

Abstract
Nonviral, DNA-mediated gene transfer is an alternative to viral delivery systems for expressing new genes in cells and tissues. The Sleeping Beauty (SB) transposon system combines the advantages of viruses and naked DNA molecules for gene therapy purposes; however, efficacious delivery of DNA molecules to animal tissues can still be problematic. Here we describe the hydrodynamic delivery procedure for the SB transposon system that allows efficient delivery to the liver in the mouse. The procedure involves rapid, high-pressure injection of a DNA solution into the tail vein. The overall procedure takes <1 h although the delivery into one mouse requires only a few seconds. Successful injections result in expression of the transgene in 5−40% of hepatocytes 1 d after injection. Several weeks after injection, transgene expression stabilizes at ∼1% of the level at 24 h, presumably owing to integration of the transposons into chromosomes.
INTRODUCTION
Gene delivery in mice
The mouse is the most used model system for developing tools and techniques for human gene therapy. Several types of genetic disease are caused by a lack of enzymes that either normally exist in the blood or can be supplied to various tissues through the circulatory system. For expressing genes whose protein products can be secreted into the circulatory system, the liver is a particularly favored organ because of its large size and its ability to secrete many polypeptides.
There are two approaches to introduce genes into organs and tissues in mice. The first method employs viral vectors that either may have preferences for specific organs or tissues, for example, hepatitis viruses that infect hepatocytes in the liver, or may have greater ranges of infectivity, for example, adenoviruses and adeno-associated viruses (AAVs) that have multiple serotypes and lesser preferences for various cell types. The second method of gene delivery is using plasmids to carry DNA into cells. Viral vector preparations from cultured mammalian cells come with the risk of contamination through a variety of different infectious agents, including replication-competent virus generated by recombination between virus vector and packaging constructs1. Moreover, the viral vector can be toxic; for example, adenoviruses can stimulate acute immune/inflammatory response following transduction in the liver2. The risks of DNA-mediated delivery, by comparison, are limited to those associated with plasmid preparation from bacterial extracts (endotoxin, etc.) and whatever chemical component is conjugated with the DNA for the purpose of delivery. A further complication in the use of retroviruses, lentiviruses and AAV may come from their preference for integrating into promoters and transcriptional units, where they may have increased chances of causing adverse effects3–11. In contrast, DNA-mediated delivery systems are likely to be more stable than viral vector preparations, more amenable to pharmaceutical formulation and do not target genes for integration. However, introducing plasmids that carry transposons into tissues of live animals is difficult.
This protocol is based on previously validated methods12–18 and describes hydrodynamic delivery of Sleeping Beauty (SB) transpo-sons to mice and means for determining the extent of gene delivery. In mice, rapid, high-volume infusion of naked plasmid DNA, called hydrodynamic delivery by the practitioners, is the most effective method for in vivo gene transfer. The method is well-established in mice, in which up to 40% of the hepatocytes in test animals take up the transgenic DNA. The liver accounts for >95% of the expressed transgenic DNA following hydrodynamic delivery. The mechanism of DNA uptake is poorly understood but appears to involve expanding liver endothelium, which in mice heals within 24−48 h19–25. The method has been adapted to larger animals15, including porcine26,27 and rabbit liver28 as well as muscles of larger animals29. The DNA should be ‘naked’, that is, not complexed with condensing polymers or other agents30.
SB as a vector for gene delivery
The SB transposon system is a nonviral vector system designed to deliver specific genetic cargos to vertebrate chromosomes31,32. The SB system consists of two components: a transposon (T), which carries a DNA sequence that is often comprised of a gene of interest (GOI) behind a promoter (the combination is referred to as a gene cassette), and an SB transposase that can transpose the transposon from a carrier plasmid (or other donor DNA) to a target DNA such as a mouse chromosome (Fig. 1). The SB system has been used for two primary purposes—gene delivery and gene discovery32–35; here we discuss only its role in gene delivery. Transposons containing a transcriptional regulatory cassette and a transgene of interest within a plasmid carrier are delivered to target cells, after which SB transposase cleaves the transposon from the donor plasmid and reinserts the transposon at a site in a chromosome for long-term (often the lifetime of the cell) expression of the transgene. In mice injected with SB transposons and transposase encoded on a plasmid, transposition occurs over a period of ∼4 d16,36.
Figure 1.

Schematic of Sleeping Beauty (SB) transposition. DNA transposition consists of a cut-and-paste reaction in which a transposon containing a gene of interest (GOI, shown in blue, with its promoter shown in magenta) is cut out of a plasmid and inserted into another DNA molecule, in this case a mouse chromosome. The cleavage reaction occurs at the ends of the inverted terminal repeats (inverted set of double arrowheads in the diagram) of the SB transposon31. SB transposons integrate only into TA-dinucleotide basepairs (∼200 million in mammalian genomes). The inverted terminal repeats are the only DNA sequences required by the transposase enzyme for transposition. The transposase gene can be on the same plasmid (cis configuration as shown) or delivered on a separate plasmid (trans configuration, see Fig. 3) to permit greater flexibility in experimental design. (a) The plasmid carrying the SB transposase gene and transposon enters a cell (large back oval) as a result of hydrodynamic delivery and proceeds through the nuclear membrane (dotted hexagon) by a poorly understood process. (b) The SB transposase gene (SB, green) is transcribed from a promoter (magenta arrowhead) and the mRNA translated in the cytoplasm to give an appropriate level of enzyme (green circles). (c) The SB transposase molecules enter the nucleus and bind to the transposon, two at each end, to bring the transposon ends together (shown in detail in Fig. 3b). (d) Four transposase enzymes work in concert to cleave the plasmid at the termini of the transposon and paste it (dotted lines) into chromosomal DNA (green tangled lines). (e) A plasmid excision product is left behind in this reaction (the site whence the transposon left is marked by an X). (f) Integration into a chromosome can confer long-term expression of the GOI that is contained within the transposon.
The SB system is particularly well suited for gene delivery because it does not preferentially insert transposons into active genes37–39. There are ∼200 million potential sites in the mouse genome into which an SB transposon can insert, which means that tracking transposition activity by cataloging integration sites is essentially impossible. However, transposition can be quantified using an excision assay (Fig. 2) in which, regardless of where it integrates, the transposon leaves a plasmid with a definitive ‘footprint’ that can be quantified by PCR36. The excision assay is relatively simple and can be used to determine the activity of the transposon system quantitatively whenever there is a question about its activity, for example, either under novel conditions or with constructs that have not been validated in tissue-cultured cells.
Figure 2.

Excision assay. (a) Theory of the excision assay. The first step of transposition is precise cleavage on both sides of the transposon (excision step, step d in Fig. 1). The gap in the donor-plasmid left by the transposon is repaired (step e in Fig. 1) to reform a circular plasmid lacking the transposon. The re-ligation step often leaves a footprint of an extra 5 bp (either TCTGA or TCAGA) at the site of the original transposon36. As a result of ligation of the plasmid, primers (red arrows in the bottom right corner of panel a) complementary to plasmid sequences located on either side of the site whence the transposon originated can bind to the repaired plasmid. PCR-amplification from the primers leads to a DNA sequence of predictable length and sequence (except for the footprint), which we refer to as the excision product (EP) (red line at the bottom right of panel a). The power of the assay is that regardless of where the transposon integrates, the EP from each event is the same thereby allowing quantification of the totality of transposition events. Thus, whereas identification of integration at any of 2 × 108 TA sites can only be determined through analysis of genomic DNA, the intensity of the PCR product from the repaired plasmid can be used to access the overall extent of transposition. The red line represents the EP from PCR and the red arrow indicates its location on the gel in panel c. Primer sites are chosen to be 100−200 bp outside of the inverted repeats of the transposon to ensure that the PCR product is efficiently synthesized, that the EP is distinguishable from the primers (and ‘primer dimers’ sometimes seen in gels of PCRs), and that the EP is distinguishable from the PCR amplification across the transposon from unexcised plasmids. (b) The level of transposition can be estimated through conventional PCR and analysis by agarose-gel electrophoresis. The gel shows a single band amplified from a genomic sequence (e.g., β-glucuronidase) in a liver sample from a sham-treated control mouse that received an injection of lactated Ringer's solution without DNA and an additional band representing the amplified EP (red EP) from a mouse injected with a transposon-containing plasmid. (c) The amount of EP can be quantitatively determined by real-time PCR using any of several instruments (the data here were generated using an iCycler (Bio-Rad Laboratories) with their IQ SYBR green supermix according to their guidelines). The data show the copy numbers of excision plasmid product (EP) as a function of the PCR threshold cycle (Ct) number when SYBR Green fluorescence is first detectable. The standard curve indicates that the signal is proportional to the input level of repaired plasmids after excision (open boxes). Triplicate samples were assayed and overlapped for all samples except that from the sham-treated mouse (two triangles; the bottom triangle represents two results) and the untreated mouse (black circles with two of the samples overlapping at the bottom). Greater variation between samples is sometimes seen with the least concentrated samples because the slightest contamination can lead to a measurable signal. The gel at the top shows the final products from real-time PCRs that were resolved by electrophoresis using 1% agarose gel. Panel a is adapted from Figure 2 of Liu et al. (2004)36 and panels b and c are from Figure 6 of Aronovich et al. (2007)16. GOI, gene of interest; SB, Sleeping Beauty.
