

Abstract
Glioma cells show up-regulation and constitutive activation of erbB2, and its expression correlates positively with increased malignancy. A similar correlation has been demonstrated for the expression of gBK, a calcium-sensitive, large-conductance K+ channel. We show here that glioma BK channels are a downstream target of erbB2/neuregulin signaling. Tyrphostin AG825 was able to disrupt the constituitive erbB2 activation in a dose-dependent manner, causing a 30-mV positive shift in gBK channel activation in cell-attached patches. Conversely, maximal stimulation of erbB2 with a recombinant neuregulin (NRG-1β) caused a 12-mV shift in the opposite direction. RT-PCR studies reveal no change in the BK splice variants expressed in treated glioma cells. Furthermore, isolation of surface proteins through biotinylation did not show a change in gBK channel expression, and probing with phospho-specific antibodies showed no alteration in channel phosphorylation. However, fura-II Ca2+ fluorescence imaging revealed a 35% decrease in the free intracellular Ca2+ concentration after erbB2 inhibition and an increase in NRG-1β-treated cells, suggesting that the observed changes most likely were due to alterations in [Ca2+]i. Consistent with this conclusion, neither tyrphostin AG825 nor NRG-1β was able to modulate gBK channels under inside-out or whole-cell recording conditions when intracellular Ca2+ was fixed. Thus, gBK channels are a downstream target for the abundantly expressed neuregulin-1 receptor erbB2 in glioma cells. However, unlike the case in other systems, this modulation appears to occur via changes in [Ca2+]i without changes in channel expression or phosphorylation. The enhanced sensitivity of gBK channels in glioma cells to small, physiological Ca2+ changes appears to be a prerequisite for this modulation.
Keywords: growth factor, potassium channel, brain tumor, patch clamp, neuregulin
Voltage-dependent, large-conductance, calcium-activated potassium channels (BK) differ from many other ion channels in that, as the name implies, they are activated by two physiological processes; a change in voltage and changes in intracellular Ca2+ concentration. Under physiological conditions, Ca2+ binding is required for channel activation. BK channels are expressed in a broad spectrum of cells and tissue types and across many species. In excitable cells, BK channel expression and activity persist throughout the life of the cell. BK channels play important roles in mediating different phases of hyperpolarization and afterhyperpolarization after an action potential and are involved in neurotransmitter release and muscle relaxation (for review see Sah, 1996; Gribkoff et al., 2001; Sah and Faber, 2002; Calderone, 2002). For nonexcitable cells, the role that these channels play in cell biology is less well understood. In glial cells, the highest channel activity is observed in undifferentiated, proliferating cells, whereas channels are largely absent in differentiated astrocytes (Puro et al., 1989; Sontheimer et al., 1989; Bringmann et al., 1999, 2000; Kodal et al., 2000; Moll et al., 2002). Furthermore, BK channels are significantly up-regulated in astrocytes that have undergone neoplastic transformation, and their expression levels appear to correlate positively with tumor grade (Manor and Moran, 1994; Bordey et al., 1996; Ransom and Sontheimer, 2001; Liu et al., 2002; Ransom et al., 2002). We have previously demonstrated that astrocyte-derived tumor cells (glioma) express a novel isoform of a BK channel (gBK), which contains a 34-amino-acid insert at splicing site 2, conferring an increased sensitivity to intracellular calcium ions at physiological calcium concentrations (Liu et al., 2002).
In addition to being activated by increases in intra-cellular Ca2+ concentrations and changes in voltage, BK channel activity and expression are known to be modulated by growth factor receptor activation, a phenomenon also observed in other ion channels (for review see Chew and Gallo, 1998). For example, in developing chick parasympathetic neurons, BK currents (IKCa) are not expressed at normal levels until contact occurs with target tissues (Subramony et al., 1996), where activation of IKCa appears to involve the action of several diffusible factors derived from the target tissue, including transforming growth factor-β (TGFβ; Cameron et al., 1998) and β-neuregulin-1 (Cameron et al., 2001). These studies suggest that growth factor receptor activation leads to plasma membrane insertion of BK channels (Lhuillier and Dryer, 2002). Alternatively, several groups have demonstrated that growth factor receptor modulators alter BK currents as a direct result of channel de/phosphorylation (Hall and Armstrong, 2000).
We and others have shown that glioma cells also express, at relatively high levels, a phosphorylated form of the erbB2 receptor (Kristt and Yarden, 1996; Ritch et al., 2005). erbB2 is a receptor tyrosine kinase in a family composed of four members (erbB1—erbB4). Upon presence of the proper ligand, the family of erbB receptors forms both homo- and heterodimers. erbB2 is the preferred partner of all erbBs, although no specific ligand for erbB2 has yet been identified. Analogously to the activation of many other growth factor receptors, initial activation of the erbB2 receptor causes increases in intra-cellular calcium ([Ca2+]i; Tsunoda, 1998). Functionally, erbB2 receptor activation stimulates proliferation and survival and enhances cell motility. Not surprisingly, levels of active erbB2 correlate positively with the degree of malignancy (Kristt and Yarden, 1996).
In light of the abundant expression of gBK and erbB2, we sought to determine whether gBK channels in glioma cells were a downstream target for erbB2 receptor activation. Indeed, we report here that gBK channel activity in human glioma cells is modulated by changes in the level of erbB2 receptor activation and that these effects are mediated through changes in [Ca2+]i.
MATERIALS AND METHODS
Cell Culture
All experiments were performed on the glioma cell line D54-MG [glioblastoma multiforme (GBM), World Health Organization (WHO) grade IV; a gift from Dr. D. Bigner (Duke University)]. Cells were cultured in DMEM/F12 (Invitrogen, Grand Island, NY) supplemented with 7% fetal calf serum (FCS; Hyclone, Logan, UT) and 2 mM glutamine (MediaTech, University of Alabama at Birmingham Media Preparation Facility). Recombinant Nrg-1β was kindly provided by Dr. S. Carroll (University of Alabama at Birmingham). The specific erbB2 inhibitor tyrphostin AG825 (TyrAG825; Calbiochem, La Jolla, CA) was dissolved in dimethyl sulfoxide and stored at -20°C. Except where otherwise mentioned, cells were treated as follows: after 48–72 hr in culture, serum-containing medium was removed from cells and replaced with serum-free medium + 0.1% fatty acid free bovine serum albumin (FAF-BSA), serum-free + medium 0.1% FAF-BSA with 50 μM TyrAG825, or serum-free + medium + 0.1% FAF-BSA with 80 nM recombinant Nrg-1β.
Electrophysiology
For these experiments, cells were plated on glass cover-slips (12 mm, round; Macalaster Bicknell, New Haven, CT) in 24-well plates (Becton Dickinson, Lincoln Park, NJ) and treated as described above. Whole-cell voltage-clamp and single-channel recordings were obtained via standard methods (Hamill et al., 1981). Patch pipettes were made from thin-walled (outer diameter 1.5 mm, inner diameter 1.12 mm) borosilicate glass (TW150-4; World Precision Instruments, Sarasota, Fl) and had resistances of 3–5 MΩ. Recordings were made on the stage of an inverted Nikon diaphot microscope equipped with Hoffman modulation contrast optics. Current recordings were obtained with an Axopatch 200A amplifier (Axon Instruments, Union City, CA). Current signals were low-pass filtered at 1 kHz and were digitized online at 10 kHz, by using a Digidata 1200 digitizing board (Axon Instruments) interfaced with an IBM-compatible computer (Dell XPS R400). Data acquisition and storage were conducted with the use of pClamp 8.2 (Axon Instruments). Resting membrane potentials, cell capacitances, and series resistances were measured directly from the amplifier, the upper limit for series resistance being 10 MΩ and series resistance compensation adjusted to 80% to reduce voltage errors.
