

Abstract
Membranes of Rhizobium leguminosarum contain a 3-deoxy-D-manno-octulosonic acid (Kdo)-activated lipid A 4′-phosphatase required for generating the unusual phosphate-deficient lipid A found in this organism. The enzyme has been solubilized with Triton X-100 and purified 80-fold. As shown by co-purification and thermal inactivation studies, the 4′-phosphatase catalyzes not only the hydrolysis of (Kdo)2-[4′-32P]lipid IVA but also the transfer the 4′-phosphate of Kdo2-[4′-32P]lipid IVA to the inositol headgroup of phosphatidylinositol (PtdIns) to generate PtdIns-4-P. Like the 4′-phosphatase, the phosphotransferase activity is not present in Escherichia coli, Rhizobium meliloti, or the nodulation-defective mutant 24AR of R. leguminosarum. The specific activity for the phosphotransferase reaction is about 2 times higher than that of the 4′-phosphatase. The phosphotransferase assay conditions are similar to those used for PtdIns kinases, except that ATP and Mg2+ are omitted. The apparent Km for PtdIns is ~500 μM versus 20–100 μM for most PtdIns kinases, but the phosphotransferase specific activity in crude cell extracts is higher than that of most PtdIns kinases. The phosphotransferase is absolutely specific for the 4-position of PtdIns and is highly selective for PtdIns as the acceptor. The 4′-phosphatase/phosphotransferase can be eluted from heparin- or Cibacron blue-agarose with PtdIns. A phosphoenzyme intermediate may account for the dual function of this enzyme, since a single 32P-labeled protein species (Mr ~68,000) can be trapped and visualized by SDS gel electrophoresis of enzyme preparations incubated with Kdo2-[4′-32P]lipid IVA. Although PtdIns is not detected in cultures of R. leguminosarum/etli (CE3), PtdIns may be synthesized during nodulation or supplied by plant membranes, given that soybean PtdIns is an excellent phosphate acceptor. A bacterial enzyme for generating PtdIns-4-P and a direct link between lipid A and PtdIns-4-P biosynthesis have not been reported previously.
Lipopolysaccharide (LPS)1 is a major component of the outer membranes of Gram-negative bacteria (1–5). The lipid A moiety of LPS makes up much of the outer monolayer of the outer membrane (1–5). LPS acts as barrier to antibiotics (6, 7) and helps bacterial cells resist complement-mediated lysis (8). The lipid A portion of LPS is essential for bacterial viability (9–11), and it is also the active component of LPS responsible for many of the pathophysiological effects associated with Gram-negative infections in animals, including septic shock (3, 4, 11, 12).
In certain plant systems LPS is required for the establishment of symbiosis between nitrogen-fixing strains of Rhizobium and their hosts (13, 14). Rhizobium leguminosarum mutants with truncated LPS structures lacking O-antigens are defective in generating functional nodules within root cells (15–19). Changes in LPS structure are also associated with the physiological adaptation to the symbiotic microenvironment (20, 21). For instance, developmentally regulated expression of distinct LPS epitopes can be demonstrated in planta by immunostaining (22). Whether or not the lipid A moiety of R. leguminosarum LPS plays a role in infection and nodulation is unknown, since defined mutants in the lipid A pathway are not yet available. A complete understanding of the structure, biosynthesis, and molecular genetics of lipid A in R. leguminosarum is a prerequisite for defining its functions during symbiosis.
The structure of lipid A in R. leguminosarum is strikingly different from that of E. coli (23–26) (Fig. 1A). R. leguminosarum lipid A lacks the 1- and 4′-phosphate groups found in the lipid A of most other Gram-negative bacteria (23, 24). A galacturonic acid residue is incorporated in place of the 4′-phosphate, and the proximal glucosamine 1-phosphate unit of Escherichia coli lipid A may be replaced with an aminogluconate moiety (23, 24) (Fig. 1A). R. leguminosarum lipid A also lacks the laurate and myristate residues present in E. coli lipid A (3, 27) but is acylated with an unusual 28-carbon chain (23, 24, 28). Despite these differences, both E. coli and R. leguminosarum employ the same seven enzymes to generate the conserved, phosphate-containing precursor, Kdo2-lipid IVA (Fig. 1B) (29). Several distinct enzymes must therefore exist in R. leguminosarum that catalyze the conversion of Kdo2-lipid IVA to R. leguminosarum lipid A. We have recently discovered a 4′-phosphatase (30), a 1-phosphatase (31), a long chain acyltransferase (32), and a mannosyl transferase (31, 33, 34) that are involved in the processing of Kdo2-lipid IVA in extracts of R. leguminosarum but not of E. coli. Many additional enzymes unique to R. leguminosarum remain to be found.
Fig. 1. Structures of E. coli and R. leguminosarum lipid A and of Kdo2-[4′-32P]lipid IVA.

A, the 1-, 3-, and 4′-positions of each lipid A structure are indicated. Evidence for the presence of an acyloxyacyl residue and partially deacylated species has recently been presented by Que et al. (25, 26). B, the same seven enzymes catalyze the formation of Kdo2-lipid IVA in both organisms (29). A phosphoenzyme intermediate could explain all of the reactions catalyzed by the 4′-phosphatase/phosphotransferase of R. leguminosarum.
We now report a novel, chemically specific and highly active phosphotransferase reaction associated with the 4′-phosphatase of R. leguminosarum (CE3) (Fig. 1B). In the presence of phosphatidylinositol (PtdIns), the 4′-phosphatase can transfer the 4′-phosphate of Kdo2-[4′-32P]lipid IVA (and related metabolites including E. coli lipid A) to the inositol moiety of PtdIns, generating exclusively PtdIns-4-P (Fig. 1B). The phosphotransferase and the previously described 4′-phosphatase activity (30) appear to be catalyzed by the same enzyme, given their identical behavior during purification and thermal inactivation. Although the apparent Km of the phosphotransferase for PtdIns is about 10 times higher than that of many eucaryotic PtdIns 4-kinases (35–37), its high specific activity in cell extracts and its absolute specificity for the 4-position of PtdIns suggest that it could play a significant role during symbiosis. A procaryotic pathway for the biosynthesis of PtdIns-4-P and a direct connection between lipid A formation and PtdIns phosphorylation have not been reported previously.
EXPERIMENTAL PROCEDURES
Chemicals and Materials
[γ-32P]ATP was obtained from NEN Life Science Products. Ptd-[2-3H]Ins-4-P and [1,2-3H]Ins were purchased from American Radiolabeled Chemicals, Inc. PtdIns-3-P, digalactosyl diglyceride and monogalactosyl-diglyceride were obtained from Matreya, Inc. Silica gel 60 (0.25-mm) thin layer plates were purchased from EM Separation Technology, E. Merck. Triton X-100 and bicinchoninic acid were from Pierce. Yeast extract and tryptone were obtained from Difco. Other chemicals were from Sigma or Mallinckrodt.
Bacterial Strains and Growth Conditions
R. leguminosarum biovar phaseoli CE3 (also recently classified as R. etli), mutant 24AR of R. leguminosarum biovar trifolii, R. leguminosarum biovar viciae 8401, Rhizobium meliloti 1021, and E. coli W3110 were described in previous studies (29–31, 38). All other strains of Rhizobium were purchased from the American Type Culture Collection. All Rhizobium strains were grown at 30 °C on TY medium, which contains 5 g/liter tryptone, 3 g/liter yeast extract, 10 mM CaCl2, and 20 μg/ml nalidixic acid. For the growth of strains CE3, 24AR, 1021, and 8401, streptomycin (200 μg/ml) was also added to the medium. E. coli W3110 was grown on LB broth (39) at 30 °C.