The power of the SB system to confer long-term expression of transgenes has been demonstrated in mice for several models of human disease. SB-mediated long-term expression of clotting factors such as Factor VIII and Factor IX in the liver has cured hemophilias A and B, respectively40–42. The procedure has also been used by us and others to treat mice with genetic deficiencies for fumarylacetoacetate hydrolase deficiency43–45 as well as by us for β-glucuronidase and iduronidase deficiencies associated with mucopolysaccharidosis Type VII and Type I, respectively16. Hydrodynamic delivery to mouse liver has also been used with other nonviral vectors including the ϕC31 integrase system46, ϕBT147,48 and a variety of plasmids encoding transgenic cassettes14,29 The SB system has also been used to deliver genes to other organs of the mouse including skin to treat junctional epidermolysis bullosa49, the brain to retard glioma xenographs50,51 and to lung allografts to prevent fibrosis52. A bibliography of papers on the SB transposon system is available at http://beckmancenter.ahc.umn.edu/html/data.html.
Designing an SB vector for gene delivery
A list of some of the available SB transposon constructs, and their full sequences, can be found at http://www.cbs.umn.edu/labs/perry/plasmids/plasmid.html. There are four aspects that should be considered in designing SB transposon vectors for gene delivery.
The expression cassette
This consists of transcriptional regulators (hereafter for convenience referred to as the promoter) and a gene or sequence that is to be expressed. The desired biological effect determines the composition of the expression cassette. The choice of promoter is dependent on several variables, including promoter strength, duration of expression, stringency of expression in certain cell types, and overall length. For example, the CAGGS promoter53, which is a fusion of transcriptional elements from the β-actin gene and the cytomegalovirus (CMV) early promoter is considered to be an exceptionally strong, fairly ubiquitous driver of transcription in many cell types54,55. In contrast to the early CMV promoter, which is often shut off in liver cells after a few days or weeks56,57, the CAGGS promoter is active over months if not the lifetime of the cell in which it resides58,59. However, owing to the promiscuous expression of the CMV and CAGGS promoters, they also will drive expression in other cell types, including stem cells60 and antigen-presenting cells that could exacerbate immunological responses to the transgene product that will abolish its overall presence in the animal16. Consequently, a combination of tissue-specific promoters, insulator or boundary elements61, and mir motifs for regulation of translation by miRNAs62 or addition of siRNA genes63 have been used to limit expression to liver cells.
Transposase source and plasmid configuration
As noted earlier, the SB system is comprised of the transposon and the SB transposase enzyme. In most cases, the transposase source is an expression cassette on a plasmid and the transposon is carried on another plasmid (Fig. 3a, trans). However, the SB expression plasmid can be included on the transposon plasmid (Fig. 3a, cis). The advantage of the cis configuration is that only a single plasmid preparation, with appropriate quality control for elimination of bacterial endotoxins, is required and every cell is guaranteed a constant ratio of the SB transposase gene and the transposon with its gene/sequence of interest. The advantage of the trans configuration is that there is more flexibility for varying the ratio of the transposase gene and the transposon. For most initial studies, the trans-delivery is the preferred choice.
Figure 3.

Design of Sleeping Beauty (SB) transposons. (a) The cis and trans configurations for delivery of the SB system are shown with transcriptional regulatory sequences represented by the magenta arrowheads as described in Figure 1. In the trans-delivery mode, the transposase gene can be omitted as a negative control. An alternative trans-delivery is possible wherein the SB mRNA is delivered rather than its gene. (b) The phenomenon of overproduction inhibition is illustrated. The rate of transposition depends on the binding of four transposase enzymes to the transposon [SB]optimal. If there are fewer than four transposase molecules [SB]low, transposition will not occur and if there are more, then the additional transposase molecules will quench the reaction by competing with bound enzymes to prevent the bringing of the transposon ends together [SB]high. (c) The effects of cargo (expression cassette) size on gene delivery as determined by long-term gene expression in dividing cells. The cause for the apparent decrease in transposition as a function of size is poorly understood because the length of the plasmid carrier sets the linear distance between the termini of the transposon once the expression cassette is longer than ∼2 kb. Factors that may influence the efficiency of gene delivery include propagation through the plasma and nuclear membranes, where effects of DNA length have not been carefully evaluated. The diagrams in panels b and c are adapted, with permission, from Figure 3 of Geurts et al. (2003)65 and Figure 2 of Hackett (2007)67,respectively.
An alternative to trans-delivery of the SB system with an SB transposase-expressing plasmid is injection of SB transposase mRNA with the transposon plasmid. This method has not been widely tested, but it works, albeit at a lower rate of transposition than is generally achieved through trans- or cis-delivery of the SB gene17,45. This alternative has the advantage of limiting transposition to a 1- or 2-d period before the transposase mRNA and protein decay, but the inherent disadvantage is the exceptional instability of mRNA both in most solutions and in the vascular system.
Molecular stoichiometry of SB transposase to the transposon
Optimal transposition rates depend on having a ratio of four transposase enzymes per transposon—less than four transposase molecules will not support transposition, and excessive levels of transposase will quench transposition by a process called over-production inhibition (Fig. 3b)64–67. The relative levels of transposase and transposons can be manipulated by the ratio of transposase genes to transposon and the relative strength of the promoter driving the SB transposase gene. Thus, delivery of the SB system in the trans configuration allows the transposase to transposon ratios to be finely adjusted by modifying the ratio of the plasmids; in the cis configuration, a promoter of appropriate strength in the target cell must be experimentally determined and then incorporated into the final cis-plasmid. The study by Mikkelsen et al. is the most complete report of the effects of a variety of promoters used for the SB gene in a cis configuration66.
Transposon size
This aspect is poorly understood because most end-points for determining effectiveness of delivery involve a screen for gene expression or simple insertion of the gene into a chromo-some. However, both assays depend not only on transposition rate, but also on the efficiency of uptake of the plasmids into the nuclei of cells and stability of expression of the transgene in the transposon. The efficiency of gene expression, which is taken as a measure of efficiency of gene delivery and transposition, decreases with size of the transposon-plasmid. As shown in Figure 3c, an SB transposon of ∼6 kb, which would accommodate an expression cassette of ∼5.5 kb, has ∼50% the efficiency of gene transfer as an optimally sized transposon of 2.0−2.5 kb. A size limitation has not been observed for the Tol2 transposon system in the single report of effects of size44. Accordingly, if size of the transposon or its plasmid is an issue, then trans-delivery is the preferred method.
Since its first appearance in 1997 (ref. 31), the SB system has been further developed to give higher transpositional activities. Improvements in the transposon inverted terminal repeats have been made36,52,68,69 and the original SB10 transposase has also been improved65,70,71. More recently, efforts to construct SB transposases that can target specific integration sites have been deployed72–74.
Hydrodynamic injection
The original conditions for hydrodynamic delivery were worked out in the labs of Jon Wolff and Dexi Liu12,13. In the mouse, hydrodynamic delivery of DNA requires injection of a large volume (10%, vol/wt) of the mouse body weight of DNA, in a saline solution that is isotonic with blood and suitable for intravenous administration, through the tail vein in <10 s (Fig. 4). Accordingly, depending on the experience of the person doing the injections, DNA can be delivered to the livers of one dozen mice or more in an hour if the materials are organized and the mice are healthy.
Figure 4.

Hydrodynamic injection procedure of a transposon containing a gene of interest and a source of Sleeping Beauty (SB) transposase encoded by an SB gene. The components of the SB system are the same as in Figures 1 and 3. Trans- and cis-deliveries are performed in the same way. (a) The desired amount of DNA (generally between 0.1 and 50 μg, although higher and lower amounts can be injected) is diluted into a volume of lactated Ringer's solution that is equivalent to 10% the mouse weight. (b) The DNA solution is injected into the tail vein of the mouse; injections that take 4−7 s are optimal. (c) Results from hydrodynamic delivery can be obtained as soon as 30 min after injection, depending on the gene of interest (GOI) and the assay.
Control plasmids
Depending on the experiment, certain control injections may be necessary. These may include a ‘no transposase’ control, in which case the trans-configuration of delivery the plasmid with the SB transposase gene is replaced with a plasmid that lacks the SB gene expression cassette or has the inactive mutant ΔDDE gene31. In the cis-configuration of delivery, the SB gene is omitted from the vector. A control for delivery of any expression cassette that may be difficult to evaluate could include adding a small (e.g., 0.1−1 μg) amount of a CMV-luciferase or CAGGS-luciferase plasmid for bioluminescent imaging; this slight amount of luciferase plasmid should be sufficient to produce 109−1010 photons s−1 (Fig. 5).
