yields of cross-coupled product obtained using THF or DME instead were significantly lower (20–25%, entries 2 and 3); however, addition of methanol (10% by volume) to the DMF solution of the indium reagent just prior to the cross-coupling reaction had no effect on the yield of 3a11 obtained (entry 4). Furthermore, it was discovered that the time for efficient formation of the indium reagent can be reduced to 2 h when the reaction temperature is raised to 100 °C. Aryl bromides were also suitable substrates for the indium insertion, although they were significantly less reactive than aryl iodides: methyl 4-bromobenzoate required heating with In•2LiCl at 100 °C for 12 h to achieve >90% consumption of the starting material, and the subsequent palladium-catalyzed cross-coupling reaction furnished 3a in only 45% yield, indicating that reagent degradation may be taking place during the prolonged heating at elevated temperature (entry 6). Utilizing InI instead of indium metal gave ~55% insertion and a 42% yield of 3a; substituting InCl2 for indium metal gave very poor insertion (<20% by GC–MS analysis) and <10% yield of cross-coupled product.
Table 1.
Exploring Optimal Conditions for Formation and Cross Coupling of Organoindium Reagents
 | ||||||
|---|---|---|---|---|---|---|
| Entry | M | X | solvent | T(°C) | time (h) | % yield 3aa |
| 1 | In | I | DMF | 80 | 12 | 85 |
| 2 | In | I | THF | 65 | 12 | 20 |
| 3 | In | I | DME | 80 | 12 | 25 |
| 4 | In | I | DMFb | 80 | 12 | 83 |
| 5 | In | I | DMF | 100 | 2 | 80 |
| 6 | In | Br | DMF | 100 | 12 | 45 |
| 7 | InI | I | DMF | 80 | 12 | 42 |
| 8 | InCl2 | I | DMF | 80 | 12 | <10 |
Refers to isolated yields after column chromatography.
Methanol (10% by volume) was added to the DMF solution of the indium reagent just prior to the cross-coupling reaction.
We next examined the ability of aryl iodides with different substitution patterns to undergo the two-step insertion/cross-coupling process. We surveyed the reactivity of both electron-deficient (Table 2, entries 1–14) and electron-rich (entry 15) aryl iodides 1 toward indium metal. Paralleling Knochel’s findings on the reactivity of aryl halides toward zinc insertion,6a we discovered that electron-poor substrates such as 4-iodoacetophenone, 3-and 4-iodobenzonitrile, and methyl 4-iodobenzoate undergo >95% insertion of indium metal at 80 °C in DMF for 12 h. In contrast, the electron-rich 4-iodoanisole required heating at 120 °C for 12 h to effect complete consumption of the starting material (entry 15). Subsequent cross-coupling with aryl iodides12 in the presence of Cl2Pd(ddpf) as catalyst gave the desired biaryls 3b–p in 35–89% yields. Unsurprisingly, reactions requiring higher temperatures to effect the insertion and/or cross-coupling led to lower yields of biaryl products (entries 9, 12 and 15); we suspect that steric issues necessitate the higher temperatures for cross-couplings with 2-iodotoluene and 2-iodoanisole. Furthermore, in several cases where lower yields were obtained (entries 6, 10, 12, and 15), significant amounts of homodimer 4 (presumably arising from palladium(II)-mediated dimerization of the indium reagent) were also isolated from the reaction mixture. To assess if the indium metal was reducing the aryl iodides to the parent aromatic compounds (R1-H) prior to the cross-coupling reaction, we performed a TLC assay on the low-yielding reactions and found that no significant amounts of reduced product were evident in the reaction mixtures.
Table 2.