Solutions
Our standard KCl pipette solution contained (in mM) 145 KCl, 1 MgCl2, 10 EGTA, 10 Hepes sodium salt, pH adjusted to 7.3 with Tris base. CaCl2 (0.2mM) was added to the pipette solution just before recording, resulting in a free calcium concentration of 1.9 nM. Cells were continuously perfused at room temperature with a saline solution containing (in mM) 125 NaCl, 5.0 KCl, MgSO4, 1.0 CaCl2, 1.6 Na2HPO4, 0.4 Na2H2PO4, 10.5 glucose, and 32.5 Hepes acid, pH adjusted to 7.4 with NaOH; osmolarity of this solution was ∼300 mOsm. For inside-out recording experiments, patches were superfused with the standard pipette solution with the Ca2+ omitted.
Calcium Imaging
For the calcium imaging experiments, cells were plated directly into an eight-well chambered coverglass system (Nalge Nunc International, Naperville, IL) and treated as described above. The cells were then rinsed twice with serum-free, gluta-mine-free media and then incubated with the membrane per-meant form of the ratiometric dye fura-2AM (Molecular Probes, Eugene, OR; final concentration 5 μM) for 45 min in a 37°C incubator. The dye was rinsed with serum-free, gluta-mine-free media, and the cells were placed back in the incubator for 30 min to allow for deesterification. The chamber slide was then placed on the stage of a Nikon Diaphot inverted epiflourescence microscope with a Nikon ×20 Flour objective. The cells were alternately excited at 340 and 380 nM with a Lambda 10-2 Optical Filter Changer (Sutter Instrument Co., Novato, CA), and images were acquired with a Sensys digital camera (Roper Scientific, Duluth GA). Image acquisition was controlled via Axon Imaging Workbench 4.0 (Axon Instruments). Image ratios were converted to absolute concentrations according to the following equation: [Ca2+]free = Kd × [(R – Rmin/Rmax – R)] × (F380max/F380min). The values for Kd, Rmin, Rmax, F380min, and F380max were determined by using an in vitro Fura-2 Calcium Imaging Calibration Kit (Molecular Probes), where Kd is the dissociated constant for fura-2 (378 nM), Rmax (6.3) is the ratio level at saturating Ca2+ levels, Rmin (0.69) is the ratio at nominally free Ca2+ concentrations, F380max/F380min (4.3) is the ratio of free over bound form of the dye when excited at 380 nM, and R is the fluorescence ratio that was determined during the experiment.
RT-PCR
mRNA was isolated from cultured D54-MG glioma cells by using the RNAqueous kit from Ambion (Austin, TX). To remove contaminating DNA from the mRNA preparation, a DNA removal kit, DNA-Free (Ambion), was used. RT-PCR was performed with a Qiagen One-Step RT-PCR Kit (Qiagen, Valencia, CA). The protocol suggested by Qiagen, including reaction components, was followed. An Eppendorf Mastercylcer Grandient Thermo Cycler was used (Eppendorf, Hamburg, Germany) with an annealing temperature of 58°C for 35 cycles. Common splicing sites had previously been identified (Tseng-Crank et al., 2004); primers for splice sites 1, 2/3, and 4 had previously been designed in our laboratory, and primers for gBK have been published previously (Liu et al., 2002). All primer sets used are given in Table I and were manufactured by Invitrogen (Carlsbad, CA). In the lanes demonstrating negative controls, samples were set on ice during the initial reverse transcription reaction.
TABLE I.
Primer Sets Used for RT-PCR of the C-Terminus of gBK
| Region | Primer sequence | Size (bp) |
|---|---|---|
| Site 1 | 5′ CCCTACTGTTTGTGAGCTGTGTTTTGTGAA 3′ | 202 |
| 5′ CATCATGACAGGCCTTGCAGTAAAAAAAT 3′ | ||
| Site 2/3 | 5′ ATTTTTTTACTGCAAGGCCTGTCATGATG 3′ | 230 |
| 5′ CACCAGTGAAACATCCCAGTAGAGTCGTA 3′ | ||
| Site 4 | 5′ TGTGCGTTATCCTGTCAGCCAATCA 3′ | 275 |
| 5′ CAGGGTCATCATCGTCTTGGTCCA 3′ | ||
| gBK | 5′-GTTGGGAAGAACATTGTGCTTTGTGG-3′ | 150 |
| 5′-ATTTAGGTGACACTATAGAAGTGGACTTTGACAGAGAAAGTTTG-3′ |
Biotinylation, Immunoprecipitation, and Western Blot Analysis
Cells were grown to confluence and then treated as described above. The cells were washed twice with phosphate-buffered saline (PBS) and then biotinylated (Pierce, Rockford, IL) for 30 min at 4°C (biotin 1.5 mg/ml bath solution, pH 8.0, with 1 mM Ca2+). The biotin reaction was quenched by washing the cells with 100 mM glycine added to bath solution, pH 8.0, three times for 5 min each. The cells were washed one final time with bath solution, pH 8.0, and then lysed using RIPA buffer [50 mM TrisCl, pH 8.0, 150 mM NaCl, 1% Nondet P-40 (NP-40), 0.5% sodium deoxycholate, 1% sodium dodecyl sulfate (SDS)] for 30 min, supplemented with protease inhibitor cocktail obtained from Sigma (St. Louis, MO). Homogenates were centrifuged for 5 min at 12,000g at 4°C. Protein quantification was performed on the supernatant by using a DC protein assay kit from Bio-Rad (Hercules, CA). At this point, an aliquot was taken and labeled as total protein. From the remainder of sample, an equal amount of protein from each of three treatment conditions (500 μg to 1 mg) was then added to 200 μl of streptavidin beads (Pierce) and incubated with rocking for 3 hr at 4°C. The beads were spun down, and the supernatant was removed. The beads were then washed three times in RIPA buffer. We found that gBK did not tolerate boiling, so, to remove biotinylated samples from the streptavidin beads, 40 μl of 100 mM glycine, pH 2.8, was added to the beads for 2 min. The fluid was then removed and to it an equal volume of Laemmli-SDS 2× sample buffer containing 600 mM β-mercaptoethanol was added. Equal amounts of protein were loaded into each lane of a 7.5% precast acrylamide SDS-PAGE gel (Bio-Rad). Proteins were separated at 120 V constant. Gels were transferred onto PVDF paper (Millipore, Bedford, MA) at 200 mA constant for 2 hr at room temperature, and membranes were blocked in blocking buffer (BB; 5% nonfat dried milk, 2% BSA, and 2% normal goat serum in TBS plus 0.1% Tween 20 (TBST). The BK antibody (anti-mbr5), kindly provided by Dr. Zhou (University of Alabama at Birmingham), was generated against mbr5, amino acids 972–1,135 and used at a concentration of 0.5–1.0 μg/μl; alternatively, a second BK antibody was purchased from Chemicon (Temecula, CA). The membranes were then rinsed three times for 10 min each and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 90 min. Blots were once again washed three times for 10 min and developed with enhanced chemiluminesence (ECL; Amersham, Arlington Heights, IL) on Hyperfilm (Amersham). Actin and secondary HRP-conjugated antibodies were purchased from Sigma. Secondary HRP-conjugated mouse secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
For erbB2 immunoprecipitation experiments, cells were serum starved overnight. The cells were placed in serum-free media with 0.1% FAF-BSA, and then TyrAG825 was added. After 30 min, cells were lysed with RIPA, and the protein was quantified as described above. The lysates were then diluted to an equal concentration (1 mg/ml). The samples were precleared with 5 μl of rabbit serum plus 50 μl of protein A beads (Roche Applied Science, Indianapolis, IN) for 30 min. Anti-erbB2 was added to the precleared lysates at 2 μg/ml, and this was incubated overnight at 4°C. On the next day, 50 μl of protein A beads was added to each sample for an additional 2 hr at 4°C. Immunoprecipitates were pelleted at low speed and washed three times in RIPA buffer. To release the immunoprecipitates from the beads, 2× sample buffer was added, and samples were boiled for 5 min. Blots were run as described above. Anti-erbB2 (Santa Cruz Bio-technology) for blotting was used at 1 μg/ml, and the anti-phosphotyrosine antibody (anti-pY99) was used at 1:1,000. BK immunoprecipitation experiments were performed similarly except cells were treated overnight, identically to electro-physiology experiments. The Chemicon BK antibody was used at a concentration of 4 μg/ml to immunoprecipitate. Note that we were not able to boil any protein samples when probing for BK (using either antibody), in Western blotting, biotinylations, or immunoprecipitations. Boiling the protein caused a high-molecular-weight band (>200 kD) that was not present in the negative Hek cell control, which we presumed to be an aggregate.
