

Abstract
Inwardly rectifying potassium (Kir) channels are a prominent feature of mature, postmitotic astrocytes. These channels are believed to set the resting membrane potential near the potassium equilibrium potential (EK) and are implicated in potassium buffering. A number of previous studies suggest that Kir channel expression is indicative of cell differentiation. We therefore set out to examine Kir channel expression in malignant glia, which are incapable of differentiation. We used two established and widely used glioma cell lines, D54MG (a WHO grade 4 glioma) and STTG-1 (a WHO grade 3 glioma), and compared them to immature and differentiated astrocytes. Both glioma cell lines were characterized by large outward K+ currents, depolarized resting membrane potentials (Vm)(-38.5 ± 4.2 mV, D54 and -28.1 ± 3.5 mV, STTG1), and relatively high input resistances (Rm) (260.6 ± 64.7 MΩ, D54 and 687.2 ± 160.3 MΩ, STTG1). These features were reminiscent of immature astrocytes, which also displayed large outward K+ currents, had a mean Vm of -51.1 ± 3.7 and a mean Rm value of 627.5 ± 164 MΩ. In contrast, mature astrocytes had a significantly more negative resting membrane potential (-75.2 ± 0.56 mV), and a mean Rm of 25.4 ± 7.4 MΩ. Barium (Ba2+) sensitive Kir currents were >20-fold larger in mature astrocytes (4.06 ± 1.1 nS/pF) than in glioma cells (0.169 ± 0.033 nS/pF D54, 0.244 ± 0.04 nS/pF STTG1), which had current densities closer to those of dividing, immature astrocytes (0.474 ± 0.12 nS/pF). Surprisingly, Western blot analysis shows expression of several Kir channel subunits in glioma cells (Kir2.3, 3.1, and 4.1). However, while in astrocytes these channels localize diffusely throughout the cell, in glioma cells they are found almost exclusively in either the cell nucleus (Kir2.3 and 4.1) or ER/Golgi (3.1). These data suggest that mislocalization of Kir channel proteins to intracellular compartments is responsible for a lack of appreciable Kir currents in glioma cells.
Keywords: brain tumor, astrocyte, patch-clamp, potassium channel, cell proliferation
INTRODUCTION
Glial cells express a variety of voltage-gated ion channels (Verkhratsky and Steinhauser, 2000). Particular attention has been paid to the family of inwardly rectifying potassium channels (Kir). These channels are responsible for the very negative resting membrane potential that is characteristic of most glial cells (Newman, 1993; Ransom and Sontheimer, 1995). Moreover, Kir channels are thought to participate in extracellular K+ homeostasis (Newman, 1995; Walz, 2000). These suspected functions have been substantiated in retinal glial cells through the use of knock-out mice in which Kir4.1, the primary Kir gene in Müller glial cells, was genetically eliminated (Kofuji et al., 2000).
Interestingly, several studies have demonstrated profound changes in Kir channel expression in glial cells during development (Kressin et al., 1995; Bordey and Sontheimer, 1997; Bringmann et al., 1999a; MacFarlane and Sontheimer, Bringmann et al., 2000a). Kir currents appear to be absent in immature cells and begin to appear gradually in glial cells as they elaborate their processes. More detailed studies suggest that Kir channel expression correlates positively with cell differentiation, and Kir activity appears to be absent in proliferating glial cells. This relationship of Kir channel expression and cell differentiation has been demonstrated in astrocytes (MacFarlane and Sontheimer, 1997), Schwann cells (Wilson and Chiu, 1990; Konishi, 1994), Müller cells (Bringmann et al., 2000), and oligodendrocytes (Sontheimer et al., 1989; Barres et al., 1990) and thus appears to be a general feature of all glial cells. Intriguingly, changes in the expression and/or activity of Kir channels occur irrespective of how the change in differentiation state is initiated. For example, in Müller cells of the retina, spontaneous cell maturation causes an increase in Kir channel activity (Bringmann et al., 1999b), but this can be reversed upon cell injury or gliosis, which causes downregulation of channel activity in concert with enhanced glial proliferation (Bringmann et al., 2000). Similarly, acute injury of spinal cord astrocytes leads to a loss of Kir channel activity and more depolarized resting membrane potentials (MacFarlane and Sontheimer, 1997). Interestingly, in spinal cord astrocytes, expression of Kir channels can be induced prematurely by exposure of cells to the differentiation agent retinoic acid (MacFarlane and Sontheimer, 2000a). Alternatively, pharmacological inhibition of Kir channels retards cell differentiation (MacFarlane and Sontheimer, 2000a). These data therefore suggest a reciprocal and possibly functional relationship between cell differentiation and Kir channel expression.
To examine this interdependence of Kir channel expression and cell differentiation further, we turned to glial-derived tumors cells (glioma) as a model system. Gliomas are characterized by defects in genes involved in cell cycle regulation (Von Deimling et al., 1995; Tang et al., 1997) and have lost the ability to differentiate spontaneously (Schmidt et al., 2000). Some of the electrophysiological characteristics of gliomas appear to be similar to those of undifferentiated or immature glial cell. For example, glioma cells both in vitro and in situ are characterized by depolarized resting membrane potentials (Bordey and Sontheimer, 1998; Ullrich et al., 1998) and express large outwardly rectifying potassium currents (Ransom and Sontheimer, 2001; Ransom et al., 2002). These electrophysiological traits typify those described in proliferating astrocytes (MacFarlane and Sontheimer, 2000a), in immature, proliferating, or diseased Müller cells (Puro et al., 1989; Francke et al., 1997; Bringmann et al., 1999b), and in oligodendrocyte precursor cells (Sontheimer et al., 1989; Williamson et al., 1997). These macroscopic features would also predict an absence of Kir channels in glioma cells.
In this study, we used Kir-specific antibodies to compare the expression and localization of Kir channels in glioma cells to that of immature, undifferentiated and fully differentiated astrocytes. Using Western blot analysis and immunocytochemistry, we demonstrate that glioma cells express several of the inwardly rectifying potassium channels previously described in astrocytes, including Kir 4.1. However, in stark contrast to mature astrocytes, in which these channels localize to the plasma membrane, Kir channels are mislocalized to the cell nucleus of glioma cells. These observations explain the inability to record significant Kir currents in glioma cells.