Reaction of Diverse Aryl Iodides with In•2LiCl: Scope and Cross-Coupling Reactions
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R1 | % conv. 1a | R2 | T (°C) | time (h) | 3 | % yield 3b |
| 1 | 4-CO2Me- C6H4 | 97 | 4-OMe | 80 | 2 | b | 75 |
| 2 | 4-CO2Me-C6H4 | 4-Ac | 80 | 4 | c | 65 | |
| 3 | 4-CO2Me- C6H4 | 4-CN | 80 | 3 | d | 59 | |
| 4 | 4-CO2Me-C6H4 | 2-CO2Me | 80 | 2 | e | 55 | |
| 5 | 4-CO2Me- C6H4 | H | 80 | 3 | f | 63 | |
| 6 | 4-Ac-C6H4 | 98 | 4-Me | 80 | 3 | g | 50d,e |
| 7 | 3-CN-C6H4 | 98 | 4-Ac | 80 | 6 | h | 89d |
| 8 | 3-CN-C6H4 | 4-OMe | 80 | 6 | i | 75d | |
| 9 | 3-CN-C6H4 | 2-Me | 100 | 12 | j | 62d | |
| 10 | 4-CN-C6H4 | 97 | 4-Cl | 80 | 6 | k | 50d,e |
| 11 | 4-CN-C6H4 | 2-F | 80 | 3 | l | 88d,f | |
| 12 | 4-CN-C6H4 | 2-OMe | 100 | 3 | m | 37e | |
| 13 | 4-Cl-C6H4 | 87 | 4-Ac | 80 | 12 | n | 61d |
| 14 | 2-Ac-C6H4 | 88 | 4-CN | 90 | 15 | o | 72d |
| 15 | 4-OMe-C6H4 | 94c | 4-Ac | 100 | 5 | p | 35e |
For the insertion reaction, conversions were assayed by GC–MS analysis of an aliquot quenched with HCl/H2O.
Refers to isolated yields after column chromatography, unless otherwise indicated.
Insertion reaction performed at 120 °C for 12 h.
Yield determined by GC–MS analysis.
Significant amounts of homodimer 4 also produced.
Catalyst for cross-coupling was Cl2Pd(PPh3)2.
We wished to probe the identity of the organoindium intermediate being formed in the insertion reaction. Araki13 has reported that reactions of indium metal with allyl halides in organic solvents such as DMF produce indium(III) sesquihalide intermediates; Chan14 has demonstrated that the same reaction in water proceeds via formation of an allylindium(I) species. We prepared a series of arylindium reagents by reaction of phenylmagnesium bromide (3 M in diethyl ether) with 1 equiv of InI, InCl2, or InCl3, respectively, in THF (0 °C, 1 h), and then cross-coupled each with 1 equiv of methyl 4-iodobenzoate in the presence of 5 mol % Cl2Pd(dppf) (DMF, 80 °C, 2 h). To our surprise, we found that the reagents derived from both InI and InCl2 gave >80% yield of cross-coupled product 3f; in contrast, PhInCl2 gave only ~55% yield of 3f. The lower reactivity of mono- and diorganoindiums versus triorganoindiums in transition-metal-mediated coupling reactions has been previously noted.15 Interestingly, Ph2In, prepared from InCl2 by reaction with 2 equiv of PhMgBr, reacted with 2 equiv of methyl 4-iodobenzoate to furnish 3f in 83% yield, indicating that both phenyl groups on the In(II) reagent are transferable. When we repeated the above experiments but added methanol (10% by volume) to the indium reagent solution prior to the cross-coupling reaction, we discovered that >5% 3f was produced in the reaction involving the reagent prepared from InI and PhMgBr. However, the reagents derived from addition of 1 equiv of PhMgBr to InCl2 and InCl3 furnished 3f in 85% and 50% yields, respectively (Scheme 1). On the basis of these experiments, we can conclude that both the indium(II) and indium(III) oxidation states may be viable candidates for the intermediate formed in the insertion reaction.
Scheme 1.
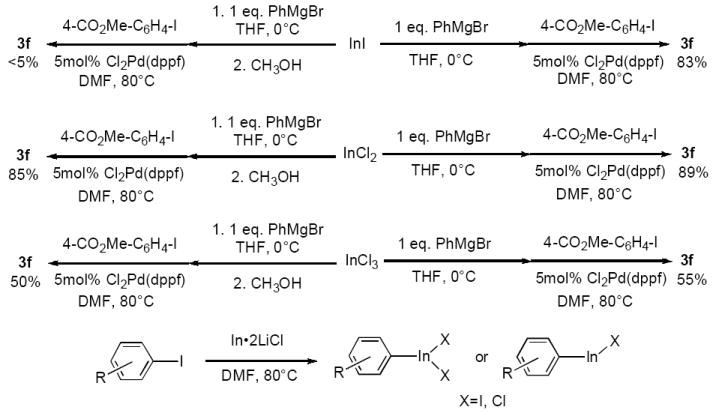
Probing the Identity of the Arylindium Intermediate.
