

Abstract
Platelets express TLR4 receptors, but its ligand lipopolysaccharide (LPS) does not directly activate thrombotic functions nor, obviously, transcription by these anucleate cells. Platelets, however, store information that changes their phenotype over a few hours in the form of unprocessed RNA transcripts. We show even low concentrations of LPS in the presence of soluble CD14 initiated splicing of unprocessed IL-1β RNA, with translation and accumulation of IL-1β protein. LPS was a more robust agonist for this response than thrombin. Platelets also contained cyclooxygenase-2 pre-mRNA, which also was spliced and translated after LPS stimulation. Flow cytometry and immunocytochemistry of platelets extensively purified by negative immunodepletion showed platelets contained IL-1β, and quantitative assessment of white blood cell contamination by CD14 real time PCR confirms that leukocytes were not the IL-1β source, nor were they required for platelet stimulation. LPS did not initiate rapid platelet responses, but over time did prime platelet aggregation to soluble agonists, induced actin rearrangement, and initiated granule secretion with P-selectin expression that resulted the coating of quiescent leukocytes with activated platelets. LPS is a direct agonist for platelets that allows these cells to directly participate in the innate immune response to bacteria.
Keywords: Lipopolysaccharide, cell activation, inflammation
Introduction
Lipopolysaccharide stimulates gene transcription in nucleated inflammatory cells through its interaction with extracellular LPS-binding protein (LBP), the CD14 displayed on the cell surface, and the transmembrane signaling receptor TLR4(1). Platelets express TLR4(2–4) in sufficient quantities to physically bind LPS(5), but they do not express CD14. This need not exclude potential platelet responses to LPS because plasma contains microgram levels of soluble CD14(6) that also can present LPS to TLR4. In vivo experiments show that LPS promotes platelet function(2, 4, 7, 8), induces thrombocytopenia (4), and expands the inflammatory reaction through TNFα expression (4). Whether these events result from a direct effect on platelets is debated because LPS neither enhances rapid homotypic platelet aggregation(7, 9, 10) nor induces surface P-selectin expression(2, 7, 10) by isolated platelets. LPS does enhance platelet adhesion to fibrinogen under flow(2), although it is less effective at this than thrombin, and it enhances surface expression of GPIIb/GPIIIa and CD40L through TLR4(5). LPS, it is clear, is a poor agonist for typical ex vivo platelet responses, although this may reflect the type of LPS employed because LPS from enterohemorrhagic E. coli are potent platelet agonists(5).
Platelets lack nuclei and so cannot transcribe new mRNA to alter their proteome, but they do contain a select group of mRNAs(11–14) that are translated to new protein in a highly regulated manner(11, 15–17). Platelets also contain unprocessed RNA transcripts, and they possess a functional, activation-dependent spliceosome that converts these transcripts to translatable mRNAs(18–20). One of these platelet pre-mRNA that is spliced and translated to new protein after thrombin stimulation encodes the multi-faceted inflammatory cytokine IL-1β(18). Thrombin activated platelets move IL-1β to their surface(21, 22) where it participates in juxtacrine stimulation of endothelial cells(21), and they secrete small amounts of mature IL-1β(23) that participates in the inflammatory process. These observations have now been challenged with the suggestion that even low numbers of monocytes in platelet preparations are sufficient to account for thrombin-stimulated IL-1β production(24). Assessing contaminating nucleated cells in platelet preparations is difficult(25) when their abundance needs to be less than one per hundred thousand platelets(24). This is an important topic given that extreme leukoreduction, on the order of 1 leukocyte per million platelets, is a critical issue in platelet transfusion medicine ((26), and references therein). Purifying platelets to this extent without activation is problematic, so these opposing views regarding the direct effects of LPS on platelets have yet to be reconciled.