MATERIALS AND METHODS
Cell Culture
Experiments were performed on two glioma cell lines, rat cultured spinal cord and rat cultured cortical astrocytes. D54-MG cells (glioblastoma multiforme [GBM], World Health Organization [WHO] grade IV) were a gift from Dr. D. Bigner (Duke University), and STTG-1 cells (anaplastic astrocytoma, WHO grade III), were obtained from the American Type Tissue Collection (ATCC, Rockville, MD). Cells were cultured in Dulbecco’s modified Eagle medium/F12 (DMEM/F12) (Gibco, Grand Island, NY) supplemented with 7% fetal calf serum (FCS) (Hyclone, Logan, UT) and 2 mM glutamine (Gibco). Primary cultures of spinal cord or cortical astrocytes were obtained by slightly modified methods described previously (MacFarlane and Sontheimer, 1997). Briefly, the spinal cord or cortex was dissected into ice-cold serum-free EMEM (Gibco) plus 20 mM glucose, meninges were stripped, and tissue was minced and placed into an O2-saturated papain solution (Worthington, Lakewood, NJ)) for 20 min. The tissue was washed twice with spinal cord astrocyte media (EMEM supplemented with 10% FCS, 20 mM glucose and penicillin/streptomycin) and then triturated. Cells were plated at a density of 1.0 × 10-6 cells/ml on polyornithine- and laminin-coated cover-slips (12-mm round, Macalaster Bicknell, New Haven, CT). The media was changed every day for the first 3 days and then every fourth day thereafter. Spinal cord astrocytes were designated as immature or mature based on two criteria: time in culture and cell morphology. Proliferation of spinal cord astrocytes in culture follows a specific temporal pattern, with high proliferation rates up to day 4 and a rapid decline in cell proliferation as cells in the culture dish approach confluency (as determined by 3H-thymidine and bromodeoxyuridine [BrdU] incorporation) (Ullrich et al., 1994). Cell morphology changes are also indicative of astrocyte maturation or differentiation. Cell processes become longer, more numerous and more complex as astrocytes differentiate. These changes have been characterized in culture and in situ upon spontaneous differentiation (Bignami and Dahl, 1974; Duffy, 1983; Tang et al., 2002) as well as upon drug-induced or serum withdrawal induced differentiation (Moonen et al., 1975; Couchie et al., 1985; MacFarlane and Sontheimer, 2000a).
Electrophysiology
Whole-cell voltage-clamp recordings were obtained by standard methods (Hamill et al., 1981). Patch pipettes were made from thin-walled (outer diameter 1.5 mm, inner diameter 1.12 mm) borosilicate glass (TW150F-4; WPI, Sarasota, FL) and had resistances of 3–5 MΩ. Recordings were made on the stage of an inverted Nikon diaphot microscope equipped with Hoffman Modulation Contrast Optics. Current recordings were obtained with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA). Current signals were low-pass filtered at 2 kHz and were digitized on-line at 10–20 kHz, using a Digidata 1200 digitizing board (Axon Instruments interfaced with an IBM-compatible computer (Dell XPS R400). Data acquisition and storage were conducted with the use of pClamp 8.2 (Axon Instruments). Resting membrane potentials, cell capacitance, and series resistances were measured directly from the amplifier, with the upper limit for series resistance being 10 MΩ, and series resistance compensation adjusted to 80% to reduce voltage errors. D54 and STTG1 cells were plated on glass coverslips in a 24-well plate and recordings were made 2–5 days after plating; spinal cord astrocytes were recorded from either 2–3 days for immature astrocytes or >6 days for mature astrocytes.
Solutions
The standard KCl pipette solution contained (in mM): 145 KCl, 1 MgCl2, 10 EGTA, and 10 Hepes sodium salt, pH adjusted to 7.3 with Tris-base. CaCl2 (0.2 mM) was added to the pipette solution just before recording resulting in a free calcium concentration of 1.9 nM. Cells were continuously perfused at room temperature with a saline solution containing (in mM): 125 NaCl, 5.0 KCl, MgSO4, 1.0 CaCl2, 1.6 Na2HPO4, 0.4 NaH2PO4, 10.5 glucose, and 32.5 Hepes acid. The pH was adjusted to 7.4 with NaOH and the osmolarity of this solution was ∼300 mOsm. For the 20 mM high K+ solution, 15 mM NaCl was omitted from the bath solution. Drugs were added directly to these solutions and, unless stated otherwise, all drugs were purchased from Sigma (St. Louis, MO). When BaCl2 was added to the bath solution, phosphates and sulfates were omitted to prevent precipitation of barium phosphate and barium sulfate.
Western Blot Analysis
Cells were lysed using RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40 (NP-40), 0.5% sodium deoxycholate, 1% sodium dodecyl sulfate [SDS]) for 30 min supplemented with protease inhibitor cocktail obtained from Sigma (St. Louis, MO). Cells were sonicated for 10 s; homogenates were then centrifuged for 5 min at 12,000g at 4°C. Protein quantification was performed on the supernatant using a DC protein assay kit from Bio-Rad (Hercules, CA). Protein was boiled in Laemmli-SDS sample buffer containing 600 mM β-mercaptoethanol for 5 min. Equal amounts of protein were loaded into each lane of a 4–20% gradient pre-cast acrylamide SDS-polyacrylamide gel electrophoresis (PAGE) gel (Bio-Rad). Proteins were separated at 120 V constant. Gels were transferred onto polyvinylidine difluoride (PVDF) paper (Millipore, Bedford, MA) at 200 mA constant for 2 h at room temperature; membranes were blocked in blocking buffer (BB, 5% nonfat dried milk, 2% bovine serum albumin (BSA) and 2% normal goat serum (NGS) in TBS plus 0.1% Tween 20; TBST). All antibodies were obtained from Alomone (Jerusalem, Israel) and were incubated in primary antibody according to the manufacturer’s instructions. The membranes were then rinsed three times for 10 min and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 90 min. Blots were once again washed three times for 10 min and developed with enhanced chemiluminesence (ECL; Amersham, Arlington Heights, IL) on Hyperfilm (Amersham). Actin and secondary HRP-conjugated antibodies were purchased from Sigma.
Immunocytochemistry
Cells plated on coverslips were washed twice with phosphate-buffered saline (PBS) and fixed with 4.0% paraformaldehyde for 15 min. Cells were then washed two more times with PBS and then permeabilized in PBS, 0.3% Triton X-100, and 3% goat serum (NGS) for 30 min. Primary Kir antibodies and glial fibrillary acidic protein (GFAP; Chemicon, Temecula, CA) was diluted in PB and added according to manufacturer’s recommendations overnight at 4°C. Phalloidin (Molecular Probes, Eugene OR) was incubated with the secondary antibody. The cells were washed three times in PBS before adding a fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit secondary antibody (Molecular Probes) diluted at 1:500 in PB for 1 h at room temperature. Cells were then washed twice with PBS, washed once with DAPI (10-4 mg/ml, Sigma), diluted in PBS for 5 min. DAPI was rinsed off with PBS; cells were then mounted onto clean coverslips with Gel/ Mount (Biomedia, Foster City, CA). 1000× fluorescent images were acquired with a Zeiss Axiovert 200M (München, Germany).