Figure 5.

Hydrodynamic delivery to liver. (a) The graph shows the reproducibility of delivery of a luciferase expression cassette. Seventy-five percent of the C57BL/6 mice express a luciferase transgene within a tenfold range (2 × 109 to 2 × 1010 relative light units). (b) The image shows the cellular distribution of expression of human β-glucuronidase (dark purple cells) in liver sections from a C57BL/6 mouse 24 h after hydrodynamic injection of transposons carrying the gene controlled by a hybrid cytomegalovirus (CMV)/β-actin promoter (yellow arrows show some of the cells expressing β-glucuronidase). (c) The image shows the cellular distribution of expression of human β-glucuronidase (dark purple cells) in liver sections from a β-glucuronidase-deficient C57BL/6 mouse 24 h after injection following hydrodynamic injection of the same transposon vector as in panel b. All of the cells are stained because those that do not express the enzyme take up enzyme produced by those cells that are expressing the transgene, a process called ‘cross-correction’. Panel c is adapted from Figure 1 of Aronovich et al. (2007)16.
In this protocol, we present a step-by-step procedure for hydrodynamic delivery of SB transposon-containing plasmids to mouse liver cells. We include methods for assaying the effectiveness of delivery and the effectiveness of transposition of expression cassettes, or other genetic cargo, from the carrier plasmid to a recipient DNA molecule, which is generally a chromosome.
MATERIALS
REAGENTS
Mice We routinely use 8−10 week-old C57BL/6 mice, available from the National Cancer Institute (NCI, Frederick, MD). We have successfully used other strains from other sources (Balb/c, FVB/N, NOD.129(B6)-Prkdcscid IDUAtm1 Clk/J, and various congenic substrains) ! CAUTION Mice should be handled as per local Institutional Animal Care and Use Committee and Institutional Biosafety Committee approved protocols.
Anesthetic cocktail: ketamine HCL (Phoenix Pharmaceutical Incorporated); acepromazine maleate (Phoenix Pharmaceutical Incorporated); and butorphanol tartrate (Fort Dodge Animal Health) (see REAGENT SETUP)
Lactated Ringer's (LR) solution (Henry Schein Company, cat. no. 1533592)
D-Luciferin firefly luciferase substrate, potassium salt (for in vivo imaging; Xenogen Corporation, cat. no. XR-1001). ▲ CRITICAL Store at −20 °C.
0.5% (vol/vol) Bleach solution [sodium hypochlorite (NaOCl)]
Vector DNA containing gene for delivery (see REAGENT SETUP)—see http://www.cbs.umn.edu/labs/perry/plasmids/plasmid.html for details of vectors available from authors on request
Primers (see Table 1)
Invitrogen Optimized Buffer A Kit (Invitrogen; cat. no. K1220−02A)
TABLE 1.
Primers used in this protocol.
| Oligo name | Sequence (5′ to 3′) | Comments |
|---|---|---|
| EP-F1 | TGACGTTGGAGTCCACGTTC | Forward primer for detection of excision product (EP; Step 24) |
| EP-R1 | GGCTCGTATGTTGTGTGG | Reverse primer for detection of EP for plasmids lacking an SB gene (Step 24) |
| EP-F2 | CTGGAACAACACTCAACCCT | Forward primer for detection of EP (Step 24) |
| EP-R2 | CCCAAGGTTTGAACTAGCTC (cis) CACACAGGAAACAGCTATGA (trans) | Reverse primers for detection of EPs for vectors in either the cis, or trans configured SB gene (Step 24) |
| GAPDH-F | TGTCTCCTGCGACTTCAACAGC | Forward primer for detection of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene used as an internal control (Step 24) |
| GAPDH-R | TGTAGGCCATGAGGTCCACCAC | Reverse primer for detection of GAPDH gene used as an internal control (Step 24) |
| GUSB-F | CTTCGACTGGCCGCTGCT | Forward primer for detection of GUSB gene |
| GUSB-R | GGCCACAGACCACATCAC | Reverse primer for detection of GUSB gene |
EQUIPMENT
Eye drops
Mouse restrainer (Braintree Scientific, cat. no. TV-150) or equivalent
Recovery cage(s) (Extra-clean; Allentown Micro-VENT Mouse cage) for the injected mice—often we inject more than one mouse in a single session and so the recovery cage is not the same as the cage containing the mice pre-injection.
45−50 °C waterbath and/or heat lamp
Heating pad
Scale for weighing mice (Ohaus compact electronic scale; Fisher Scientific, cat. no. 01−919−33)
Bleach
Alcohol pads
Box of 1-ml TB syringes
Box of 3-ml syringes
Box of 18-gauge needles
Box of 27-gauge butterfly needles
Shaver to remove mouse hair
REAGENT SETUP
Anesthetic cocktail
In a 10-ml sterile vacutainer tubes (Becton Dickenson, cat. no. BD 2006−12), add 9 ml normal saline, 0.8 ml stock ketamine HCl (100 mg ml−1), 0.1 ml butorphanol (1 mg ml−1) and 0.1 ml acepromazine (10 mg ml−1) to give a final anesthetic mixture of 8 mg ml−1 ketamine HCl, 0.1 mg ml−1 acepromazine maleate and 0.01 mg ml−1 butorphanol tartrate. This drug cocktail is premixed and is stable for 4 weeks at room temperature (20−25 °C). Protect anesthetic mixture from light.
Luciferin stock solution
Prepare a stock solution of luciferin (28.5 mg ml−1) by dissolving 1 g substrate in 35 ml sterile Dulbecco's PBS (Cambrix Bio Science, cat. no. 17−512F). Filter-sterilize solution through a 0.22-μm sterile filter. Store reconstituted substrate in 1 ml aliquots at −20 °C. Protect substrate from light. Shake well before use.
Preparation of vector DNA
Vector DNA can be prepared with an Endo-Free kit, for example, Maxi Kit (Qiagen, cat. no. 12362), or can be commercially prepared (e.g., Aldevron Inc.). We store the DNA at ∼1−2 μg μl−1 in 10 mM Tris–HCl, pH 7.2, 0.1 mM EDTA at 4 °C.
Preparation of samples for numerical evaluation of transgenes, genome copy number and EPs in liver (Step 23)
DNA is isolated from ∼50 mg liver specimens using a kit for isolation of total cellular DNA from mammalian tissues, for example, the DNeasy Tissue Kit (Qiagen, cat. no. 69504) according to manufacturer's instructions.
Preparation of samples for EP standard curve (Step 24)
A standard curve for the plasmid EP that relates the copy number of excision plasmid product (EP) as a function of the PCR threshold cycle (Ct) number has to be obtained (Fig. 2). For this, a defined number of copies of the reference DNA sequence (mockEP, a plasmid that lacks the complete transposon and therefore mirrors the EP) in a defined number of genomes is prepared by diluting a known amount of plasmid into a known amount of genomic DNA and calculating the relative copy numbers using the known molecular weights of the plasmid and the genomic DNA (≈2.2 × 1012 g mol−1 for the mouse genome). Experimentally, the standard curve for EP is obtained through serial dilutions of known quantities of plasmid containing the mockEP in genomic DNA prepared from mouse liver. Alternatively, a standard EP curve can be obtained through serial dilutions of genomic DNA prepared from livers of mice 24 h after injection with 25 mg (the same amount as the transposon plasmid with GOI) of the mockEP (∼7 × 1012 molecules in the case of a pT2/BH-based mockEP plasmid; see Plasmid Info at http://www.cbs.umn.edu/labs/perry/). Twenty-five microgram of transposon plasmids represents a delivery of ∼20,000−40,000 DNA molecules (depending on transposon size) per hepatocyte. On the basis of quantitative PCR (qPCR) curves that detect down to 0.05 copy per cell at their limit, we have estimated that ∼1,000 plasmids are associated with each liver cell (diploid genome) 1 d after hydrodynamic delivery16. The mockEP used in our studies was pT2/BH-based in which the transposon was deleted between two flanking BamHI sites. The number of EP molecules can be standardized to the number of genomes by quantifying the number of genomic copies of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene (Step 23A). In this case, a standard curve of GAPDH is obtained through serial dilution of the genomic liver DNA into TE buffer (10 mM Tris pH 7.6, 1 mM EDTA). Figure 2c shows the type of curve that can be obtained.
