

Abstract
An unusual feature of the lipid A from the plant endo-symbionts Rhizobium etli and Rhizobium leguminosarum is the presence of a proximal sugar unit consisting of a 2-amino-2-deoxy-gluconate moiety in place of glucosamine. An outer membrane oxidase that generates the 2-amino-2-deoxy-gluconate unit from a glucosamine-containing precursor is present in membranes of R. leguminosarum and R. etli but not in S. meliloti or Escherichia coli. We now report the identification of a hybrid cosmid that directs the overexpression of this activity by screening 1800 lysates of individual colonies of a R. leguminosarum 3841 genomic DNA library in the host strain R. etli CE3. Two cosmids (p1S11D and p1U12G) were identified in this manner and transferred into S. meliloti, in which they also directed the expression of oxidase activity in the absence of any chromosomal background. Subcloning and sequencing of the oxidase gene on a 6.5-kb fragment derived from the ~20-kb insert in p1S11D revealed that the enzyme is encoded by a gene (lpxQ) that specifies a protein of 224 amino acid residues with a putative signal sequence cleavage site at position 28. Heterologous expression of lpxQ using the T7lac promoter system in E. coli resulted in the production of catalytically active oxidase that was localized in the outer membrane. A new outer membrane protein of the size expected for LpxQ was present in this construct and was subjected to microsequencing to confirm its identity and the site of signal peptide cleavage. LpxQ expressed in E. coli generates the same products as seen in R. leguminosarum membranes. LpxQ is dependent on O2 for activity, as demonstrated by inhibition of the reaction under strictly anaerobic conditions. An ortholog of LpxQ is present in the genome of Agrobacterium tumefaciens, as shown by heterologous expression of oxidase activity in E. coli.
As demonstrated in the accompanying article (1), the outer membranes of Rhizobium leguminosarum and Rhizobium etli contain an unusual oxidase that converts the proximal glucosamine unit of 1-dephosphorylated lipid A (or related molecules) to a novel 2-aminogluconate moiety. The membranes of Sinorhizobium meliloti and Escherichia coli do not normally contain such an oxidase activity. Although the function of this unusual covalent modification of lipid A is unknown (2–4), the existence of an oxidative enzyme in the outer membrane of a Gram-negative bacterium is without precedent. The few outer membrane enzymes described to date are all phospholipases (5, 6), acyltransferases (7, 8), or proteases (9). Other characterized outer membrane proteins function either as porins or specialized transporters (10–12). The presence of the oxidase in outer membranes of certain strains of Rhizobium suggests that lipid A oxidation (1), when it occurs, is a late event in lipopolysaccharide assembly.
Given the considerable progress that has recently been made with the structural biology of outer membrane proteins by x-ray crystallography (12) and NMR spectroscopy (8, 13) and the great interest in lipid A modifications in the context of microbial pathogenesis (14–16), we now report the expression cloning of a novel R. leguminosarum gene, designated lpxQ, that encodes the oxidase. Hydropathy analysis predicts that LpxQ is a typical outer membrane protein of 224 amino acid residues with a leader sequence that is cleaved during export. The R. leguminosarum oxidase is properly localized to the outer membrane when expressed in E. coli behind the T7lac promoter. The only other bacterial genomes that contain clear-cut orthologs of LpxQ are those of two Agrobacterium tumefaciens strains, the sequences of which were recently reported (17, 18). Although 2-aminogluconate has not been described as a component of lipid A in A. tumefaciens, we show that A. tumefaciens LpxQ is in fact fully active as a lipid A oxidase when overexpressed in E. coli. The availability of the lpxQ genes of R. leguminosarum, R. etli, and A. tumefaciens should enable purification to homogeneity and mechanistic studies of the oxidase and should also facilitate genetic studies of 2-aminogluconate function during plant infection and symbiosis.
EXPERIMENTAL PROCEDURES
Materials
Glass-backed 0.25-mm Silica Gel 60 thin layer chromatography plates were from Merck. Chloroform, ammonium acetate, and sodium acetate were obtained from EM Science. Pyridine, methanol, and formic acid were from Mallinckrodt. [U-14C]acetate was purchased from Amersham Biosciences, and 14C-labeled component B was prepared as described in the accompanying article (1).
Bacterial Growth Conditions
Briefly, R. leguminosarum 3855 was grown at 30 °C in TY broth (5 g of tryptone and 3 g of yeast extract/liter) supplemented with 10 mM CaCl2. Unless otherwise indicated, R. etli CE3 (19, 20) and S. meliloti 1021 (21, 22) were grown in TY broth supplemented with 10 mM CaCl2, 20 μg/ml nalidixic acid, and 200 μg/ml streptomycin sulfate. E. coli strains were generally grown at 37 °C in LB broth (23) with one of the following antibiotics, depending on the resistance markers of the plasmid that the strain harbors: ampicillin (100 μg/ml), tetracycline (15 μg/ml), and kanamycin (25 μg/ml). Table I describes the various plasmids and bacterial strains used in this study. Cell-free extracts and washed membranes were prepared as described in the accompanying article (1).
Table I.
Bacterial strains and plasmids used in this study
| Name | Description | Source |
|---|---|---|
| Plasmid | ||
| pRK404a | Shuttle vector, Tetr | Ref. 32 |
| pLAFR-1 | Shuttle vector, Tetr | Ref. 25 |
| p1S11D | ≈20-kb cosmid in pLAFR-1 | this work |
| p1E11D | ≈20-kb cosmid in pLAFR-1 | this work |
| p1U12G | ≈20-kb cosmid in pLAFR-1 | this work |
| pQN208 | ≈3.9-kb EcoRI fragment in pRK404a | from p1S11D insert |
| pQN209 | ≈4.7-kb HindIII fragment in pRK404a | from p1S11D |
| pQN210 | ≈6.5-kb HindIII fragment in pRK404a | from p1S11D |
| pQN211 | ≈4-kb HindIII fragment in pRK404a | from p1S11D |
| pQN212 | ≈3.5-kb HindIII fragment in pRK404a | from p1S11D |
| pQN214 | ≈1.3-kb HindIII fragment in pRK404a | from p1S11D |
| pQN215 | ≈0.5-kb PstI fragment in pRK404a | from p1S11D |
| pQN217 | ≈6.5-kb HindIII fragment in pRK404a | from p1U12G |
| pQN231 | IpxQ in pRK404a | this work |
| pQN233 | IpxQ in pET21a(+) | this work |
| pQN234 | IpxQ in pET28b(+) with N-terminal His tag | this work |
| pQN235 | IpxQ in pET21b(+) with C-terminal His tag | this work |
| pQN240 | A. tumefaciens IpxQ in pET21b+ | this work |
| pET21a+ and pET21b+ | T7lac expression vector, Ampr | Novagen |
| pET28b+ | T7lac expression vector, Kanr | Novagen |
| Rhizobium strains | ||
| R. etli CE3 | Derivative of CFN42, Nalr, Strr | Refs. 19 and 20 |
| R. leguminosarum 3855 | Lipid A structure similar to R. etli CE3 | Ref. 52 |
| S. meliloti 1021 | Contains phosphorylated lipid A, Nalr, Strr | Ref. 21 |
| R. leguminosarum 24AR | LPS mutant with phosphorylated lipid A | Ref. 53 |
| E. coli strains | ||
| HB101 | recA, Strr | BRL/88 |
| Novablue (DE3) | K-12 strain, DE3 lysogen | Novagen |
| BL21 (DE3)/pLysS | B strain, DE3 lysogen, contains pLysS | Novagen |
| BLR (DE3)/pLysS | B strain, DE3 lysogen, contains pLysS | Novagen |
| XL1-Blue | K-12 strain | Stratagene |
Quantitative Assay for Measuring the Conversion of [14C]B to [14C]-D-1
As described fully in the accompanying article (1), the standard reaction mixture (10 μl) contained (unless otherwise indicated) 10 μM [14C]B (~500 cpm/tube), 0.5–1.0 mg/ml membrane protein, 0.1% Triton X-100, 1 mM gCl2, and 50 mM MES1 buffer, pH 6.5. The reactions were incubated under aerobic conditions at 30 °C and terminated at the indicated times by spotting 4- or 5-μl samples onto a 20 × 20-cm silica gel TLC plate. The spots were dried for 30 min with a cold air stream, and the plate was then developed in the solvent CHCl3, MeOH, H2O, pyridine (40:25:4:2, v/v). The remaining substrate and product(s) were detected with a Molecular Dynamics Storm PhosphorImager equipped with ImageQuant software. Enzyme specific activity (usually expressed as nmol/min/mg) was calculated based on the percentage of conversion of substrate to product(s).
Anaerobic Assay Conditions
For demonstrating oxygen dependence, an assay system consisting of 25 μM [14C]B, 50 mM MES buffer, pH 6.5, 0.1% Triton X-100, 1 mM MgCl2, and 0.1 mg/ml BLR(DE3)/pLysS/pQN235 membranes was set up in an anaerobic chamber. The reactions were started by the addition of membranes as the enzyme source after equilibration of all tubes in the absence of oxygen for ~30 min. To remove traces of oxygen, glucose oxidase (20 μg/ml), catalase (2 μg/ml), and glucose (0.1%) were included in some cases, as indicated. After 30, 60, or 90 min, portions of the reaction mixtures were spotted onto a silica gel TLC plate, which was developed as described above. A parallel set of reaction mixtures was assayed simultaneously under ambient aerobic conditions.
Expression Cloning of the R. leguminosarum Lipid A Oxidase Gene
A genomic DNA library of the R. leguminosarum strain 3841 was obtained from Dr. J. Downie of the John Innes Institute (Norwich, UK). This library was constructed by the ligation of ~20 –25-kb fragments of genomic DNA (24), generated by partial EcoRI digestion, into the cosmid pLAFR-1 (25). E. coli 803 was employed as the host strain (26).
The library was transferred from E. coli 803 into R. etli CE3 by tri-parental mating (27, 28) with E. coli MT616 as the helper (28). Three thousand individual R. etli CE3 colonies harboring random fragments of the library were picked into microtiter dishes containing (per well) 150 μl of TY medium with 20 μg/ml nalidixic acid, 200 μg/ml streptomycin, 12 μg/ml tetracycline, and 10 mM CaCl2 and then were grown at 30 °C with shaking at 225 rpm to stationary phase. The cultures were stored at −80 °C as master stocks in the same dishes after adjusting the medium to 20% glycerol by addition of an appropriate volume of 60% glycerol. To prepare fresh lysates for screening purposes, 1800 colonies were taken out of storage and regrown in 96-well microtiter plates. Each well was inoculated with a 5-μl portion of the master glycerol stock culture into 150 μl of TY broth supplemented with 10 mM CaCl2, 20 μg/ml nalidixic acid, 200 μg/ml streptomycin sulfate, and 12 μg/ml tetracycline. The microtiter plates were then shaken at 225 rpm in a 30°C incubator for ~24 h or until the OD550 reached ~0.6, as measured with a microtiter plate reader. (Back-up glycerol stocks of these master plates were made by transferring 50 μl from each well into a new microtiter plate, in which each well contained 25 μl of 60% glycerol. The new stocks were stored at −80 °C.)