Statistical Analysis
Current responses to varied voltage steps and ramps were analyzed and measured in Clampfit (Axon Instruments); the resulting raw data were graphed and plotted in Origin 6.0 (MicroCal, Northampton, MA). Unless otherwise stated, all values are reported as mean ±SE, with n equal to the number of cells sampled.
RESULTS
Expression of Outward and Inward K+ Currents in Astrocytes and Glioma Cells
As a first step toward the characterization of Kir channels in glioma cells, we obtained whole-cell recordings from two widely used glioma cell lines, D54-MG (WHO grade 4) and STTG-1 (WHO grade 3), and compared them to cultured astrocytes that were either immature (see Materials and Methods) and actively dividing or fully differentiated astrocytes. Representative recordings are illustrated in Figure 1. Both glioma cell lines showed large outward currents that could be attributed to the activation of large conductance Ca2+-activated potassium channels (Fig. 1A), as previously shown (Ransom and Sontheimer, 2001; Ransom et al., 2002). Indeed, these currents activated at very positive potentials (Fig. 1C) and were steeply voltage dependent. Moreover, outward currents in glioma cells were sensitive to TEA, charybdotoxin, and iberiotoxin, but were insensitive to Ba2+ and 4-aminopyridine (not shown). At the macroscopic level, inward K+ currents in both glioma cell lines were negligible (Fig 1A,C). Immature, undifferentiated spinal cord astrocytes (<4 days in culture [DIC]) similarly showed only outward K+ currents, which activate at potentials >-50 mV (Fig. 1B,C), and which are sensitive to 4-AP and margatoxin. These currents are largely mediated by Kv1.5, as previously described (MacFarlane and Sontheimer, 2000b). In contrast, more mature, differentiated spinal cord astrocytes express both outward and inward K+ currents (Fig. 1B, right) and consequently show significant K+ conductance at essentially all membrane potentials (Fig. 1B,C).
Fig. 1.

Whole-cell currents in malignant and non—malignant glia. A: Representative examples of whole-cell current in D54 and STTG1 cells that were evoked with voltage steps from -160 to +160 mV from a holding potential of -40 mV. Traces demonstrate the primary current in both STTG1 and D54 glioma cells is an outward current. B: Representative examples of whole-cell current in an immature and mature spinal cord astrocyte that were evoked with voltage steps from -160 to +160 mV from a holding potential of -80 mV. Traces demonstrate the primary current in an immature astrocyte is an outward current while mature astrocytes express large inward currents as well. C: An I-V curve constructed from the traces in A and B compares the difference in total current between both glioma cells, an immature astrocyte and a differentiated astrocyte.
Since each of these cells has a number of K+ channels, and possibly other ion channels contributing to whole-cell currents, we chose to isolate the contribution of Kir channels pharmacologically from the total current. Specifically, we defined the Kir current as the component of the current that was inhibited by 200 μM Ba2+, which has been shown to block nearly all Kir subunits (Coetzee et al., 1999). We also observed an increase in the inward currents when exposed to a higher extracellular K+ concentration ([K+]o). It has been well established Kir currents are dependent on [K+]o, whereby the inward current increases with increasing [K+]o concentrations (current proportional to the square root of [K+]o) (Hagiwara and Takahashi, 1974; Newman, 1993; Sakmann and Trube, 1984). Figure 2 demonstrates representative whole-cell recordings in response to voltage-ramps again comparing the two glioma cell lines, STTG-1 and D54, to immature astrocytes and differentiated astrocytes. Recordings were obtained under control conditions (5 mM [K+]o), after application of Ba2+ (200 μM), and after application of a bath solution containing high [K+]o (20 mM). Application of Ba2+ showed only a very modest reduction of inward currents in STTG1 glioma cells and immature astrocytes, and had essentially no effect on D54 glioma cells. The average decrease in inward current was 36.2 ± 6.0% (n = 10), 25.6 ± 13.6 (n = 7), and 3.9 ± 2.8 (n = 6), respectively. In contrast, inward currents in mature spinal cord astrocytes were largely blocked in Ba2+ (80.8 ± 6.2%, n = 6).
Fig. 2.
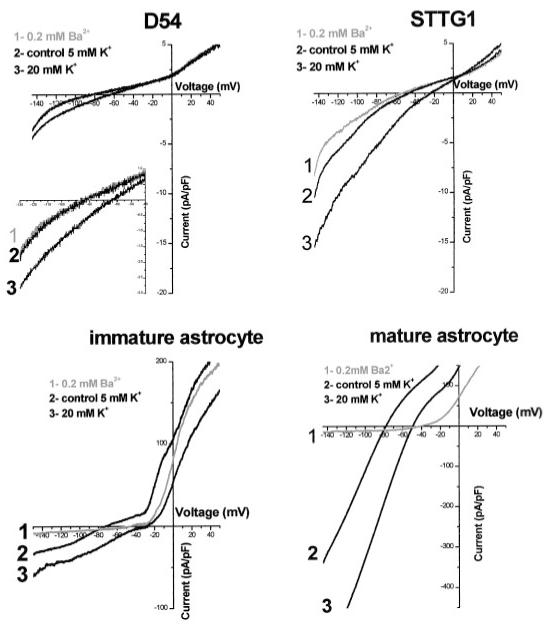
Representative examples of inward currents observed in STTG1 cells, D54 cells, immature astrocytes, and mature astrocytes. Currents were elicited by a linear voltage-ramp protocol (-145 mV to +50 mV, holding at -40 mV) and were normalized to cell size (pA/pF). Trace 1, after 200 μM Ba2+; trace 2, control (5 mM [K+]o); trace 3, high or 20 mM [K+]o). The inset magnifies the inward current in the D54 glioma cells. Note the differences in current amplitude when comparing traces.