EQUIPMENT SETUP
General setup considerations
The necessary materials should be set up in a biosafety cabinet or appropriate hood, as shown in Figure 6. Place sufficient 1- and 3-ml syringes, 18-gauge needles, 27-gauge butterfly needles and alcohol pads on table for the entire procedure. If using a water bath: wipe the water bath with a 0.5% (vol/vol) bleach solution. Turn heat control for water bath on high and fill 600-ml beaker with 0.5% bleach solution. Put beaker on top of heat block and read temperature using a thermometer. When solution temperature is ∼35−40 °C, turn heat to low. Residual heat will raise temperature to 45−50 °C. If using a heat lamp: Turn on heat lamp and place first cage under it to warm mice
Figure 6.

Layout of equipment for hydrodynamic injection. The items, except for a marker pen in middle are numbered for easy identification: 1) Bleach bucket, for washing items and hands that are used in the hood in accordance with specific-pathogen free (SPF) procedure. 2) Bleach Spray bottle to facilitate washing in accordance with SPF procedure. 3) Lactated Ringer's solution for dilution and injection of transgenic DNA. 4) Three milliliter syringes for hydrodynamic injections. 5) Butterfly needles for hydrodynamic injections; the needle attaches to the 3-ml syringe. 6) Rack with samples that contain the DNA solutions for injection. 7) One-milliliter syringe with anesthetic drug already drawn up; it is covered because the anesthetic is sensitive to visible light. 8) Alcohol pads to sanitize tails. 9) Timer to measure duration of each injection. 10) Scale for weighing mice. 11) Mouse restrainer to contain mouse and reduce its activity during the injection. 12) Top of mouse cage set at an angle for better viewing. 13) Bottom of mouse cage for animal housing. 14) Heat lamp and ring stand to dilate veins in mice and keep them warm. The vertical rod in the stand is ∼50 cm tall.
Recovery cage
Turn on heating pad to medium for several minutes and then turn down to low. Place half of recovery cage on the warmed (40−50 °C) pad to provide a temperature gradient in the cage, which allows the animal to find its preferred warmth.
PROCEDURE
Prepare mice for injection • TIMING 1 min
1| Anesthetize 25−30 g mice by administering 50 μl of anesthetic cocktail i.p., prepared as indicated in REAGENT SETUP. We inject 25−30 μl of anesthetic cocktail to mice weighing <20 g to avoid rendering them unconscious.
▲ CRITICAL STEP The mouse should NOT be unconscious but moving slower (as if it were ‘drunk’). The hydrodynamic procedure will increase the effect of the anesthetic. If the mouse is unconscious, there is a significant chance of animal death if injected hydrodynamically.
? TROUBLESHOOTING
2| Weigh mouse. Weighing is easier and thereby more accurate when the mouse is ‘drowsy’ than when the mouse is fully active.
Prepare DNA sample for injection • TIMING 3 min
3| Before entering the mouse room (i.e., in the lab), transfer the desired amount of stock DNA solution into a 1.5-ml Eppendorf tube. Then, while the anesthetic takes effect, calculate the volume required for hydrodynamic injection based on the weight of the mouse [10% (vol/wt)]. For example: a 20 g mouse will be injected with 2 ml of a DNA solution that may range from 0.05 to 30 μg ml−1; we customarily use ∼10−12.5 μg ml−1 initially with modifications to the dose in subsequent experiments that depend on the level of gene expression attained compared to that desired.
▲ CRITICAL STEP We find that if an animal weighs >25 g and >2.5 ml is injected, the expression of the reporter genes does not increase (E. Aronovich, J. Frandsen & R.S. McIvor, unpublished data). Accordingly, we have set an upper limit for the injection volume at 2.5 ml.
4| Place an 18-gauge needle on 3-ml syringe and draw up the amount of LR calculated in Step 3 plus an additional 200 μl to take into account fluid that will remain in the 27-gauge butterfly needle line after the injection is complete.
5| Inject ∼1 ml of the LR from Step 4 into the tube and mix the DNA by withdrawing and re-injecting LR ‘up and down’ in the tube two to three times. At the end, after all of the solution in the Eppendorf tube has been recovered into the syringe, make sure that the DNA is thoroughly mixed by rocking the syringe several times using the air bubble to mix. As an example, for injection of a 20-g mouse with 25 μg DNA we would transfer 12.5 μl of stock DNA(at 2 μg μl−1) into the Eppendorf tube and mix it into 2.2 ml LR solution (2 ml + 200 μl) from Step 4; we generally store our plasmids at concentrations of 1−2 μg μl−1.
6| Remove the 18-gauge needle and replace it with a 27-gauge butterfly needle. Remove the air from the syringe by tapping it and then press plunger slowly to fill the butterfly line. Set syringe aside until injection. The DNA solution should be stable at room temperature for the duration of the injection time if properly stored in a buffer that lacks free divalent cations such as Mg2+.
Hydrodynamic injection • TIMING 1 min
7| Place the tail of the mouse under a heat lamp or in a 45−50 °C water bath for 10−20 s. This will increase the vascular volume in the tail and make the tail vein more easily visualized.
▲ CRITICAL STEP If using water bath: ensure that the temperature does not exceed 50 °C or the tail will be burned.
▲ CRITICAL STEP When using a heat lamp, avoid overheating the mice; they die quickly when heated too much or for too long. Attach the heat lamp on a ring stand rod ∼45−50 cm high. Place the cage under the lamp. The length of time that the mouse can stay under the lamp depends on how close the lamp is to the cage; we find that at a height of 45 cm the mice can rest up to 15 min, which is sufficient time to inject five mice. When a mouse is being overheated, it becomes sluggish and usually will dig into the bedding at the corners of the cage. If you notice this, either move the cage farther away from the heat lamp or turn off the lamp for 2−5 min until the mouse regains normal activity.
? TROUBLESHOOTING
8| Place the mouse in a restrainer tube (Fig. 7a) and wipe the tail with an alcohol pad.
Figure 7.

Hydrodynamic injection with mouse in restrainer. (a) The mouse is placed in a restrainer and the tail is rotated until the lateral vein faces up. In this example, the needle is in the right hand and is being placed into the tail vein for injection. (b) Detail of insertion of the needle into a tail vein. Two tail arteries run along the top and the bottom of the tail and two tail veins run on either side, midway between the arteries. The needle is inserted ∼2−3 cm from the tip of the tail (which is under the injector's thumb in this image). The needle is almost completely inserted into the vein for best results. The tip of the tail behind the injection site may be bent downward by some injectors. The tail is pulled taut, but excessive force may result in the tail coming off in young mice. If the injection fails, a new site, roughly in the middle of the tail can be tried. If this fails, one can try the other tail vein on the other side. When the needle is removed, some blood will flow from the puncture; bleeding is stopped by pressing lightly (not squeezing tightly) the wound with a finger and thumb.
? TROUBLESHOOTING
9| Place the tail with the lateral side up between the thumb and forefingers. Inject the needle into the vein ∼2−3 cm from the tip of the tail, making sure that the bevel of the needle is up (Fig. 7b). Once in the vein, you may see a little backflow of blood into the tubing of the butterfly needle. In white or nude mice, you can actually see the needle inside the vein.
▲ CRITICAL STEP As there are several variations for mouse tail-vein injections, an experienced person should use his/her normal injection procedure. A butterfly needle is not necessary, but we find it is easier to control the tip of the needle during the injection.
10| Press on plunger of the syringe with even force. The plunger will move smoothly with ease. Do not force it. If the plunger will not move easily, remove the needle and reinsert it anterior to the previous injection site about midway between the first injection site and the body of the mouse. If a third attempt is made, it can be done using the other vein on the opposite side of the tail.
▲ CRITICAL STEP If the plunger stops moving, you have blown the vein. Usually, the mouse is removed from the experiment. You can retry repositioning the needle. However, we have found that often delivery is compromised and not as efficient in these animals.
11| Inject the entirety of the DNA sample into the tail vein within a period of 4−7 s.
▲ CRITICAL STEP This short period of time, from start to finish, is critical. An optimal duration of the injection is ∼4−7 s. Injections lasting 8−10 s will have moderate success and injections taking > 10 s are considered unsuccessful and are not included in test groups.
? TROUBLESHOOTING
12| When finished, remove needle and, if there is any bleeding or blood, apply light fingertip pressure to the injection site until the bleeding stops.
Recovery phase • TIMING 60 min for full recovery
13| After injection, remove the mouse from the restrainer as quickly as possible and place eye drop in its eyes to prevent them from drying out owing to anesthesia. We hold the mouse until its respiration rate returns to approximately the normal rate.
14| Return the mouse to the recovery cage, half of which should be on a heating pad set at low heat (see EQUIPMENT SETUP). The mouse is considered to have recovered when it is up and back to normal activity. The mice may be sufficiently stable for the first blood drawing or other tests, such as in vivo bioluminescence, as soon as 30 min after hydrodynamic injection.