Preparation of Radiolabeled Substrates
The substrate [4′-32P]lipid IVA was generated using [γ-32P]ATP, tetraacyldisaccharide 1-phosphate acceptor (DS-1-P) (40), and membranes of E. coli BLR(DE3)/ pLysS/pJK2, which contain large amounts of the 4′-kinase (41). Kdo2-[4′-32P]lipid IVA could then be prepared from the [4′-32P]lipid IVA using the purified Kdo transferase (38, 42). Alternatively, Kdo2-[4′-32P]lipid IVA was also prepared directly from DS-1-P and [γ-32P]ATP without purification of the [4′-32P]lipid IVA intermediate. Briefly, a 100-μCi portion of [γ-32P]ATP (3000 Ci/mmol) was dried in a 1.5-ml polypropylene microcentrifuge tube under a stream of N2. Next, the following components were added: (a) 25 μl of a mixture of DS-1-P and cardiolipin, prepared by mixing 0.5 mg of DS-1-P and 4 mg of cardiolipin in 1 ml of water, followed by ultrasonic dispersion; (b) 5 μl of 10% Nonidet P-40, a nonionic detergent; and (c) 10 μl of 25 mM MgCl2. The 4′-kinase reaction was started by adding 5 μl of membranes (500 μg/ml in 50 mM Hepes, pH 7.5) of E. coli strain BLR(DE3)/pLysS/pJK2 and was held at room temperature. After 10 min, a second 5-μl portion of the enzyme stock was added, and the incubation was continued for another 10 min. The Kdo transferase reaction was then carried out in the same tube by adding the following: 2 μl of 5 mM lipid IVA, 25 μl of 2% Triton X-100, 50 μl of 100 mM CTP in 0.5 M Hepes, pH 7.5, 50 μl of 40 mM Kdo, 50 μl of 100 mM MgCl2, 50 μl of purified CMP-Kdo synthase (0.03 total units) (42, 43), and 25 μl of purified Kdo transferase (38) (from a stock solution at 80 μg/ml). The final reaction volume was 500 μl. After incubation at 30 °C for 30 min, 80–90% of the [γ-32P]ATP was incorporated into the Kdo2-[4′-32P]lipid IVA, as judged by analytical thin layer chromatography and autoradiography.
Next, the reaction mixture was carefully spotted across the origin of a 20 × 20-cm silica gel 60 thin layer plate. The plate was dried under a cold air stream and developed in the solvent chloroform/pyridine/88% formic acid/water (30:70:16:1, v/v/v/v). The plate was again dried under a cold air stream and exposed to x-ray film for 30–60 s to locate the Kdo2-[4′-32P]lipid IVA band. The appropriate region was scraped and transferred to a 30-ml sintered glass filter funnel. The silica powder was washed with 10 ml of chloroform, and the 32P-labeled product was then eluted with 24 ml of a single phase Bligh-Dyer mixture (44), consisting of CHCl3/methanol/50 mM ammonium acetate in water adjusted to pH 1.5 with HCl (1:2:0.8, v/v/v). Three 8-ml fractions were collected in glass tubes. CHCl3 (2.1 ml) and 50 mM ammonium acetate in water adjusted to pH 1.5 with HCl (2.1 ml) were added to each 8-ml fraction to form three two-phase systems. The lower phases were removed with a Pasteur pipette and pooled into a single 15-ml Corex glass tube equipped with a Teflon cap. Six drops of HPLC grade pyridine were added, and the solution was dried under a stream of N2 at room temperature. While the drying was in progress, two additional extractions of the remaining three upper phases were carried to optimize product recovery. Each upper phase was extracted twice with 2 ml of a fresh lower phase from a pre-equilibrated two-phase Bligh-Dyer system, consisting of CHCl3/methanol/50 mM ammonium acetate in water adjusted to pH 1.5 with HCl (1:1:0.9, v/v/v). The lower phases derived from these additional extractions were pooled into the same 15-ml Corex tube used for the initial lower phases, and the resulting solution was again dried under a stream of N2. The final dried Kdo2-[4′-32P]lipid IVA preparation was dissolved in 2 ml of CHCl3/MeOH (1:1, v/v) with vortexing and sonic irradiation, and the material was divided equally among four 1.5-ml polypropylene microcentrifuge tubes. These aliquots were dried immediately under a stream of N2. Additional residual Kdo2-[4′-32P]lipid IVA was recovered by washing the Corex tube two more times with 2-ml portions of CHCl3/MeOH (1:1, v/v) and distributing the solution equally among the four microcentrifuge tubes. Following the final drying under N2, the four portions of Kdo2-[4′-32P]lipid IVA were each suspended by sonic irradiation in 500 μl of 10 mM Tris-HCl, pH 7.8, containing 1 mM EDTA and 1 mM EGTA, and they were stored at −20 °C. The entire extraction process took about 5 h. A typical final yield of Kdo2-[4′-32 P]lipid IVA (specific activity ~8 μCi/nmol) following these extractions was ~30% of the input [γ-32P]ATP.
Kdo-[4′-32P]lipid IVA was prepared with the monofunctional Kdo transferase from Hemophilus influenzae (45). The substrate [4′-32P]lipid A was prepared from compound 505 by the action of the highly overexpressed 4′-kinase (41). The [4′-32P]lipid IVA, Kdo-[4′-32P]lipid IVA, Kdo2-[4′-32P]lipid IVA, and [4′-32P]lipid A were purified by thin layer chromatography (38, 42, 45) (or see above). These substances were stored as aqueous dispersions at −20 °C in 10 mM Tris-HCl, pH 7.8, containing 1 mM EDTA and 1 mM EGTA. Prior to use, all of the substrates were dispersed again by sonic irradiation for 1 min in a bath sonicator.
Assays of the 4′-Phosphatase and the Phosphotransferase Reactions
Standard assay conditions for the 4′-phosphatase were as follows. The reaction mixture (10–20 μl) contained 50 mM MES, pH 6.5, 0.1% Triton X-100, 2 mM dithiothreitol, 2 mM EDTA, 10 mM potassium phosphate, and 10 μM Kdo2-[4′-32P]lipid IVA (20,000 cpm/nmol). PtdIns (usually 1 mg/ml or at the indicated concentrations) was added as the acceptor substrate in the phosphotransferase reactions. When PtdIns was included as the phosphate acceptor, a 1-μl solution of PtdIns in chloroform/methanol (4:1, v/v) was transferred to each reaction tube, and the solvent was removed with a stream of N2. Next, the Triton X-100 and the other reaction components were added. The reaction mixture was subjected to sonic irradiation to disperse the phospholipids into the detergent-containing buffer. Reactions were initiated with enzyme. The reaction mixtures were incubated at 30 °C for 15 min or as indicated. Reactions were terminated by spotting 2-μl samples onto a silica gel 60 thin layer chromatography plate, which was developed in the solvent chloroform/pyridine/88% formic acid/water (30:70:16:10; v/v/v/v). The remaining Kdo2-[4′-32P]lipid IVA, the 32Pi, and the PtdIns-4-32P were quantified on the plate using a Molecular Dynamics model 425S PhosphorImager, equipped with ImageQuant software. The percentage conversion of substrate to product(s) was calculated for each reaction, and the specific activities were expressed as nmol/min/mg, as indicated.
Partial Purification of the 4′-Phosphatase and Its Associated Phosphotransferase Activity
All enzyme preparations were carried out at 0–4 °C. Protein was determined by the bicinchoninic acid method (46), using bovine serum albumin as a standard. R. leguminosarum CE3 cells in late logarithmic phase (A550 = 1.2–1.4) were harvested from 5 liters of culture by centrifugation (8000 × g, 15 min) and resuspended in 50 mM Hepes, pH 7.5, to give a final protein concentration of 8–10 mg/ml. Cells were broken by two passages through a French pressure cell at 18,000 p.s.i., and the debris was removed by centrifugation at 8000 × g for 15 min. Membranes were prepared by ultracentrifugation at 149,000 × g for 60 min. The membrane pellet was resuspended in Buffer A (50 mM Hepes, pH 7.5, 3% glycerol, and 10 mM potassium phosphate) at ~4–5 mg/ml. Solubilization was carried out by the addition of 10% Triton X-100 (reduced) to yield a protein:detergent ratio of 1:2 (~0.9% final detergent concentration), followed by stirring for 2 h, and centrifugation at 149,000 × g for 60 min. The supernatant, which contained over 95% of both activities, was collected and stored at −80 °C.
Next, a Q-Sepharose (Amersham Pharmacia Biotech) column (30 ml) was equilibrated with Buffer B (20 mM Tris-HCl, pH 7.8, 0.15% Triton X-100 (reduced), 2 mM EDTA, 3% glycerol, and 10 mM potassium phosphate). Solubilized membranes (30 ml, 3.1 mg/ml protein) were loaded onto the column. Unbound proteins were washed out with Buffer C (50 mM MES, pH 6.5, 0.15% reduced Triton X-100, 2 mM EDTA, 3% glycerol, and 10 mM potassium phosphate containing 100 mM NaCl) until A280 returned to base line. Elution was carried out with a 400-ml linear gradient from 0.1 to 1.0 M NaCl in Buffer C. The column fractions (8 ml) were assayed for the 4′-phosphotransferase and 4′-phosphatase. The active fractions (~80 ml total volume) were pooled, desalted, and concentrated (~3-fold) by ultrafiltration through an Amicon (30K) filter. The concentrated material was then diluted to the original volume with Buffer C containing no detergent. The cycle was repeated three times. The final concentrated active fractions (~25 ml) from the Q-Sepharose column were then loaded onto a heparin-agarose Type 1 (Sigma) column (15 ml) equilibrated with Buffer C. Next, the column was washed with equilibration buffer containing 100 mM NaCl. The 4′-phosphatase/phosphotransferase activities were eluted with a 200-ml linear gradient from 0.1 to 1.5 M NaCl in Buffer C. The column fractions (5 ml) were assayed for both the phosphotransferase and the 4′-phosphatase. The active fractions were pooled, concentrated (3 fold), and desalted as above.