Analysis
For whole-cell recordings, current responses to varied voltage steps and ramps were analyzed and measured in Clampfit (Axon Instruments); the resulting raw data were graphed and plotted in Origin 6.0 (MicroCal, Northampton, MA). For single-channel recordings, data were leak subtracted offline in Clampfit and then analyzed with Origin 6.0. We calculated conductance of the macropatches as follows: g = area/[(Vm – EK) × t], where Vm is the membrane potential and EK is the potassium equilibrium potential (0), assuming equal potassium concentrations on both sides of the membrane. The data were analyzed by computing and comparing total current flow determined by integrating the area (pA x ms) under the curve over time (t). Unless otherwise stated, all values are reported ±SE, with n being the number of cells sampled. Two-tailed t-tests were performed to evaluate statistical significance, and P values are given in Results (Origin).
RESULTS
erbB2 Receptors in Human Glioma Cells are Constitutively Active
The relentless growth of astrocyte-derived tumors is exacerbated by the up-regulation or high levels of basal activation of several growth factor receptors, including erbB2 (Schwechheimer et al., 1994). Indeed, erbB2 receptor expression correlates positively with the degree of tumor malignancy and higher levels of receptor phosphorylation (Kristt and Yarden, 1996). As a model system for study, we used a glioma cell line, D54-MG, derived from a WHO grade 4 glioblastoma, to examine the functional role of erbB2 as a potential regulator of BK channels in glioma cells. As a first step, we set out to confirm that D54-MG glioma cells demonstrate the constitutive activation of the erbB2 receptor. We refer to constitutive activation as high levels of phosphorylation under basal conditions, because we have no evidence that the receptor is mutated in any way. To demonstrate this, we immuno-precipitated glioma cell lysates by using erbB2-specific antibodies and subsequently probed Western blots with phosphotyrosine-specific antibodies. Even when serum starved overnight (Fig. 1, control), glioma cells showed significant erbB2 phosphorylation. Indeed, our lab has shown that six glioma cell lines demonstrated basal level of tyrosine phosphorylation in the absence of exogenous growth factors (Ritch et al., 2005). This basal activation was attributed to the fact that each of the cell lines tested expressed and secreted neuregulin isoforms, a potent erbB2 activator. We were able to disrupt this constitutive activation with a tyrosine kinase inhibitor, TyrAG825, which is specific for erbB2 (Osherov et al., 1993; Fernandes et al., 1999). By immunoprecipitating with an erbB2 antibody and blotting with phosphotyrosine, we demonstrated that increasing concentrations of TyrAG825 caused a dose-dependent decrease in erbB2 receptor tyro-sine phosphorylation, with almost complete inhibition at 50 μM (Fig. 1A). Conversely, immunoprecipitating with a phosphotyrosine antibody and then blotting against erbB2 in a second glioma cell line demonstrates similar results (Fig. 1B).
Fig. 1.
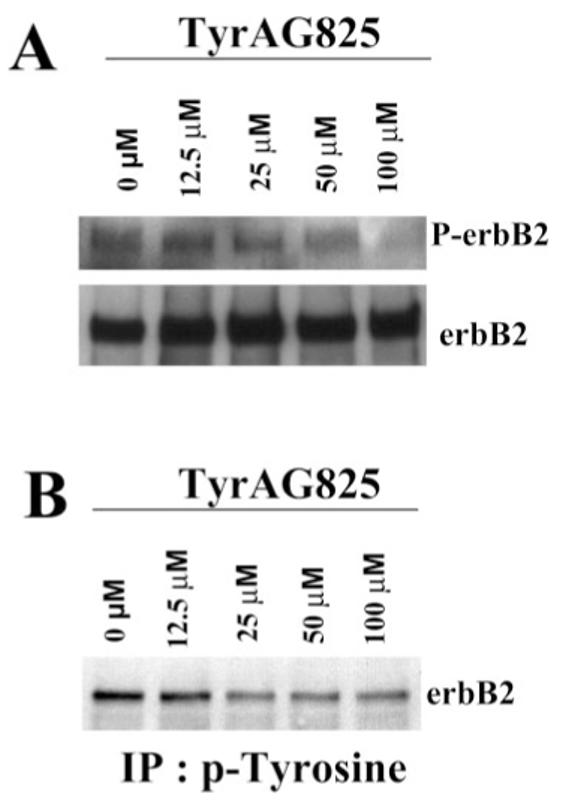
TyrAG825 decreases erbB2 phosphorylation. A: D54 glioma cells were treated with increasing concentrations (0–50 μM) of TyrAG825. Cell lysates were immunoprecipitated with erbB2-specific antibodies, and then blots were probed with a phosphotyrosine antibody. These blots were then stripped and reprobed with erbB2 as a loading control. Bands on this blot correspond to a molecular weight at ∼190 kD. B: Similarly, when treated cell lysates were immunoprecipitated with a phosphotyrosine antibody and blots probed with erbB2, a similar decrease in tyrosine phosphorylated erbB2 was observed.
gBK Channel Activity is Altered by Changes in erbB2 Receptor Activation
We next sought to examine whether erbB2 receptor activity modulates glioma BK channels. Therefore, we incubated glioma cells for 12 hr with TyrAG825 to disrupt the constitutive activation of erbB2 (see Fig. 1) and compared them with cells maintained in serum-free medium for the same length of time. Cell-attached patch-clamp recordings were obtained by using 140 KCl pipette solution, and the membrane potential was gradually stepped to achieve the potentials indicated in Figure 2. As discussed previously (Ransom et al., 2002), the high density of BK channels that is typical of glioma cells precluded activation of single channels and instead led to the concurrent activation of tens of channels. As a consequence, analysis of this data was limited to the analysis of macropatches containing multiple channels. When cells with similar resting membrane potentials were investigated (Rm within 2–3 mV of each other, as determined by the entrance potential), treatment with TyrAG825 resulted in markedly decreased macroscopic channel activity at depolarized potentials (Fig. 2A). When conductances were plotted as a function of applied voltage, these I-V curves revealed a rightward shift (Fig. 2B) compared with control. Indeed, the mean activation threshold was shifted by about 30 mV, from 56 mV under control conditions to 86 mV after TyrAG825 treatment. To show regulation by erbB2 in the opposite direction, we incubated cells for the same period of time with 80 nM recombinant neuregulin (Nrg-1β) in an attempt to activate erbB2 receptors maximally. As evident from the representative examples shown in Figure 2A, BK channels activated at more negative potentials after cells were exposed to Nrg-1β, and the macroscopic currents mediated by these channels were larger. These data show a leftward shift in the conductance-voltage curve with an activation threshold that was about 12 mV more negative after treatment with Nrg-1β (Fig. 2B). These experiments demonstrate that, when the cytosolic composition of the cell is unaltered, as is the case in cell-attached patches, changes in erbB2 receptor activation levels lead to a pronounced modulation of BK channel activity.