The remaining 100 μl in each well of the microtiter plate was centrifuged at 3,660 × g for 5 min at 4 °C. The cell pellets were washed with 50 μl of 50 mM HEPES, pH 7.5, and centrifuged as above. After decanting the supernatant, the cell pellets were resuspended in 10 μl of lysis buffer, which consists of 100 mg/ml lysozyme in 50 mM HEPES, pH 7.5. The plates were sealed, and the contents were vigorously mixed for a minute. Lysis was allowed to proceed at room temperature for 30 min. The lysates were frozen by placing the plate into a −80 °C freezer for several hours, after which they were thawed. Each 96-well microtiter plate contained eight rows (rows A–H) of 12 wells (wells 1–12). To facilitate the screening, each row was further grouped into four pools of three lysates. Row A, for example, was grouped into pools A(1–3), A(4–6), A(7–9), and A(10–12). The pooled lysates were assayed for their ability to convert [14C]B to [14C]D-1. A portion of each pool (9 μl) was mixed with 2 μl of a concentrated reaction buffer (consisting of 5 mM l of 0.05 MgCl2, 250 mM MES, pH 6.5, and 0.5% Triton X-100) and 0.5 μl of 0.05μM [14C]B (~600 cpm/reaction tube). After 30 and 60 min of incubation at 30 °C, 5-μl portions of each reaction mixture were spotted onto a 20 × 20-cm silica TLC plate. Negative (no enzyme) and positive (wild type R. leguminosarum 3855 membranes) controls were included for each 96-well microtiter plate of pooled lysates that were screened. The amount of [14C]D-1 formed in each reaction tube was quantified with a PhosphorImager.
Three apparently positive pools of lysates were identified and subjected to further analysis. Membranes from each of the nine cosmid-bearing strains contributing to the three positive pools were prepared and assayed individually for overproduction of oxidase activity versus membranes of the control CE3 strain. Next, the hybrid cosmids of the three strains that directed overexpression of oxidase activity were isolated, transformed into E. coli HB101, and then transferred into S. meliloti via tri-parental mating (27, 28). Finally, membranes of S. meliloti cells harboring these cosmids were assayed for their ability to express oxidase activity to verify the presence of the lipid A oxidase gene on the cosmid inserts. Membranes of the S. meliloti 1021 host strain (22) used for this purpose (unlike CE3) lack background oxidase activity.
Recombinant DNA Techniques
Protocols for handling of DNA samples were those of Sambrook et al. (29). Competent cells of E. coli were prepared by the CaCl2 method (29, 30). Cosmids and plasmids were purified using Qiagen DNA purification kits. Double-stranded DNA sequencing was performed with an ABI Prism 377 instrument at the Duke University DNA Analysis Facility. Primers were purchased from Invitrogen. DNA sequence analysis and searches for open reading frames were performed with GCG and ORF Finder (31).
Subcloning and Sequencing of the DNA Inserts in Cosmids p1S11D and p1U12G
Digestion of the cosmid p1S11D with HindIII generated fragments of ~0.2, 1.2, 3.5, 4, and 6.5 kb, as well as a large band (~21 kb) corresponding to the vector pLAFR-1. The insert-derived fragments were ligated into the shuttle vector pRK404a digested with HindIII (32), and the desired constructs were transferred into S. meliloti via tri-parental mating. Membranes of S. meliloti cells harboring each of these subclones were prepared and assayed for oxidase activity. Only one hybrid plasmid, pQN210, which contains the 6.5-kb HindIII fragment of p1S11D, directed the expression of oxidase activity. The same 6.5-kb fragment was also present in the DNA insert of p1U12G and was active when expressed on the hybrid plasmid pQN217 (Table I). The 6.5-kb HindIII fragment of pQN210 was sequenced using a primer walking strategy (29). The DNA sequencing was facilitated by subcloning of several EcoRI fragments derived from the 6.5-kb insert present in pQN210 into the vector pRK404a. None of these subclones directed the expression of oxidase activity, given that an EcoRI site is present in the oxidase structural gene lpxQ (see below).
Construction of Plasmids That Express R. leguminosarum lpxQ behind the lac or the T7lac Promoters
The lpxQ gene was cloned into several vectors, which were tested for their ability to direct the overexpression of oxidase activity. The coding region of lpxQ was amplified by PCR from pQN210 with various primers to optimize restriction enzyme sites, translational start sites, or translational stop sites.
For all of the primers, restriction enzyme sites are italicized, and a G/C clamp is incorporated at their 5′-ends. The 5′-lpxQ/1 primer (5′-C-GCGCAAGCTTAGGAGGAATTTAAAATGACATATGCGCTGCGTT-CTTCCG-3′) was designed for ligation of lpxQ into lac promoter-driven vectors, such as the shuttle vector pRK404a. This primer also incorporates a HindIII site (in italics), a ribosome-binding site (underlined sequence), and a translational spacer element between the ribosome-binding site and the ATG start codon (33). The sequences in bold type correspond to the N-terminal coding region of lpxQ.
An internal NdeI site (CATAT9G) (in which the superscript reflects the number of bases from the A of the ATG start codon) is present in the coding sequence of lpxQ. The primer 5′-lpxQ/2 (5′-CGCGCCATATGA-CATAC9GCGCTGCGTTCTTCCG-3′) was designed to eliminate this internal site and to facilitate the insertion of lpxQ into pET vectors behind the T7lac promoter via an engineered NdeI site (in italics). The internal NdeI site in the coding region of lpxQ was removed by mutating T9 to C9 in 5′-lpxQ/2. This base substitution does not alter the amino acid sequence of LpxQ. Sequences in bold type correspond to the N-terminal coding region of lpxQ.
The primer 3′-lpxQ/1 (5′-GCGCAAGCTTTTACACATATTCCCTGACGATAGCAGGC-3′) contains an engineered HindIII site (in italics). The underlined sequence corresponds to a region of DNA that is 96 bases downstream of the termination codon of lpxQ. Lastly, The primer 3′-lpxQ/2 (5′-GCGCAAGCTTCCAGTGGAATGAAACGCCGACGTT-GA-3′) was designed for ligation of lpxQ into pET21b+, whereby a protein is made with a His6 tag fused to the C terminus of LpxQ. The sequence in bold type corresponds to the coding region at the C-terminal end of lpxQ less the termination codon.
The following combinations of primers were used for PCRs and subsequent ligations. The PCR product of 5′-lpxQ/1 and 3′-lpxQ/1 was digested with HindIII and ligated into the shuttle vector pRK404a to yield pQN231. The lpxQ gene amplified with primers 5′-lpxQ/2 and 3′-lpxQ/1 was digested with NdeI and HindIII and then ligated into pET21a+, yielding pQN233. The DNA generated with 5′-lpxQ/2 and 3′-lpxQ/2 was digested with NdeI and HindIII and then ligated into pET21b+, yielding pQN235.
A typical PCR mixture contained 100 ng of template DNA (pQN210), 1× Pfu polymerase buffer (Stratagene), 10% Me2SO, 1% glycerol, 200 μM of each of the dNTPs, 125 ng of each primer, and 2.5 units of Pfu polymerase (Stratagene) in a total volume of 100 μl. The reaction was placed in a DNA thermal cycler and subjected to 5 min of denaturation at 94 °C, followed by five cycles consisting of denaturation (1 min, 95 °C), annealing (1 min, 55 °C), and extension (2 min, 72 °C). Full amplification was then achieved with 25 cycles of the following: denaturation (1 min, 95 °C), annealing (1 min, 68 °C), and extension (2 min, 72°C). After the 30th overall cycle, an additional 6-min extension at 72 °C was performed. The reaction was terminated by cooling the tubes to 4 °C. In each case, the product was analyzed on a 0.8% agarose gel, excised and purified using the Gene Clean II gel DNA purification kit (Bio 101), digested with the appropriate restriction enzymes as indicated above, and ligated into a vector that had been similarly digested. The ligation mixtures that resulted in the construction of pQN233 and pQN235 were first transformed into competent E. coli XL-1Blue cells. The transformants were screened for the desired insert by restriction enzyme digestion. The insert and flanking regions of pQN233 were confirmed by DNA sequencing. The final recombinant plasmids were then transformed, as indicated below, into competent E. coli cells of strain BLR(DE3)/pLysS or Novablue(DE3) (Novagen) to evaluate the overexpression of LpxQ upon induction of mid-log phase cells at 37 °C with 1 mM IPTG.
The ligation mixture that yielded pQN231 was first transformed into competent cells of E. coli HB101. The four candidate plasmids containing the proper insert were then moved into S. meliloti 1021 by the tri-parental mating procedure. Of these four hybrid plasmids, only two were found to direct the expression of LpxQ activity and were designated pQN231, presumably reflecting the proper orientation of the lpxQ gene for expression behind the lac promoter. The membranes for assay of these constructs were prepared as described in the accompanying article (1).
PCR Amplification and Cloning of an A. tumefaciens lpxQ Ortholog
The A. tumefaciens lpxQ gene was amplified by PCR followed by ligation into the pET21b+ vector. The N-terminal primer for the PCRs had the following sequence: 5′-CGCGCATATGTCGCGCCATACTTTACTT-GTTTGC-3′. This primer was designed with a G/C clamp, an NdeI restriction site (in italics) that overlaps with the initiation codon ATG and the first 26 base pairs of the A. tumefaciens lpxQ gene coding sequence (in bold type). The C-terminal primer was designed with a G/C clamp, a BamHI site (in italics), and sequence (in bold type) corresponding to a region that is 52 base pairs downstream of the lpxQ TAA termination codon. The sequence of the C-terminal primer was: 5′-GCGCGGATCCTGGAACTGCTGGACTGGGCTTATGG-3′.
A. tumefaciens C58 genomic DNA purchased from the American Type Culture Collection (Manassas, VA) was used as the template. The PCR mixture (100 μl) contained 250 ng of template DNA, 1× Pfu polymerase buffer (Stratagene), 10% of the Me2SO, 1% glycerol, 200 μM of each dNTPs, 125 ng of each primer, and 2.5 units of Pfu polymerase (Stratagene). The reaction was placed in a DNA thermal cycler and subjected to a 5-min denaturation at 94 °C, followed by five cycles consisting of denaturation (1 min, 95 °C), annealing (1 min, 55 °C), and extension (2 min, 72 °C). Full amplification was then achieved with 25 cycles of the following: denaturation (1 min, 95 °C), annealing (1 min, 65 °C), and extension (2 min, 72 °C). After the 30th overall cycle, an additional 6-min extension at 72 °C was performed. The PCR was terminated by cooling the tubes to 4 °C.
The PCR product was analyzed on a 0.9% agarose gel and was gel-purified with the GeneClean DNA system (see above). The purified lpxQ gene, and the vector were both digested with NdeI and BamHI and then ligated at 16 °C overnight. The ligation mixture was transformed into competent E. coli XL-1Blue cells (Stratagene). Several ampicillin-resistant colonies were picked, and the purified plasmids were screened for the desired insert by digestion with NdeI and BamHI. The construct harboring the A. tumefaciens lpxQ gene was designated pQN240 and was transformed into BLR(DE3)/pLysS (Novagen) for T7lac-directed overexpression.