For a more quantitative comparison of the relative expression of Kir currents between the proliferating cells (D54, STTG1, and SCA <3 DIC) and the mature astrocytes, we calculated the specific conductance of the Ba2+ sensitive inward current (GIR) for each cell and plotted mean values (±SEM) for each cell type in Figure 3A. These values were independent of cell size, as cell conductance was normalized to membrane capacitance (nS/pF). These data clearly illustrate the almost complete absence of significant Kir conductance from proliferating glioma cells or immature astrocytes and prominent expression in mature astrocytes. To assess more quantitatively the relative contribution of the Kir conductance to the overall conductance of the cells, we determined the ratio of the conductance associated with inward current (GKin) to that associated with outward K currents (GKout) (Fig. 3B). These data illustrate that in mature astrocytes, the conductance associated with Kir channels predominates by far over that associated with outwardly rectifying K+, while the reverse is true for immature astrocytes and for glioma cells. Note, however, that the outward currents in glioma cells are mediated by Ca2+ activated potassium channel (gBK) (Liu et al., 2002), whereas outward K+ currents in immature astrocytes are mediated largely by Kv1.5 (MacFarlane and Sontheimer, 2000b). To assess the possible effect that these conductances might have on input resistance (resistance of the whole cell near the resting potential) and resting membrane potential, we also plotted these parameters for comparison in Figure 3C,D (note in Fig. 3D that the more hyperpolarized potentials are toward the top of the y-axis). In previous studies, the relative expression of Kir channels has correlated well with the cells resting membrane potential. For example, O2A progenitor cells or immature astrocytes and Müller cells have low Kir channel densities and have depolarized (∼-40 to -60 mV) resting membrane potentials (Sontheimer et al., 1989; Ransom and Sontheimer, 1995; Williamson et al., 1997; Bringmann et al., 1999a). Differentiated astrocytes and retinal Müller cells express Kir channels at relatively high densities and have Vm values near EK (Ransom and Sontheimer, 1995; Kofuji et al., 2000). Consistent with these studies, both the glioma cell lines and immature spinal cord astrocytes had resting membrane potentials that were depolarized relative to spinal cord astrocytes (Fig. 3C; -38.5 ± 4.2 mV, D54 (n = 11) and -28.1 ± 3.5 mV, STTG1 (n = 13), -51.1 ± 3.7 mV, immature spinal cord astrocyte (n = 9) and -75.2 ± 0.56, mature spinal cord astrocyte (n = 6). Kir channels show a large open probability near or negative of EK. As would be expected, the spinal cord astrocytes, which express a high density of Kir channels, also have a relatively low input resistance (Rm) (25.4 ± 7.4, n = 6) (Fig. 3D). In contrast, D54, STTG1 and immature astrocytes, which express smaller Kir currents, have high Rm values, 302.7 ± 63.6, 687.2 ± 160.3, and 627.5 ± 164.3, respectively.
Fig. 3.

Summary of electrophysiological properties in D54 cells, STTG1 cells, immature and mature astrocytes. A: The specific conductance of the Ba2+-sensitive inward current (nS/pF) in each cell type was plotted (D54, n = 6, STTG1, n = 10; SCA <4 days in culture [DIC], n = 7; SCA >8 DIC n = 5). B: Plot of the ratio of inward versus outward current (nS/pF) in all four cell types (D54, n = 19; STTG1, n = 18; SCA <4 DIC, n = 12; SCA >8 DIC n = 6). C: Plot of the mean input resistance (MΩ) in each cell type (D54, n = 21; STTG1, n = 19; SCA <4 DIC, n = 11; SCA >8 DIC n = 6). D: The mean resting membrane potential (mV) of each cell type (D54, n = 11; STTG1, n = 13; SCA <4 DIC, n = 9; SCA >8 DIC n = 6) is shown. Note that more hyperpolarized potentials are toward the top of the y-axis.
Glioma Cells Express Several Kir Channel Proteins
While the above data suggest the possible absence of Kir channel proteins in glioma cells, it is conceivable that channels are expressed but are not functional under our recording conditions. Kir channel activity is modulated by kinases, pH, cAMP, G-proteins, interactions with lipids, and other proteins (for review, see Ruppersberg, 2000; Yi et al., 2001; Nichols and Lopatin, 1997; Isomoto et al., 1997). We therefore decided to examine Kir channel expression at the protein level by performing Western blot analysis on both glioma cell lysates (D54 and STTG1) and spinal cord astrocytes using antibodies specific for Kir2.1, 2.3, 3.1, and 4.1. We also included cortical astrocytes in our Western blots to ensure that we were not limiting our comparison to astrocytes from a single brain region. Surprisingly, our Western blots show expression of several Kir channel proteins that have previously been described in astrocytes and Müller cells (Kofuji et al., 2000; Raap et al., 2002; Schroder et al., 2002; Li et al., 2001) in both glioma cell lines (Fig. 4). Specifically, Figure 4A demonstrates a lack of Kir2.1 in glioma cells although protein expression was high in both spinal cord and cortical astrocytes. The opposite was observed for Kir2.3; glioma cells express relatively high levels, while astrocyte expression was low (Fig. 4B). Bands corresponding to Kir3.1 and 4.1 were prominent in both astrocytes and human glioma cells (Fig. 4C,D). All blots were stripped and re-probed with actin as a loading control.
Fig. 4.
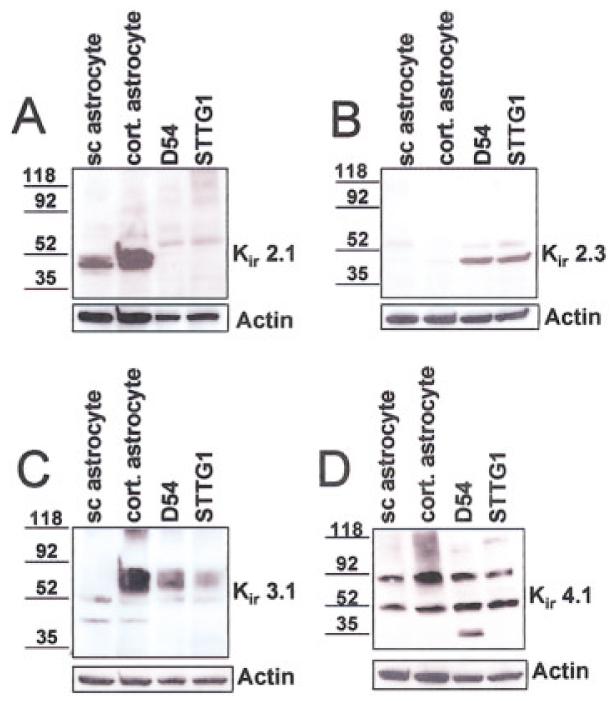
Western blot analysis demonstrating expression of Kir 2.1, 2.3, 3.1 and 4.1 in human glioma cells and in both spinal cord and cortical astrocytes. A: Anti-Kir 2.1 recognizes a prominent band at ∼50 kD in both spinal cord (sc) and cortical astrocytes, while only faint bands were detected in the D54 and STTG1 lanes. B: The opposite is observed for Kir 2.3; strong bands are detected in the two glioma cell lanes, with faint bands detected in the spinal cord and cortical astrocyte lanes. C: Anti-Kir 3.1 recognizes a faint band at ∼50 kD for each lane and a stronger band at a slightly higher molecular weight in the cortical astrocytes as well as both the glioma cell lines. D: Two prominent bands were detected with anti-Kir 4.1 in each lane, one at the appropriate molecular weight ∼50 KD and a higher band, possibly a multimer. All blots were stripped and re-probed with actin (Sigma) as a loading control.