▲ CRITICAL STEP Monitor breathing to verify that the mouse survived the injection. The mouse's breathing may slow to one breath every 1 or 2 s. After 5−15 s the breathing rate should increase to near normal, 94−163 breaths per min. If the mouse stops breathing or gasps, chest massage may be required to promote breathing and recovery.
? TROUBLESHOOTING
15| Repeat the entire procedure with the next mouse. In general, a cohort of several mice is injected one after the other, with occasional inspection of the recovering mice to ensure they are not in distress. The only limitation on the number of mice injected over a single period is the investigator's ability to avoid confusing various DNA solutions that are being injected, keep accurate records that include injection times and gross physiological responses to the injection procedure and all the while monitor the health of the recovering mice. We have injected up to 50 mice in a day but generally inject only ∼20 in a single session. When a large number of animals are injected, a team of two investigators is best to ensure all the tasks can be accomplished while maintaining vigilance over mouse activities.
Evaluation of the efficiency of the hydrodynamic injection by in vivo bioluminescent imaging • TIMING 10 min or more per image set (depending on exposure of images)
16| The effectiveness of nucleic acid delivery can be evaluated as soon as 30 min after hydrodynamic injection through bioluminescent imaging59. Weigh mice and use Table 2 to determine the appropriate dose of anesthesia.
TABLE 2.
Doses for anesthetizing mice for imaging.
| Animal weight (g) | Dose (ketamine HCl:acepromazine malieate: butorphanol tartrate) |
|---|---|
| <15 | 150 μl Anesthetic cocktail (1.2 mg:15 μg:1.5 μg) |
| 15−20 | 200 μl Anesthetic cocktail (1.6 mg:20 μg:2.0 μg) |
| 20−25 | 250 μl Anesthetic cocktail (2.0 mg:25 μg:2.5 μg) |
| >25 | 300 μl Anesthetic cocktail (1.4 mg:30 μg:3.0 μg) |
▲ CRITICAL STEP If imaging equipment is not available, blood can be drawn from mice using standard methods and assayed for activity of the GOI.
17| Inject anesthetic i.p. Wait 2−5 min, until the mouse is fully anesthetized. Note that the level of anesthesia is higher than that used in Step 1 because for this procedure the mouse must be completely anesthesized for compliance with animal care regulations.
▲ CRITICAL STEP Do not overdose the mouse with anesthetic—if the mouse goes into respiratory distress it will probably die. Owing to the variation in responses to anesthetic by various mouse strains and mutant lines, there is no simple method to determine the anesthetic dose other than by experimentation. Check with appropriate animal care and veterinary staff for any guidelines they can provide for particular lines of mice.
? TROUBLESHOOTING
18| If necessary, shave the ventral area over the liver on the underside of the mouse. Generally, only dark-coated mice must be shaved for acceptable imaging.
19| Inject 100 μl luciferin i.p. and wait at least 5 min before imaging; or inject 50 μl i.v. and image immediately. For detection of firefly luciferase activity, either i.p. or i.v. delivery of luciferin can be employed. For detection of Renilla luciferase, substrate should be delivered i.v.
▲ CRITICAL STEP For the i.p. luciferin injections, several mice can be injected for imaging after hydrodynamic DNA delivery. Imaging should be carried out between 5 and 25 min after administration of the luciferin; after 30 min the level of luciferase activity drops significantly and the assay will be compromised.
▲ CRITICAL STEP Sometimes, imaging multiple animals at one time results in one animal apparently lacking expression in terms of the color-coding. This could be an artifact owing to strong signals from one animal interfering with detection of the image from another animal that has a lesser signal. Accordingly, in this circumstance, mice that do not show a signal should be imaged separately to ensure that their signals are appropriately measured.
20| Place the animal on its back onto black construction paper (Fig. 8). If the animal cannot be positioned stably on its back with its underside fully exposed, tape down its legs.
Figure 8.

Typical results from in vivo bioluminescence imaging of mice 24 h after hydrodynamic injection. Both mice are on black construction paper, which may appear differently according to background illumination. Color-coded luminescence scales for the circled regions are shown on the right. Exposures are 1 s. The imaging identifies the liver as the primary site of expression of a luciferase expression cassette. The mouse on the left is a C57BL/6 mouse (Imaged for 0.5 s) and the mouse on the right is a NOD.129(B6)-PrkdcscidIduatm1Clk (imaged for 1 s). ROI, region of interest.
21| Image the mice according to manufacturer's instructions. We use the Living Image program for which the basic guidelines are provided in Box 1.
BIOLUMINESCENT IMAGE ACQUISITION.
We have experience only with Xenogen's IVIS Imaging System with Living Image program.
Initialize the system according to manufacturer's instructions.
Set stage at A or B (depending on number of mice imaged simultaneously). Use stage A for one to three mice imaged at the same time (depending on size of mice). Use stage B for three to five mice imaged at the same time (depending on size of mice).
Set imaging parameters as follows: Exposure time: 0.5 s to 5 min (duration depends on expression cassette and the time after injection). Binning—medium (we use most commonly 4 × 4 pixels setting—binning can be decreased if the image saturates). F-stop—1 (increasing the F-stop reduces saturation of the image).
Take a photo to verify position.
Select lumination overlay key.
Acquire image.
▲ CRITICAL STEP Exposures should not exceed 5 min—good delivery of a transgenic cassette with a promoter that provides robust expression in the liver usually requires ∼1 s exposure for saturation (Fig. 8).
? TROUBLESHOOTING
22| Return mice to their cage (see EQUIPMENT SETUP) to recover until they are up and moving around.
▲ CRITICAL STEP Avoid hypothermia. Use heating pad set on low (warm) to keep mouse from shivering. The heating pad will help keep the mouse's internal temperature from dropping too low and causing hypothermia.
Excision assay for transposition efficiency • TIMING 2−3 d
23| After bioluminescent imaging, the animals may be killed and the excision assay performed. The excision assay16,36 is the most convenient and comprehensive assay for transposition of SB transposons in a multicellular organ like the liver. Isolate ∼0.1 mg total DNA from ∼50 mg liver specimens using a kit for isolation of total cellular DNA from mammalian tissues, for example, the DNeasy Tissue Kit according to manufacturer's instructions.
24| Quantify DNA copy number by real-time qPCR (option A) or approximate copy number by conventional PCR (option B).
▲ CRITICAL STEP All PCR experiments require appropriate controls. Suitable negative controls should include samples that lack any DNA and samples that specifically lack the target DNA sequence. A positive control should be included when possible, but a plasmid source that might lead to contamination of the experimental sample should not be used. Internal PCR controls, for example, β-glucuronidase in Figure 2b, serve to validate the reaction mix and conditions of amplification.
(A) Determining DNA copy number by real-time qPCR
- Set up a 25 μl reaction mixture for each sample, as tabulated below. All qPCRs should be run in triplicate and include appropriate controls and standard curve samples (see REAGENT SETUP). We use mouse single-copy gene sequences, such as GAPDH, as an internal control of genomic DNA content. A standard curve of GAPDH is obtained through serial dilution of genomic liver DNA from the hydrodynamically injected mice into TE buffer (10 mM Tris pH 7.6, 1 mM EDTA) and measuring the Ct numbers using the GAPDH-F and GAPDH-R primers (Table 1)16.
Component Amount Final concentration Liver DNA 500 ng 20 ng μl−1 (≈ 0.01 pM GAPDH gene) GAPDH-F and -R (Table 1) 5 pmol each 200 nM each 2× IQ SYBR green supermix 12.5 μl 1×
- Carry out thermocycling. Typical conditions are tabulated below. We use an iCycler instrument (Bio-Rad).
Cycle number Denature Anneal Extend 1 95 °C 2:00 min — — 2−41 95 °C 0:40 min 58 °C 0:40 min 72 °C 1:00 min 42 — — 72 °C 5:00 min The DNA content (GAPDH gene number, y-axis) for each dilution is plotted as a function Ct value (x-axis) to obtain a standard curve for genomic DNA copy number. Figure 2c shows the type of curve that can be obtained.
(B) Conventional PCR for qualitative assessment of EP formation
- Excision analysis using conventional PCR can be used to back up, at lower resolution, the qPCR results. Conventional PCR can also be used to compare transposition rates qualitatively in various reactions where the absolute numbers of events is not required. Our conventional PCR protocol requires two rounds of amplification. We use a dNTP mix, PCR buffer and DMSO from the Invitrogen Optimized Buffer A Kit. For the first round, set up a 50 μl reaction mixture for each sample (including controls), as tabulated below.
Component Amount Final concentration Liver DNA 100 ng 2 ng μl−1 (≈ 30,000 haploid genomes) EP-F1 and EP-R1 primers 10 pmol each 200 nM dNTPs (A, G, C, T) 10 nmol each 200 μM 5× Optimized Buffer A 10 μl 1× Optimized Buffer A DMSO 2.5 μl 100% stock 5% (vol/vol) Glycerol 2.5 μl 100% stock 5% (vol/vol) Taq DNA polymerase 5 U 0.1 U μl−1 - For the first amplification, we use the following conditions.