Preparation of E. coli Membranes Containing A. tumefaciens LpxQ for Assay
For this purpose, a single colony of BLR(DE3)/pLysS/pQN240 was used to inoculate 5 ml of LB broth containing 100 μg/ml ampicillin and 20 μg/ml chloramphenicol. After shaking at 225 rpm for 16 h at 37 °C, a portion of this overnight culture was used to inoculate 50 ml of LB broth containing ampicillin and chloramphenicol at an A600 of 0.01. The cells were grown in shaking culture (225 rpm) at 37 °C until the A600 had reached ~0.5, at which time 1 mM IPTG was added. After another 3 h of growth at 37 °C, the cells were harvested by centrifugation at 3660 × g for 15 min at 4 °C. The cells were washed once with 10 ml of 50 mM HEPES, pH 7.5, and again collected by centrifugation. The cell pellet was then resuspended in 5 ml of 50 mM HEPES, pH 7.5, and stored at −80 °C prior to preparation of membranes. Two other E. coli host strains, BL21(DE3)/pLysS and Novablue(DE3), were tested in the same manner for their ability to express the lpxQ gene product. The empty vector pET21b+ was also transformed into each of the above host strains in parallel.
Subcellular Localization of LpxQ Expressed in E. coli
The subcellular localization of the R. leguminosarum LpxQ oxidase expressed in E. coli Novablue(DE3)/pQN233 was determined using a protocol similar to that described by Trent et al. (34) and in the accompanying article (1). Briefly, a 500-ml culture of Novablue(DE3)/pQN233 was grown with shaking (225 rpm) at 37 °C to A600 = 0.5 and then induced with 1 mM IPTG. After 3 h of further growth, the culture was centrifuged at 6,000 × g for 10 min at 4 °C. The cell pellet was resuspended in 10 ml of 50 mM HEPES, pH 7.5, and the cells were broken by two passages through a French press cell at 10,000 p.s.i. Unbroken cells and large debris were removed by centrifugation at 12,100 × g for 10 min at 4 °C. The membranes were prepared and washed once as described in the accompanying article (1). The washed membranes were homogenized with a 25-gauge 1/2 syringe needle in a total volume of 2.5 ml of 50 mM HEPES, pH 7.5, containing 0.5 mM EDTA. After layering on top of a seven-step isopycnic sucrose gradient Guy-Caffey et al. (35), the inner and outer membranes were separated by centrifugation in a Beckman SW41 swinging bucket rotor at 155,000 × g for 18 h at 4 °C. Fractions of 0.5 ml were collected, and their protein content was determined using the bicinchoninic method (36). The following volumes of each fraction were used without dilution for the various enzyme assays: 5 μl for LpxQ, 50 μl for the NADH oxidase, and 10 μl for phospholipase A (34).
The inner and outer membranes were pooled separately. Each pool was diluted 4-fold with 50 mM HEPES, pH 7.5, and the membrane fragments were collected by centrifugation at 149,000 × g for an hour. The membranes were resuspended in 0.15 ml of 50 mM HEPES, pH 7.5, and the protein concentrations were determined by the bicinchoninic acid assay (36). Control membranes from a 500-ml culture of Novablue(DE3) cells containing the empty vector pET21a+ were prepared in parallel.
Protein Microsequencing of LpxQ
Inner and outer membranes of Novablue(DE3)/pET21a+ and Novablue(DE3)/pQN233 were analyzed by SDS-PAGE on a Bio-Rad Protean II XI apparatus with a 12% polyacrylamide, 1.5-mm thick gel (37). A band of the size expected for LpxQ was observed in the outer membranes only. A piece of the gel containing LpxQ was excised. The proteins were blotted onto an Immobilon-P polyvinylidene fluoride membrane (Millipore) equilibrated in 10 mM CAPS, pH 11, containing 10% methanol, at 15 V for 30 min, using a Bio-Rad Semi-Dry Transfer apparatus. To visualize the transferred proteins, the blot was immersed in 0.1% Ponceau S in 1% acetic acid for 1 min, followed by destaining with 1% acetic acid for 10 min. The band corresponding to LpxQ was excised, rinsed three times with distilled water, and analyzed by high sensitivity protein microsequencing on ABI model 492A Sequencer at the University of Massachusetts Medical School Proteomic Mass Spectrometry Laboratory (Worcester, MA).
Mass Spectrometry
Matrix-assisted laser desorption ionization/time of flight (MALDI/TOF) mass spectra were acquired on a Kompact MALDI 4 from Kratos Analytical (Manchester, UK), equipped with a nitrogen laser (337 nm), 20 kV extraction voltage, and time-delayed extraction, as described in the accompanying article (1).
RESULTS
Expression Cloning of the Lipid A Oxidase of R. leguminosarum
The gene encoding the lipid A oxidase was found by assaying ~600 pools of three individual lysates of an R. leguminosarum genomic DNA library harbored in R. etli CE3 for their ability to convert [14C]B to [14C]D-1 (Fig. 1A). A basal level of oxidase activity was present in all of the pools because of the chromosomal copy of the oxidase gene present in the CE3 host. As shown in Fig. 1B, this background activity was quantified at about 11% conversion of [14C]B to [14C]D-1 in 60 min under the assay conditions employed. Occasional samples, such as the one indicated by the arrow, derived from the pool of wells 10 –12 from row D of plate 1S (Fig. 1B), catalyzed about 2.5-fold more rapid conversion of [14C]B to [14C]D-1 than did the others. The lysates making up these active pools were analyzed individually (data not shown). In this manner, three positive cosmids (p1U12G, p1S11D, and p1E11D) capable of directing the overexpression of oxidase activity in CE3 were identified. Because p1E11D and p1S11D contained exactly the same inserts (data not shown), only p1S11D and p1U12G were further characterized.
Fig. 1. Expression cloning of the R. leguminosarum lipid A oxidase in R. etli.
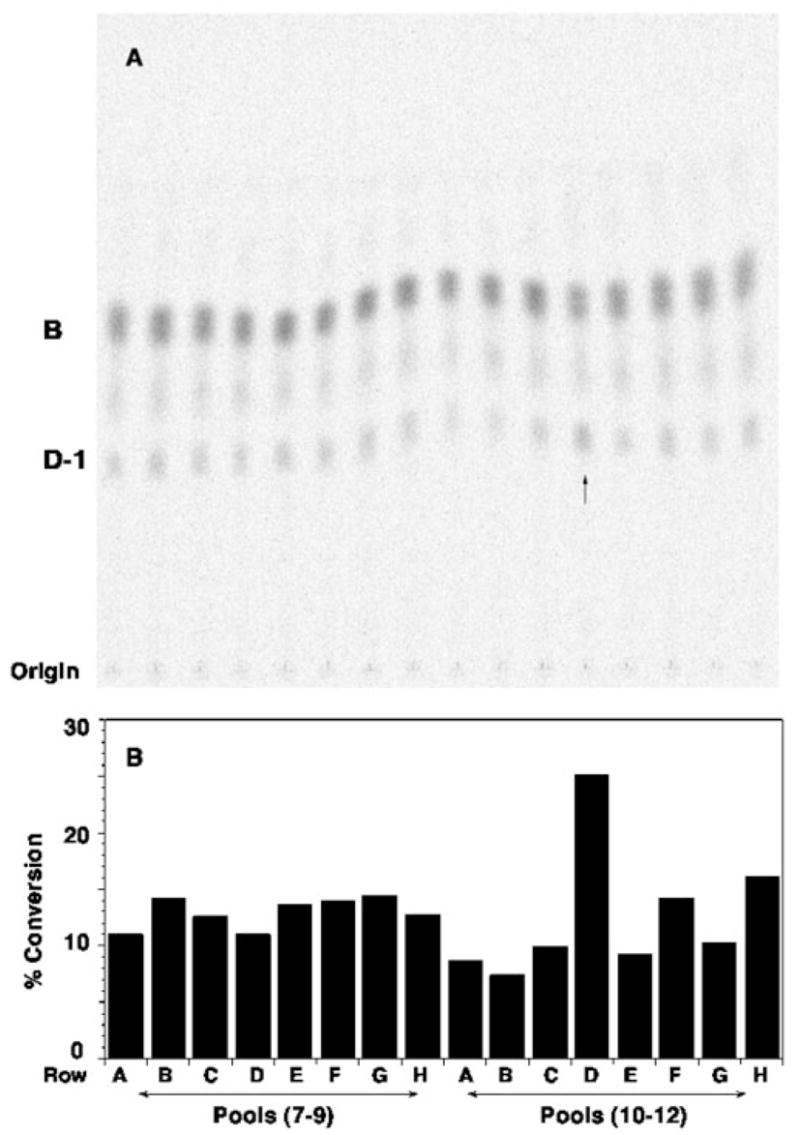
A, a genomic R. leguminosarum DNA library in cosmid pLAFR-1 (24, 32, 44) was transferred into R. etli CE3. Cells from ~1800 cosmid-containing colonies were grown up individually in 96-well microtiter plates. As described under “Experimental Procedures,” the lysates prepared by lysozyme treatment were pooled into groups of three and assayed for overexpression of the lipid A oxidase activity, as judged by the conversion of ~0.003 μM [14C]B (~600 cpm/reaction tube) to [14C]D-1 after 60 min. The arrow indicates a possible pool with elevated oxidase activity. B, the calculated percentage of conversion of [14C]B to [14C]D-1 is shown for each lane. The activity in the pool from row D (wells 10 –12 from plate 1S containing lysates 1S10D, 1S11D, and 1S12D) is approximately twice that of the other pools. Assays of the individual lysates (data not shown) revealed that only 1S11D contained high levels of oxidase activity. Among the ~1800 colonies tested, only three positive cosmids (p1S11D, p1E11D, and p1U12G) were identified in this manner. Based on restriction enzyme digests, p1S11D and p1E11D contained the same insert.
The active cosmids p1U12G and p1S11D were transferred via tri-parental mating from an E. coli HB101 stock culture into S. meliloti 1021. The latter does not contain endogenous oxidase activity and lacks the 2-aminogluconate unit in its lipid A. S. meliloti expresses R. leguminosarum genes very effectively from their native promoters, whereas E. coli does not. As shown in Fig. 2, no background oxidase activity is present in cell extracts or membranes of the control strain S. meliloti/pLAFR-1 (lanes 2 and 5). In contrast, robust conversion of [14C]B to [14C]D-1 is observed in both cell extracts (lanes 3 and 4) and membranes (lanes 6 and 7) of S. meliloti/p1S11D and S. meliloti/p1U12G. These results provide compelling evidence for the presence of the lipid A oxidase gene on the inserts in each of the above cosmids.
Fig. 2. Transfer of the R. leguminosarum cosmids expressing the lipid A oxidase activity into S. meliloti.