Cibacron Blue Column Chromatography and Substrate Elution
A Cibacron blue 3GA Type 300 (Sigma) column (3 ml) was equilibrated with Buffer C (see above). Partially purified enzyme from the heparin-agarose step (5 ml, 0.3 mg/ml protein) was loaded onto the column and was recirculated through the column two more times. The column was then washed successively with 5 ml of Buffer C, 15 ml of Buffer C containing 0.5 M NaCl, 15 ml of Buffer C containing 0.5% Triton X-100 (reduced), and 10 ml of Buffer C. Finally, the elution of the 4′-phosphatase/phosphotransferase was carried out with 35 ml of Buffer C containing 0.4 mg/ml PtdIns and 0.2 M NaCl. The column fractions (2.5 ml) were assayed for both the phosphotransferase and the 4′-phosphatase activities.
Separation of Inner and Outer Membranes
R. leguminosarum (CE3) membranes were separated by isopycnic sucrose gradient centrifugation as described previously (33). The turbidity (A600) of each fraction was determined to confirm the presence of membrane fragments, and each fraction was assayed for NADH oxidase as the inner membrane marker and phospholipase A as the outer membrane marker (33). The protein content of each fraction was determined by the bicinchoninic acid assay (46) using bovine serum albumin as the standard. Last, each fraction was assayed for both phosphotransferase and 4′-phosphatase activities under the standard assay conditions described above.
Deacylation and HPLC Analysis of the 32P-Labeled PtdIns Derivative Generated from Kdo2-[4′-32P]lipid IVA
Prior to HPLC analysis, the product of the phosphotransferase reaction and the appropriate lipid standards were deacylated. This was accomplished by treating the compounds with methylamine reagent (47). Briefly, a 0.5-ml portion of methylamine reagent, prepared by bubbling methylamine (Fluka, Buchs, Switzerland) at 0 °C through a mixture in which it is very soluble (6.2 ml of methanol, 4.6 ml of deionized water, and 1.5 ml of 1-butanol) to give a final volume of 20 ml was added to the dried lipids (see Fig. 10 legend). The mixture was incubated at 53 °C for 40 min. Cold 1-propanol (0.2 ml) was added to the sample. After mixing, the sample was dried and resuspended in 0.2 ml of water. This aqueous dispersion was extracted three times with 0.5-ml portions of butanol/ petroleum ether/ethyl formate (20:4:1, v/v/v). The lower aqueous phase was dried, and deacylation products were redissolved in 200 μl of deionized water for HPLC analysis. Anion exchange HPLC was performed using a Partisil SAX column (Whatman) (4.6 × 250 mm) (47). The deacylation products were eluted (flow rate of 1 ml/min) with a linear gradient from 10 to 510 mM NH4H2PO4 (pH 3.8) in 50 min. Effluent from the HPLC column flowed directly into a BetaRAM™ in-line continuous flow scintillation detector (INUS, Tampa, FL).
Fig. 10. Selective transfer of the 4′-phosphate group of Kdo2-[4′-32P]lipid IVA to the 4-position of the inositol moiety of PtdIns.

Three reactions (10 μl each) were carried out under standard assay conditions with 10 μM Kdo2-[4′-32P]lipid IVA (20,000 cpm/nmol) as donor. A, with enzyme (10 μg/ml heparin-agarose step) but without PtdIns; B and C, with enzyme (10 μg/ml heparin-agarose step) and with PtdIns (1 mg/ml) as the acceptor. After incubation for 40 min, internal standards consisting of PtdIns and/or its phosphorylated derivatives in 30 μl of chloroform/methanol (1:1, v/v) were added to each tube. A [1,2-3H]inositol-labeled mixture (40,000 cpm) of PtdIns and phosphorylated PtdIns derivatives isolated from yeast cells was used as the internal standard for reactions A and B, while Ptd-[2-3H]Ins-4-P (2000 cpm) was used as the internal standard for tube C. Each sample was then dried by vacuum centrifugation, followed by deacylation with methylamine and HPLC analysis of the water-soluble deacylation products (47). The elution of the 3H-labeled deacylated internal standards (broken lines) and of the 32P-labeled enzymatic reaction products (solid lines) was monitored by in-line liquid scintillation counting. The elution profiles for reactions A (no PtdIns, yeast standards), B (with PtdIns, yeast standards), and C (with PtdIns, PtdIns-4P standard) are shown in the respective panels. The peaks for internal standards (glycerophosphoinositol (GPI), glycerophosphoinositol 3-phosphate (GPI-3P), glycerophosphoinositol 4-phosphate (GPI-4P), and the 4′-phosphatase/ phosphotransferase products (32Pi and [32P]phosphotransferase product) are indicated. No 32 Pi or [32P]phosphotransferase product was seen without enzyme (not shown). The slight decrease in the retention times for the 32Pi and the GPI-4P in A versus C probably is caused by aging of the column following multiple cycles of chromatography.
Trapping and Detection of a [32P]Phosphoenzyme Intermediate Generated by Incubating Enzyme with Kdo2-[4′-32P]lipid IVA
The enzyme (2 μg purified through the heparin-agarose step or 6 μg purified through the Q-Sepharose step as indicated) was mixed with (Kdo)2-[4′-32P]lipid IVA (25,000 cpm at 8 μ Ci/nmol) in a reaction buffer containing 50 mM MES (pH 6.5), 0.1% Triton X-100, and 2 mM EDTA. The reaction mixture (10 μl) was incubated at 30 °C for 5 min. The reaction was quenched by rapidly mixing with 10 μl of water and 10 μl of 0.3 M CAPS buffer (pH 10), containing 12% SDS, 60% glycerol, and bromphenol blue dye. The sample was directly loaded, without heating, onto a SDS-polyacrylamide gel (10%) to separate the denatured proteins. The gels were dried and subjected to PhosphorImager analysis for visualization and quantification.
For pulse-chase experiments, three reactions (final volume of 70 μl each) were set up in the presence of 2 μM(Kdo)2-[4′-32P]lipid IVA (175,000 cpm/reaction) in the same buffer as described above. Phosphoenzyme formation was initiated by adding 8.4 μg of concentrated heparin-agarose enzyme in 3 μl to each tube. After 5 min at 30 °C, the phosphoenzyme was chased by the addition of either 5 μl of reaction buffer or unlabeled (Kdo)2-lipid IVA in reaction buffer to yield final concentrations of 10 or 50 μM. The incubations were continued at 30 °C. Finally, 10-μl portions of the reaction mixtures were withdrawn at different times after the chase (0.5, 2, 5, and 10 min), and the remaining [32P]phosphoenzyme was trapped and detected as described above.
RESULTS
A Phosphotransferase in Membranes of R. leguminosarum That Uses Kdo2-[4′-32P]lipid IVA to Phosphorylate PtdIns
The 4′-phosphatase found in membranes of R. leguminosarum (biovars trifolii, phaseoli, and viciae) but not of R. meliloti or E. coli catalyzes the release of inorganic phosphate from the 4′-position of the lipid A disaccharide precursor Kdo2-[4′-32P]lipid IVA (30). In the presence of phosphatidylinositol, however, which was initially tested as a stabilizing agent, the same membranes that dephosphorylate Kdo2-[4′-32P]lipid IVA also catalyze transfer of 32P from Kdo2-[4′-32P]lipid IVA onto PtdIns (Fig. 2, lanes 3, 5, and 6). This phosphotransferase activity is not detected in membranes of the nodulation-deficient mutant 24AR of R. leguminosarum, which lacks the 4′-phosphatase (30) and possesses a 4′-phosphate moiety on its lipid A (48). As with the 4′-phosphatase (30), no phosphotransferase activity is seen in the cytosol.
Fig. 2. Presence of a PtdIns-dependent phosphotransferase in membranes of R. leguminosarum.

Membranes of different strains were assayed for phosphotransferase activity using the standard assay. A protein concentration of 0.2 mg/ml was used, and the incubation was carried out for 20 min at 30 °C. The thin layer chromatographic analysis of the reaction products generated from Kdo2-[4′-32P]lipid IVA and PtdIns are shown. Lane 1, no membranes; lane 2, E. coli W3110; lane 3, R. leguminosarum biovar etli CE3; lane 4, R. leguminosarum biovar trifolii 24AR; lane 5, R. leguminosarum biovar viciae 8401; lane 6, R. leguminosarum biovar trifolii ATCC 14479; lane 7, R. meliloti 1021. No PtdIns-4-P was formed in the absence of added PtdIns.