Since these data fail to explain our inability to measure Kir current in glioma cells, we next performed immunocytochemistry to examine the cellular and sub-cellular distribution of these Kir channel proteins (Kir2.1, 2.3, 3.1, and 4.1) in the human glioma cells and astrocytes (green). Representative examples are illustrated in Figure 5. DAPI was used as a nuclear counterstain throughout (blue), and the cells’ cytoskeleton was labeled with GFAP in the case of astrocytes and with phalloidin in the case of STTG1 glioma cells (red). In agreement with our Western blot analyses, expression of Kir2.1 was not detectable in STTG1 cells. Astrocyte labeling was strong but was confined to the cell nucleus. Kir2.3, which was highly expressed in both glioma cells lines based on our Western blots, predominantly labeled the nucleus of STTG1 cells, while expression in spinal cord astrocytes was diffuse, but faint. Kir3.1 antibodies labeled the ER/Golgi region in both STTG1 and spinal cord astrocytes. Kir4.1, the major astrocytic Kir channel protein, localized primarily to the cell nucleus in STTG1 cells. This was in sharp contrast to the punctuate plasma membrane labeling in spinal cord astrocytes. D54 cells displayed staining patterns similar to STTG1 cells (data not shown). We subsequently labeled STTG1 cells with a panel of six other Kir antibodies (Kir1.1, 2.2, 3.2, 3.3, 4.2, and 6.2), all of which were negative, except Kir6.2, which also localized to the cell nucleus (not shown).
Fig. 5.

Immunoreactivity for Kir 2.1, 2.3, 3.1, and 4.1 in STTG1 cells and mature spinal cord astrocytes. A: STTG1 cells stained with the four different Kir antibodies. B: Merged image of STTG1 cells with the four Kir antibodies (green), phalloidin (red), and DAPI (blue). C: Spinal cord astrocytes at 9 days in culture (DIC) with the four Kir antibodies. D: Merged image of the spinal cord astrocytes with the four Kir antibodies (green), with GFAP (red) and DAPI (blue). Scale bar = 20 μm.
Figure 6 demonstrates the difference in cellular localization of one Kir channel, Kir4.1, between STTG1 cells, immature and differentiated spinal cord astrocytes. Although both immature astrocytes and glioma cells lack significant Kir currents, the mechanism is likely different in the two cell types. In the glioma cells, channel localization is predominantly nuclear. In the immature astrocyte, the labeling appears diffuse throughout the cell body, with most of the labeling being perinuclear, suggesting that channel protein may be synthesized and processed for insertion into the plasma membrane. Indeed, the same cells will show prominent membrane labeling after an additional 3–6 “days in culture” (Fig. 6).
Fig. 6.
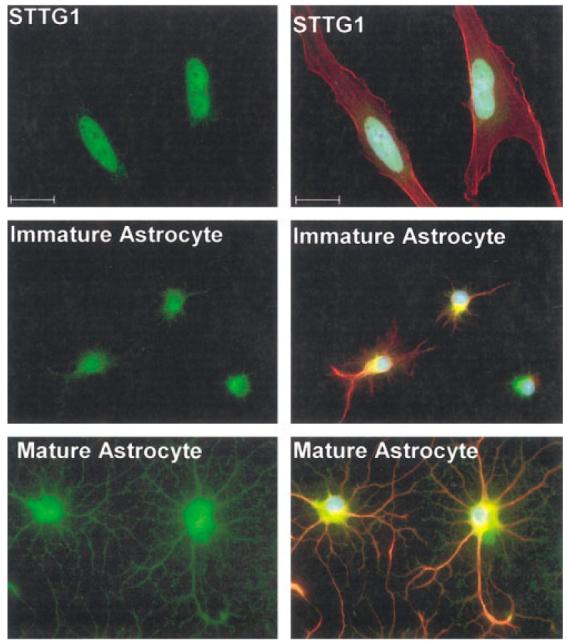
Comparison of Kir4.1 immunoreactivity in STTG1 cells, immature astrocytes and mature or differentiated astrocytes. STTG1 cells (A) demonstrate nuclear localization in contrast to immature (B) or mature astrocytes (C). Immature astrocytes express Kir4.1 albeit at lower levels than a differentiated astrocyte, and expression is more confined to the cell body, rather than the processes. Green, Kir4.1 immunoreactivity; blue, DAPI; red top, phalloidin; red middle and bottom, glial fibrillary acidic protein [GFAP]). Scale bar = 20 μm.
Thus, taken together, our biochemical and immunocytochemical data suggest that glioma cells express several of the inward rectifier potassium channels that are also expressed in astrocytes; however, the mislocalization of these channels to intracellular compartments may explain why Kir currents are rarely observed during electrophysiological recordings.
DISCUSSION
It has been known for some time that tumor cells (Cone Jr., 1970), including gliomas (Picker et al., 1981; Bordey and Sontheimer, 1998; Ullrich et al., 1998), have very depolarized resting membrane potentials. In nonmalignant astrocytes, the activity of Kir channels contributes to a very negative resting potential that is often close to the equilibrium potential for K+ ions (Dennis and Gerschenfeld, 1969; Ransom and Sontheimer, 1995). Indeed, the progressive increase in Kir expression during development correlates with a gradual negative shift in resting potential of astrocytes (Bordey and Sontheimer, 1997). In light of these findings, we were not surprised to find a near complete absence of Kir current in two glioma cell lines studied. We were surprised, however, that Western blot analyses demonstrate significant expression of at least 3 Kir channel proteins, including Kir4.1, the major Kir channel in retinal glial cells (Kofuji et al., 2000). Indeed, Kir4.1 was found at comparable expression levels in both glioma cells and astrocytes. The discrepancy between our electrophysiological observation and the Western blots was largely explained by immunocytochemical studies that revealed a mislocalization of these Kir channel proteins in glioma cells. Channels appear to be retained in intracellular membranes rather than being inserted into the plasma membrane. Although we refer to this as mislocalization, it is possible that Kir channels serve a function currently unknown in the glioma cells. Similarly, it is unclear whether these channels could be recruited to the cell membrane. It is conceivable that an appropriate extracellular stimulus may recruit these channels to the cell surface. Nuclear localization of an inward rectifier potassium channel has been observed previously. Although the physiological relevance is not understood, Kir2.2 has been demonstrated to be localized to both the cell nucleus and the plasma membrane in sections of rat hindbrain and dorsal root ganglia tissue (Stonehouse et al., 2003).
We (MacFarlane and Sontheimer, 1997, 2000a) and others (Wilson and Chiu, 1990; Konishi, 1994; Bringmann et al., 1999b, 2000) have previously proposed that Kir channel activity correlates with the differentiation of astrocytes and their exit from the cell cycle. Treatment of immature astrocytes with the differentiation agent retinoic acid leads to a premature differentiation (MacFarlane and Sontheimer, 2000a) and also induces enhanced membrane expression of Kir channels. Unfortunately, retinoic acid is not able to differentiate glioma cells in culture (Bouterfa et al., 2000), and indeed, this inability to differentiate appropriately appears to be one of the important differences between glioma cells and normal astrocytes (Linskey, 1997; Schmidt et al., 2000).