Platelets express cyclooxygenase-1 that produces thromboxane A2 that enhances homotypic aggregation(27). Cyclooxygenase-1 is irreversibly inhibited by aspirin, while the cyclooxygenase-2 isoform—which can be induced by inflammatory stimuli in a variety of cells—is less sensitive to this inhibitor(28). Because this might contribute to the clinical and laboratory phenomenon of aspirin resistance(29), the presence of cyclooxygenase-2 in platelets has been well studied. Although cyclooxygenase-2 occasionally has been reported in platelets(30, 31), potentially remaining from megakaryopoiesis(32), it is not apparent that mature platelets express cyclooxygenase-2(33, 34). Platelets stimulated over a short time by thrombin also do not express functional cyclooxygenase-2(33).
Platelets have a role in inflammation beyond their initial, rapid response(18–20, 35), and here we show that LPS is a potent agonist for prolonged platelet responses. We generated highly purified platelets by negative immunoselection and then quantified monocyte contamination by quantitative RT-PCR for CD14 mRNA. This allowed us to show that LPS acts directly on platelets to induce RNA processing and translation, including cyclooxygenase-2 RNA processing and expression, functionally altered platelet physiology, and initiated platelet coating of quiescent leukocytes.
Materials and Methods
Cell isolation
Human blood in a protocol approved by the Cleveland Clinic IRB was drawn into acid-citrate-dextrose (ACD) and centrifuged (200×g, 20 min) to obtain platelet-rich plasma. All centrifugations were without braking. Platelet-rich plasma was filtered through two layers of 5 μ mesh (BioDesign, Carmel, NY) to remove nucleated cells and recentrifuged (500×g, 20 min) in the presence of 100 nM prostaglandin E1 (PGE1). The pellet was resuspended in 50 ml Pipes/saline/glucose (5 mM Pipes, 145 mM NaCl, 4 mM KCl, 50 μM Na2HPO4, 1 mM MgCl2, and 5.5 mM glucose) containing 100 nM of PGE1. These cells were centrifuged (500×g, 20 min), resuspended in AutoMACS sample buffer, 5 μL anti-CD45-, anti-CD15-, anti-CD14 and anti-glycophorin-coated magnetic beads (Miltenyi) per 109 cells for 25 min with constant rotation before purification in an AutoMACS magnetic separator (Miltenyi). For some experiments, this negative microbead selection was repeated. Light microscopy was used to confirm the cells had a discoidal, unactivated shape. Recovered platelets were centrifuged (500×g, 20 min) and resuspended in HBSS/A (0.5% human serum albumin in HBSS) at 4 × 108 cells/ml for confocal microscopy, aggregation and FACS analysis, and at 8 × 108 platelets/ml for all other uses. Platelet activation was induced by 100 ng/ml of LPS or Kdo2-Lipid A, 10 μg/ml Pam3Cys-SK4, or 0.1U thrombin for the stated time. Autologous serum (0.5%) or recombinant soluble CD14 and LPS-binding protein (final concentration 0.15 μg/ml and 0.1 μg/ml, respectively) was added with TLR4 agonists. Density purified monocytes were isolated as before (36).
RNA isolation and Real Time RT-PCR
Total RNA from 2.4 × 109 platelets or 107 monocytes or PMN was isolated using RnEasy Mini Kit (Qiagen Inc., Valencia, CA) and treated with RNase-free DNase (Qiagen Inc.). Total RNA was quantitated by NanoDrop and used to normalize PCR samples. Real time reverse transcriptase-polymerase chain reaction primers for: IL1β mRNA, sense 5′-AAACCTCTTCGAGGCACAAG-3′ (exon 1), antisense 5′-GTTTAGGGCCATCAGCTTCA-3′ (exon 3); COX-2 mRNA, sense - 5′-TGAAACCCACTCCAAACACA-3′ (exon 3) and antisense – 5′-GAGAAGGCTTCCCAGCTTTT-3′ (exon 4); CD14 mRNA, sense 5′-GGGTTCACAGAGGAGGGAAC-3′ (exon 1), antisense 5′-CGCGCTCCATGGTCGATA-3′ (the junction between exon 1 and 2). Conditions for IL-1β were: reverse transcription (30′, 50°C); PCR 94°C (15″), 61°C (30″), 72°C (30″), data collection (80°C, 15″), 40–45 cycles with SYBR Green I in either an Applied Biosystems ABI Prism 7700 Sequence Detection System or a MyCycler. This amplification across intron 2 does not detect unprocessed IL-1β RNA. Conditions for COX-2 and CD14 were the same, with the exception that annealing temperature was 60 °C and 61.9°C, respectively, and data collection for COX-2 was at 75.5°C. Products were analyzed by melting curve, gel electrophoresis, and sequencing. DNAse I treatment did not affect IL-1β mRNA expression, while RNAse I and no RT abolished amplification.