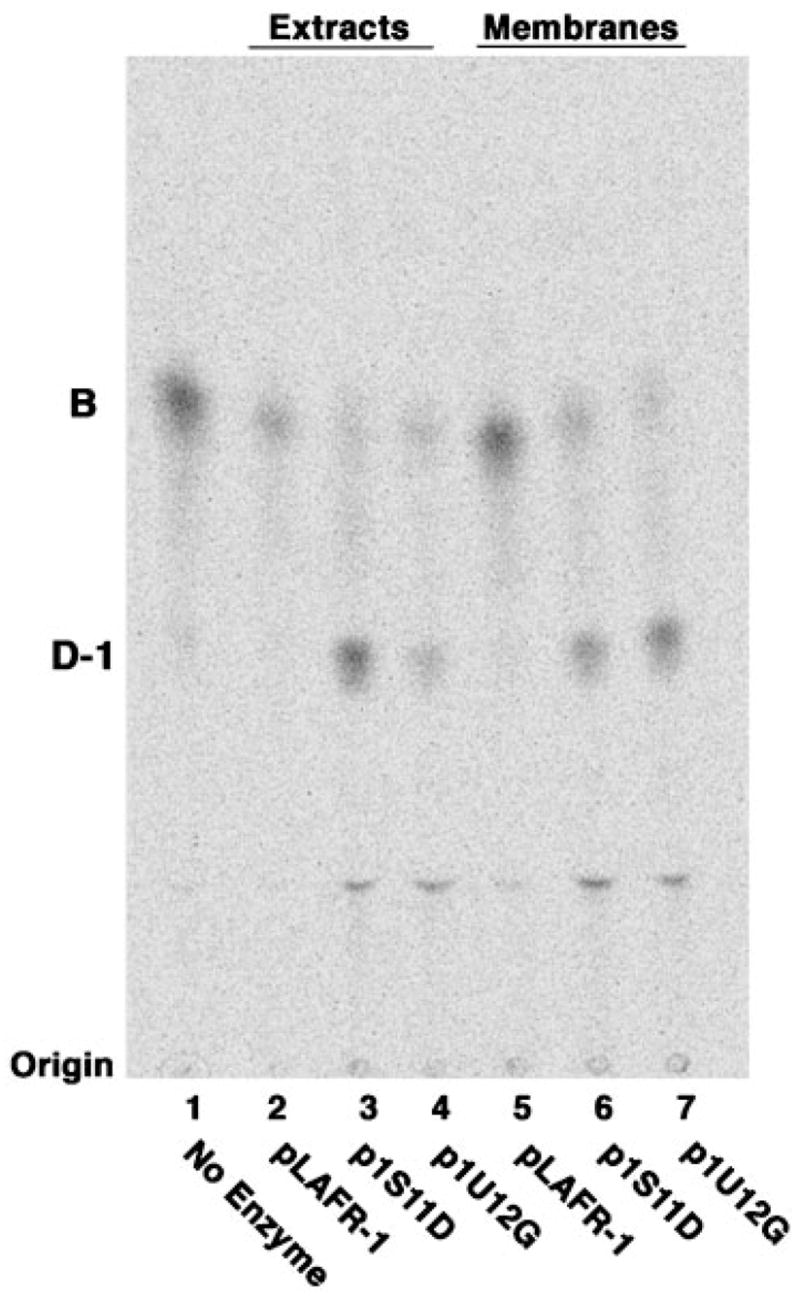
The active cosmids identified in Fig. 1 and the empty vector pLAFR-1 were transferred into S. meliloti 1021 via tri-parental mating (27, 28). Cell-free extracts and membranes derived from late log phase cells containing these cosmids were prepared and assayed for oxidase activity using 10 μM [14C]B (600 cpm/reaction tube) as the substrate. Lanes 2–4 show the results with 0.5 mg/ml crude extracts, whereas lanes 5–7 show assays with 0.5 mg/ml washed membranes. Lane 1, no enzyme control; lane 2, pLAFR-1; lane 3, p1S11D; lane 4, p1U12G; lane 5, pLAFR-1; lane 6, p1S11D; lane 7, p1U12G. The reaction mixtures were incubated overnight at 30 °C. Similar results were obtained when [4′-32P]1-dephospholipid IVA was used as the substrate (data not shown).
Subcloning of p1S11D Localizes the Oxidase Gene to a 6.5-kb HindIII Fragment
To determine the exact location of the oxidase gene, the DNA inserts in both p1S11D and p1U12G were digested with EcoRI and HindIII (Fig. 3), as well as with PstI (not shown). The resulting DNA fragments were ligated into pRK404a and transformed into E. coli HB101. The subclones obtained in this manner were transferred into S. meliloti 1021 by tri-parental mating (Fig. 3 and Table I). Upon assaying membranes of the various constructs, only pQN210, which contains a ~6.5-kb Hind III fragment (Table I) from p1S11D, directed expression of oxidase activity (Fig. 4). None of the fragments recovered from the EcoRI or PstI digestions of p1S11D were active.
Fig. 3. Digestion of cosmids p1S11D, p1U12G, and pRK404a with EcoRI and HindIII.
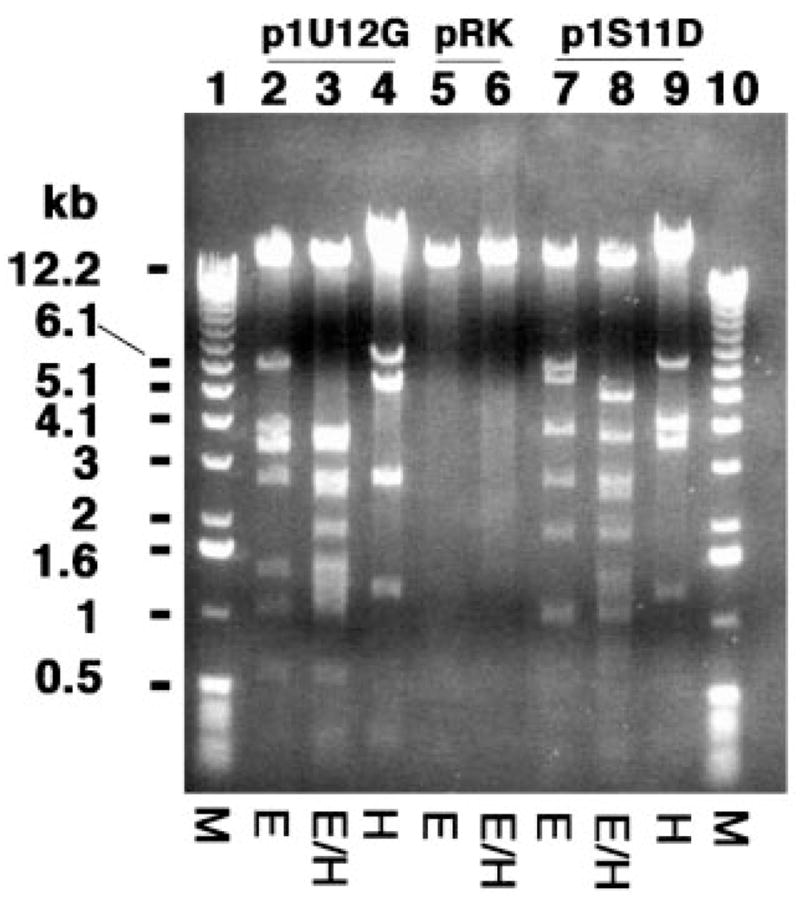
Cosmids p1U12G (lanes 2–4) and p1S11D (lanes 7–9) were digested with EcoRI (E), HindIII (H), or with both EcoRI and HindIII (E/H). The vector pRK404a was digested in parallel (lanes 5 and 6). The HindIII fragment migrating near 6.5 kb (indicated by the thin line) directed the overexpression of oxidase activity (see below).
Fig. 4. A 6.5-kb HindIII digestion fragment of p1S11D directs the overexpression of the oxidase.
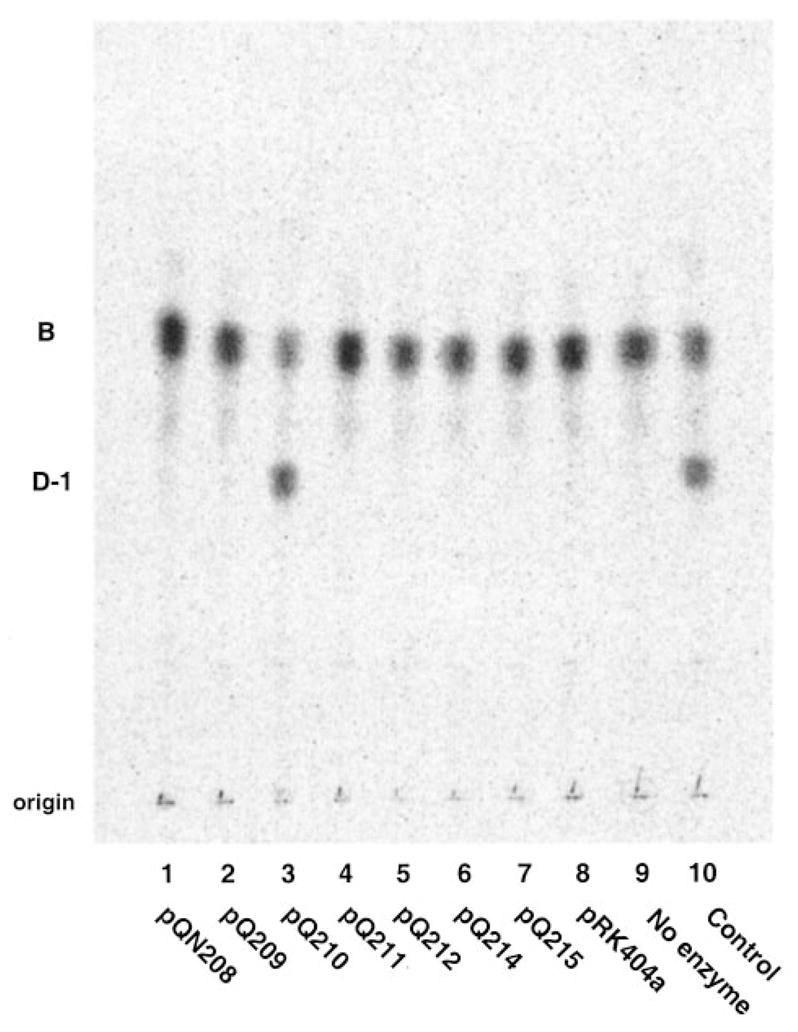
The cosmid p1S11D harbors ~20 kb of R. leguminosarum 8401 genomic DNA. Subclones of the insert were constructed by restriction enzyme digestion and ligation of the fragments into pRK404a. Plasmids pQN209 –pQN214 were derived from various HindIII fragments of the p1S11D insert. pQN208 contains a 3.9-kb EcoRI fragment of the insert, and pQN215 contains a ~0.5-kb PstI fragment. All of the constructs were transferred into S. meliloti 1021 by tri-parental mating (27, 28). Only pQN210, which contains the ~6.5-kb HindIII fragment of the p1S11D insert (see Fig. 3) directed the overexpression of oxidase activity in S. meliloti, as shown by assaying 0.5 mg/ml washed membranes from cells harboring the various constructs with 5 μM [14C]B for 30 min at 30 °C.