Because of the expense of indium metal (~$7/g), we were interested in recovering/regenerating the indium used in our experiments. When the indium metal is separated and weighed after the insertion reaction (following water/acetone rinse and drying in vacuo), we observed that for every millimole of aryl iodide employed, between 0.83 and 1.0 mmol of indium is consumed. The aqueous layer derived from workup of the cross-coupling reaction was filtered, neutralized, and then treated with ~1.5 equiv of zinc dust while stirring. Within minutes, clumps of indium metal formed and aggregated in the bottom of the flask; after removal of the water, rinsing with acetone, and drying in vacuo, we found that, in total, approximately 85% of indium metal originally employed could be recovered.16
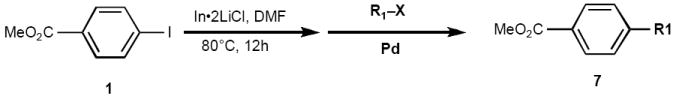
Finally, we evaluated the ability of the intermediate arylindium species to react with electrophiles other than aryl iodides. Acyl halides and allylic acetates and carbonates were selected as coupling partners for the in-situ generated indium reagent. As can be seen from Table 3, moderate yields were obtained for acyl substitution reactions involving acid chlorides under Pd(PPh3)4 (5 mol %) catalysis (entries 1, 2, and 3). Although there have been recent successful examples of transition-metal-catalyzed cross-couplings between acyl fluorides and arylzinc reagents,17 as well as between thioesters and arylindium reagents,18 we found that acid chlorides consistently gave the highest yields of arylketones. For example, employing hydrocinnamoyl fluoride instead of the corresponding chloride (entry 2) gave only 10% yield of 7b; using S-phenyl hex-5-ene thioate instead of hex-5-enoyl chloride (entry 3) gave <5% 7c. The use of other palladium catalysts such as Cl2Pd(dppf) or Pd(OAc)2/PPh3 gave negligible amounts of ketone products. Cross-coupling with the allylic electophiles cinnamyl acetate (entry 4) and methyl cyclohex-2-enyl carbonate (entry 5) under Pd(dba)2 (10 mol %) catalysis furnished substitution products 7d and 7e in 48% and 51% yields, respectively; several unidentified side products and well as homodimer 4a were also evident in the reaction mixtures. These results are most likely due to the increased stability of the intermediate aryl-palladium(II)-π allyl complex and thus the decreased rate of reductive elimination. Furthermore, the low reactivity of PhInCl2 in allylic substitution reactions has been previously noted.19 The addition of phosphine ligands such as PPh3, P(t-Bu)3, or dppf to the palladium catalyst led to no measurable improvement in the yields of product obtained.
Table 3.
Reaction of in Situ Generated Organoindium Reagents with Acyl and Allylic Electrophiles.a
 | |||||
|---|---|---|---|---|---|
| Entry | R1 | X | Pdb | 7 | % yield 7c |
| 1 | 4-Me-PhCO | Cl | A | 7a | 40 |
| 2 | PhCH2CH2CO | Cl | A | 7b | 45 |
| 3 | CH2CH(CH2)3CO | Cl | A | 7c | 40 |
| 4 |
|
OAc | B | 7d | 48 |
| 5 |
|
OCO2Me | B | 7e | 51 |
Typical reaction conditions: a DMF solution of the indium reagent (~0.81 M) was added to R1-X (1.0 equiv) and palladium catalyst (5–10 mol %) and heated to 80 °C under argon for 2 h, at which time complete consumption of R1-X was observed by TLC.
A= 5 mol % Pd(PPh3)4; B= 10 mol % Pd(dba)2.
Refers to isolated yields after column chromatography.