Fractionation of R. leguminosarum CE3 membranes by sucrose density gradient centrifugation (33) revealed that both the 4′-phosphatase and the phosphotransferase are localized in the inner membrane (Fig. 3). The distribution of NADH oxidase activity served as the inner membrane marker in the same gradient fractions. Phospholipase A indicated the presence of outer membrane fragments (Fig. 3).
Fig. 3. Inner membrane localization of the 4′-phosphatase and the PtdIns-dependent phosphotransferase of R. leguminosarum.

The inner and outer membranes of strain CE3 were separated by isopycnic sucrose density gradient centrifugation, and ~0.4-ml fractions were collected. NADH oxidase and phospholipase A activities were assayed to locate inner and outer membrane fragments, respectively. A, NADH oxidase and phospholipase A activities in each fraction are expressed as a percentage of the total activity across the entire gradient. B, the 4′-phosphatase and the phosphotransferase activities were assayed in each fraction.
The Solubilized 4′-Phosphatase and Phosphotransferase Activities Co-purify
The possibility that the same enzyme might be catalyzing the 4′-phosphatase and the phosphotransferase reactions was investigated by partially purifying the 4′-phosphatase. Isolation of membranes as the first step resulted in a 2.8-fold increase in 4′-phosphatase-specific activity (Table I). Quantitative solubilization of both activities was achieved with Triton X-100 (reduced) at a detergent:protein ratio of 2:1, as described under “Experimental Procedures.” In Table I, the -fold purification and yields were calculated based on the 4′-phosphatase activity, but the pattern was the same if the phosphotransferase data was used (not shown).
Table I. Solubilization and partial purification of the 4′-phosphatase/phosphotransferase.
The activities were assayed with 50 μM Kdo2-[4′-32P]lipid IVA, either with or without 1 mg/ml PtdIns. The -fold purification and yield after each step were calculated using the 4′-phosphatase activity data. Within the experimental error of the assays, the ratio of the phosphotransferase specific activity divided by the 4′-phosphatase specific activity was the same after each step following solubilization.
| Purification step | Total volume | Protein | 4′-Phosphatase specific activity | Phosphotransferase specific activity | Ratio | Purification | Yield |
|---|---|---|---|---|---|---|---|
| ml | mg/ml | nmol/min/mg | nmol/min/mg | -fold | % | ||
| Crude extract | 60 | 8.2 | 0.65 | 1.0 | 1.5 | 1 | 100 |
| Membranes | 40 | 4.4 | 1.82 | 3.27 | 1.8 | 2.8 | 100 |
| Solubilized membranes | 30 | 3.1 | 3.52 | 7.74 | 2.2 | 5.4 | 120 |
| Q-Sepharose | 25 | 0.46 | 21.4 | 51.36 | 2.4 | 32.9 | 77 |
| Heparin-agarose | 15 | 0.3 | 53.2 | 117.04 | 2.2 | 81.8 | 75 |
Several ion exchange resins were surveyed for their ability to bind the solubilized 4′-phosphatase. The enzyme displayed strong, reversible affinity for Q-Sepharose and heparin-agarose. About 80% of both the 4′-phosphatase and the phosphotransferase bound to Q-Sepharose, and both were quantitatively eluted from the column at ~0.3–0.4 M NaCl (Fig. 4A). The pooled peak fractions were purified about 33-fold over the cell extract based on the specific activity of the 4′-phosphate (Table I). Next, column chromatography on heparin-agarose (Fig. 4B) resulted in an additional 2.5-fold purification of the 4′-phosphatase with an overall yield of about 75% (Table I). Importantly, the peaks of 4′-phosphatase and the phosphotransferase activities in both of these chromatograms (Fig. 4, A and B) overlapped each other within experimental error. Following solubilization of the membranes, the ratio of the specific activity of the phosphotransferase to the 4′-phosphatase remained constant at each step of the purification (2.2–2.4) (Table I). The identical chromatographic behaviors of both activities (Fig. 4) and the constant ratios of specific activities (Table I) indicate that a single enzyme species probably catalyzes both the 4′-phosphatase and the phosphotransferase reactions. However, the heparin-agarose fractions are yet not homogeneous, as judged by SDS gel electrophoresis (not shown).
Fig. 4. Identical chromatographic behaviors of the 4′-phosphatase and the phosphotransferase.
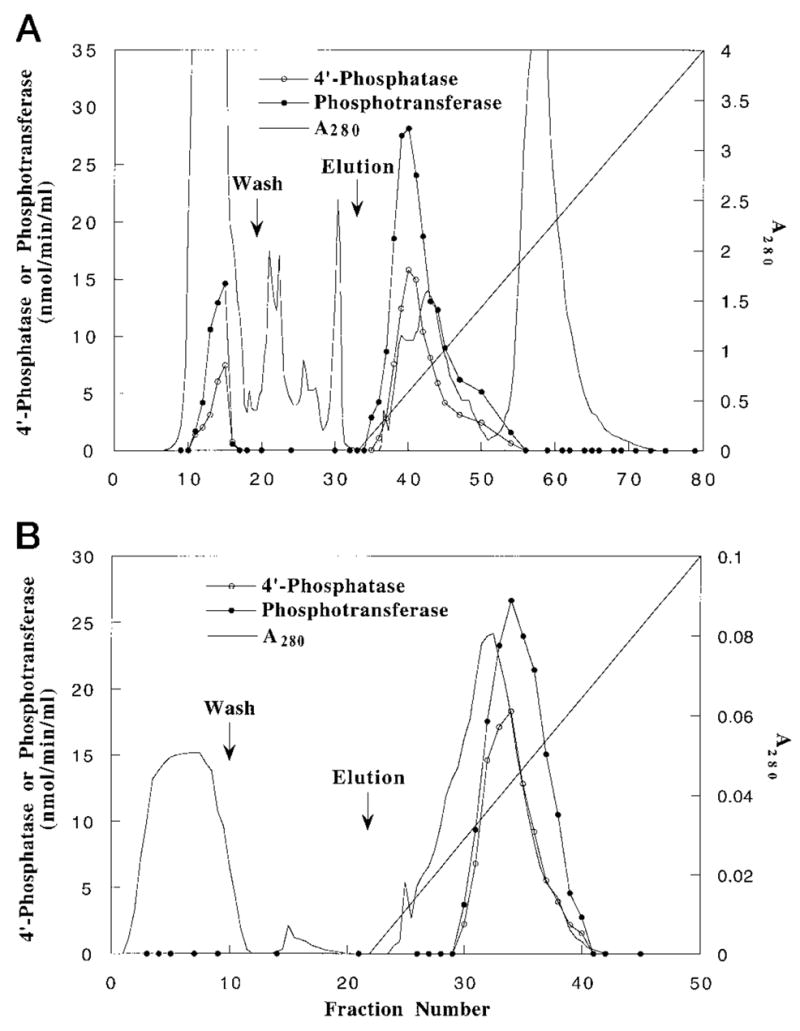
A, co-elution of 4′-phosphatase and phosphotransferase activities from Q-Sepharose. B, co-elution of 4′-phosphatase and phosphotransferase activities from heparin-agarose.
Following solubilization and purification, both the 4′-phosphatase and the phosphotransferase activities are linear with time for up to 20 min at 30 °C (Fig. 5).
Fig. 5. Linearity of the partially purified 4′-phosphatase and phosphotransferase reactions with time.

Partially purified enzyme (heparin-agarose step) was assayed for the 4′-phosphatase activity in the absence of PtdIns (open circles and squares) or for the phosphotransferase activity in the presence of PtdIns (closed circles and squares). Protein concentrations of 0.6 μg/ml (circles) or 1.2 μg/ml (squares) were used, and the assays were incubated at 30 °C for the indicated times under standard conditions (30 μl final volume). At every time point indicated, a 2-μl portion of the reaction mixture was withdrawn and analyzed by thin layer chromatography and PhosphorIm-ager analysis, as described under “Experimental Procedures.” The 4′-phosphatase activity is expressed as the amount of inorganic phosphate released per ml of reaction mixture in absence of PtdIns, while the phosphotransferase activity is expressed as the amount of PtdIns-4-P formed in the presence of PtdIns.
Thermal Inactivation and Affinity Elution Studies
When the heparin-agarose-purified material was preincubated at various temperatures (30, 37, or 45 °C), the 4′-phosphatase and the phosphotransferase activities were inactivated at the same rates (Fig. 6), further supporting the view that the same protein may be catalyzing both reactions. Concurrent inactivation of both of the activities was also observed in the presence of higher inorganic phosphate concentrations (40 mM) or orthovanadate (1 mM) (data not shown).