Although the DNA inserts in p1U12G and p1S11D are not identical, their restriction enzyme digestion patterns suggested that they share a common ~9-kb segment of DNA (data not shown). In fact, the insert in p1U12G contains a ~6.5-kb Hin-dIII fragment (Fig. 3) that appears to be identical to the one from p1S11D, as judged by the fact that it also can direct the expression of oxidase activity (data not shown). Thus, p1U12G and p1S11D appear to share overlapping DNA segments containing the oxidase structural gene.
Identification of orfE (lpxQ) as the Structural Gene for the Oxidase
Based upon DNA sequencing, at least nine complete or partial open reading frames were tentatively identified on the 6.5-kb HindIII fragment present in pQN210 (Fig. 5). Sequence similarity searches with the BLASTx program indicated that orfA and orfM (Fig. 5) encode glycolate oxidase subunits. However, when orfA was cloned into pRK404a and then transferred into S. meliloti, no lipid A oxidase activity was observed in cell extracts (data not shown).
Fig. 5. Order of the R. leguminosarum genes present on the DNA insert in plasmid pQN210.

Open reading frames contained within the 6.5-kb insert present in pQN210 were identified based on analysis of the DNA sequence (accession number AY228164) with the program ORF Finder (31) and compared with the nonredundant data base with BLASTx (54). The most plausible candidate for the oxidase is the lpxQ gene. H, HindIII; B, BamHI; Sm, SmaI; Sl, SalI; P, PstI; E, EcoRI.
Both OrfC and OrfD (Fig. 5) share significant sequence similarity with a set of hypothetical membrane proteins of unknown function found in various members of the Rhizobiacea and other bacteria, including R. leguminosarum, Mesorhizobium loti, and S. meliloti. Expression cloning of orfC and orfD (either separately or together) in pRK404a and subsequent transfer into S. meliloti failed to induce oxidase activity in cell extracts (data not shown). A similar hybrid plasmid harboring orfB, which encodes a protein with strong similarity to DNA-3-methyladenine glycosylase I, was likewise inactive in S. meliloti.
Although OrfE does not show strong similarity to any functionally assigned protein in the NCBI data base, it does display weak similarity to an outer surface protein of Wolbachia and to the outer membrane ferripyoverdine receptor of Pseudomonas. As discussed further below, a significant OrfE ortholog of unknown function is present in the plant pathogen A. tumefaciens (Fig. 6). Interestingly, extracts of S. meliloti/pQN231, which contains R. leguminosarum orfE behind a lac promoter on pRK404a, display robust oxidase activity (Fig. 7). Additional constructs containing orfE in various T7lac promoter-driven pET vectors (designated pQN233 through pQN235 in Table I) direct high levels of oxidase expression in E. coli cell extracts and membranes (Fig. 7), providing unequivocal evidence that orfE is the oxidase structural gene. Given its unique function in lipid A modification, we suggest that orfE be renamed lpxQ in accordance with the nomenclature used for other genes encoding lipid A biosynthetic enzymes (16, 38, 39).
Fig. 6. Sequence comparison of LpxQ from R. leguminosarum and Agrobacterium tumefaciens.

The predicted amino acid sequence of the R. leguminosarum lipid A oxidase LpxQ (accession number AY228164) is compared with an ortholog of unknown function from A. tumefaciens (17, 18). The order and sequence of the other genes around LpxQ is likewise conserved in both organisms. No other proteins with significant similarity to LpxQ are present in the NCBI data base.
Fig. 7. Heterologous expression of the lpxQ encoded oxidase in S. meliloti and E. coli.
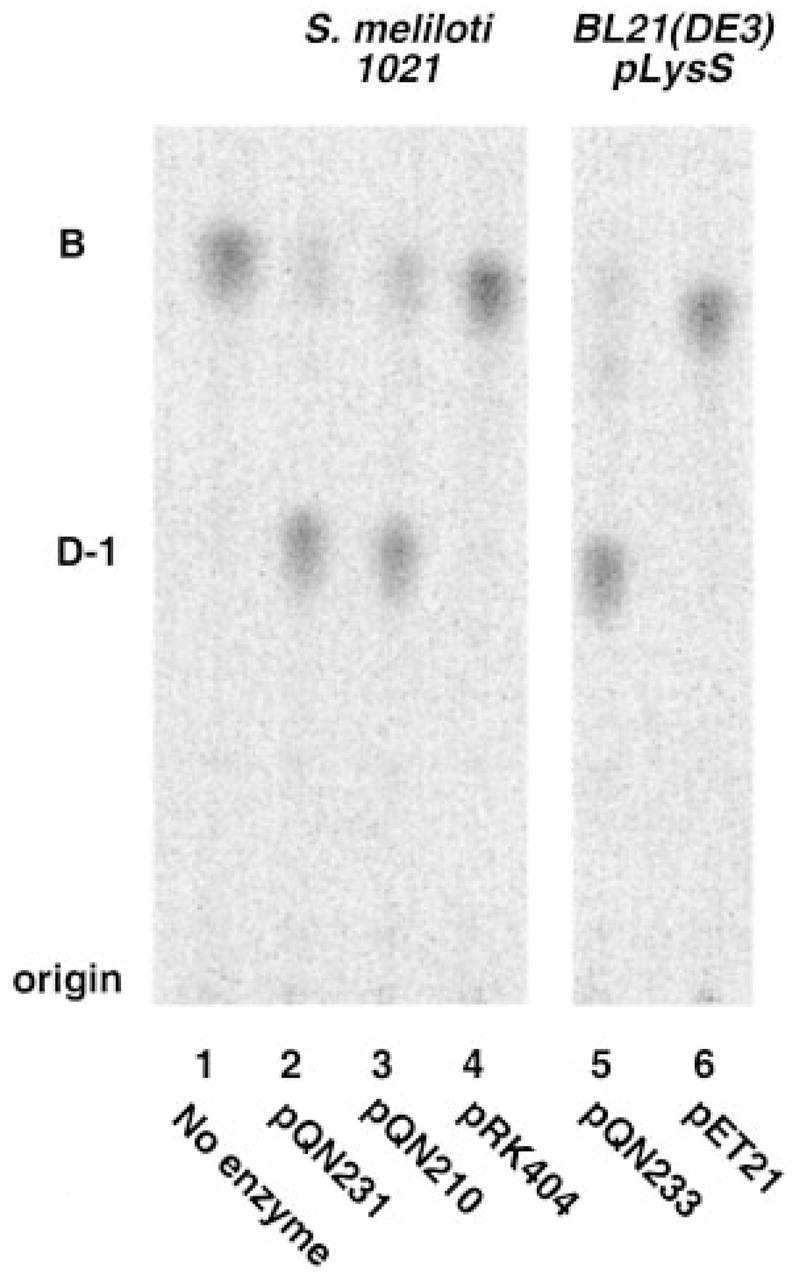
The lpxQ gene was amplified by PCR and ligated into both the shuttle vector pRK404a and into the T7-promoter based vector pET21a+. The resulting constructs, pQN231 and pQN233, were transferred into S. meliloti 1021 and E. coli BL21(DE3)/pLysS, respectively. S. meliloti/pQN231 membranes were prepared from late log phase cells. Membranes of BL21(DE3)/pLysS/pQN233 were obtained from mid log phase cells induced with 1 mM IPTG for 3 h. The membranes (0.5 mg/ml) were assayed for 120 min at 30 °C for their ability to convert 10 μM [14C]B to [14C]D-1 under standard oxidase assay conditions.
Expression and Function of the lpxQ Ortholog of A. tumefaciens in E. coli
As shown in Fig. 6, the genomes of both sequenced strains of A. tumefaciens encode a protein that is ~59% identical and ~77% similar to R. leguminosarum LpxQ (17, 18). Expression of the putative A. tumefaciens lpxQ gene behind the T7lac promoter in two strains of E. coli resulted in the appearance of significant lipid A oxidase activity in cell extracts, as judged by the conversion of [14C]B to [14C]D-1 (Fig. 8). This finding suggests that A. tumefaciens may be able to synthesize 2-aminogluconate containing lipid A under some conditions.
Fig. 8. The lpxQ homolog of A. tumefaciens directs the expression of oxidase activity in E. coli.
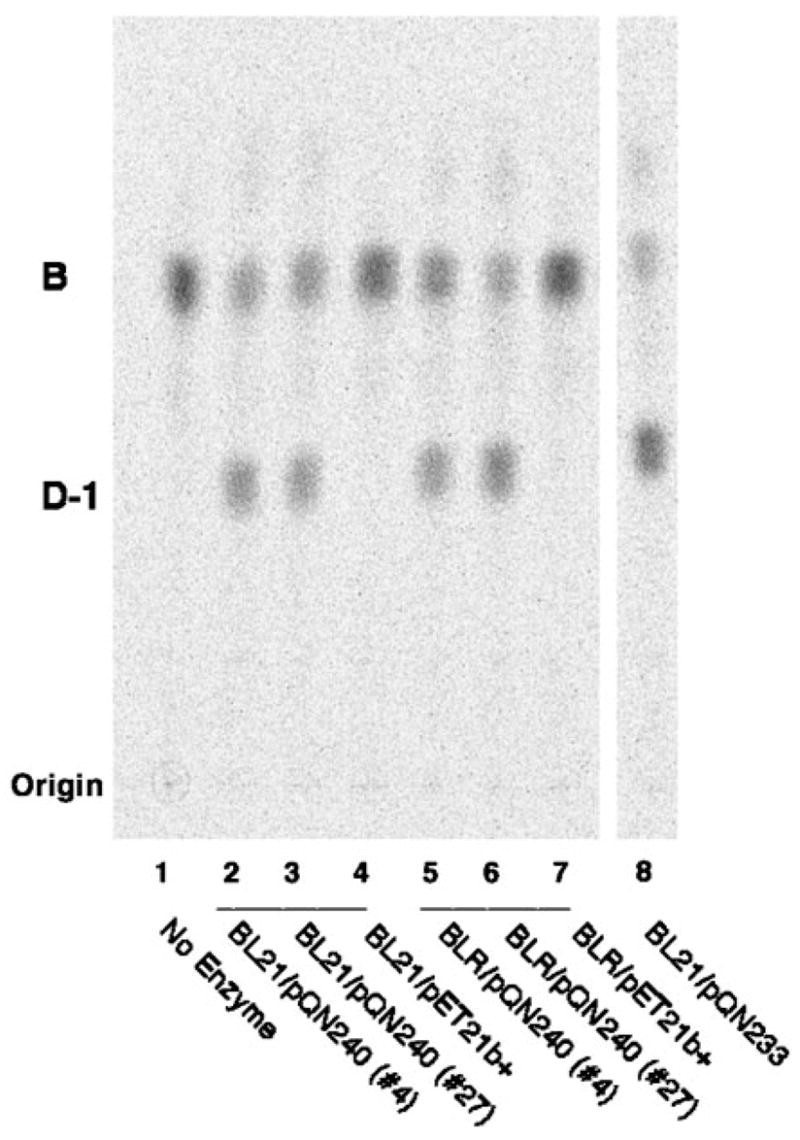
The lpxQ homolog of A. tumefaciens was amplified by PCR and cloned into pET21b+. The resulting hybrid plasmid is designated pQN240. Membranes of E. coli BL21(DE3)/pLysS/pQN240 (lanes 2 and 3) or BLR(DE3)/pLysS/pQN240 (lanes 5 and 6), grown and induced as described in the legend to Fig. 7, were assayed under standard conditions for 15 min. The no enzyme control is shown in lane 1. Membranes derived from cells containing the vector were assayed in parallel (lanes 4 and 7). A positive control (i.e. membranes of the R. leguminosarum lpxQ overexpressing strain E. coli BL21(DE3)/pLysS/pQN233) is shown in lane 8.