As part of our program directed toward the synthesis of β-C-aryl glycosides,20 we demonstrated the utility of our acyl substitution products by transforming aryl ketone 7c into tetrahydropyran 8 in three steps (Upjohn dihydroxylation,21 primary alcohol silylation, and triisopropylsilane-mediated ketol reduction22) in 62% overall yield (Scheme 2). The silane reduction proceeded with >10:1 stereoselectivity, and the 2,6-syn stereochemistry of the product was indicated by the C.1 proton–C.5 proton cross-peaks in the NOESY spectrum of 8 (see Supporting Information for details).
Scheme 2.

Synthesis of cis-2,6-Disubstituted Pyran 8 from Aryl Ketone 7c.
In summary, we have demonstrated that indium reagents capable of participating in transition-metal-catalyzed cross-coupling and substitution reactions can be formed by direct treatment of aryl iodides with In•2LiCl. Some noteworthy characteristics of this process are that minimal activation of the indium is required, biaryls are formed efficiently even in the presence of protic solvents, and the indium used can be regenerated by reduction of the residual indium salts with zinc metal. Further experiments to extend the scope and delineate the mechanism of this process are underway and will be reported in due course.
Experimental Section
General Procedure for the Synthesis of 3a–p
LiCl (84 mg, 2mmol) was heated at 150 °C for 20 min. Then indium powder (containing 0.1% Mg)10 (114 mg, 1 mmol) was added and the solids were heated with stirring in vacuo at 110 °C for an additional 20 min. The mixture was cooled to room temperature, and the aryl iodide (0.66 mmol) was added. Then DMF (0.5 mL) was added, and the mixture was stirred at 80°C under argon for 14 hours, at which time complete consumption of the iodide was observed by TLC. An additional portion of DMF (0.5 mL) was added, and the amber solution was withdrawn from the flask by syringe and added to a mixture of aryl iodide or bromide (0.44 mmol, 0.67 equiv) and Cl2Pd(dppf) (18.0 mg, 0.022 mmol, 5 mol %). The reaction was stirred at 80 °C under argon for 1–3 h, at which time TLC indicated complete conversion. The reaction was cooled to room temperature, and then diluted with ether (20 mL) and washed with 1 N HCl (2 × 20 mL). The combined aqueous phases were back-extracted with ether (1 × 50 mL). The combined organic extracts were then dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography (silica gel, hexane/Et2O = 99:1 to 95:5).
Methyl 4’-Methylbiphenyl-4-carboxylate (3a)
Following the general procedure, methyl 4-iodobenzoate (173 mg, 0.66 mmol) and 4-iodotoluene (96 mg, 0.44 mmol) were combined to provide 3a as a white solid (84 mg, 0.37 mmol, 85%). 1H NMR (400 MHz, CDCl3): δ 8.09 (d, J= 8.4 Hz, 2H); 7.64 (d, J= 8.4 Hz, 2H); 7.52 (d, J= 8.0 Hz, 2H); 7.27 (d, J= 8.4 Hz, 2H); 3.94 (s, 3H); 2.41 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 144.4; 142.5; 128.4; 127.5; 125.8; 109.3; 80.6; 43.0. GC/MS m/z=226 (M+).
Supplementary Material
Detailed experimental procedures, spectroscopic data, and 1H NMR spectra for all compounds in Tables 2 and 3 and Scheme 2, as well as NOESY data for compound 8. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the National Institutes of Health (SC2 GM081064-01), the ACS Petroleum Research Fund (No. PRF 45277-B1), Research Corporation (No. CC3343), and the Henry-Dreyfus Teacher-Scholar Award for their generous support of our research program. We also thank Dr. Paul Shin (CSUN) for assistance in obtaining high-resolution mass spectra.