Fig. 2.
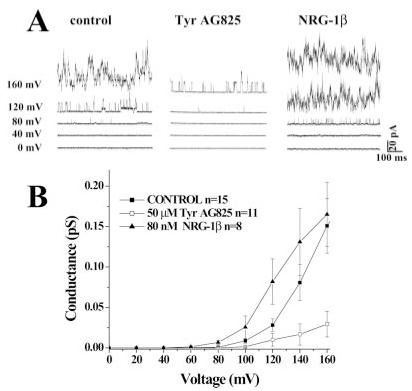
TyrAG825 and Nrg-1β shift the activation threshold of BK channels in glioma cells. A: Current responses at five different voltages from representative cell-attached patches of control, TyrAG825-treated (50 μM), and Nrg-1β-treated (80 nM) cells. B: The mean conductance from several cells under each condition as a function of voltage. Data points are the mean ± SE, and n = the number of cells under each condition.
We next sought to understand better the mechanisms that underlie this modulation of gBK by erbB2. Three distinct possibilities exist for the changes observed: 1) a change in the number of functional channels in the membrane; this has been demonstrated for increased BK channel activity in parasympathetic ciliary ganglion neurons upon acute treatment of TGFβ-1 (Lhuillier and Dryer, 2002); 2) a change in channel activity resulting from channel phosphorylation, as has been demonstrated in multiple systems by both kinases and kinase inhibitors (Hall and Armstrong, 2000); or 3) a change in [Ca2+]i, which would alter the channel voltage dependence of activation (Ransom et al., 2002). An assumption with this third mechanism is that the specific BK splice variant expressed in glioma cells, which is uniquely calcium sensitive, remains unchanged.
To investigate the first possibility, namely, that membrane expression of BK channels was altered, we isolated BK channel protein biochemically after treatment of cells with either Nrg-1β or TyrAG825. Figure 3 shows a Western blot in which total cell lysates were probed with BK antibodies (Fig. 3, lanes 1–3) under the three different treatment conditions. These data suggest that total BK channel protein was unaltered by the activation status of erbB2. Human embryonic kidney (HEK) cells that lack BK channels served as a negative control for these studies, and antibodies to actin served as loading control for total cell lysates. To specifically evaluate changes in expression levels of channels expressed in the plasma membrane, we used a surface biotinylation approach. Again, surface BK protein (Fig. 3, lanes 5–7) appeared unchanged in the TyrAG825- and Nrg-1β-treated cells (MW ∼120 kD). The relative expression of actin and the Na+K+/ATPase allowed us to validate the success of our surface biotinylation approach. As would be expected, the surface (bound) fractions did not express actin, which is an intracellular protein but showed prominent expression of the Na+K+/ATPase, which is a membrane protein. This blot is representative of five independent experiments.
Fig. 3.
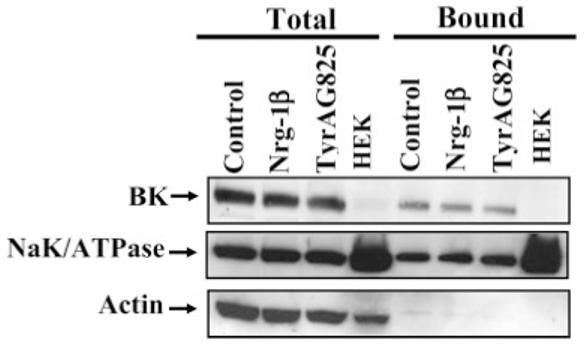
Biotinylation experiments demonstrate that total and surface expression of BK channels in glioma cells is unchanged. D54 glioma cells were treated with vehicle (serum-free media and 0.1% DMSO), Nrg-1β or TyrAG825 for 12 hr. The cells were biotinylated and lysates collected. The same amount of protein in each treatment condition was incubated with strepavidin beads (surface protein). Blots were probed with an anti-BK antibody and then stripped and reprobed with Na-K-ATPase antibody and an anti-actin antibody to demonstrate surface expression and a lack of intracellular proteins in surface lanes.
Because changes in cell surface expression could not account for the observed modulation of BK channel activity, we next examined the possibility that channels show differential modulation by channel phosphorylation (Fig. 4). To examine this possibility, we performed immunoprecipitations for the three treatment conditions, i.e., control, Nrg-1β, and TyrAG825, and then probed these blots with each of the phospho-antibodies and a BK antibody to serve as a loading control. With each treatment condition, we were able to precipitate BK channels, as evident by the prominent bands at approximately 120 kD (Fig. 4A). However, this suggests that there is no change in the tyrosine phosphorylation status of BK under any treatment condition. Similar results were observed when these blots were probed with the phosphoserine antibody and phosphothreonine antibody (Fig. 4A). This result is typical of four independent experiments. It has been demonstrated in various other cell types expressing BK channels that there can be a large intracellular population of channels (Lhuillier and Dryer, 2002). For this reason, we cannot exclude the possibility that there is a change in the phosphorylation status of a small portion of membrane BK channels that we would be unable to detect with this biochemical approach. It does appear that we have successfully immunoprecipitated all of the BK protein from each sample. An equal amount of protein was loaded in the total and unbound lanes, yet BK was absent from the unbound fractions (Fig. 4B, lanes 5–7).
Fig. 4.