Fig. 6. Thermal inactivation of the 4′-phosphatase and the phosphotransferase.

Samples of the partially purified enzyme (heparin-agarose step) were preincubated at various temperatures (30, 37, or 45 °C) for the indicated times. A portion of the incubation was then used to assay for remaining 4′-phosphatase and phosphotransferase activities. The percentage of the activity remaining is normalized to a control sample of the enzyme held on ice.
Last, a physical association of the enzyme with PtdIns was shown by the ability of PtdIns to elute both the 4′-phosphatase and the phosphotransferase from a Cibacron blue-agarose column (Fig. 7) or from a heparin-agarose column (not shown). The 4′-phosphatase/phosphotransferase bound very strongly to Cibacron blue-agarose (type 300), since it could not be eluted with buffers containing 0.5% Triton X-100 (reduced) and 0.5 M NaCl. Buffer containing PtdIns (0.4 mg/ml) and 0.2 M NaCl was effective in co-eluting both activities (Fig. 7). However, the resulting preparation still contained several protein species, as judged by gel electrophoresis (data not shown).
Fig. 7. Co-elution of the 4′-phosphatase and the phosphotransferase activities from Cibacron blue (type 300) by PtdIns.

The chromatography was carried out as described under “Experimental Procedures.”
Kinetic Studies and Substrate Specificity of the 4′-Phosphatase and the Phosphotransferase
In crude extracts, the 4′-phosphatase functions optimally when a Kdo disaccharide is part of the substrate, as in Kdo2-lipid IVA (30). We confirmed the Kdo dependence of both the 4′-phosphatase and the phosphotransferase with the 80-fold purified enzyme (heparin-agarose step). The apparent Km for lipid IVA was about 5 times higher than that for Kdo2-lipid IVA (Table II) with both activities, but Kdo-[4′-32P]lipid IVA prepared with the monofunctional Kdo transferase of H. influenzae was a good substrate as well (Table II). Either one or two Kdo moieties also increased the Vmax by 2–3-fold in comparison with lipid IVA in each case (Table II). The Vmax for the phosphotransferase, using PtdIns as the acceptor, was 3 times higher than that of the 4′-phosphatase with the partially purified enzyme, using Kdo2-lipid IVA as the substrate (Table II).
Table II. Kinetic analysis of the partially purified 4′-phosphatase and phosphotransferase.
The kinetic constants for the 4′-phosphatase and the phosphotransferase reactions were determined from assays at varying concentrations of Kdo2-[4′-32P]lipid IVA, Kdo-[4′-32P]lipid IVA, or [4′-32P]lipid IVA in the presence or absence of PtdIns (1 mg/ml). The apparent Km and Vmax were then estimated from double reciprocal plots. The Vmaxof the phosphotransferase reaction is higher than that of the 4′-phosphatase with each substrate.
| Donor substrate | Acceptor substrate | Apparent Km | Vmax |
|---|---|---|---|
| μM | nmol/min/mg | ||
| Kdo2-lipid IVA | Water | 30.4 | 73.9 |
| PtdIns | 32.1 | 170.0 | |
| Kdo-lipid IVA | Water | 40.5 | 55.6 |
| PtdIns | 38.2 | 122.3 | |
| Lipid IVA | Water | 144.6 | 30.0 |
| PtdIns | 150.4 | 57.0 |
The ability of the phosphotransferase to tolerate structural modifications of the donor substrate was investigated. Kdo2-[4′-32P]1-dephospholipid IVA (38), galactose-mannose-Kdo2-[4′-32P]lipid IVA (34), mannose-Kdo2-[4′-32P]lipid IVA (34), lauroyl-Kdo2-[4′-32P]lipid IVA (49), and [4′-32P]lipid A (41) were tested in both assays (not shown). These substances were all utilized efficiently. Hexa-acylated [4′-32P]lipid A was utilized at about the same rate as tetra-acylated [4′-32P]lipid IVA. Neither Kdo2-[1-32P]lipid IVA nor [1-32P]lipid IVA (31) supported the generation of PtdIns-4-32P (data not shown). This finding indicates that the 1-phosphatase of R. leguminosarum (31) does not function as a phosphotransferase to PtdIns. Furthermore, [32P]phosphatidic acid was inactive as a donor for the biosynthesis of PtdIns-4-32P under these conditions (data not shown), supporting the proposal that the 4′-phosphatase is highly selective for lipid A-like molecules as donor substrates.
The acceptor substrate specificity of the phosphotransferase was studied with various inositol-containing substances and other membrane lipids (Fig. 8, A and B). The phosphotransferase exhibits a very high degree of selectivity toward PtdIns. PtdIns from bovine liver (containing mainly stearate and arachidonate) and PtdIns from soybean (containing mostly palmitate and linoleate) were equally active as acceptors (Fig. 8A). No other inositol-containing compound served as an acceptor (Fig. 8A), including PtdIns-4-P, which is actually an inhibitor (not shown), PtdIns-3-P, lysophosphatidylinositol, or myo-inositol. Among the other membrane lipids tested, inefficient transfer of the 4′-phosphate of Kdo2-[4′-32P]lipid IVA to phosphatidylglycerol (PtdGro) was detected (Fig. 8B, lane 4). However, the rate of transfer of 32P to PtdGro was 50 times slower than to PtdIns (Fig. 9) over a wide range of acceptor concentrations.
Fig. 8. Acceptor substrate specificity of the phosphotransferase of R. leguminosarum.

Compounds were tested as acceptors for the phosphotransferase under standard assay conditions using partially purified (heparin-agarose) enzyme (10 μg/ml). Incubations were carried out for 40 min at 30 °C. A, thin layer analysis of the reaction products obtained with different inositol-containing compounds as acceptor. The lipid acceptor substrates were used at 1 mg/ml, and inositol was used at 10 mM. Lane 1, no enzyme; lane 2, no acceptor; lane 3, PtdIns (bovine liver); lane 4, PtdIns (soybean); lane 5, PtdIns-4-P; lane 6, PtdIns-3-P; lane 7, lysophosphatidylinositol; lane 8, inositol. B, thin layer analysis of the reaction products obtained with different phospholipids as acceptors, each at 1 mg/ml. Lane 1, no enzyme; lane 2, no acceptor; lane 3, PtdIns (bovine liver); lane 4, PtdGro; lane 5, phosphatidylethanolamine; lane 6, phosphatidylcholine; lane 7, phosphatidylserine; lane 8, cardiolipin; lane 9, phosphatidic acid; lane 10, diacylglycerol.
Fig. 9. Acceptor substrate specificity of the phosphotransferase for PtdIns versus PtdGro.

The effects of PtdIns and PtdGro concentrations on phosphotransferase specific activity were measured in the presence of excess of Kdo2-[4′-32P]lipid IVA (50 μM). Heparin-agarose-purified enzyme (10 μg/ml) was used, and the incubation was carried out for 10 min at 30 °C.
Since glycosylated diacylglycerols are major components of R. leguminosarum membranes, commercially available di-galactosyl diacylglycerol and monoglactosyl diacylglycerol were tested. However, neither compound was active as a phosphate acceptor (not shown).
As shown in Fig. 9, the apparent Km for PtdIns is approximately 500 μM. This is 10-fold higher than what is typically seen with eucaryotic PtdIns kinases (35–37), but the specific activity of the phosphotransferase in membranes is higher than that of many eucaryotic PtdIns kinases assayed under comparable conditions. The relatively high apparent Km for PtdIns therefore does not eliminate the possibility of a physiological role for the phosphotransferase. Indeed, many key enzymes of glycerophospholipid and lipid A biosynthesis display even higher apparent Km values for their physiological substrates (40, 50).
Selective Transfer of the 4′-Phosphate Group of Kdo2-Lipid IVA to the Inositol 4-Position of PtdIns
To determine the site of phosphorylation on PtdIns, the 32P-labeled phosphotransferase product was characterized by chromatography and by chemical degradation. Migration of the 32P-labeled PtdIns-dependent product with authentic PtdIns-4-P was demonstrated using silica gel 60 thin layer chromatography plates developed with the solvents chloroform/pyridine/88% formic acid/water (30:70:16:10, v/v/v/v) (Fig. 8) or chloroform/methanol/acetic acid (65:35:10, v/v/v) (not shown). This procedure does not distinguish PtdIns-4-P from PtdIns-3-P. Accordingly, the 32P-labeled PtdIns-dependent product was deacylated by mild alkaline hydrolysis, followed by anion exchange HPLC of the water-soluble 32P-labeled material(s) on a Partisil SAX column (47), as described in detail in the legend to Fig. 10. No 32P-labeled peak was detected in the control incubated without enzyme (not shown). The deacylated 32P-labeled PtdIns-dependent product eluted with glycerophosphoinositol 4-phosphate (Fig. 10, B and C), which is clearly separated from glycerophosphoinositol 3-phosphate (and other possible isomers) in this system (47). The 32Pi produced by the 4′-phosphatase from Kdo2-[4′-32P]lipid IVA either in the presence (Fig. 10, B and C) or absence of PtdIns (Fig. 10A) elutes 3–4 min before glycerophosphoinositol 3-phosphate. The HPLC analysis of the phosphotransferase reaction product therefore provides unequivocal evidence that the 4′-phosphate of Kdo2-lipid IVA is transferred only to the 4-position of the inositol moiety of PtdIns. This remarkable specificity shows that the enzyme cannot transfer the 4′-phosphate of Kdo2-[4′-32P]lipid IVA to any other hydroxyl group present on the surface of the Triton X-100/PtdIns mixed micelle, since alternative phosphorylated products, like PtdIns-3-32P or 32P-Triton X-100, are not detected.