Cycle number Denature Anneal Extend 1 95 °C 5:00 min — — 2−41 95 °C 0:40 min 58 °C 0:40 min 72 °C 1:00 min 42 — — 72 °C 5:00 min - Set up the second, nested round of amplification reactions as tabulated below.
Component Amount Final concentration PCR mixture from reaction 1 5 μl Not applicable EP-F2 and EP-R2 primers 10 pmol each 200 nM dNTPs (A, G, C, T) 10 nmol each 200 μM 5× Optimized Buffer A 10 μl 1× Optimized Buffer A DMSO 2.5 μl 100% stock 5% (vol/vol) Glycerol 2.5 μl 100% stock 5% (vol/vol) Taq DNA polymerase 5 U 0.1 U μl−1 - Use the following conditions for second round amplification.
Cycle number Denature Anneal Extend 1 95 °C 5:00 min — — 2−31 95 °C 0:40 min 58 °C 0:40 min 72 °C 1:00 min 32 — — 72 °C 7:00 min We run 8 μl PCR product on an 1% (wt/vol) agarose gel. An EP band of ∼460 bp is expected on the gel (Fig. 2b). In this example, the murine β-glucuronidase (GUSB) housekeeping gene served as an internal control that was amplified in the same PCR using primers GUSB-F and GUSB-R (Table 1). PCR amplification yields a GUSB PCR product of 358 bp.
• TIMING
It takes ∼6−10 min per mouse from anesthesia to completion of the hydrodynamic injection although a group of 10−15 mice can be performed in <1.5 h as mice can be injected while others recover Steps 1−6, preparing mice and DNA solutions for injection, 4 min (per mouse—cohorts may take less time since the preparation of the DNA solution is a single event for all mice in a given group)
Steps 7−12, hydrodynamic injection, 1 min (per mouse)
Steps 13−15, recovery phase, 60 min for full recovery; 30 min for sufficient recovery for bioluminescent imaging
Steps 16−22, evaluation of the efficiency of the hydrodynamic injection: in vivo bioluminescent imaging, 10 min or more per image set (depending on exposure of images)
Steps 23−24, excision assay for evaluation of transposition efficiency, 2−3 d after harvesting of liver
? TROUBLESHOOTING Troubleshooting advice can be found in Table 3.
TABLE 3.
Troubleshooting table.
| Step | Problem | Possible reason | Proposed solution |
|---|---|---|---|
| 1: Anesthetic dosing | Mouse appears fully anesthesized | Overdose of anesthetic | Reduce dosing to account for variety in mouse strains |
| 7: Heating mice | Vein not visible | Underheating | Heat tail longer |
| Tails fall off or the mouse dies | Overheating | Watch mouse closely; mouse should slowly move around cage | |
| 8: Loading mouse into restrainer | Mouse not breathing | Mouse wedged into restrainer | Do not cover nose of mouse |
| 11: Hydrodynamic injection | Injection takes longer than 10 s | Injection too slow | Omit mouse from test group |
| Blown tail vein | Poor placement of needle; injection too rapid | Omit mouse from test group | |
| Mouse suffers cardiac arrest or develops fluid in its lungs | Unknown | Massage chest to assist labored blood flow and/or help clear lungs | |
| 14: Recovery | Death | Hypothermia | Place subsequent mice on heating pad set on low heat until they are moving |
| Troubled breathing | Unknown | Massage chest | |
| 17: Imaging | Mouse dies | Overdose of anesthetic | Reduce dose in subsequent mice to account for strain differences |
| Mouse is too active for imaging | Underdose of anesthetic | Increase anesthetic. The mouse may need to be taped down | |
| 21: Imaging | Signals not distinct | Overexposure | Reduce exposure time |
| Image only one mouse at a time |
ANTICIPATED RESULTS
A natural question is the reliability and extent of gene delivery using the hydrodynamic procedure. In our experiments, we inject ∼25 mg of DNA, which depending on the size of the plasmid, corresponds to ∼2−4 × 1012 plasmids per injection. Biodistribution studies have shown that, based on a CMV promoter that transiently expresses well in many cell types, >95% of the expressing plasmids are in liver, and 0.1−1% in other organs12,75. Assuming that immediately after injection ∼1% of the injected DNA remains in the liver, which contains ∼108 hepatocytes as well as Kupffer and sinusoidal epithelial cells in an adult mouse76, there would be on the order of 1,000 transposon-containing plasmids per cell.
Figure 2 shows examples of expected results for assay of the first step of transposition—the excision of the transposon from its carrier plasmid. The excision assay can quantify EPs down to the level of one excised transposon plasmid per 10,000 cells36.
Figure 5 shows the reproducibility of hydrodynamic delivery to the liver and an example of the distribution of cells that take up transgenic DNA for expression in the liver. The efficiency of delivery to liver varies about tenfold from mouse to mouse as measured by expression of luciferase from a transposon-containing plasmid 24 h after injection (Fig. 5a). The duration of transposition following hydrodynamic delivery to the liver appears to be relatively short, ∼3−4 d if a short-duration promoter like the CMV promoter is used to direct transcription of transposase77. Within the first day, as many as 40%, but more often 10−20% of hepatocytes (Fig. 5b and c)takeup ∼100 transposon-containing plasmids each16 but only express those which are able to enter the nucleus for transcription. In ∼1−5% of cells, a transposition event occurs that moves the transposon into a mouse chromosome16,40,43. After ∼2−4 weeks, Southern blots indicate that there is <1 copy of transgenic DNA per liver cell18. Thus, of the 1012 injected transposons, we estimate that ∼106 (10−4%) actually integrate into liver cells, which in mice can be sufficient to treat diseases effectively such as hemophilia and lessen the effects of other diseases such as lysosomal storage disorders. The retention of plasmids may be dependent on their CpG contents and the strain of mouse.
Figure 8 shows examples of two mice imaged 1 d after hydrodynamic injection. A hybrid CMV/β-actin promoter was used to regulate expression of the firefly luciferase gene. The bioluminescence imaging allows estimation of luciferase gene delivery in vivo.
Figure 9 shows the first reported example of hydrodynamic delivery of the SB system into mice40. In this example, the human Factor IX gene (hFIX) behind an EF1α promoter was delivered with either a functional SB10 transposase or the inactive mutant form in cis. Expression of hFIX at therapeutic levels was achieved over the lifetime of the mice when functional transposase was co-delivered but not when the mutant transposase was delivered.
Figure 9.
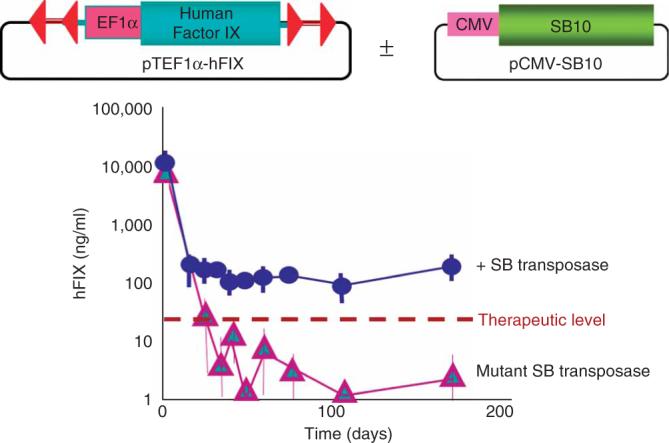
Example of long-term gene expression following hydrodynamic delivery into mice. Results of delivery of the human clotting Factor IX (hFIX) gene in a Sleeping Beauty (SB) transposon into FIX-deficient mice. Long-term expression depended on the presence of a functional transposase (SB10 regulated by a cytomegalovirus (CMV) promoter). The top portion of the figure shows the constructs delivered in cis. The graphs show the levels of hFIX in the mice as a function of time after hydrodynamic injection relative to normal enzyme amounts (therapeutic level). This figure is adapted from Figure 4 of Yant et al. (2000)40.
ACKNOWLEDGMENTS
We thank the Arnold and Mabel Beckman Foundation for support of our work and all members of the Beckman Center for Transposon Research for a long history of contributions of ideas and results. We appreciate the help of Mr. Joel Frandsen for teaching us the intricacies of hydrodynamic injection and our veterinarian technician, Brenda Koniar. The authors were partially supported by National Institutes of Health grant 1PO1 HD32652-07.
Footnotes
COMPETING INTERESTS STATEMENT The authors declare competing financial interests (see the HTML version of this article for details).