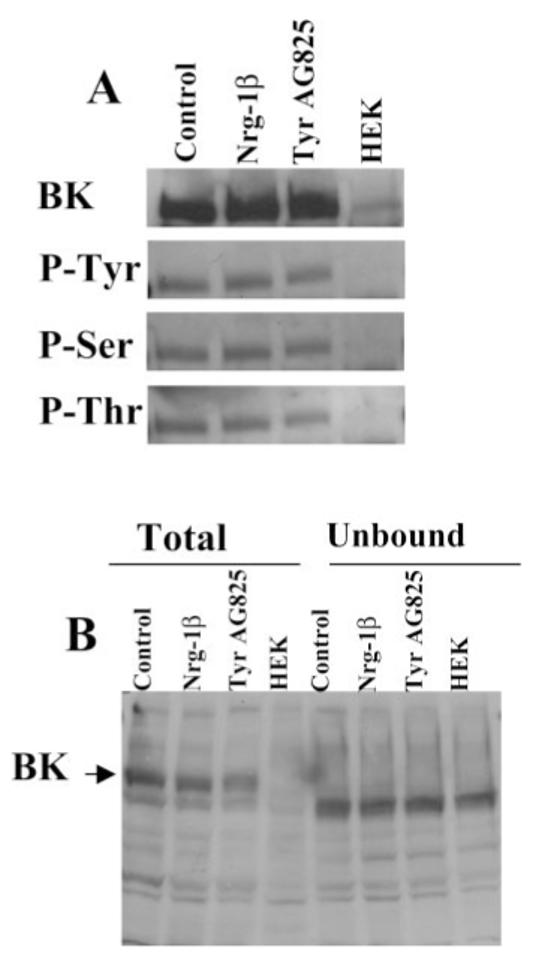
Tyrosine, serine, and threonine phosphorylation status of BK channels is unchanged in Nrg-1β- or TyrAG825-treated cells. D54 cells were treated overnight with vehicle, Nrg-1β, or TyrAG825, and cells were lysed and protein harvested. A: Cell lysates were immunoprecipitated with an anti-BK antibody (Chemicon) and then probed with an anti-BK (top), antiphosphotyrosine (top, middle), antiphosphothreonine (bottom, middle), and antiphosphothreonine (bottom). Each band in this figure had an approximate molecular weight of 120 kD. B: Equal amounts of protein from whole-cell lysates and the unbound fractions were analyzed by Western blot analysis to demonstrate a complete immunoprecipitation of the BK channel from our samples. Note that both antibodies used in this study consistently recognized a band slightly lower than the band at 120 kD (BK); this band was always present in the HEK cell lanes, and we presume it to be nonspecific binding.
[Ca2+]i Appears Necessary and Sufficient for Changes in Glioma BK Channel Activity
One of the earliest events to occur following growth factor binding is often an increase in intracellular Ca2+ caused either by a release from intracellular stores and by influx of Ca2+ from calcium-permeable channels. It is thought that the release of Ca2+ from intracellular stores triggers a relatively short-lived rise in Ca2+, whereas the increase from plasma membrane channels can be long lived, lasting for minutes to hours (Munaron, 2002). Once activated, erbB2 initiates a multitude of signaling events, which include, but are not limited to, activation of DAG and PI3 kinase and may ultimately culminate in a rise in [Ca2+]i (Kassis et al., 1999). The enhanced calcium sensitivity of gBK makes it an attractive target for modulation by signaling events that change intracellular Ca2+ concentration, e.g., activation or inhibition of erbB receptors. For this reason, we next focused our studies on changes that might occur to intracellular Ca2+ in glioma cells when either exogenous growth factor was applied or constitutively active erbB2 receptors were inhibited. An inherent assumption of these experiments is that the enhanced calcium sensitivity conferred by the unique 34-amino-acid insert found in gBK channels remains unchanged when cells are incubated in serum-free medium or serum-free medium plus TyrAG825 and in the presence of neuregulin (serum-containing media). To test this, we performed RT-PCR at several different splicing sites in the C-terminus, the most highly spliced region of the BK channel (Fig. 5). As a first step, we confirmed the presence of the 34-amino-acid insert that makes the glioma BK channel uniquely sensitive to intracellular calcium. The primer sets used and with the expected molecular weight of the product are given in Table I. As is demonstrated in Figure 5A, the gBK insert of approximately 128 bp is present under each of the three treatment conditions. Furthermore, splicing site 2/3, which contains the gBK 34-amino-acid insert (Fig. 5B), also contains only one band at the expected molecular weight. Had the insert that confers the high calcium sensitivity on gBK channels been spliced out, a product of approximately 100 bp less would have been observed. For completeness, we also performed RT-PCR on splice site 1 and splice site 4. Our PCR results indicate only one band at each splice sight at the expected molecular weight, once again suggesting that treatment with either TyrAG825 or Nrg-1β does not alter the splice variants expressed in gBK channels.
Fig. 5.
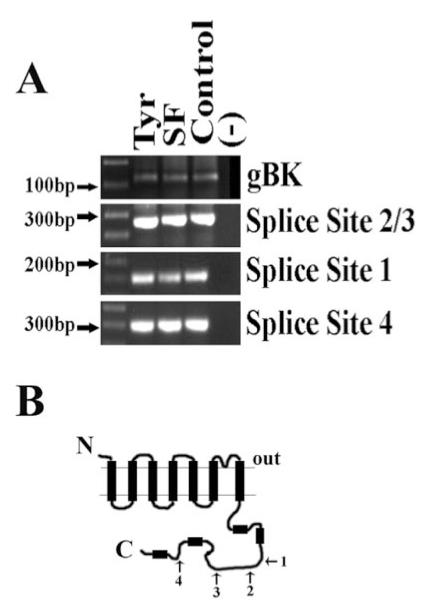
RT-PCR demonstrates that the gBK insert is present in TyrAG825-treated cells as well as cells in the presence of endogenous neuregulin. A: D54 cells were incubated overnight in normal serum-containing medium (contains endogenous neuregulin), serum-free medium, or serum-free medium with 50 μM TyrAG825. mRNA was collected and RT-PCR performed. Primers were designed to target the specific gBK insert and the four common splicing sites located in the C-terminal tail of the protein shown in B. B: Schematic of the BK channel α-subunit detailing the relative location of each of the four splicing sites targeted with RT-PCR.
These data suggest that the switch in activation threshold of the channel is not due to a switch in channel isoform expression, and the increased calcium activity of gBK remains unaltered. We next employed Fura-2, Ca2+ fluorescence imaging, to investigate changes in [Ca2+]i under conditions identical to those used in cell-attached patch recording. Data from well over 100 cells under each condition from four independent experiments suggest a free Ca2+ concentration of 89 ± 1.1 nM in untreated cells compared with 58 nM ± 0.8 nM in TyrAG825-treated cells (P < 0.001) and 95 ± 0.9 nM (P < 0.05) in Nrg-1β-treated cells (Fig. 6A—C). The overlay of the bar graphs demonstrates the changes in intra-cellular free Ca2+ under the different treatment conditions (Fig. 6D). We are aware that there are inherent limitations and difficulties in determining absolute intra-cellular free calcium concentrations. For this reason, we chose to examine each treatment condition each time the experiment was performed to minimize daily experimental variability. Additionally, we believe that we may be underestimating the actual absolute values of intracellular free calcium and instead wish to emphasize the relative differences in intracellular calcium. Importantly, intracellular calcium is decreased by more than one-third in TyrAG825-treated cells, and a smaller but significant increase was observed in cells treated with Nrg-1β. We propose that the less pronounced shift in the Nrg-1β-treated cells compared with TyrAG825-treated cells (also observed in the cell-attached patches) was due to the constitutive or baseline activation of the receptor in the absence of exogenously applied growth factor (see Fig. 1). Furthermore, this type of imaging allows for the investigation of global changes in calcium rather than increases in local changes that may occur very near the channel itself either through calcium channels or through a release from intracellular stores. The overlay of the bar graphs demonstrates the changes in intracellular free Ca2+ under the different treatment conditions (Fig. 6D).
Fig. 6.
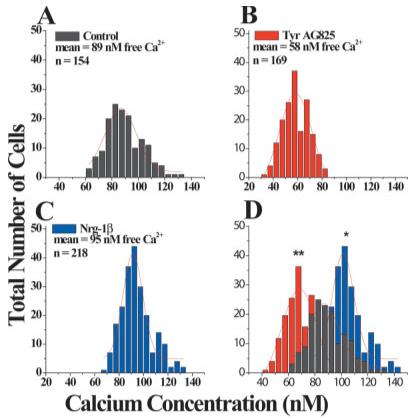
Fura-2 imaging demonstrates that intracellular free Ca2+ is decreased in TyrAG825- and increased in Nrg-1β-treated cells. A: D54 cells treated with vehicle for 12 hr have mean resting free intra-cellular calcium concentrations of 89 ± 1.1 nM (n = 154). B: D54 cells treated overnight with TyrAG825 (50 μM) had a mean resting calcium concentration of 58 ± 0.8 nM (n = 169). C: D54 cells treated overnight with Nrg-1β (80 nM) had a mean intracellular free calcium concentration of 95 ± 0.9 nM (n = 218). D: Overlay of the bar graphs in A—C demonstrate the shift in mean intracellular calcium concentrations in the three different treatment conditions. *P < 0.05, **P < 0.001.