A similar study has examined Kir4.1 expression in neuroblastoma × glioma cells (NG108-15), which by polymerase chain reaction (PCR) show expression of Kir4.1. mRNA levels increase as these cells differentiate in culture; however, as in glioma cells, Kir currents remain absent (Ma et al., 1999). The authors of this study suggest that these tumor cells may express “silent” Kir channels that may have a genetic defect.
While the vast majority of studies examining biophysical properties of glioma cells report an absence of Kir channels and very depolarized membrane potentials, one group has reported on Kir channel expression in several glioma cell lines (Brismar and Collins, 1988, 1989). These cell lines displayed biophysical properties more reminiscent of astrocytes leading the authors to suggest their use as a model system to study astrocytes. We have confirmed the near absence of Kir channels in two other widely studied and commercially available glioma cell lines (U-251MG, U-373MG) and have examined acute patient biopsies extensively to corroborate that these cell lines show properties identical to those of human tumors in situ (Bordey and Sontheimer, 1998).
We went to great length to ensure that technical issues may not account for the observed channel mislocalization. Indeed, a number of factors could affect ion channel expression in cultured cells. For example, different sera have been shown to influence Ca2+ channel expression in cultured glial progenitor cells (Barres et al., 1989), and the extracellular matrix molecule laminin has been shown to affect localization and expression of Kir4.1 in Müller cells (Ishii et al., 1997). We have used the identical serum for astrocyte and glioma cultures and always observed prominent Kir expression in astrocytes. We also varied the extracellular matrix on which glioma cells were plated. Indeed, the same laminin-polyornithine extracellular matrix used in our astrocyte cultures failed to induce Kir current in glioma cells. Similarly, passage number did not affect Kir channel expression. Finally, we believe that the strongest support for the validity of these observations comes from our previous comparison to patient biopsies in which Kir currents were completely absent (Bordey and Sontheimer, 1998). Taken together, we find it highly unlikely that the specific culture condition used here have artifactually suppressed the expression of functional Kir channels in glioma membranes and instead suggest that their absence from the cell membrane is a true biological difference to astrocytes.
Functional Implications
Buffering of extracellular K+ and establishment of a very negative resting potential remain the two most frequently discussed roles for inward rectifier potassium channels (Orkand et al., 1966). It is widely believed that one of the primary functions of glial cells involves potassium uptake from the extracellular space following neuronal discharge. K+ ions are subsequently redistributed toward areas in which [K+]o is lower, possibly involving diffusion from cell to cell through the coupled glial network or syncytium. Ultimately, K+ may be released through the end-feet of astrocytes into blood vessels (Newman, 1985). Potassium buffering is essential to maintain neuronal firing, as elevated extracellular K+ concentrations would depolarize neurons, rendering them unable to fire action potentials. By all accounts, glial Kir channels appear to be perfectly suited for the task of taking up extracellular K+; they have a large open probability at the resting potential (Newman, 1993) and their conductance increases with increasing extracellular K+. The small Kir currents in glioma cells likely render these cells deficient of significant K+ uptake and clearance, possibly exacerbating symptoms associated with their malignancy. Accumulations of extracellular K+ occur in conjunction with epileptic seizures (Traynelis and Dingledine, 1988), and peritumoral seizures are common in glioma patients. The absence of Kir channels from glioma cells certainly would be ill suited to ameliorate any contributions from excess extracellular K+ to this condition. On the other hand, one may argue that few if any neurons survive in or around glioma cells, in light of the recently reported neurotoxicity due to glutamate release from these tumors (Ye and Sontheimer, 1999; Takano et al., 2001). Therefore, the need for K+ clearance due to activity-dependent K+ release may be diminished.
A second important function of Kir channels relates to their ability to establish and maintain a very negative resting potential in astrocytes and other glial cells. This in turn establishes the Na+ gradient across the membrane which is then used as the driving force for several important transporters, most notably Na+-dependent glutamate transporters. The depolarized potential of glioma cells by comparison would result in a greatly reduced ability for these transporters to function. Two recent studies suggest that Na+-dependent glutamate uptake is not operative in glioma cells (Ye et al., 1999; Ye and Sontheimer, 1999). Indeed, as with Kir channels, glutamateaspartate transporter (GLAST), one of the astrocytic glutamate transporters (Rothstein et al., 1994; Torp et al., 1994), mislocalizes to the cell nucleus of glioma cells permitting little Glu uptake into glioma cells (Ye et al., 1999). Instead, glioma cells release Glu via a cystine-Glu exchanger (Ye and Sontheimer, 1999). The latter is an electro-neutral transporter and hence is not affected by the depolarized resting potential of glioma cells. Thus, it appears unlikely that glioma cells participate in extracellular glutamate homeostasis.
Finally, another important difference between astrocytes and glioma cells pertains to their cellular coupling. Astrocytes appear to couple extensively to neighboring cells via gap-junctions (Kettenmann and Ransom, 1988; Binmoller and Muller, 1992) and establish a large functional syncytium whereby cells share a common cytoplasm. This allows a more effective dilution of amino acids and ions transported into their cytoplasm. By contrast, glioma cells show an almost complete lack of gap junctions, as determined by Western blot and functional studies (Soroceanu et al., 2001). Taken together, these studies suggest that glioma cells are ill-equipped to participate effectively in ion and amino acid homeostasis, those important “altruistic” tasks performed by their nonmalignant counterparts. Instead, these cells may be more concerned about their relentless growth and invasive migration.
ACKNOWLEDGMENTS
The authors acknowledge the valuable input provided by Amy K Weaver.
Grant sponsor: National Institutes of Health (NIH); Grant number: RO1-NS36692; Grant number: RO1-NS31234.