Confocal microscopy
Platelets were incubated for the stated times in gelatin-coated 4-well borosilicate chambered coverslips. Adherent cells were washed with endotoxin-free PBS and fixed with 2% paraformaldehyde for 30 min. Globular G-actin was detected using Alexa594-DNAse I, and polymerized F-actin with Alexa488-phalloidin. IL-1β expression was visualized with anti-IL-1β antibody and Alexa488-conjugated goat-anti-mouse. Plasma membranes were stained with Alexa594-wheat germ agglutinin. White blood cell nuclei were stained with DAPI. Microscopy used a Plan Apochromat 63X, N.A. 1.4 oil objective and processed with Leica confocal software.
Nucleation assay
Platelets were incubated in gelatin-coated 4-well borosilicate chambered coverslips as above. After 3h of incubation, cells were permeabilized with saponin and challenged with 2 μM rhodamine G-actin for 5 min. Cells were then fixed with 2% paraformaldehyde, blocked by 10 min incubation in 0.1M glycine, washed five times in PBS and counterstained with Alexa488-phalloidin. Samples were mounted in Vectashield (Vector Laboratories, Inc., Burlingame, CA) and examined under epifluorescence. The intensity of G-actin staining was analyzed using Image-Pro software (Media Cybernetics, Inc., Silver Spring, MD).
Flow Cytometry
FACScan analysis used the same anti-IL-1β as above and PerCP-conjugated secondary goat-anti-mouse antibodies. Cells were washed, permeabilized using BD Pharmingen Cytofix/Cytoperm kits. Platelets were gated by forward and side scatter that defined a CD42b+ population.
Aggregometry
ADP initiated aggregation in platelet-rich plasma, assessed by transmittance (Chrono-Log, Havertown, PA). Platelets from different donors demonstrated broad variation in their agonist sensitivity, ranging from 2–10 μg/ml (N = 13), so a threshold concentration was determined for every donor.
Western blotting and mass spectrometry
Monocytes or platelets were incubated with buffer or LPS and MG132, lysed in RIPA buffer, the proteins resolved by SDS-PAGE before the resulting blot was stained with anti-cycloxygenase-2 monoclonal antibody and goat-anti-mouse-HRP tagged secondary antibody. Actin staining of a duplicate blot used mouse-anti-actin monoclonal antibody followed by goat-anti-mouse-HRP tagged secondary antibody. The unique cyclooxygenase-2 peptide LILIGETIK was detected by mass spectrometry after the appropriate sized band was excised from a 1D SDS-PAGE, reduced, alkylated, and digested with trypsin. The digests were analyzed on an LTQ linear ion trap instrument (ThermoFinnigan) with selected reaction monitoring.