If, indeed, the changes seen in gBK channel activity were caused by changes in [Ca2+]i, one would expect that modulation by erbB2 activation status would disappear under conditions when intracellular Ca2+ is well controlled, as is the case in both whole-cell and inside-out patch-clamp recordings. This was indeed the case. Representative whole-cell recordings are shown in Figure 7A, which shows examples of families of current traces from each of the three different treatment conditions in response to a series of depolarizing voltage steps ranging from –20 to 160 mV. These traces demonstrate that, macroscopically, whole-cell currents in the treated cells were similar to control. Furthermore, when the currents were normalized to cell size (pA/pF) there was no difference in current amplitude or the threshold of channel activation (Fig. 7B). Mean current densities of both the TyrAG825- and the Nrg-1β-treated cells were similar to the density of control cells, suggesting that there was no change in cell size (Fig. 7C). Finally, cell resting membrane potential, as determined directly from the entrance potential, did not differ under either treatment condition compared with control (Fig. 7D). The whole-cell data, when considered together, suggest that treatment with TyrAG825 and Nrg-1β does not change the channel population on the plasma membrane. This includes both expression level and any channel modulation. These data correlate well with our biotinylation data as well as our phosphorylation data.
Fig. 7.
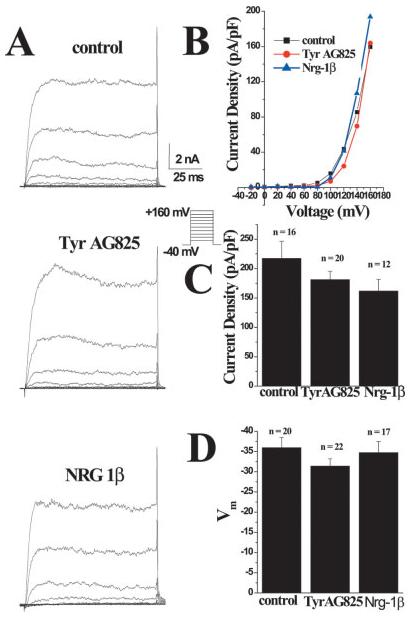
Whole-cell recording parameters are unchanged in TyrAG825- and Nrg-β1-treated cells. A: Representative examples of whole-cell currents evoked from stepping the membrane from 0 to 100 mV from a holding potential of –40 mV under each treatment condition. B: I-V plot of peak current in each treatment condition. C: Plot of the mean current density (pA/pF) of control, Nrg-1β and TyrAG825-treated cells. D: Plot of the mean resting membrane potential (mV) of cells in each treatment condition. Note that the more hyperpolarized potentials are toward the top of the graph.
Finally, the role of Ca2+ as a mediator of erbB2 activation was investigated by inside-out, patch-clamp recordings in which channels were exposed to nominally Ca2+ - free bath solution (0 Ca2+/5 mM EGTA) and current responses were recorded at five different voltages. The motivation for these experiments was that, if the results we observe are due largely to relative changes in intracellular calcium, we should be able to reverse the results if we control the calcium concentration (similar to the whole-cell recording conditions). Representative examples are shown in Figure 8A and demonstrate that there is no shift in channel activation under the three different treatment conditions. Indeed, a plot of the mean conductance (pS) from several cells under each condition shows that channel activation is nearly identical when Ca2+ is controlled (Fig. 8B). These results are in contrast to the results for cell-attached patches (Fig. 2), which demonstrate profound differences in activation in both the TyrAG825- and the Nrg-1β-treated cells and once again suggest calcium as a key mediator in the changes we observe. The process of pulling a patch of membrane from the cell leads to the loss of many intracellular factors and signaling parameters that may lead to the loss of treatment effects we observed in the inside-out patch-clamp configuration. However, these signaling parameters should be intact during our whole-cell recordings, and, under this recording condition, the same loss of treatment effects was observed. Indeed, many of whole-cell recordings were performed on cells that had demonstrated large shifts in channel activation during the cell-attached patch configuration.
Fig. 8.
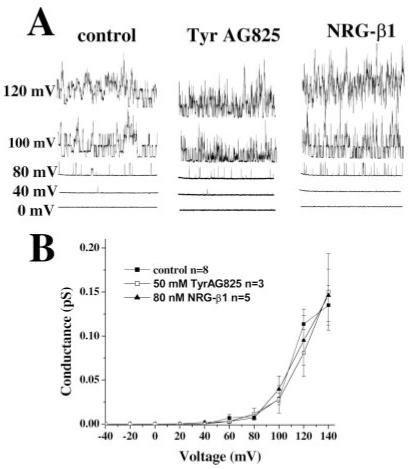
The shift in BK activation threshold in treated cells is lost when [Ca2+]i is controlled. A: Current responses at five different voltages from representative inside-out patch recordings of control, TyrAG825-treated (50 μM), and Nrg-1β-treated (80 nM) cells in the absence of extracellular Ca2+. B: Plot of the mean conductance (pS) from several cells under each condition as a function of voltage (mV). Data points are the mean ± SE, and n = the number of cells under each condition.
Taken together, our data indicate that, in glioma cells, BK channel activity is modulated by erbB2 receptor signaling. Unlike previous studies in other systems, this modulation does not involve transcriptional or translational regulation of the channel protein, nor does it involve channel phosphorylation. Instead, these changes appear to be mediated via changes in intracellular calcium.
DISCUSSION
Several studies have implicated Ca2+-activated K+ channels in the growth control of keratinocytes, fibro-blasts, T lymphocytes, Müller glial cells, and astrocytomas (Khanna et al., 1999; Pena and Rane, 1999; Kodal et al., 2000; Basrai et al., 2002; Koegel et al., 2003). A significant body of literature supports the notion that growth factors, including those acting through the erbB2 receptor, stimulate the growth of many of these cells. In light of the correlated up-regulation of the erbB2 receptors and gBK channels in glioma cells, our motivation for this study was to link these two proteins functionally. The findings presented here suggest that BK channels in glioma cells are indeed modulated by neuregulin acting through erbB2 receptors, which show a high level of constitutive activation in glioma cells. erbB2 activity in turn modulates [Ca2+]i, which is the principle mediator of gBK channel modulation. The most surprising aspect of our study was the sole dependence of this modulation on [Ca2+]i as a signal, insofar as previous examinations in other biological systems have led to different conclusions. For example, BK channels in ciliary ganglia of developing chick show a long-term modulation in which channels are synthesized de novo in response to TGFβ and an acute modulation in which channels are translocated from the cytoplasm to the membrane (Lhuillier and Dryer, 2002). In contrast, the activity of BK channels in vascular smooth muscle cells is modulated via tyrosine kinase phosphorylation (Rezzonico et al., 2002). We are quite convinced that we have established that the regulation of gBK in glioma cells is different again and occurs predominantly via [Ca2+]i. Rather than relying on immunohistochemical approaches, we pursued a rigorous biochemical examination to rule out transcriptional regulation of BK channels. Indeed, our Western blots, with controls for antibody specificity and protein loading, did not reveal any change in total cell protein. In light of the now commonly observed trafficking of channel protein from intracellular compartments to the cell membrane, we separated surface proteins from total cell proteins through a well-established cell surface biotinylation approach that we (Ye et al., 1999) have employed successfully in the past. These studies allowed us reliably to separate the actin-containing intracellular membrane fraction from the Na+/K+-ATPase-containing surface membrane fraction. However, no change in surface expression could be demonstrated in five examples of such experiments. Finally, by using affinity pull-down experiments with BK-channel-specific antibodies, we were able conclusively to rule out changes in BK channel phosphorylation upon erbB2 signaling events. This left only [Ca2+]i as a viable signal, and converging experimental evidence in our study unequivocally supported its role in this process. First and foremost, Fura-II Ca2+ imaging revealed significant changes in resting [Ca2+]i upon erbB2 activation or inhibition, and the directionality of these changes was consistent with the biophysical changes in BK channel activity observed. Importantly, all recording conditions that dialyzed the cytoplasmic, Ca2+-sensing side of the BK channel with defined and well-controlled [Ca2+] eliminated any regulatory effect of erbB2, leaving us convinced that [Ca2+]i is the principle mediator of gBK modulation by erbB2 in glioma cells.