References
- 1.Broeke AV, Burny A. Retroviral biosafety: lessons from sheep. J. Biomed. Biotech. 2003;1:9–12. doi: 10.1155/S1110724303209128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muruve DA, Barnes MJ, Stillman IE, Libermann TA. Adenoviral gene therapy leads to rapid induction of multiple chemokines and acute neutrophil-dependent hepatic injury in vivo. Hum. Gene Ther. 1999;10:965–976. doi: 10.1089/10430349950018364. [DOI] [PubMed] [Google Scholar]
- 3.Schroder AR, et al. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 4.Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- 5.Nakai H, et al. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat. Genet. 2003;34:297–302. doi: 10.1038/ng1179. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell RS, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PloS Biol. 2004;2:1127–1136. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laufs S, et al. Insertion of retroviral vectors in NOD/SCID repopulating human peripheral blood progenitor cells occurs preferentially in the vicinity of transcription start regions and in introns. Mol. Ther. 2004;10:874–881. doi: 10.1016/j.ymthe.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 8.De Palma M, et al. Promoter trapping reveals significant differences in integration site selection between MLV and HIV vectors in primary hematopoietic cells. Blood. 2005;105:2307–2315. doi: 10.1182/blood-2004-03-0798. [DOI] [PubMed] [Google Scholar]
- 9.Ciuffi A, et al. Integration site selection by HIV-based vectors in dividing and growth-arrested IMR-90 lung fibroblasts. Mol. Ther. 2006;13:366–373. doi: 10.1016/j.ymthe.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Bushman F, et al. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 2005;3:848–858. doi: 10.1038/nrmicro1263. [DOI] [PubMed] [Google Scholar]
- 11.Donsamte A, et al. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:477. doi: 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]
- 12.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 13.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum. Gene Ther. 1999;10:1735–1737. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- 14.Al-Dosari MS, Knapp JE, Liu D. Hydrodynamic delivery. Adv. Genet. 2005;54:65–82. doi: 10.1016/S0065-2660(05)54004-5. [DOI] [PubMed] [Google Scholar]
- 15.Herweijer H, Wolff JA. Gene therapy progress and prospects: hydrodynamic gene delivery. Gene Ther. 2007;14:99–107. doi: 10.1038/sj.gt.3302891. [DOI] [PubMed] [Google Scholar]
- 16.Aronovich EL, et al. Prolonged expression of a lysosomal enzyme in mouse liver after Sleeping Beauty transposon-mediated gene delivery: implications for non-viral gene therapy of mucopolysaccharidoses. J. Gene Med. 2007;9:403–415. doi: 10.1002/jgm.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilber A, et al. RNA as a source of transposase for Sleeping Beauty-mediated gene insertion and expression in somatic cells and tissues. Mol. Ther. 2006;13:625–630. doi: 10.1016/j.ymthe.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 18.Score PR, et al. Sleeping Beauty-mediated transposition and long-term expression in vivo: use of the LoxP/Cre recombinase system to distinguish transposition-specific expression. Mol. Ther. 2006;13:617–624. doi: 10.1016/j.ymthe.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 19.Suda T, Gao X, Stolz DB, Liu D. Structural impact of hydrodynamic injection on mouse liver. Gene Ther. 2007;14:129–137. doi: 10.1038/sj.gt.3302865. [DOI] [PubMed] [Google Scholar]
- 20.Crespo A, et al. Hydrodynamic liver gene transfer mechanism involves transient sinusoidal blood stasis and massive hepatocyte endocytic vesicles. Gene Ther. 2005;12:927–935. doi: 10.1038/sj.gt.3302469. [DOI] [PubMed] [Google Scholar]
- 21.Budker VG, et al. Mechanism of plasmid delivery by hydrodynamic tail vein injection. II. Morphological studies. J. Gene Med. 2006;8:852–873. doi: 10.1002/jgm.921. [DOI] [PubMed] [Google Scholar]
- 22.Sebestyen MG, et al. Mechanism of plasmid delivery by hydrodynamic tail vein injection. I. Hepatocyte uptake of various molecules. J. Gene Med. 2006;8:852–873. doi: 10.1002/jgm.921. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi N, Nishikawa M, Hirata K, Takakura Y. Hydrodynamics-based procedure involves transient hyperpermeability in the hepatic cellular membrane: implication of a nonspecific process in efficient intracellular gene delivery. J. Gene Med. 2004;6:584–592. doi: 10.1002/jgm.541. [DOI] [PubMed] [Google Scholar]
- 24.Andrianaivo F, Lecocq F, Wattiaux-De Coninck S, Wattiaux R, Jadot M. Hydrodynamics-based transfection of the liver: entrance into hepatocytes of DNA that causes expression takes place very early after injection. J. Gene Med. 2004;6:877–883. doi: 10.1002/jgm.574. [DOI] [PubMed] [Google Scholar]
- 25.Lecocq M, et al. Uptake by mouse liver and intracellular fate of plasmid DNA after a rapid tail vein injection of a small or a large volume. J. Gene Med. 2003;5:142–146. doi: 10.1002/jgm.328. [DOI] [PubMed] [Google Scholar]
- 26.Yoshino H, Hashizume K, Kobayashi E. Naked plasmid DNA transfer to the porcine liver using rapid injection with large volume. Gene Ther. 2006;13:1696–1702. doi: 10.1038/sj.gt.3302833. [DOI] [PubMed] [Google Scholar]
- 27.Aliño SF, Herrero MJ, Noguera I, Dasí F, Sánchez M. Pig liver gene therapy by noninvasive interventionist catheterism. Gene Ther. 2007;14:334–343. doi: 10.1038/sj.gt.3302873. [DOI] [PubMed] [Google Scholar]
- 28.Eastman SJ, et al. Development of catheter-based procedures for transducing the isolated rabbit liver with plasmid DNA. Hum. Gene Ther. 2002;13:2065–2077. doi: 10.1089/10430340260395910. [DOI] [PubMed] [Google Scholar]
- 29.Wolff JA, Budker V. Hydrodynamic delivery. Adv. Genet. 2005;54:3–20. doi: 10.1016/S0065-2660(05)54001-X. [DOI] [PubMed] [Google Scholar]
- 30.Rossmanith W, Chabicovsky M, Herkner K, Schulte-Hermann R. Cellular gene dose and kinetics of gene expression in mouse livers transfected by high-volume tail-vein injection of naked DNA. DNA Cell. Biol. 2002;21:847–853. doi: 10.1089/104454902320908496. [DOI] [PubMed] [Google Scholar]
- 31.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 32.Hackett PB, Ekker SC, Largaespada DA, McIvor RS. Sleeping Beauty transposon-mediated gene therapy for prolonged expression. Adv. Genet. 2005;54:187–229. doi: 10.1016/S0065-2660(05)54009-4. [DOI] [PubMed] [Google Scholar]
- 33.Ivics Z, Izsvak Z. Transposable elements for transgenesis and insertional mutagenesis in vertebrates: a contemporary review of experimental strategies. Meth. Mol. Biol. 2004;260:255–276. doi: 10.1385/1-59259-755-6:255. [DOI] [PubMed] [Google Scholar]
- 34.Hackett PB, Ekker SE, Essner JJ. Applications of transposable elements in fish for transgenesis and functional genomics. In: Gong Z, Korzh V, editors. Fish Development and Genetics Chapter. 16. World Scientific, Inc.; New Jersey, USA and Singapore: 2004. pp. 532–580. [Google Scholar]
- 35.Ivics Z, Izsvak Z. Transposons for gene therapy! Curr. Gene Ther. 2006;6:593–607. doi: 10.2174/156652306778520647. [DOI] [PubMed] [Google Scholar]
- 36.Liu G, Aronovich EL, Cui Z, Whitley CB, Hackett PB. Excision of Sleeping Beauty transposons: parameters and applications to gene therapy. J. Gene Med. 2004;6:574–583. doi: 10.1002/jgm.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yant SR, et al. High-resolution genome-wide mapping of transposon integration in mammals. Mol. Cell. Biol. 2005;25:2085–2094. doi: 10.1128/MCB.25.6.2085-2094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berry C, Hannenhalli S, Leipzig J, Bushman FD. Selection of target sites for mobile DNA integration in the human genome. PLoS Comp. Biol. 2006;2:e157. doi: 10.1371/journal.pcbi.0020157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hackett CS, Geurts AM, Hackett PB. Predicting preferential DNA vector insertion sites: implications for functional genomics and gene therapy. Genome Biol. 2007;8(Suppl 1):S12. doi: 10.1186/gb-2007-8-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yant SR, et al. Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat. Genet. 2000;25:35–41. doi: 10.1038/75568. [DOI] [PubMed] [Google Scholar]