Evidence for a Phosphoenzyme Intermediate
When either 6 μg of Q-Sepharose or 2 μg of heparin-agarose 4′-phosphatase/ phosphotransferase (Table I) was incubated with Kdo2-[4′-32P]lipid IVA for 5 min, followed by the addition of buffer containing SDS at pH 10, a single 32P-labeled 68-kDa protein was detected by SDS-gel electrophoresis and PhosphorImager analysis (Fig. 11A, lanes 2 and 3). The intensity of the radio-labeled band increased with increasing protein concentrations (not shown). Heat inactivation of the enzyme preparation prior to incubation with the Kdo2-[4′-32P]lipid IVA abolished the incorporation of 32P (Fig. 11A, lane 1). The formation of the phosphoprotein was transient, since maximum labeling was observed after only 2–5 min of incubation with Kdo2-[4′-32P]lipid IVA (not shown). Thereafter, the intensity of the band steadily decreased until it was virtually undetectable after 90 min (not shown).
Fig. 11. Trapping and detection of a 68-kDa phosphoprotein formed by incubating Kdo2-[4′-32P]lipid IVA with partially purified 4′-phosphatase/phosphotransferase.

A, a specific 32P-labeled phosphoprotein, generated during a 5-min incubation of Kdo2-[4′-32P]lipid IVA with enzyme (as described under “Experimental Procedures”) is resolved by SDS-polyacrylamide gel electrophoresis and detected by PhosphorImager analysis of the dried gel. Partially purified enzyme from either the Q-Sepharose step (lane 2, 6 μg of protein) or the heparin-agarose step (lanes 1 and 3; 2 μg of protein) was used. The enzyme for the reaction shown in lane 1 was first heated to 65 °C for 15 min. B, PtdIns reduces the level of the phosphoprotein generated during a 5-min incubation of Kdo2-[4′-32P]lipid IVA with enzyme. Reactions were performed with identical amounts (2 μg) of heparin-agarose enzyme and Kdo2-[4′-32P]lipid IVA in the absence (lane 1) or in the presence (lane 2) of PtdIns (0.5 mg/ml). After 5 min, the reactions were quenched with SDS and subjected to gel electrophoresis.
The addition of PtdIns to the enzyme preparation together with Kdo2-[4′-32P]lipid IVA resulted in decreased labeling of the 68-kDa protein (Fig. 11B, lane 2). Faster turnover of the proposed phosphoenzyme intermediate in the presence of PtdIns (Fig. 1) may account for this observation, since PtdIns is a better acceptor than water. The simultaneous presence of PtdIns-4-P during the reaction of the enzyme with Kdo2-[4′-32P]lipid IVA also resulted in decreased 32P labeling of the protein (not shown). Dilution of the specific radioactivity of the 32P-labeled phosphoenzyme intermediate, caused by the reverse reaction (Fig. 1) with excess unlabeled PtdIns-4-P, would explain this effect.
A pulse-chase experiment (Fig. 12) confirmed that the loss of 32P label from the 68-kDa phosphoprotein was dependent both upon time and the concentration of the unlabeled Kdo2-lipid IVA used to dilute the specific radioactivity. The formation and turnover of the 32P-labeled phosphoprotein are consistent with the proposed mechanism involving a phosphoenzyme intermediate (Fig. 1).
Fig. 12. Turnover of the labeled phosphoprotein following a chase with unlabeled Kdo2-lipid IVA.

The enzyme preparation (heparin-agarose step) was labeled for 5 min with Kdo2-[4′-32P]lipid IVA as described under “Experimental Procedures.” A, B, and C are the images of the SDS-polyacrylamide gels used to resolve the remaining 32P-labeled phosphoprotein present at different times after being chased with 0, 10, or 50 μM nonradioactive (Kdo)2-lipid IVA, respectively. The percentage of the 32P-labeled phosphoprotein remaining at various times after the start of the chase (indicated by the arrow) is shown in D.
The chemical stability of the 32P-labeled protein species visualized in Figs. 11 and 12 was explored to determine the nature of the covalent linkage that might be involved. Treatment with 8 M urea or 200 mM dithiothreitol did not reduce the intensity of the labeled band (data not shown). The 32P-labeled protein was very labile under acidic conditions, but it was stable under mild basic or at neutral conditions in the presence of SDS. The stability of the protein-bound 32P actually increased under mild alkaline conditions. This pattern is consistent with a phosphohistidine residue (51, 52). Isolation and analysis of the active site peptide labeled with 32P will be necessary to confirm this proposal.
DISCUSSION
The Kdo-dependent 4′-phosphatase of R. leguminosarum catalyzes a key reaction in generating a phosphate-deficient variant of lipid A (30). Here we show that the same enzyme preparations can also transfer the 4′-phosphate group from lipid A and certain lipid A precursors to PtdIns. An enzymatic reaction that directly links lipid A biosynthesis with PtdIns-4-P production has not been described previously. This unique R. leguminosarum phosphotransferase is the first example of procaryotic enzyme capable of generating PtdIns-4-P, a molecule that plays a central role in eucaryotic signal transduction (53, 54).
The current studies strongly suggest that one enzyme catalyzes both the 4′-phosphatase and the phosphotransferase reactions. Like the 4′-phosphatase (30), the phosphotransferase is present in wild type strains of R. leguminosarum but not in E. coli, R. leguminosarum mutant 24AR, or R. meliloti. In R. leguminosarum (CE3), both activities are localized in the inner membrane (Fig. 3). Both activities display similar chromatographic properties on Q-Sepharose (Fig. 4), heparin-agarose (Fig. 4) and Cibacron blue-agarose (Fig. 7), and their thermal inactivation profiles are identical (Fig. 6). Furthermore, both activities co-purify (Table I) and show the same donor substrate specificity (Table II).
The ability of one enzyme to catalyze both the 4′-phosphatase and the phosphotransferase reaction could be explained by a mechanism involving a covalent phosphoenzyme intermediate (Fig. 1B). The intermediate would be generated by the initial transfer of the 4′-phosphate moiety of Kdo2-lipid IVA to the enzyme. The intermediate could transfer the phosphate group either to water or to the 4-OH of PtdIns. The direct demonstration (Figs. 11 and 12) of a 32P-labeled phosphoprotein, obtained by incubation of the 80-fold purified enzyme with Kdo2-[4′-32P]lipid IVA and chased by nonradioactive Kdo2-lipid IVA (Fig. 12) or PtdIns (Fig. 11B), strongly supports this scheme.
Our studies show that the phosphotransferase, like the 4′-phosphatase (30), functions more efficiently in the presence of at least one Kdo residue on the donor substrate (Table II). The enzyme is less sensitive to other structural modifications of Kdo2-[4′-32P]lipid IVA, such as removal of the 1-phosphate, the addition of extra sugars, or further acylation. Such a dependence on Kdo may be necessary, given that the Kdo transferase requires the presence of the 4′-phosphate residue for catalysis (38, 42). Premature removal of the 4′-phosphate might inhibit LPS maturation. Although the 4′-phosphatase/phosphotransferase is associated with the inner membrane, its active site might face the periplasm, further limiting access to LPS precursors lacking Kdo to the 4′-phosphatase (3).
A 1-phosphatase, distinct from the 4′-phosphatase and capable of cleaving several precursors of lipid A including Kdo2-lipid IVA, is present in membranes of R. leguminosarum (31). Using Kdo2-[1-32P]lipid IVA (31) as the donor, however, transfer of the 1-phosphate to PtdIns was not observed in R. leguminosarum membranes (data not shown). Thus, phosphotransferase activity appears to be associated only with the 4′-phosphatase of R. leguminosarum. In the presence of [γ-32P]ATP and Mg2+, no transfer of 32P to PtdIns was observed when either R. leguminosarum CE3 membranes or partially purified heparin-agarose fractions were employed (data not shown). Hence, the R. leguminosarum phosphotransferase reaction is not attributable to a PtdIns kinase (35–37).