References
- 1.(a) Perez I, Perez Sestelo J, Sarandeses LA. Org Lett. 1999;1:1267. doi: 10.1021/ol990878z. [DOI] [PubMed] [Google Scholar]; (b) Perez I, Perez Sestelo J, Sarandeses LA. J Am Chem Soc. 2001;123:4155. doi: 10.1021/ja004195m. [DOI] [PubMed] [Google Scholar]; (c) Lee PH, Sung S-Y, Lee K. Org Lett. 2001;3:3201. doi: 10.1021/ol016532h. [DOI] [PubMed] [Google Scholar]; (d) Lee K, Lee J, Lee PH. J Org Chem. 2002;67:8265. doi: 10.1021/jo026121u. [DOI] [PubMed] [Google Scholar]; (e) Lehmann U, Awasthi S, Minehan T. Org Lett. 2003;5:2405. doi: 10.1021/ol0345428. [DOI] [PubMed] [Google Scholar]; (f) Rodriguez D, Perez Sestelo J, Sarandeses LA. J Org Chem. 2003;68:2518. doi: 10.1021/jo0265939. [DOI] [PubMed] [Google Scholar]; (g) Baker L, Minehan T. J Org Chem. 2004;69:3957. doi: 10.1021/jo0357162. [DOI] [PubMed] [Google Scholar]; (h) Rodriguez D, Perez Sestelo J, Sarandeses LA. J Org Chem. 2004;69:8136. doi: 10.1021/jo0491511. [DOI] [PubMed] [Google Scholar]; (i) Riveiros R, Rodriguez D, Perez Sestelo J, Sarandeses LA. Org Lett. 2006;8:1403. doi: 10.1021/ol060192o. [DOI] [PubMed] [Google Scholar]
- 2.Takami K, Yorimitsu H, Shinokobu H, Matsubara S, Oshima K. Org Lett. 2001;3:1997. doi: 10.1021/ol015975i. [DOI] [PubMed] [Google Scholar]
- 3.Yus M, Foubelo F. In: Handbook of Functionalized Organometallics. Knochel P, editor. Vol. 1. Wiley-VCH; Weinheim, Germany: 2005. p. 7. [Google Scholar]
- 4.Recently, a directed ortho-lithiation/transmetallation sequence was used in the preparation of triarylindium reagents: Pena MA, Perez Sestelo J, Sarandeses LA. J Org Chem. 2007;72:1271. doi: 10.1021/jo062148s.
- 5.Lian Y, Hinkle RJ. J Org Chem. 2006;71:7071. doi: 10.1021/jo060738k. [DOI] [PubMed] [Google Scholar]
- 6.Zinc: Krasovskiy A, Malakhov V, Gavryushin A, Knochel P. Angew Chem Int Ed. 2006;45:6040. doi: 10.1002/anie.200601450.Boudet N, Sase S, Sinha P, Liu C-Y, Krasovskiy A, Knochel P. J Am Chem Soc. 2007;129:12358. doi: 10.1021/ja074060h. Magnesium: Knochel P, Dohle W, Gommermann N, Kniesel FF, Kopp F, Korn T, Sapountzis I, Vu VA, editors. Angew Chem Int Ed. Vol. 42. 2003. p. 4302.Krasovskiy A, Knochel P, editors. Angew Chem Int Ed. Vol. 43. 2004. p. 3333.Ila H, Baron O, Wagner AJ, Knochel P. Chem Lett. 2006;35:2. doi: 10.1039/b510866g.Ila H, Baron O, Wagner AJ, Knochel P. Chem Commun. 2006;6:58. doi: 10.1039/b510866g.Krasovskiy A, Straub BF, Knochel P, editors. Angew Chem Int Ed. Vol. 45. 2006. p. 159.Knochel P, Krasovskiy A, Sapountzis I. In: Handbook of Functionalized Organometallics. Knochel P, editor. Vol. 1. Wiley-VCH; Weinheim, Germany: 2005. p. 109.
- 7.Knochel P, Gavryushin A, Malakhov VA. DE 102006015378. German Patent. 2007 In the preparation of this manuscript, we became aware of a manuscript in review (J. Am. Chem. Soc.) by Knochel and Chen detailing the direct insertion of indium into aryl and heteroaryl iodides and the cross-coupling of the so-formed indium reagents with aryl halide to form biaryls.