- 41.Ohlfest JR, et al. Phenotypic correction and long-term expression of factor VIII in hemophilic mice by immunotolerization and nonviral gene transfer using the Sleeping Beauty transposon system. Blood. 2005;105:2691–2698. doi: 10.1182/blood-2004-09-3496. [DOI] [PubMed] [Google Scholar]
- 42.Liu L, Mah C, Fletcher BS. Sustained FVIII expression and phenotypic correction of hemophilia A in neonatal mice. Mol. Ther. 2006;13:1006–1015. doi: 10.1016/j.ymthe.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 43.Montini EP, et al. In vivo correction of murine tyrosinemia type I by DNA-mediated transposition. Mol. Ther. 2002;6:759–769. doi: 10.1006/mthe.2002.0812. [DOI] [PubMed] [Google Scholar]
- 44.Balciunas D, et al. Harnessing an efficient large cargo-capacity transposon for vertebrate gene transfer applications. PLoS Genet. 2006;4 doi: 10.1371/journal.pgen.0020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilber A, et al. Messenger RNA as a source of transposase for Sleeping Beauty transposon-mediated correction of hereditary tyrosinemia type I. Mol. Ther. 2007;15:1280–1287. doi: 10.1038/sj.mt.6300160. [DOI] [PubMed] [Google Scholar]
- 46.Calos MP. The γC31 integrase system for gene therapy. Curr. Gene Ther. 2006;6:633–645. doi: 10.2174/156652306779010642. [DOI] [PubMed] [Google Scholar]
- 47.Chen L, Woo SL. Complete and persistent phenotypic correction of phenylketonuria in mice by site-specific genome integration of murine phenylalanine hydroxylase cDNA. Proc. Natl. Acad. Sci. USA. 2005;102:15581–15586. doi: 10.1073/pnas.0503877102. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Chen L, Thung SN, Woo SL. Metabolic basis of sexual dimorphism in PKU mice after genome-targeted PAH gene therapy. Mol. Ther. 2007;15:1079–1085. doi: 10.1038/sj.mt.6300137. [DOI] [PubMed] [Google Scholar]
- 49.Ortiz S, et al. Sustainable correction of junctional epidermolysis bullosa via transposon-mediated nonviral gene transfer. Gene Ther. 2003;10:1099–1104. doi: 10.1038/sj.gt.3301978. [DOI] [PubMed] [Google Scholar]
- 50.Ohlfest JR, Lobitz PD, Perkinson SG, Largaespada DA. Integration and long-term expression in xenografted human glioblastoma cells using a plasmid-based transposon system. Mol. Ther. 2004;10:260–268. doi: 10.1016/j.ymthe.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 51.Ohlfest JR, et al. Combinatorial anti-angiogenic gene therapy by nonviral gene transfer using the Sleeping Beauty transposon causes tumor regression and improves survival in mice bearing intracranial human glioblastoma. Mol. Ther. 2005;12:778–788. doi: 10.1016/j.ymthe.2005.07.689. [DOI] [PubMed] [Google Scholar]
- 52.Liu H, Liu L, Fletcher BS, Visner GA. Sleeping Beauty-based gene therapy with indoleamine 2,3-dioxygenase inhibits lung allograft fibrosis. FASEB J. 2006;20:2384–2386. doi: 10.1096/fj.06-6228fje. [DOI] [PubMed] [Google Scholar]
- 53.Akagi Y, et al. Transcriptional activation of a hybrid promoter composed of cytomegalovirus enhancer and beta-actin/beta-globin gene in glomerular epithelial cells in vivo. Kidney Int. 1997;51:1265–1269. doi: 10.1038/ki.1997.172. [DOI] [PubMed] [Google Scholar]
- 54.Xu L, et al. CMV-beta-actin promoter directs higher expression from an adeno-associated viral vector in the liver than the cytomegalovirus or elongation factor 1 alpha promoter and results in therapeutic levels of human factor X in mice. Hum. Gene Ther. 2001;12:563–573. doi: 10.1089/104303401300042500. [DOI] [PubMed] [Google Scholar]
- 55.Chu Q, Joseph M, Przybylska M, Yew NS, Scheule RK. Transient siRNA-mediated attenuation of liver expression from an alpha-galactosidase A plasmid reduces subsequent humoral immune responses to the transgene product in mice. Mol. Ther. 2005;12:264–273. doi: 10.1016/j.ymthe.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 56.Hodges BL, Taylor KM, Joseph MF, Bourgeois SA, Scheule RK. Long-term transgene expression from plasmid DNA gene therapy vectors is negatively affected by CpG dinucleotides. Mol. Ther. 2004;10:269–278. doi: 10.1016/j.ymthe.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 57.Herweijer H, et al. Time course of gene expression after plasmid DNA gene transfer to the liver. J. Gene Med. 2001;3:280–291. doi: 10.1002/jgm.178. [DOI] [PubMed] [Google Scholar]
- 58.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 59.Wilber AC, et al. Dynamic gene expression following systemic delivery of plasmid DNA as determined by in vivo bioluminescence imaging. Hum. Gene Ther. 2005;16:1325–1332. doi: 10.1089/hum.2005.16.1325. [DOI] [PubMed] [Google Scholar]
- 60.Tolar J, et al. Real-time in vivo imaging of stem cells following transgenesis by transposition. Mol. Ther. 2005;12:42–48. doi: 10.1016/j.ymthe.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 61.Manno CS, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 62.Brown BD, Venneri MA, Zingale A, Sergi GS, Naldini L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat. Med. 2006;5:585–591. doi: 10.1038/nm1398. [DOI] [PubMed] [Google Scholar]
- 63.Grimm D, Kay MA. Combinatorial RNAi: a winning strategy for the race against evolving targets? Mol. Ther. 2007;15:878–888. doi: 10.1038/sj.mt.6300116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hartl DL, Lohe AR, Lozovskaya ER. Regulation of the transposable element mariner. Genetica. 1997;100:177–184. [PubMed] [Google Scholar]
- 65.Geurts AM, et al. Gene transfer into genomes of human cells by the Sleeping Beauty transposon system. Mol. Ther. 2003;8:108–117. doi: 10.1016/s1525-0016(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 66.Mikkelsen JG, et al. Helper-independent Sleeping Beauty transposon-transposase vectors for efficient nonviral gene delivery and persistent gene expression in vivo. Mol. Ther. 2003;8:654–665. doi: 10.1016/s1525-0016(03)00216-8. [DOI] [PubMed] [Google Scholar]
- 67.Hackett PB. ntegrating DNA vectors for gene therapy. Mol. Ther. 2007;15:10–12. doi: 10.1038/sj.mt.6300065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cui Z, Guerts AM, Liu G, Kaufman CD, Hackett PB. Structure-function analysis of the inverted terminal repeats of the Sleeping Beauty transposon. J. Mol. Biol. 2002;318:1221–1235. doi: 10.1016/s0022-2836(02)00237-1. [DOI] [PubMed] [Google Scholar]
- 69.Zayed H, Izsvak Z, Walisko O, Ivics Z. Development of hyperactive Sleeping Beauty transposon vectors by mutational analysis. Mol. Ther. 2004;9:292–304. doi: 10.1016/j.ymthe.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 70.Yant SR, Park J, Huang Y, Mikkelsen JG, Kay MA. Mutational analysis of the N-terminal DNA-binding domain of Sleeping Beauty transposase: critical residues for DNA binding and hyperactivity in mammalian cells. Mol. Cell Biol. 2004;24:9239–9247. doi: 10.1128/MCB.24.20.9239-9247.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baus J, Liu L, Heggestad AD, Sanz S, Fletcher BS. Hyperactive transposase mutants of the Sleeping Beauty transposon. Mol. Ther. 2005;12:1148–1156. doi: 10.1016/j.ymthe.2005.06.484. [DOI] [PubMed] [Google Scholar]
- 72.Yant SR, Huang Y, Akache B, Kay MA. Site-directed transposon integration in human cells. Nucleic Acids Res. 2007;35:e50. doi: 10.1093/nar/gkm089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ivics Z, et al. Targeted Sleeping Beauty transposition in human cells. Mol. Ther. 2007;15:1137–1144. doi: 10.1038/sj.mt.6300169. [DOI] [PubMed] [Google Scholar]
- 74.Wilson MH, Kaminski JM, George AL., Jr. Functional zinc finger/sleeping beauty transposase chimeras exhibit attenuated overproduction inhibition. FEBS Lett. 2005;579:6205–6209. doi: 10.1016/j.febslet.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 75.Herweijer H, Wolff JA. Progress and prospects: naked DNA gene transfer and therapy. Gene Ther. 2003;10:453–458. doi: 10.1038/sj.gt.3301983. [DOI] [PubMed] [Google Scholar]
- 76.Ponder KP, et al. Mouse hepatocytes migrate to liver parenchyma and function indefinitely after intrasplenic transplantation. Proc. Natl. Acad. Sci. USA. 1991;88:1217–1221. doi: 10.1073/pnas.88.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bell JB, et al. Duration of expression of Sleeping Beauty transposase in mouse liver following hydrodynamic delivery. Mol. Ther. 2006;13(Suppl):S150. doi: 10.1038/mt.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]