Mass Spectrometry of D-1 Synthesized in Vitro by Recombinant LpxQ
The membranes of E. coli that express R. leguminosarum LpxQ were used to convert component B to a substance resembling D-1, as judged by TLC. The latter product was then separated from residual B and purified by DEAE cellulose column chromatography, as described in the accompanying article (1) for D-1 synthesized by membranes of R. leguminosarum 3855. MALDI/TOF mass spectrometry of the product in the positive mode (Fig. 9B) reveals ions at m/z 1996.2 and 2024.6, which are interpreted as [M+Na]+ of D-1 species differing in acyl chain length. Fig. 9A shows the positive mode spectrum of an authentic sample of D-1 isolated from R. etli CE3 (3), which gives rise to [M +Na]+ ions at m/z 1996.2 and 2024.6, both of which are 16 atomic mass units larger than the corresponding species in component B (spectrum not shown). The D-1 standard and the material synthesized by LpxQ under these conditions both possess identical ions near m/z 1299 (Fig. 9), indicating that the distal lipid A unit is not modified by recombinant LpxQ. Interestingly, in the E. coli system (Fig. 9B) additional peaks are observed at m/z 2234.8 and 2261.8 that are interpreted as arising from the further incorporation of a palmitate moiety into the proximal unit of D-1 by the E. coli acyltransferase PagP (7).
Fig. 9. Positive ion MALDI/TOF mass spectrum of D-1 generated by membranes of BL21(DE3)/pLysS/pQN233.

A reaction mixture (5 ml) containing 50 μM B was incubated for 16 h at 30 °C with 0.5 mg/ml membranes of BL21(DE3)/pLysS/pQN233 under standard conditions, as described in the accompanying article (1). Following partial purification of the D-1-like reaction product by ion exchange chromatography on a small column of DEAE-cellulose, MALDI/TOF mass spectrometry was performed in the positive ion mode (1). Panel A shows the spectrum of a standard preparation of component D-1 isolated from R. etli (3). Panel B shows the spectrum of the partially purified in vitro reaction product. In addition to confirming the incorporation of an oxygen atom into the proximal unit of B, this spectrum also reveals that about half of the D-1 was further modified with a palmitate residue, presumably because of the PagP acyltransferase activity that is present in membranes of the E. coli host strain (7).
Disappearance of LpxQ Activity under Anaerobic Conditions in Vitro
As discussed in the accompanying article (1), a plausible mechanism for the oxidation B to D-1 might involve oxygen as the electron accepting substrate. Although it has not yet been possible to demonstrate stoichiometric hydrogen peroxide formation concomitant with D-1 synthesis, the results of Fig. 10 clearly demonstrate that LpxQ activity is strictly dependent upon the presence of oxygen. The further addition of glucose oxidase and catalase did not alter the rate of D-1 formation. Even after an overnight incubation, no detectable product was observed during oxygen deprivation (data not shown). The apparent requirement of recombinant LpxQ for molecular oxygen provides the strongest evidence presently available that this enzyme might be a novel type of oxidase.
Fig. 10. The lipid A oxidase encoded by lpxQ is dependent upon atmospheric oxygen.

The oxidase assay was performed either in an anaerobic chamber or under ambient atmospheric conditions, using membranes of an E. coli strain expressing a C-terminal His-tagged version of LpxQ. The conversion of [14C]B to [14C]D-1 was determined after 30, 60, or 90 min at 30 °C, using 0.1 mg/ml membranes from strain BLR(DE3)/pLysS/pQN235 that was induced with IPTG as in Fig. 5. The presence of glucose oxidase and catalase had little or no effect on the rate of the reaction or the extent of conversion.
LpxQ Localizes to the Outer Membrane when Expressed in E. coli
Isopycnic sucrose density gradient centrifugation of membranes prepared from induced cells of Novablue(DE3)/pQN233 was used to evaluate the subcellular localization of the recombinant LpxQ oxidase. The inner and outer membranes of the induced construct were well separated, as shown in Fig. 11A, by assay of the marker enzymes NADH oxidase and phospholipase A, respectively. Most of the lipid A oxidase is associated with the outer membrane in this construct (Fig. 11B), as in R. leguminosarum 3855 (1).
Fig. 11. Outer membrane localization of LpxQ oxidase activity in E. coli Novablue(DE3)/pQN233.

Washed membranes obtained from induced cells of E. coli Novablue(DE3)/pQN233 were separated by isopycnic sucrose density gradient centrifugation. Marker enzymes localized in the outer or inner membranes (phospholipase A and NADH oxidase respectively) were assayed to evaluate the extent of separation. A, phospholipase A and NADH oxidase activity. B, LpxQ oxidase activity and protein concentration.
A protein corresponding in size to that expected for LpxQ (~23 kDa) is present in the outer membranes of Novablue(DE3)/pQN233 (Fig. 12), as judged by SDS-PAGE, but not in the outer membranes of the vector control Novablue(DE3)/pET21a+. The program SignalP (40) does in fact predict that LpxQ is an outer membrane protein with an N-terminal signal peptide that may be cleaved between Ala27 and Glu28. Signal sequences of this kind are present in virtually all outer membrane proteins of Gram-negative bacteria, because they are essential for proper translocation (41). The N-terminal sequence of the first 10 amino acids of the putative mature LpxQ protein band present in the outer membranes of Novablue(DE3)/pQN233 was determined as EDLQFSIYGG. This result corresponds precisely to the predicted cleavage site and establishes conclusively that LpxQ is a genuine outer membrane protein.
Fig. 12. Outer membrane localization of overexpressed LpxQ protein in E. coli Novablue(DE3)/pQN233.
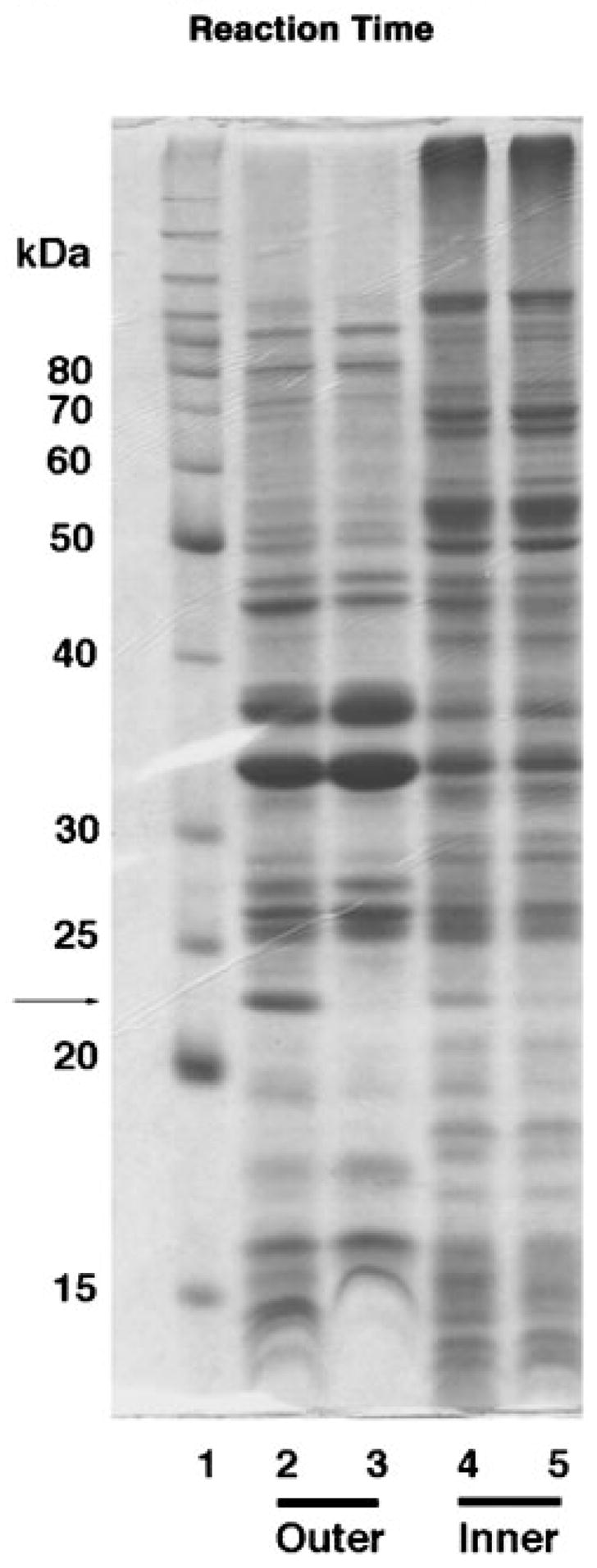
Membranes of E. coli Novablue(DE3)/pQN233 and Novablue(DE3)/pET21a+ were fractionated by isopycnic sucrose gradient centrifugation as described under “Experimental Procedures.” Peak fractions of outer and inner membranes were pooled separately, and recovered by ultracentrifugation. The membranes were subjected to SDS-PAGE (12% gel and 40 μg of protein/lane). A band of the size expected for LpxQ is seen only in the outer membranes derived from E. coli Novablue(DE3)/pQN233, as indicated by the arrow. Lane 1, Molecular weight marker (Benchmark Protein Ladder from Invitrogen); lane 2, outer membranes of Novablue(DE3)/pQN233; lane 3, outer membranes of Novablue(DE3)/pET21a+; lane 4, inner membranes of Novablue(DE3)/pQN233; lane 5, inner membranes of Novablue(DE3)/pET21a+. The putative LpxQ band, indicated by the arrow, was excised and subjected to N-terminal microsequencing.
DISCUSSION
All Gram-negative bacteria synthesize the lipid A component of lipopolysaccharide by a means of a constitutive seven-step pathway that starts with UDP-GlcNAc and proceeds via the tetra-acylated precursor, Kdo2-lipid IVA, as indicated schematically in Fig. 13 (16, 42). At least one additional secondary acyl chain is usually added after Kdo incorporation, most commonly at the 2 ′position (16, 42). In the case of R. leguminosarum, this additional acyl chain is 28 carbon atoms long and is hydroxylated at position 27 (Fig. 1) (1, 43, 44). A special acyl carrier protein is required for 27-OH-C28 synthesis and transfer to lipid A (Fig. 13) (43, 44). In E. coli a secondary laurate chain is added at the 2 ′ position, and myristate is added at 3 ′ (1, 45–48). All of the reactions leading to Kdo2-lipid IVA are cytosolic or associated with the inner membrane, as is the incorporation of 27-OH-C28 (Fig. 13) in R. leguminosarum or laurate and myristate in E. coli (16, 42).