- 8.(a) Knochel P, Millot N, Rodriguez AL. Org React. 2001;58:417. [Google Scholar]; (b) Knochel P, Singer RD. Chem Rev. 1993;93:2117. [Google Scholar]; (c) Knochel P, editor. Handbook of Functionalized Organometallics. Vol. 1. Wiley-VCH; Weinheim, Germany: 2005. p. 109. [Google Scholar]
- 9.(a) Li C-J. Chem Rev. 1993;93:2023. [Google Scholar]; (b) Cintas P. Synlett. 1995:1087. [Google Scholar]; (c) Li C-J. Tetrahedron. 1996;52:5643. [Google Scholar]; (d) Li C-J, Chan T-H. Organic Reactions in Aqueous Media. Wiley; New York: 1997. [Google Scholar]; (e) Li C-J, Chan T-H. Tetrahedron. 1999;55:11149. [Google Scholar]; (f) Podlech J, Maier TC. Synthesis. 2003:633–655. [Google Scholar]; (g) Li C-J. Chem Rev. 2005;105:3095. doi: 10.1021/cr030009u. [DOI] [PubMed] [Google Scholar]
- 10.Indium containing 0.1 % Mg was prepared my mixing 102.6 mg indium metal with 11.4 mg of commercially available indium containing 1% Mg (Aldrich).
- 11.For a previous preparation of 3a, see: So CM, Lau CP, Kwong FY. Org Lett. 2008;9:2795. doi: 10.1021/ol070898y.
- 12.Electron-deficient aryl bromides were also suitable partners for the cross-coupling reaction with the in situ formed arylindium reagents, furnishing biaryl products 3 in similar or slightly lower yields when compared to the corresponding aryl iodides. Electron-rich aryl bromides, however, gave very poor yields (5–20%) of cross-coupled products.
- 13.Araki S, Ito H, Butsugan Y. J Org Chem. 1988;53:1831. [Google Scholar]
- 14.Chan TH, Yang Y. J Am Chem Soc. 1999;121:3228. [Google Scholar]
- 15.(a) Rodriguez D, Perez Sestelo J, Sarandeses LA. J Org Chem. 2004;69:8136. doi: 10.1021/jo0491511. [DOI] [PubMed] [Google Scholar]; (b) Wilkinson G, Stone FGA, Abel EW. Comprehensive Organometallic Chemistry. I. Pergamon Press; New York: 1982. p. 711. [Google Scholar]
- 16.While the indium metal recovered after the insertion reaction (30% of the total used) could be directly reused in insertion and cross-coupling reactions with minimal (<10%) decrease in yield, the indium metal recovered after zinc-mediated reduction of the residual aqueous indium salts (50–60% of the total used) was a soft, ball-like clump that could not be reused directly because of its decreased surface area compared to indium powder. Work on preparing this material for reuse in insertion and cross-coupling reactions is currently in progress.
- 17.Zhang Y, Rovis T. J Am Chem Soc. 2004;126:15964. doi: 10.1021/ja044113k. [DOI] [PubMed] [Google Scholar]
- 18.Fausett BW, Liebeskind LS. J Org Chem. 2005;70:4851. doi: 10.1021/jo050110u. [DOI] [PubMed] [Google Scholar]
- 19.See reference 15a and also: Metza JT, Terzian RA, Minehan TG. Tetrahedron Lett. 2006;47:8905.
- 20.See ref 1e and also: Price S, Edwards S, Wu T, Minehan T. Tetrahedron Lett. 2004;45:5197.Pham M, Allatabakhsh A, Minehan TG. J Org Chem. 2008;73:741. doi: 10.1021/jo7016857.
- 21.(a) Van Rheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976;17:1973. [Google Scholar]; (b) Van Rheenen V, Cha DY, Hartley WM. Org Synth Coll. 1988;6:342. [Google Scholar]
- 22.Ellsworth BA, Doyle AG, Patel M, Caceres-Cortes J, Meng W, Deshpande PP, Pullockaran A, Washburn WN. Tetrahedron Asymmetry. 2003;14:3243. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed experimental procedures, spectroscopic data, and 1H NMR spectra for all compounds in Tables 2 and 3 and Scheme 2, as well as NOESY data for compound 8. This material is available free of charge via the Internet at http://pubs.acs.org.