Chemicals and Reagents
Chemicals and reagents were purchased from the following sources: sterile filtered HBSS and M199, BioWhittaker (Walkersville, MD); sterile tissue culture plates, Falcon Labware (Lincoln Park, NJ); human serum albumin, Baxter Healthcare (Glendale, CA); endotoxin-free PBS, phenol-extracted LPS (Escherichia coli O111:B4) that is free of lipoprotein contamination, List Biological Laboratories, Inc. (Campbell, CA); Kdo2-Lipid A, Avanti Polar Lipids (Alabaster, AL); Pam3Cys-SK4 and Cdc2-like kinase (Clk-1) inhibitor TG003, Calbiochem (San Diego, CA); 4-well Lab-Tek® II Chamber Slide System, Nalge Nunc International (Naperville, IL); Alexa594 chicken anti-rabbit IgG (H + L), Alexa488-phalloidin, Alexa594-wheat germ agglutinin, Alexa594-DNAse I, and DAPI, Molecular Probes (Eugene, OR); recombinant soluble CD14, LPS-binding protein, IL-1β ELISA kit, ELISpot kit, IL-1β antibody, R&D Systems (Minneapolis, MN); PerCP-conjugated goat-anti-mouse, FITC-mouse IgG1, phycoerythrin-anti-human CD42b, and phycoerythrin-mouse IgG1κ BD Pharmingen (San Diego, CA); anti-cyclooxygenase-2 and cognate peptide, Cayman Chemical (Ann Arbor, MI); rhodamine-labeled G-actin (Cytoskeleton Inc. Denver, CO). Other chemicals were from Sigma or BIOMOL Research Laboratories (Plymouth Meeting, PA).
Expression of data and statistics
Experiments were performed at least three times with cells from different donors, and all assays were performed in triplicate. The mean ± SE from all experiments are presented. Representative experiments are shown in some cases because we observed significant interassay variation in IL-1β expression from different donors. ANOVA was used to determine differences among the groups. If significant differences were found, a Newman-Keuls post hoc procedure was used to determine the location of the difference. P < 0.05 was considered statistically significant.
Results
LPS stimulates IL-1β transcript processing
Thrombin rapidly stimulates platelet processing of IL-1β pre-mRNA to functional, intronless mRNA(18), which we confirmed using real time PCR with primers anchored in exon 1 and 3 that do not amplify unprocessed message (Fig. 1A). The progression curves of the real time PCR show that E. coli LPS also induced platelet IL-1β RNA processing, and was significantly better at this than thrombin. The thousand-fold increase in spliced IL-1β RNA after LPS exposure was time-dependent (Fig 1B); maximal accumulation of processed IL-1β RNA occurred 3 h after stimulation, but with significant accumulation of spliced message 1 h after LPS exposure. Accumulation of processed IL-1β RNA in response to LPS developed more slowly than in response to the complete platelet agonist thrombin, but was both more effective and more prolonged (Fig. 1B). The amount of processed RNA after stimulation with either agonist gradually declined, although not to background levels, by 20 h post stimulation. The loss of processed RNA was not due to a loss of cell viability or functionality because platelets, even after 20 h of continuous LPS stimulation, remained metabolically competent with functional, polarized mitochondria that could be depolarized with the protonophore CCCP (Fig. 1C).
Figure 1. LPS induces IL-1β RNA processing in purified platelets.