Fig. 13. Proposed compartmentalization of R. leguminosarum lipid A biosynthesis and function of LpxQ.
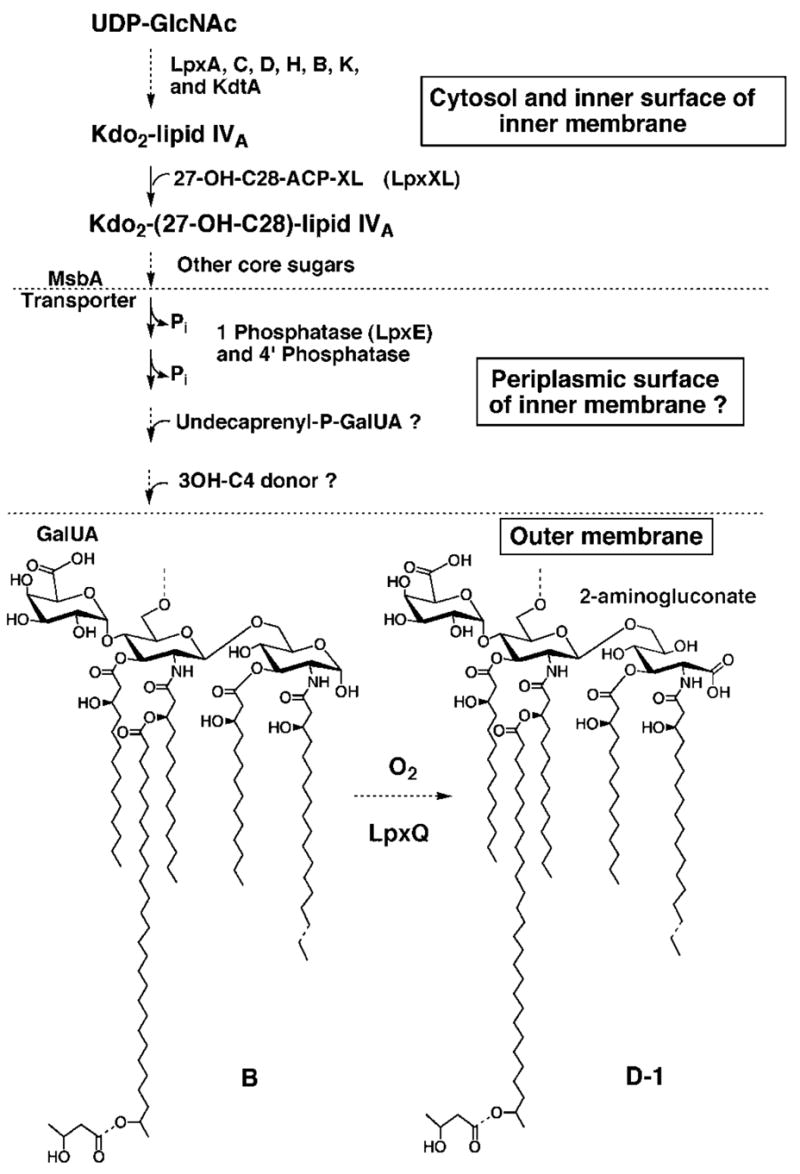
Almost all of the enzymes needed to generate component B have been detected in extracts of R. leguminosarum and R. etli (43, 44, 55–57). The only exceptions are the reactions that incorporate the 4 ′-galacturonic acid and the β-hydroxybutyrate residues. Although the hypothetical glycolipid, undecaprenylphosphate-GalUA, has not actually been isolated from cells or confirmed as the GalUA donor substrate in vitro, recent work in our laboratory suggests that GalUA transfer to R. leguminosarum lipopolysaccharide precursors requires a membrane-bound donor (S. S. Basu, M. Kanipes, and C. R. H. Raetz, unpublished results). The lpxE gene, which was recently found by expression cloning,2 encodes the 1-phosphatase, and it is predicted to have a periplasmic active site. The gene encoding the 4 ′-phosphatase is unknown. The ABC transporter MsbA is proposed to catalyze the flip-flop of the nascent lipid A 1,4 ′ bis-phosphate with attached core sugars across the inner membrane (58–60), thereby presenting this intermediate to the periplasmic lipid A modification enzymes. Following the formation of component B and transport to the outer membrane by unknown mechanisms, the LpxQ oxidase converts the proximal glucosamine residue to the 2-aminogluconate unit in an oxygen-dependent manner. If the oxidation reaction proceeds through a lactone intermediate (as discussed in the accompanying article (1)), an additional lactonase (not shown) might be needed to generate D-1. However, hydrolysis of such a lactone could be catalyzed by LpxQ itself or be nonenzymatic.
An emerging theme in the enzymatic assembly of lipid A is that additional (often regulated) covalent modifications of the conventional lipid A disaccharide 1, 4 ′-bis-phosphate backbone may occur beyond the cytoplasm (16). In polymyxin-resistant mutants of E. coli and S. typhimurium, the 4-amino-4-deoxy-L-arabinose (L-Ara4N) substituent is attached to the 4 ′ phosphate moiety of lipid A on the outer surface of the inner membrane (49, 50). The proposal is supported by the recent discovery that the L-Ara4N donor substrate is the novel lipid, undecaprenyl phosphate-L-Ara4N, and by the predicted topography of the enzyme (ArnT) that transfers the L-Ara4N unit to lipid A (49, 50). Some covalent modifications of lipid A can even occur in the outer membrane. The addition of a secondary palmitate chain at the 2 position of lipid A is catalyzed by the outer membrane acyltransferase PagP in S. typhimurium and E. coli, which uses a phospholipid as its acyl donor (7, 8, 51). The recent NMR studies of PagP have localized the active site of the enzyme to the outer surface of the outer membrane (8). Finally, S. typhimurium also contains PagL, a specific lipid A 3-O deacylase that is localized in the outer membrane (34). Unlike the constitutive (cytoplasmic) enzymes in the lipid A pathway (Fig. 13), which is highly conserved in virtually all Gram-negative bacteria (16), the lipid A-modifying enzymes found in the outer layers of the cell envelope are variable and relatively restricted in their distribution.
The data presented in this and the accompanying article (1) demonstrate conclusively that LpxQ is a novel type of outer membrane enzyme catalyzing an unusual oxidative modification of lipid A molecules that are dephosphorylated at the 1 position (Fig. 13). Heterologous expression behind the T7lac promoter in E. coli provides definitive evidence that lpxQ is indeed the structural gene for the oxidase (Fig. 7). Although two other outer membrane enzymes (PagP and PagL) have recently been discovered that catalyze covalent modifications of lipid A in S. typhimurium and E. coli (7, 34), as noted above, LpxQ is the first example of an outer membrane oxidase in any bacterial system.
The primary sequence of LpxQ is distinct from all previously described sugar oxidases or dehydrogenases but is distantly similar to selected outer membrane proteins, as judge by BLASTp or PSI-BLAST analysis of the nonredundant data base. The only significant ortholog (Fig. 6) of LpxQ (55% identity and 77% similarity over the length of the protein) currently in the nonredundant data base occurs in strains of A. tumefaciens, the plant pathogen that causes Crown-Gall disease and that is used to generate transgenic and mutant plants (17, 18). The results of Fig. 8 show that A. tumefaciens LpxQ expressed in E. coli catalyzes the same oxidative lipid A modification as does R. leguminosarum LpxQ. However, the presence of lipid A species containing the 2-aminogluconate moiety have not actually been described in A. tumefaciens, although this issue has not been investigated in any depth. Interestingly, the identity and arrangement of the genes that flank LpxQ in A. tumefaciens (17, 18) is quite similar to what is seen in R. leguminosarum (Fig. 5).
The function of the 2-aminogluconate unit generated by LpxQ in R. leguminosarum is unknown. The occurrence of the lpxQ gene in two important bacterial systems that are of great interest in plant biology necessitates further studies of the significance of the 2-aminogluconate moiety. The isolation of mutants of R. leguminosarum and A. tumefaciens, in which the lpxQ gene is deleted, should provide a powerful approach to this problem, especially if such strains are viable and grow normally under laboratory conditions. These mutants could be tested for their ability to infect and interact with their plant hosts in comparison with wild type bacteria. The additional availability of the lpxE gene encoding the lipid A 1-phosphatase,2 which generates the substrate for LpxQ (Fig. 13), together with lpxQ might even enable the oxidative modification of lipid A in bacteria like S. meliloti, which do not ordinarily make the 2-aminogluconate residue. The R. leguminosarum lpxE structural gene encoding the 1-phosphatase has recently been cloned in our laboratory, and its properties will be described elsewhere.2 It is an inner membrane protein with an active site that is predicted to face the periplasm (Fig. 13).
Our results demonstrate that formation of the 2-aminogluconate unit in component D-1 occurs late in the R. leguminosarum lipid A pathway. The lipid A of R. leguminosarum/pQN210, which overexpresses lpxQ, is highly enriched in component D-1 with very little remaining B (data not shown). In contrast, the ratio between B to D-1 in wild type R. leguminosarum lipid A is usually about 1:2 (3, 4). The extra copies of the oxidase present in R. leguminosarum/pQN210 appear to deplete the available component B.
The availability of the lpxQ gene should facilitate the purification of the oxidase to homogeneity. With pure enzyme it should be possible to explore the proposed role of oxygen as the electron accepting cosubstrate in the conversion of B to D-1 and to confirm the stoichiometric formation of hydrogen peroxide. The identification of possible organic cofactors, the characterization of the catalytic mechanism, and the evaluation of the significance of the EDTA inhibition should be greatly simplified with pure protein. Finally, the structural biology of LpxQ should also be of great interest in relation to other outer enzymes of know structure, like PagP (8), OmpT (9), and PldA (6). The tertiary structure of LpxQ might reveal how the characteristic inside out β-barrel folds of outer membrane proteins have evolved to generate novel catalytic sites.
Acknowledgments
We thank Kimberly Johnson and Margo Wuebbens of the Rajagopalan laboratory at Duke University for assistance with the anaerobic chamber.
Footnotes
This work was supported by National Institutes of Health Grants R37-GM-51796 (to C. R. H. R.) and GM54882 (to R. J. C.).
The abbreviations used are: MES, 2-(N-morpholino)-ethanesulfonic acid; 2-aminogluconate, 2-amino-2-deoxy-gluconate; CAPS, 3-(cyclo-hexylamino)propanesulfonic acid; IPTG, isopropyl-1-thio-β-D-galacto-pyranoside; MALDI, matrix-assisted laser desorption ionization; TOF, time-of-flight; Kdo, 3-deoxy-D-manno-2-octulosonic acid.