REFERENCES
- Barres BA, Chun LLY, Corey DP. Calcium current in cortical astrocytes: induction by cAMP and neurotransmitters and permissive effect of serum factors. J Neurosci. 1989;9:3169–3175. doi: 10.1523/JNEUROSCI.09-09-03169.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Koroshetz WJ, Swartz KJ, Chun LLY, Corey DP. Ion channel expression by white matter glia: the O2A glial progenitor cell. Neuron. 1990;4:507–524. doi: 10.1016/0896-6273(90)90109-s. [DOI] [PubMed] [Google Scholar]
- Bignami A, Dahl D. Astrocyte-specific protein and neuroglial differentiation. An immunofluorescence study with antibodies to the glial fibrillary acidic protein. J Comp Neurol. 1974;153:27–38. doi: 10.1002/cne.901530104. [DOI] [PubMed] [Google Scholar]
- Binmoller FJ, Muller CM. Postnatal development of dye-coupling among astrocytes in rat visual cortex. Glia. 1992;6:127–137. doi: 10.1002/glia.440060207. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Postnatal development of ionic currents in rat hippocampal astrocytes in situ. J Neurophysiol. 1997;78:461–477. doi: 10.1152/jn.1997.78.1.461. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Electrophysiological properties of human astrocytic tumor cells in situ: enigma of spiking glial cells. J Neurophysiol. 1998;79:2782–2793. doi: 10.1152/jn.1998.79.5.2782. [DOI] [PubMed] [Google Scholar]
- Bouterfa H, Picht T, Kess D, Herbold C, Noll E, Black PM, Roosen K, Tonn JC. Retinoids inhibit human glioma cell proliferation and migration in primary cell cultures but not in established cell lines. Neurosurgery. 2000;46:419–430. doi: 10.1097/00006123-200002000-00029. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Biedermann B, Reichenbach A. Expression of potassium channels during postnatal differentiation of rabbit Müller glial cells. Eur J Neurosci. 1999a;11:2883–2896. doi: 10.1046/j.1460-9568.1999.00706.x. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Francke M, Pannicke T, Biedermann B, Faude F, Enzmann V, Wiedemann P, Reichelt W, Reichenbach A. Human Müller glial cells: altered potassium channel activity in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1999b;40:3316–3323. [PubMed] [Google Scholar]
- Bringmann A, Francke M, Pannicke T, Biedermann B, Kodal H, Faude F, Reichelt W, Reichenbach A. Role of glial K+ channels in ontogeny and gliosis: a hypothesis based upon studies on Müller cells. Glia. 2000;29:35–44. doi: 10.1002/(sici)1098-1136(20000101)29:1<35::aid-glia4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Brismar T, Collins VP. Inward K-current in human malignant glioma cells: possible mechanisms for K-homeostasis in the brain. Acta Physiol Scand. 1988;132:259–260. doi: 10.1111/j.1748-1716.1988.tb08326.x. [DOI] [PubMed] [Google Scholar]
- Brismar T, Collins VP. Inward rectifying potassium channels in human malignant glioma cells. Brain Res. 1989;480:249–258. doi: 10.1016/0006-8993(89)90190-x. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz dM, Rudy B. Molecular diversity of K+ channels. Ann NY Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Cone CD., Jr. Variation of the transmembrane potential level as a basic mechanism of mitosis control. Oncology. 1970;24:438–470. doi: 10.1159/000224545. [DOI] [PubMed] [Google Scholar]
- Couchie D, Fages C, Bridoux AM, Rolland B, Tardy M, Nunez J. Microtubule-associated proteins and in vitro astrocyte differentiation. J Cell Biol. 1985;101:2095–2103. doi: 10.1083/jcb.101.6.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MJ, Gerschenfeld HM. Some physiological properties of identified mammalian neuroglial cells. J Physiol. 1969;203:211–222. doi: 10.1113/jphysiol.1969.sp008860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy PE. Astrocytes: normal, reactive and neoplastic. Raven Press; New York: 1983. [Google Scholar]
- Francke M, Pannicke T, Biedermann B, Faude F, Wiedemann P, Reichenbach A, Reichelt W. Loss of inwardly rectifying potassium currents by human retinal glial cells in diseases of the eye. Glia. 1997;20:210–218. doi: 10.1002/(sici)1098-1136(199707)20:3<210::aid-glia5>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Hagiwara S, Takahashi K. The anomalous rectification and cation selectivity of the membrane of a starfish egg cell. J Membr Biol. 1974;18:61–80. doi: 10.1007/BF01870103. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Ishii M, Horio Y, Tada Y, Hibino H, Inanobe A, Ito M, Yamada M, Gotow T, Uchiyama Y, Kurachi Y. Expression and clustered distribution of an inwardly rectifying potassium channel, KAB-2/ Kir4.1, on mammalian retinal Müller cell membrane: their regulation by insulin and laminin signals. J Neurosci. 1997;17:7725–7735. doi: 10.1523/JNEUROSCI.17-20-07725.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomoto S, Kondo C, Kurachi Y. Inwardly rectifying potassium channels: their molecular heterogeneity and function. Jpn J Physiol. 1997;47:11–39. doi: 10.2170/jjphysiol.47.11. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Ransom BR. Electrical coupling between astrocytes and between oligodendrocytes studied in mammalian cell culture. Glia. 1988;1:64–73. doi: 10.1002/glia.440010108. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi T. Activity-dependent regulation of inwardly rectifying potassium currents in non-myelinating Schwann cells in mice. J Physiol. 1994;474:193–202. doi: 10.1113/jphysiol.1994.sp020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kressin K, Kuprijanova E, Jabs R, Seifert G, Steinhäuser C. Developmental regulation of Na+ and K+ conductances in glial cells of mouse hippocampal brain slices. Glia. 1995;15:173–187. doi: 10.1002/glia.440150210. [DOI] [PubMed] [Google Scholar]
- Li L, Head V, Timpe LC. Identification of an inward rectifier potassium channel gene expressed in mouse cortical astrocytes. Glia. 2001;33:57–71. doi: 10.1002/1098-1136(20010101)33:1<57::aid-glia1006>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Linskey ME. Glial ontogeny and glial neoplasia: the search for closure. J Neurooncol. 1997;34:5–22. doi: 10.1023/a:1005751018617. [DOI] [PubMed] [Google Scholar]
- Liu X, Chang Y, Reinhart PH, Sontheimer H, Chang Y. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. J Neurosci. 2002;22:1840–1849. doi: 10.1523/JNEUROSCI.22-05-01840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Grant GM, Pancrazio JJ, Kao WY, Shaffer KM, Liu QY, Barker JL, Cohen NA, Stenger DA. Kir 4.1 channel expression in neuroblastoma × glioma hybrid NG108-15 cell line. Brain Res Dev Brain Res. 1999;114:127–134. doi: 10.1016/s0165-3806(99)00015-2. [DOI] [PubMed] [Google Scholar]