Although the phosphotransferase exhibits a high degree of selectivity toward PtdIns, the latter could not be demonstrated among the membrane lipids of R. leguminosarum (CE3) by labeling cells with either 32Pi or [1,2-3H]inositol. A search for an alternative endogenous lipid acceptor among the total membrane lipids of R. leguminosarum proved unsuccessful. Gerson and Patel (55) reported PtdIns in Rhizobium loti, but other studies (56) of different strains of R. leguminosarum failed to demonstrate PtdIns. One intriguing explanation for this anomaly might be that the synthesis of PtdIns is induced in the bacteria by root cell exudates or during symbiosis. A recent report (57) showed the presence of PtdIns in membranes of free-living Bradyrhizobium japonicum grown at low oxygen concentrations, one of the environmental factors known to induce conversion of bacteria to bacteroids in root nodules (58).
An alternative possibility is that plant membranes supply PtdIns or a related compound to R. leguminosarum during nodulation. PtdIns is available in the peribacteroid membranes of soybean root nodules (58), and soybean PtdIns is an excellent acceptor substrate for the phosphotransferase (Fig. 8). In addition, Perotto et. al. (59) have demonstrated presence of other inositol-containing phosphoglycolipids in pea nodules. The close association of the bacteroid outer membrane with the plant-derived peribacteroid membrane is well documented by immunostaining and electron microscopy (60–62). Inside the symbiosome, the outer membrane of the bacteroid may actually be fragmented (61), potentially giving PtdIns access to the 4′-phosphotransferase. PtdIns-specific lipid exchange proteins are known to exist in plants and could serve as PtdIns carriers (63).
In heptose-deficient mutants of Salmonella typhimurium, small quantities of exogenous glycerophospholipids can fuse with the outer membrane and move to the inner membrane (64, 65). In the case of exogenous phosphatidylserine, the incorporated lipid is metabolized to form phosphatidylethanolamine within the inner membrane (64, 65). While retrograde transport of exogenous lipids is not sufficiently rapid in Gram-negative bacteria to enable the isolation of phospholipid auxotrophs, it is nevertheless established that such transport occurs with a variety of lipids (64, 65). In the case of minor lipids like PtdIns-4-P, which function at low concentrations in specific signal transduction pathways, the possibility that the plant provides the 4′-phosphatase/phosphotransferase with PtdIns deserves serious consideration. The PtdIns-4-P made by R. leguminosarum from exogenous PtdIns might even be secreted again by the bacteroids and made available to the plant.
Majerus and co-workers have recently reported a remarkable example of a specific interaction between an enzyme made by a Gram-negative bacterium and a eucaryotic inositol phosphate signaling system (66). Salmonella dublin secretes a virulence protein (SopB) that hydrolyzes inositol 1,3,4,5,6-pentakis-phosphate to form inositol 1,4,5,6-tetrakis-phosphate (66). The latter compound increases chloride secretion in animal cells, and the catalytic activity of SopB is necessary for the induction of fluid secretion observed in calf intestinal loops infected with S. dublin (66). SopB can also hydrolyze phosphatidylinositol 3,4,5-trisphosphate, a lipid that directly antagonizes Ca2+-dependent chloride secretion (66). Like R. leguminosarum, however, enteric Gram-negative bacteria are not thought to make their own inositol phospholipids (67). If not actually involved in making PtdIns-4-P during symbiosis, the 4′-phosphatase/phosphotransferase of R. leguminosarum might conceivably function to remove PtdIns-4-P by catalyzing the reverse of the proposed reactions (Fig. 1), a possibility that we have not yet examined.
Although the roles played by phosphorylated derivatives of PtdIns in plants have not been fully established, evidence is accumulating that signal transduction mediated by the turnover of inositol phospholipids may indeed occur (68). For instance, Ehrhardt et al. (69) observed calcium spiking, as is typically associated with inositol trisphosphate signaling in animal cell systems, when alfalfa root hairs are exposed to the R. meliloti Nod factor. In animal cells, phosphatidylinositol kinases play additional important roles in membrane biogenesis, secretion, vesicle trafficking, and regulation of the actin cytoskeleton (53, 54, 70, 71). All of these processes accompany bacterial invasion of plant root hair cells and nodule formation (58). Since the inositol phospholipid cycle begins with the phosphorylation of PtdIns to generate PtdIns-4-P, the R. leguminosarum phosphotransferase could play a key role in any of the above processes during symbiosis. Cloning of the gene encoding the 4′-phosphatase/phosphotransferase and isolation of mutants specifically defective in the enzyme should reveal the significance of the phosphate-deficient lipid A that is found in R. leguminosarum and the biological role of the phosphotransferase activity associated with the 4′-phosphatase.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant GM-51796 (to C. R. H. R.).
The abbreviations used are: LPS, lipopolysaccharide; Kdo, 3-deoxy-D-manno-octulosonic acid; Ins, inositol; PtdIns, phosphatidylinositol; PtdGro, phosphatidylglycerol; PtdIns-4-P, phosphatidylinositol 4-phosphate; PtdIns-3-P phosphatidylinositol 3-phosphate; MES, 4-morpholinoethanesulfonic acid; CAPS, 3-(cyclohexylamino)-1-propanesulfonic acid; DS-1-P, tetraacyldisaccharide 1-phosphate; HPLC, high pressure liquid chromatography.
References
- 1.Raetz CRH. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 2.Raetz CRH. J Bacteriol. 1993;175:5745–5753. doi: 10.1128/jb.175.18.5745-5753.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raetz CRH. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. 2. Neidhardt FC, editor. Vol. 1. American Society for Microbiology; Washington, D. C: 1996. pp. 1035–1063. [Google Scholar]
- 4.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zähringer U, Seydel U, Di Padova F, Schreier M, Brade H. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 5.Schnaitman CA, Klena JD. Microbiol Rev. 1993;57:655–682. doi: 10.1128/mr.57.3.655-682.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nikaido H. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. 2. Neidhardt FC, editor. Vol. 1. American Society for Microbiology; Washington, D. C: 1996. pp. 29–47. [Google Scholar]
- 7.Vaara M. Antimicrob Agents Chemother. 1993;37:2255–2260. doi: 10.1128/aac.37.11.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roantree RJ. Annu Rev Microbiol. 1967;21:443–466. doi: 10.1146/annurev.mi.21.100167.002303. [DOI] [PubMed] [Google Scholar]
- 9.Galloway SM, Raetz CRH. J Biol Chem. 1990;265:6394–6402. [PubMed] [Google Scholar]
- 10.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, Raetz CRH. Science. 1996;274:980–982. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 11.Wyckoff TJO, Raetz CRH, Jackman JE. Trends Microbiol. 1998;6:154–159. doi: 10.1016/s0966-842x(98)01230-x. [DOI] [PubMed] [Google Scholar]
- 12.Ulevitch RJ, Tobias PS. Annu Rev Immunol. 1995;13:437–457. doi: 10.1146/annurev.iy.13.040195.002253. [DOI] [PubMed] [Google Scholar]
- 13.Kannenberg EL, Brewin NJ. Trends Microbiol. 1994;2:277–283. doi: 10.1016/0966-842x(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 14.Carlson RW, Bhat UR, Reuhs B. In: Plant Bio/Technology and Development. Gresshoff PM, editor. CRC Press, Inc; Boca Raton, FL: 1992. pp. 33–44. [Google Scholar]