M. Karbarz and C. R. H. Raetz, manuscript in preparation.
References
- 1.Que-Gewirth NLS, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2003;278:12109–12119. doi: 10.1074/jbc.M300378200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhat UR, Forsberg LS, Carlson RW. J Biol Chem. 1994;269:14402–14410. [PubMed] [Google Scholar]
- 3.Que NLS, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2000;275:28006–28016. doi: 10.1074/jbc.M004008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Que NLS, Ribeiro AA, Raetz CRH. J Biol Chem. 2000;275:28017–28027. doi: 10.1074/jbc.M004009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishijima M, Nakaike S, Tamori Y, Nojima S. Eur J Biochem. 1977;73:115–124. doi: 10.1111/j.1432-1033.1977.tb11297.x. [DOI] [PubMed] [Google Scholar]
- 6.Snijder HJ, Ubarretxena-Belandia I, Blaauw M, Kalk KH, Verheij HM, Egmond MR, Dekker N, Dijkstra BW. Nature. 1999;401:717–721. doi: 10.1038/44890. [DOI] [PubMed] [Google Scholar]
- 7.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CRH. EMBO J. 2000;19:5071–5080. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CRH, Prive GG, Bishop RE, Kay LE. Proc Natl Acad Sci U S A. 2002;99:13560–13565. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vandeputte-Rutten L, Kramer RA, Kroon J, Dekker N, Egmond MR, Gros P. EMBO J. 2001;20:5033–5039. doi: 10.1093/emboj/20.18.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W. Science. 1998;282:2215–2220. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- 11.Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Nature. 2000;405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- 12.Schulz GE. Biochim Biophys Acta. 2002;1565:308–317. doi: 10.1016/s0005-2736(02)00577-1. [DOI] [PubMed] [Google Scholar]
- 13.Fernandez C, Hilty C, Bonjour S, Adeishvili K, Pervushin K, Wüthrich K. FEBS Lett. 2001;504:173–178. doi: 10.1016/s0014-5793(01)02742-9. [DOI] [PubMed] [Google Scholar]
- 14.Groisman EA. J Bacteriol. 2001;183:1835–1842. doi: 10.1128/JB.183.6.1835-1842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohl ME, Miller SI. Annu Rev Med. 2001;52:259–274. doi: 10.1146/annurev.med.52.1.259. [DOI] [PubMed] [Google Scholar]
- 16.Raetz CRH, Whitfield C. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wood DW, Setubal JC, Kaul R, Monks DE, Kitajima JP, Okura VK, Zhou Y, Chen L, Wood GE, Almeida NF, Jr, Woo L, Chen Y, Paulsen IT, Eisen JA, Karp PD, Bovee D, Sr, Chapman P, Clendenning J, Deatherage G, Gillet W, Grant C, Kutyavin T, Levy R, Li MJ, McClelland E, Palmieri A, Raymond C, Rouse G, Saenphimmachak C, Wu Z, Romero P, Gordon D, Zhang S, Yoo H, Tao Y, Biddle P, Jung M, Krespan W, Perry M, Gordon-Kamm B, Liao L, Kim S, Hendrick C, Zhao ZY, Dolan M, Chumley F, Tingey SV, Tomb JF, Gordon MP, Olson MV, Nester EW. Science. 2001;294:2317–2323. doi: 10.1126/science.1066804. [DOI] [PubMed] [Google Scholar]
- 18.Goodner B, Hinkle G, Gattung S, Miller N, Blanchard M, Qurollo B, Goldman BS, Cao Y, Askenazi M, Halling C, Mullin L, Houmiel K, Gordon J, Vaudin M, Iartchouk O, Epp A, Liu F, Wollam C, Allinger M, Doughty D, Scott C, Lappas C, Markelz B, Flanagan C, Crowell C, Gurson J, Lomo C, Sear C, Strub G, Cielo C, Slater S. Science. 2001;294:2323–2328. doi: 10.1126/science.1066803. [DOI] [PubMed] [Google Scholar]
- 19.Cava JR, Elias PM, Turowski DA, Noel KD. J Bacteriol. 1989;171:8–15. doi: 10.1128/jb.171.1.8-15.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Segovia L, Young JP, Martinez-Romero E. Int J Syst Bacteriol. 1993;43:374–377. doi: 10.1099/00207713-43-2-374. [DOI] [PubMed] [Google Scholar]
- 21.Meade HM, Long SR, Ruvkun GB, Brown SE, Ausubel FM. J Bacteriol. 1982;149:114–122. doi: 10.1128/jb.149.1.114-122.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galibert F, Finan TM, Long SR, Puhler A, Abola P, Ampe F, Barloy-Hubler F, Barnett MJ, Becker A, Boistard P, Bothe G, Boutry M, Bowser L, Buhrmester J, Cadieu E, Capela D, Chain P, Cowie A, Davis RW, Dreano S, Federspiel NA, Fisher RF, Gloux S, Godrie T, Goffeau A, Golding B, Gouzy J, Gurjal M, Hernandez-Lucas I, Hong A, Huizar L, Hyman RW, Jones T, Kahn D, Kahn ML, Kalman S, Keating DH, Kiss E, Komp C, Lelaure V, Masuy D, Palm C, Peck MC, Pohl TM, Portetelle D, Purnelle B, Ramsperger U, Surzycki R, Thebault P, Vandenbol M, Vorholter FJ, Weidner S, Wells DH, Wong K, Yeh KC, Batut J. Science. 2001;293:668–672. doi: 10.1126/science.1060966. [DOI] [PubMed] [Google Scholar]
- 23.Miller JR. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 24.Ronson CW, Astwood PM, Downie JA. J Bacteriol. 1984;160:903–909. doi: 10.1128/jb.160.3.903-909.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedman AM, Long SR, Brown SE, Buikema WJ, Ausubel FM. Gene (Amst) 1982;18:289–296. doi: 10.1016/0378-1119(82)90167-6. [DOI] [PubMed] [Google Scholar]
- 26.Wood W. J Mol Biol. 1966;16:118–133. doi: 10.1016/s0022-2836(66)80267-x. [DOI] [PubMed] [Google Scholar]
- 27.Glazebrook J, Walker GC. Methods Enzymol. 1991;204:398–418. doi: 10.1016/0076-6879(91)04021-f. [DOI] [PubMed] [Google Scholar]
- 28.Finan TM, Kunkel B, De Vos GF, Signer ER. J Bacteriol. 1986;167:66–72. doi: 10.1128/jb.167.1.66-72.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 30.Bergmans HE, van Die IM, Hoekstra WP. J Bacteriol. 1981;146:564–570. doi: 10.1128/jb.146.2.564-570.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, Tatusova TA, Wagner L. Nucleic Acids Res. 2003;31:28–33. doi: 10.1093/nar/gkg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ditta G, Stanfield S, Corbin D, Helinski DR. Proc Natl Acad Sci U S A. 1980;77:7347–7351. doi: 10.1073/pnas.77.12.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacFerrin KD, Terranova MP, Schreiber SL, Verdine GL. Proc Natl Acad Sci U S A. 1990;87:1937–1941. doi: 10.1073/pnas.87.5.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trent MS, Pabich W, Raetz CRH, Miller SI. J Biol Chem. 2001;276:9083–9092. doi: 10.1074/jbc.M010730200. [DOI] [PubMed] [Google Scholar]
- 35.Guy-Caffey JK, Rapoza MP, Jolley KA, Webster RE. J Bacteriol. 1992;174:2460–2465. doi: 10.1128/jb.174.8.2460-2465.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 37.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 38.Raetz CRH. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 39.Raetz CRH. J Bacteriol. 1993;175:5745–5753. doi: 10.1128/jb.175.18.5745-5753.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nielsen H, Brunak S, von Heijne G. Protein Engineering. 1999;12:3–9. doi: 10.1093/protein/12.1.3. [DOI] [PubMed] [Google Scholar]
- 41.Wickner W, Driessen AJ, Hartl FU. Annu Rev Biochem. 1991;60:101–124. doi: 10.1146/annurev.bi.60.070191.000533. [DOI] [PubMed] [Google Scholar]
- 42.Raetz CRH. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. 2. Neidhardt FC, editor. Vol. 1. American Society for Microbiology; Washington, D.C.: 1996. pp. 1035–1063. [Google Scholar]
- 43.Brozek KA, Carlson RW, Raetz CRH. J Biol Chem. 1996;271:32126–32136. doi: 10.1074/jbc.271.50.32126. [DOI] [PubMed] [Google Scholar]
- 44.Basu SS, Karbarz MJ, Raetz CRH. J Biol Chem. 2002;277:28959–28971. doi: 10.1074/jbc.M204525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clementz T, Bednarski JJ, Raetz CRH. J Biol Chem. 1996;271:12095–12102. doi: 10.1074/jbc.271.20.12095. [DOI] [PubMed] [Google Scholar]
- 46.Clementz T, Zhou Z, Raetz CRH. J Biol Chem. 1997;272:10353–10360. doi: 10.1074/jbc.272.16.10353. [DOI] [PubMed] [Google Scholar]
- 47.Vorachek-Warren MK, Carty SM, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2002;277:14186–14193. doi: 10.1074/jbc.M200408200. [DOI] [PubMed] [Google Scholar]
- 48.Vorachek-Warren MK, Ramirez S, Cotter RJ, Raetz CRH. J Biol Chem. 2002;277:14194–14205. doi: 10.1074/jbc.M200409200. [DOI] [PubMed] [Google Scholar]
- 49.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2001;276:43122–43131. doi: 10.1074/jbc.M106961200. [DOI] [PubMed] [Google Scholar]
- 50.Trent MS, Ribeiro AA, Doerrler WT, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2001;276:43132–43144. doi: 10.1074/jbc.M106962200. [DOI] [PubMed] [Google Scholar]
- 51.Brozek KA, Bulawa CE, Raetz CRH. J Biol Chem. 1987;262:5170–5179. [PubMed] [Google Scholar]
- 52.Ronson CW, Astwood PM, Nixon BT, Ausubel FM. Nucleic Acids Res. 1987;15:7921–7934. doi: 10.1093/nar/15.19.7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Russa R, Lüderitz O, Rietschel ET. Arch Microbiol. 1985;141:284–289. [Google Scholar]
- 54.Gish W, States DJ. Nat Genet. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- 55.Price NPJ, Kelly TM, Raetz CRH, Carlson RW. J Bacteriol. 1994;176:4646–4655. doi: 10.1128/jb.176.15.4646-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Price NJP, Jeyaretnam B, Carlson RW, Kadrmas JL, Raetz CRH, Brozek KA. Proc Natl Acad Sci U S A. 1995;92:7352–7356. doi: 10.1073/pnas.92.16.7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brozek KA, Kadrmas JL, Raetz CRH. J Biol Chem. 1996;271:32112–32118. [PubMed] [Google Scholar]
- 58.Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CRH. J Biol Chem. 1998;273:12466–12475. doi: 10.1074/jbc.273.20.12466. [DOI] [PubMed] [Google Scholar]
- 59.Doerrler WT, Reedy MC, Raetz CRH. J Biol Chem. 2001;276:11461–11464. doi: 10.1074/jbc.C100091200. [DOI] [PubMed] [Google Scholar]
- 60.Chang G, Roth CB. Science. 2001;293:1793–1800. doi: 10.1126/science.293.5536.1793. [DOI] [PubMed] [Google Scholar]