- MacFarlane SN, Sontheimer H. Electrophysiological changes that accompany reactive gliosis in vitro. J Neurosci. 1997;17:7316–7329. doi: 10.1523/JNEUROSCI.17-19-07316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane SN, Sontheimer H. Changes in ion channel expression accompany cell cycle progression of spinal cord astrocytes. Glia. 2000a;30:39–48. doi: 10.1002/(sici)1098-1136(200003)30:1<39::aid-glia5>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- MacFarlane SN, Sontheimer H. Modulation of kv1.5 currents by src tyrosine phosphorylation: potential role in the differentiation of astrocytes. J Neurosci. 2000b;15(20):5245–5253. doi: 10.1523/JNEUROSCI.20-14-05245.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonen G, Cam Y, Sensenbrenner M, Mandel P. Variability of the effects of serum-free medium, dibutyryl-cyclic AMP or theophylline on the morphology of cultured new-born rat astroblasts. Cell Tissue Res. 1975;163:365–372. doi: 10.1007/BF00219470. [DOI] [PubMed] [Google Scholar]
- Newman EA. Membrane physiology of retinal glial (Müller) cells. J Neurosci. 1985;5:2225–2239. doi: 10.1523/JNEUROSCI.05-08-02225.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Inward-rectifying potassium channels in retinal glial (Müller) cells. J Neurosci. 1993;13:3333–3345. doi: 10.1523/JNEUROSCI.13-08-03333.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Glial cell regulation of extracellular potassium. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford University Press; New York: 1995. pp. 717–731. [Google Scholar]
- Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- Picker S, Pieper CF, Goldring S. Glial membrane potentials and their relationship to [K+]o in man and guinea pig. A comparative study of intracellularly marked normal, reactive, and neoplastic glia. J Neurosurg. 1981;55:347–363. doi: 10.3171/jns.1981.55.3.0347. [DOI] [PubMed] [Google Scholar]
- Puro DG, Roberge F, Chan CC. Retinal glial cell proliferation and ion channels: a possible link. Invest Ophthalmol Vis Sci. 1989;30:521–529. [PubMed] [Google Scholar]
- Raap M, Biedermann B, Braun P, Milenkovic I, Skatchkov SN, Bringmann A, Reichenbach A. Diversity of Kir channel subunit mRNA expressed by retinal glial cells of the guinea-pig. NeuroReport. 2002;13:1037–1040. doi: 10.1097/00001756-200206120-00012. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Liu X, Sontheimer H. BK channels in human glioma cells have enhanced calcium sensitivity. Glia. 2002;38:281–291. doi: 10.1002/glia.10064. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J Neurophysiol. 1995;73:333–345. doi: 10.1152/jn.1995.73.1.333. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. BK Channels in human glioma cells. J Neurophysiol. 2001;85:790–803. doi: 10.1152/jn.2001.85.2.790. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP. Intracellular regulation of inward rectifier K+ channels. Pflugers Arch. 2000;441:1–11. doi: 10.1007/s004240000380. [DOI] [PubMed] [Google Scholar]
- Sakmann B, Trube G. Conductance properties of single inwardly rectifying potassium channels in ventricular cells from guinea-pig heart. J Physiol. 1984;347:641–657. doi: 10.1113/jphysiol.1984.sp015088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt F, Groscurth P, Dichgans J, Weller M. Human malignant glioma cell lines are refractory to retinoic acid-mediated differentiation and sensitization to apoptosis. Cell Physiol Biochem. 2000;10:159–168. doi: 10.1159/000016346. [DOI] [PubMed] [Google Scholar]
- Schroder W, Seifert G, Huttmann K, Hinterkeuser S, Steinhauser C. AMPA receptor-mediated modulation of inward rectifier K+ channels in astrocytes of mouse hippocampus. Mol Cell Neurosci. 2002;19:447–458. doi: 10.1006/mcne.2001.1080. [DOI] [PubMed] [Google Scholar]
- Sontheimer H, Trotter J, Schachner M, Kettenmann H. Channel expression correlates with differentiation stage during development of oligodendrocytes from their precursor cells in culture. Neuron. 1989;2:1135–1145. doi: 10.1016/0896-6273(89)90180-3. [DOI] [PubMed] [Google Scholar]
- Soroceanu L, Manning TJ, Jr, Sontheimer H. Reduced expression of connexin-43 and functional gap junction coupling in human gliomas. Glia. 2001;33:107–117. doi: 10.1002/1098-1136(200102)33:2<107::aid-glia1010>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Stonehouse AH, Grubb BD, Pringle JH, Norman RI, Stanfield PR, Brammar WJ. Nuclear immunostaining in rat neuronal cells using two anti-Kir2.2 ion channel polyclonal antibodies. J Mol Neurosci. 2003;20:189–94. doi: 10.1385/JMN:20:2:189. [DOI] [PubMed] [Google Scholar]
- Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat Med. 2001;7:1010–1015. doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- Tang F, Shang K, Wang X, Gu J. Differentiation of embryonic stem cell to astrocytes visualized by green fluorescent protein. Cell Mol Neurobiol. 2002;22:95–101. doi: 10.1023/a:1015397829913. [DOI] [PubMed] [Google Scholar]
- Tang P, Steck PA, Yung WKA. The autocrine loop of TGF-α/EGFR and brain tumors. J Neurooncol. 1997;35:303–314. doi: 10.1023/a:1005824802617. [DOI] [PubMed] [Google Scholar]
- Torp R, Danbolt NC, Babaie E, Bjoras M, Seeberg E, Storm-Mathisen J, Ottersen OP. Differential expression of two glial glutamate transporters in the rat brain: an in situ hybridization study. Eur J Neurosci. 1994;6:936–942. doi: 10.1111/j.1460-9568.1994.tb00587.x. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol. 1988;59:259–276. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- Ullrich N, Bordey A, Gillespie GY, Sontheimer H. Expression of voltage-activated chloride currents in acute slices of human gliomas. Neuroscience. 1998;83:1161–1173. doi: 10.1016/s0306-4522(97)00456-9. [DOI] [PubMed] [Google Scholar]
- Ullrich N, Fernandez-Marques E, Pappas CA, Sontheimer H.Ion channels and glial proliferation. II. Comparison of normal and neoplastic glia Soc Neurosci Abs 199420Suppl Pt 1/2 [Google Scholar]
- Verkhratsky A, Steinhauser C. Ion channels in glial cells. Brain Res Brain Res Rev. 2000;32:380–412. doi: 10.1016/s0165-0173(99)00093-4. [DOI] [PubMed] [Google Scholar]
- Von Deimling A, Louis DN, Wiestler OD. Molecular pathways in the formation of gliomas. Glia. 1995;15:328–338. doi: 10.1002/glia.440150312. [DOI] [PubMed] [Google Scholar]
- Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36:291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Williamson AV, Compston DAS, Randall AD. Analysis of the ion channel complement of the rat oligodendrocyte progenitor in a commonly studied in vitro preparation. Eur J Neurosci. 1997;9:706–720. doi: 10.1111/j.1460-9568.1997.tb01419.x. [DOI] [PubMed] [Google Scholar]
- Wilson GF, Chiu SY. Ion channels in axon and Schwann cell membranes at paranodes of mammalian myelinated fibers studied with patch-clamp. J Neurosci. 1990;10:3263–3274. doi: 10.1523/JNEUROSCI.10-10-03263.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59:4383–4391. [PubMed] [Google Scholar]
- Ye ZC, Rothstein JD, Sontheimer H. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci. 1999;19:10767–10777. doi: 10.1523/JNEUROSCI.19-24-10767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi BA, Minor DL, Jr, Lin YF, Jan YN, Jan LY. Controlling potassium channel activities: interplay between the membrane and intracellular factors. Proc Natl Acad Sci USA. 2001;98:11016–11023. doi: 10.1073/pnas.191351798. [DOI] [PMC free article] [PubMed] [Google Scholar]