This conclusion has several important ramifications. Our findings support the conclusion that gBK channels are a physiological target of erbB2 modulation, but these findings also suggest that other events capable of altering intracellular Ca2+ have the potential to modulate these channels. Similarly, the finding that the activation status of erbB2 leads to a sustained change in baseline [Ca2+]i indicates that it is possible, even likely, that this growth factor receptor regulates other, unrelated Ca2+-dependent processes concurrently. Therefore, it will remain unanswered whether gBK channels are destined to be a downstream target of erbB2 or rather an opportunistic target. We favor the former interpretation; we would argue that the changes in [Ca2+] observed in this study would be insufficient to modulate BK channels other than gBK, which is the only member of the channel family that shows a significantly enhanced Ca2+ sensitivity and hence appears to have properties that make it uniquely able to “listen” to the relatively small changes in Ca2+ presented.
With regard to the functional consequences of BK channel activation and its regulation by erbB2, we can only speculate, in that the role of gBK channels in glia or glioma biology is not well understood. BK channel blockers have been shown to inhibit the proliferation of glioma cells induced by elevated extracellular K+ (Basrai et al., 2002), and, in retinal Müller glial cells, BK channel inhibitions similarly cause inhibition of DNA synthesis, consistent with a role of these channels in growth control. We recently found, quite unexpectedly, that gBK channels endow glioma cell with an enhanced ability to survive under conditions of nutrient and growth factor deprivation (Weaver et al., 2004). BK channels have also been implicated in cell migration and invasion. Migrating glioma cells show spontaneous oscillations in [Ca2+]i, and these Ca2+ oscillations lead to concurrent changes in resting potential, which are caused by the activation of BK channels (Bordey et al., 2000). It is under such conditions that one might expect neuregulin/erbB2 signaling to occur quite prominently, insofar as neuregulin is thought to act primarily as a paracrine signal being presented to neighboring cells while still affiliated with the membrane of the cell that produces it. Indeed, the migration of cerebellar and cortical neurons along radial glia depends on the paracrine activation of erbB2 receptors via neuregulin (Anton et al., 1997), and erbB2 expression has been shown to be intrinsically associated with the “motogenic” migratory or invasive phenotype in many cancers (Feldner and Brandt, 2002). We have shown in glioma cells that neuregulin acting via erbB2 enhances the motility of gliomas cell (Ritch et al., 2003). In that glioma cell migration appears to recapitulate numerous features of early embryonic development, these cells may utilize mechanisms and signaling pathways that are characteristic of immature neurons in the developing brain. We are currently investigating the physiological consequences of BK channel modulation by erbB2 and its direct affect on cell proliferation and cell migration.
ACKNOWLEDGMENTS
The authors thank Dr. S. Carroll for kindly providing Nrg-1β and Dr. Y. Zhou for generously providing the anti-mbr5 antibody used in these studies.
Contract grant sponsor: NIH; Contract grant number: RO1-NS36692; Contract grant number: RO1-NS31234.
REFERENCES
- Anton ES, Marchionni MA, Lee KF, Rakic P. Role of GGF/neuregulin signaling in interactions between migrating neurons and radial glia in the developing cerebral cortex. Development. 1997;124:3501–3510. doi: 10.1242/dev.124.18.3501. [DOI] [PubMed] [Google Scholar]
- Basrai D, Kraft R, Bollensdorff C, Liebmann L, Benndorf K, Patt S. BK channel blockers inhibit potassium-induced proliferation of human astrocytoma cells. Neuroreport. 2002;13:403–407. doi: 10.1097/00001756-200203250-00008. [DOI] [PubMed] [Google Scholar]
- Bordey A, Ullrich N, Sontheimer H. Electrophysiological characterization of astrocytes and astrocytoma cells in slices from human biopsies and experimental intracranial tumors. Soc Neurosci Abstr. 1996;22:594.13. [Google Scholar]
- Bordey A, Sontheimer H, Trouslard J. Muscarinic activation of BK channels induces membrane oscillations in glioma cells and leads to inhibition of cell migration. J Membrane Biol. 2000;176:31–40. doi: 10.1007/s00232001073. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Biedermann B, Reichenbach A. Expression of potassium channels during postnatal differentiation of rabbit Muller glial cells. Eur J Neurosci. 1999;11:2883–2896. doi: 10.1046/j.1460-9568.1999.00706.x. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Francke M, Pannicke T, Biedermann B, Kodal H, Faude F, Reichelt W, Reichenbach A. Role of glial K+ channels in ontogeny and gliosis: a hypothesis based upon studies on Muller cells. Glia. 2000;29:35–44. doi: 10.1002/(sici)1098-1136(20000101)29:1<35::aid-glia4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Calderone V. Large-conductance, Ca2+-activated K+ channels: function, pharmacology and drugs. Curr Med Chem. 2002;9:1385–1395. doi: 10.2174/0929867023369871. [DOI] [PubMed] [Google Scholar]
- Cameron JS, Lhuillier L, Subramony P, Dryer SE. Developmental regulation of neuronal K+ channels by target-derived TGF beta in vivo and in vitro. Neuron. 1998;21:1045–1053. doi: 10.1016/s0896-6273(00)80622-4. [DOI] [PubMed] [Google Scholar]
- Cameron JS, Dryer L, Dryer SE. Beta-neuregulin-1 is required for the in vivo development of functional Ca2+-activated K+ channels in parasympathetic neurons. Proc Natl Acad Sci USA. 2001;98:2832–2836. doi: 10.1073/pnas.041394098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew LJ, Gallo V. Regulation of ion channel expression in neural cells by hormones and growth factors. Mol Neurobiol. 1998;18:175–225. doi: 10.1007/BF02741300. [DOI] [PubMed] [Google Scholar]
- Feldner JC, Brandt BH. Cancer cell motility—on the road from cerbB2 receptor steered signaling to actin reorganization. Exp Cell Res. 2002;272:93–108. doi: 10.1006/excr.2001.5385. [DOI] [PubMed] [Google Scholar]