- 15.Kannenberg EL, Rathbun EA, Brewin NJ. Mol Microbiol. 1992;6:2477–2487. doi: 10.1111/j.1365-2958.1992.tb01424.x. [DOI] [PubMed] [Google Scholar]
- 16.Noel KD, Vandenbosch KA, Kulpaca B. J Bacteriol. 1986;168:1392–1401. doi: 10.1128/jb.168.3.1392-1401.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Priefer UB. J Bacteriol. 1989;171:6161–6168. doi: 10.1128/jb.171.11.6161-6168.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Maagd RA, Rao AS, Mulders IH, Goosen-de Roo L, van Loosdrecht MC, Wijffelman CA, Lugtenberg BJ. J Bacteriol. 1989;171:1143–1150. doi: 10.1128/jb.171.2.1143-1150.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perotto S, Brewin NJ, Kannenberg EL. Mol Plant Microbe Interact. 1994;7:99–112. [Google Scholar]
- 20.Tao H, Brewin NJ, Noel KD. J Bacteriol. 1992;174:2222–2229. doi: 10.1128/jb.174.7.2222-2229.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sindhu SS, Brewin NJ, Kannenberg EL. J Bacteriol. 1990;172:1804–1813. doi: 10.1128/jb.172.4.1804-1813.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kannenberg EL, Perotto S, Bianciotto V, Rathbun EA, Brewin NJ. J Bacteriol. 1994;176:2021–2032. doi: 10.1128/jb.176.7.2021-2032.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhat UR, Forsberg LS, Carlson RW. J Biol Chem. 1994;269:14402–14410. [PubMed] [Google Scholar]
- 24.Forsberg LS, Carlson RW. J Biol Chem. 1998;273:2747–2757. doi: 10.1074/jbc.273.5.2747. [DOI] [PubMed] [Google Scholar]
- 25.Que NLS, Basu SS, White KA, Raetz CRH. FASEB J. 1998;12:1284. abstr. [Google Scholar]
- 26.Lin S, Woods AS, Cotter RJ, Raetz CRH, Que NLS. FASEB J. 1998;12:A14LB56. abstr. [Google Scholar]
- 27.Brozek KA, Raetz CRH. J Biol Chem. 1990;265:15410–15417. [PubMed] [Google Scholar]
- 28.Bhat UR, Mayer H, Yokota A, Hollingsworth RI, Carlson R. J Bacteriol. 1991;173:2155–2159. doi: 10.1128/jb.173.7.2155-2159.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price NJP, Kelly TM, Raetz CRH, Carlson RW. J Bacteriol. 1994;176:4646–4655. doi: 10.1128/jb.176.15.4646-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price NJP, Jeyaretnam B, Carlson RW, Kadrmas JL, Raetz CRH, Brozek KA. Proc Natl Acad Sci U S A. 1995;92:7352–7356. doi: 10.1073/pnas.92.16.7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brozek KA, Kadrmas JL, Raetz CRH. J Biol Chem. 1996;271:32112–32118. [PubMed] [Google Scholar]
- 32.Brozek KA, Carlson RW, Raetz CRH. J Biol Chem. 1996;271:32126–32136. doi: 10.1074/jbc.271.50.32126. [DOI] [PubMed] [Google Scholar]
- 33.Kadrmas JL, Brozek KA, Raetz CRH. J Biol Chem. 1996;271:32119–32125. [PubMed] [Google Scholar]
- 34.Kadrmas JL, Allaway D, Studholme RE, Sullivan JT, Ronson CW, Poole PS, Raetz CRH. J Biol Chem. 1998;273:26432–26440. doi: 10.1074/jbc.273.41.26432. [DOI] [PubMed] [Google Scholar]
- 35.Porter FD, Li YS, Deuel TF. J Biol Chem. 1988;263:8989–8995. [PubMed] [Google Scholar]
- 36.Jenkins GH, Subrahmanyam G, Anderson RA. Biochim Biophys Acta. 1991;1080:11–18. doi: 10.1016/0167-4838(91)90105-9. [DOI] [PubMed] [Google Scholar]
- 37.Flanagan CA, Thorner J. J Biol Chem. 1992;267:24117–24125. [PubMed] [Google Scholar]
- 38.Belunis CJ, Raetz CRH. J Biol Chem. 1992;267:9988–9997. [PubMed] [Google Scholar]
- 39.Miller JR. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 40.Radika K, Raetz CRH. J Biol Chem. 1988;263:14859–14867. [PubMed] [Google Scholar]
- 41.Garrett TA, Kadrmas JL, Raetz CRH. J Biol Chem. 1997;272:21855–21864. doi: 10.1074/jbc.272.35.21855. [DOI] [PubMed] [Google Scholar]
- 42.Brozek KA, Hosaka K, Robertson AD, Raetz CRH. J Biol Chem. 1989;264:6956–6966. [PubMed] [Google Scholar]
- 43.Goldman RC, Kohlbrenner WE. J Bacteriol. 1985;163:256–261. doi: 10.1128/jb.163.1.256-261.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bligh EG, Dyer JJ. Can J Biochem Physiol. 1959;37:911–918. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 45.White KA, Kaltashov IA, Cotter RJ, Raetz CRH. J Biol Chem. 1997;272:16555–16563. doi: 10.1074/jbc.272.26.16555. [DOI] [PubMed] [Google Scholar]
- 46.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 47.Stolz LE, Kuo WJ, Longchamps J, Sekhon MK, York JD. J Biol Chem. 1998;273:11852–11861. doi: 10.1074/jbc.273.19.11852. [DOI] [PubMed] [Google Scholar]
- 48.Russa R, Lüderitz O, Rietschel ET. Arch Microbiol. 1985;141:284–289. [Google Scholar]
- 49.Clementz T, Bednarski JJ, Raetz CRH. J Biol Chem. 1996;271:12095–12102. doi: 10.1074/jbc.271.20.12095. [DOI] [PubMed] [Google Scholar]
- 50.Larson TJ, Dowhan W. Biochemistry. 1976;15:5212–5218. doi: 10.1021/bi00669a003. [DOI] [PubMed] [Google Scholar]
- 51.Hultquist DE, Moyer RW, Boyer PD. Biochemistry. 1966;5:322–333. doi: 10.1021/bi00865a041. [DOI] [PubMed] [Google Scholar]
- 52.Stukey J, Carman GM. Protein Sci. 1997;6:469–472. doi: 10.1002/pro.5560060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liscovitch M, Cantley LC. Cell. 1995;81:659–662. doi: 10.1016/0092-8674(95)90525-1. [DOI] [PubMed] [Google Scholar]
- 54.Zhang X, Majerus PW. Semin Cell Dev Biol. 1998;9:153–160. doi: 10.1006/scdb.1997.0220. [DOI] [PubMed] [Google Scholar]
- 55.Gerson T, Patel JJ. Appl Microbiol. 1975;30:193–198. doi: 10.1128/am.30.2.193-198.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orgambide GG, Huang ZH, Gage DA, Dazzo FB. Lipids. 1993;28:975–979. doi: 10.1007/BF02537117. [DOI] [PubMed] [Google Scholar]
- 57.Tang Y, Hollingsworth RI. Appl Environ Microbiol. 1998;64:1963–1966. doi: 10.1128/aem.64.5.1963-1966.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whitehead LF, Day DA. Physiol plant. 1997;100:30–44. [Google Scholar]
- 59.Perotto S, Donavan N, Drobak BK, Berwin NJ. Mol Plant-Microbe Interact. 1995;8:560–568. [Google Scholar]
- 60.Bradley DJ, Butcher GW, Galfre G, Wood EA, Brewin NJ. J Cell Sci. 1986;85:47–61. doi: 10.1242/jcs.85.1.47. [DOI] [PubMed] [Google Scholar]
- 61.Bal AK, Shantharam S, Verma DP. Can J Microbiol. 1980;26:1096–1103. doi: 10.1139/m80-182. [DOI] [PubMed] [Google Scholar]
- 62.Brewin NJ, Bolanos L, Dahiya P, Gardner CD, Hernandez LE, Kardailsky IV, Rathbun EA, Sherrier DJ. In: Nitrogen Fixation: Fundamentals and Applications. Tikhonovich IA, Provorov NA, Romanov VI, Newton WE, editors. Kluwer Academic Publisher; Dordrecht, The Netherlands: 1995. pp. 455–460. [Google Scholar]
- 63.Kearns MA, Monks DE, Fang M, Rivas MP, Courtney PD, Chen J, Prestwich GD, Theibert AB, Dewey RE, Bankaitis VA. EMBO J. 1998;17:4004–4017. doi: 10.1093/emboj/17.14.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jones NC, Osborn MJ. J Biol Chem. 1977;252:7398–7404. [PubMed] [Google Scholar]
- 65.Jones NC, Osborn MJ. J Biol Chem. 1977;252:7405–7412. [PubMed] [Google Scholar]
- 66.Norris FA, Wilson MP, Wallis TS, Galyov EE, Majerus PW. Proc Natl Acad Sci U S A. 1998;95:14075–14059. doi: 10.1073/pnas.95.24.14057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xia W, Dowhan W. J Bacteriol. 1995;177:2926–2928. doi: 10.1128/jb.177.10.2926-2928.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drobak BK. Biochem J. 1992;288:697–712. doi: 10.1042/bj2880697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ehrhardt DW, Wais R, Long SR. Cell. 1996;85:673–681. doi: 10.1016/s0092-8674(00)81234-9. [DOI] [PubMed] [Google Scholar]
- 70.Pike LJ. Endocr Rev. 1992;13:692–706. doi: 10.1210/edrv-13-4-692. [DOI] [PubMed] [Google Scholar]
- 71.Hong Z, Verma DP. Proc Natl Acad Sci U S A. 1994;91:9617–9621. doi: 10.1073/pnas.91.20.9617. [DOI] [PMC free article] [PubMed] [Google Scholar]