- Fernandes A, Hamburger AW, Gerwin BI. ErbB-2 kinase is required for constitutive stat 3 activation in malignant human lung epithelial cells. Int J Cancer. 1999;83:564–570. doi: 10.1002/(sici)1097-0215(19991112)83:4<564::aid-ijc20>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK, Starrett JE, Jr, Dworetzky SI. Maxi-K potassium channels: form, function, and modulation of a class of endogenous regulators of intracellular calcium. Neuroscientist. 2001;7:166–177. doi: 10.1177/107385840100700211. [DOI] [PubMed] [Google Scholar]
- Hall SK, Armstrong DL. Conditional and unconditional inhibition of calcium-activated potassium channels by reversible protein phosphorylation. J Biol Chem. 2000;275:3749–3754. doi: 10.1074/jbc.275.6.3749. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Kassis J, Moellinger J, Lo H, Greenberg NM, Kim HG, Wells A. A role for phospholipase C-gamma-mediated signaling in tumor cell invasion. Clin Cancer Res. 1999;5:2251–2260. [PubMed] [Google Scholar]
- Khanna R, Chang MC, Joiner WJ, Kaczmarek LK, Schlichter LC. Hsk4/hik1, a calmodulin-binding KCa channel in human T lymphocytes. roles in proliferation and volume regulation. J Biol Chem. 1999;274:14838–14849. doi: 10.1074/jbc.274.21.14838. [DOI] [PubMed] [Google Scholar]
- Kodal H, Weick M, Moll V, Biedermann B, Reichenbach A, Bringmann A. Involvement of calcium-activated potassium channels in the regulation of DNA synthesis in cultured Muller glial cells. Invest Ophthalmol Vis Sci. 2000;41:4262–4267. [PubMed] [Google Scholar]
- Koegel H, Kaesler S, Burgstahler R, Werner S, Alzheimer C. Unexpected down-regulation of the hIK1 Ca2+-activated K+ channel by its opener 1-ethyl-2-benzimidazolinone in HaCaT keratinocytes. Inverse effects on cell growth and proliferation. J Biol Chem. 2003;278:3323–3330. doi: 10.1074/jbc.M208914200. [DOI] [PubMed] [Google Scholar]
- Kristt DA, Yarden Y. Differences between phosphotyrosine accumulation and Neu/ErbB-2 receptor expression in astrocytic proliferative processes. Implications for glial oncogenesis. Cancer. 1996;78:1272–1283. doi: 10.1002/(SICI)1097-0142(19960915)78:6<1272::AID-CNCR16>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Lhuillier L, Dryer SE. Developmental regulation of neuronal K(Ca) channels by TGFbeta1: an essential role for PI3 kinase signaling and membrane insertion. J Neurophysiol. 2002;88:954–964. doi: 10.1152/jn.2002.88.2.954. [DOI] [PubMed] [Google Scholar]
- Liu X, Chang Y, Reinhart PH, Sontheimer H, Chang Y. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. J Neurosci. 2002;22:1840–1849. doi: 10.1523/JNEUROSCI.22-05-01840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manor D, Moran N. Modulation of small conductance calcium-activated potassium channels in C6 glioma cells. J Membrane Biol. 1994;140:69–79. doi: 10.1007/BF00234487. [DOI] [PubMed] [Google Scholar]
- Moll V, Weick M, Milenkovic I, Kodal H, Reichenbach A, Bringmann A. P2Y receptor-mediated stimulation of Muller glial DNA synthesis. Invest Ophthalmol Vis Sci. 2002;43:766–773. [PubMed] [Google Scholar]
- Munaron L. Calcium signalling and control of cell proliferation by tyrosine kinase receptors [review] Int J Mol Med. 2002;10:671–676. [PubMed] [Google Scholar]
- Osherov N, Gazit A, Gilon C, Levitzki A. Selective inhibition of the epidermal growth factor and HER2/neu receptors by tyrphostins. J Biol Chem. 1993;268:11134–11142. [PubMed] [Google Scholar]
- Pena TL, Rane SG. The fibroblast intermediate conductance K(Ca) channel, FIK, as a prototype for the cell growth regulatory function of the IK channel family. J Membrane Biol. 1999;172:249–257. doi: 10.1007/s002329900601. [DOI] [PubMed] [Google Scholar]
- Puro DG, Roberge F, Chan CC. Retinal glial cell proliferation and ion channels: a possible link. Invest Ophthalmol Vis Sci. 1989;30:521–529. [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. BK Channels in human glioma cells. J Neurophysiol. 2001;85:790–803. doi: 10.1152/jn.2001.85.2.790. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Liu X, Sontheimer H. BK channels in human glioma cells have enhanced calcium sensitivity. Glia. 2002;38:281–291. doi: 10.1002/glia.10064. [DOI] [PubMed] [Google Scholar]
- Rezzonico R, Schmid-Alliana A, Romey G, Bourget-Ponzio I, Breuil V, Breittmayer V, Tartare-Deckert S, Rossi B, Schmid-Antomarchi H. Prostaglandin E2 induces interaction between hSlo potassium channel and Syk tyrosine kinase in osteosarcoma cells. J Bone Miner Res. 2002;17:869–878. doi: 10.1359/jbmr.2002.17.5.869. [DOI] [PubMed] [Google Scholar]
- Ritch PA, Carroll SL, Sontheimer H. Neuregulin-1 enhances motility and migration of human astrocytic glioma cells. J Biol Chem. 2003;278:20971–20978. doi: 10.1074/jbc.M213074200. [DOI] [PubMed] [Google Scholar]
- Ritch PS, Carroll SL, Sontheimer H.Neuregulin-1 enhances survival of human astrocytic glioma cells Glia (in press).2005 [DOI] [PMC free article] [PubMed]
- Sah P. Ca2+-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Schwechheimer K, Laufle RM, Schmahl W, Knodlseder M, Fischer H, Hofler H. Expression of neu/c-erbB-2 in human brain tumors. Hum Pathol. 1994;25:772–780. doi: 10.1016/0046-8177(94)90246-1. [DOI] [PubMed] [Google Scholar]
- Sontheimer H, Trotter J, Schachner M, Kettenmann H. Channel expression correlates with differentiation stage during development of oligodendrocytes from their precursor cells in culture. Neuron. 1989;2:1135–1145. doi: 10.1016/0896-6273(89)90180-3. [DOI] [PubMed] [Google Scholar]
- Subramony P, Raucher S, Dryer L, Dryer SE. Posttranslational regulation of Ca2+-activated K+ currents by a target-derived factor in developing parasympathetic neurons. Neuron. 1996;17:115–124. doi: 10.1016/s0896-6273(00)80285-8. [DOI] [PubMed] [Google Scholar]
- Tseng-Crank J, Foster CD, Krause JD, Mertz R, Godinot N, DiChiara TJ, Reinhart PH. Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron. 1994;13:1315–1330. doi: 10.1016/0896-6273(94)90418-9. [DOI] [PubMed] [Google Scholar]
- Tsunoda Y. Receptor-operated calcium influx mediated by protein tyrosine kinase pathways. J Recept Signal Transduct Res. 1998;18:281–310. doi: 10.3109/10799899809047748. [DOI] [PubMed] [Google Scholar]
- Weaver A, Liu X, Sontheimer H. The role for calcium-activated potassium channels (BK) in growth control of human malignant gliomas. J Neurosci Res. 2004;78:224–234. doi: 10.1002/jnr.20240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Rothstein JD, Sontheimer H. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodiumdependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci. 1999;19:10767–10777. doi: 10.1523/JNEUROSCI.19-24-10767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]