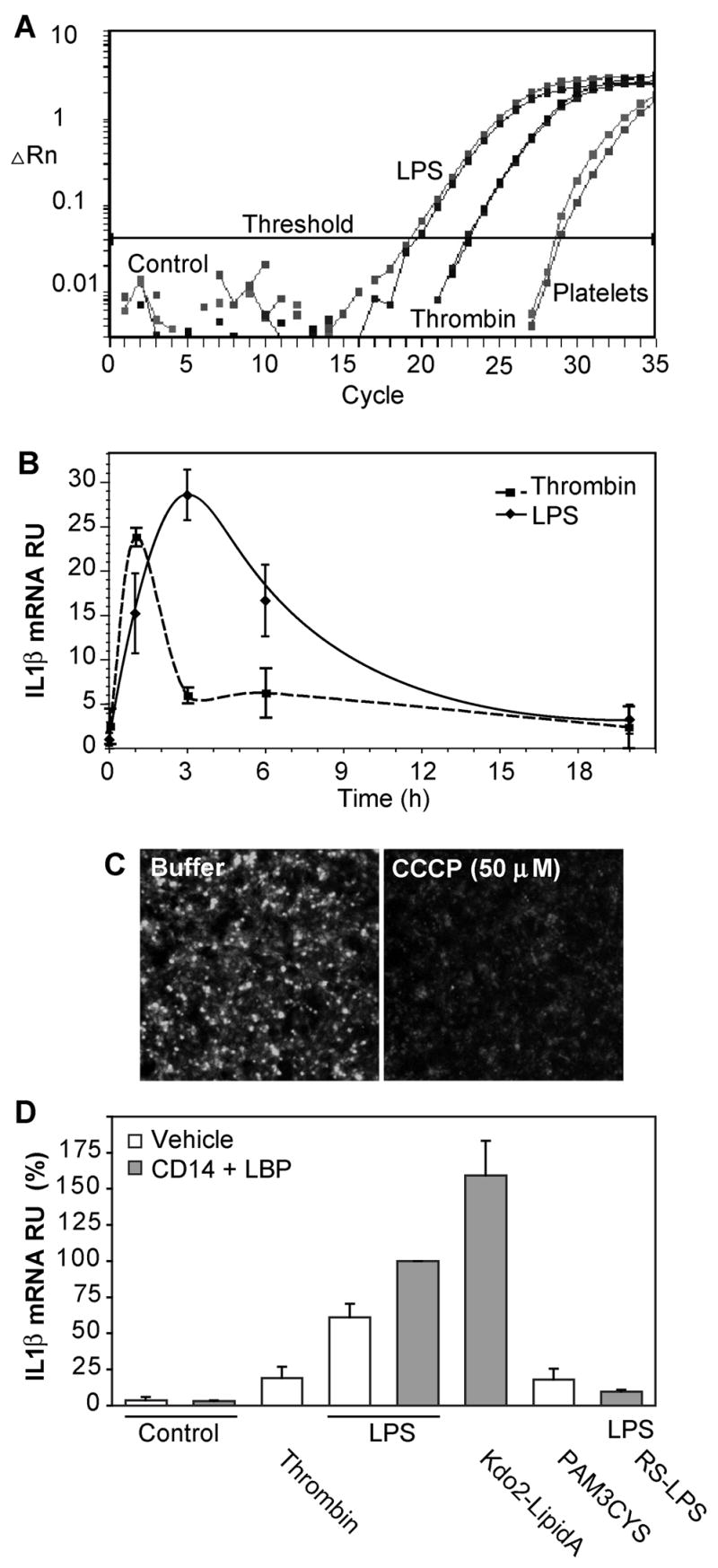
A. Real-time RT-PCR progression curves for spliced IL-1β RNA. Logarithmic accumulation of IL-1β amplicons using primers anchored in exon 1 and 3. Duplicate determinations using RNA extracted from platelets (8 ng total RNA) left untreated, or stimulated with 0.1 U thrombin for 30 min, or 100 ng/ml of LPS along with 150 ng/ml of recombinant CD14 and 100 ng/ml LPS-binding protein (LBP) for 3 hours. ▵Rn is the magnitude of the signal normalized to total RNA. B. Kinetics of IL-1β mRNA accumulation. The accumulation of spliced IL-1β RNA in purified human platelets treated with 0.1 u/ml thrombin or LPS in the presence of soluble CD14 and LBP (in three separate experiments ± SE) compared to the basal amount by quantitative RT-PCR as in the above panel. C. Platelets remain viable after prolonged stimulation. Accumulation of the cationic dye MitoTracker Red®, with or without subsequent dissipation of the mitochondrial transmembrane potential with CCCP, was imaged after 20 h of LPS exposure. D. LPS stimulation of platelet RNA splicing requires TLR4 receptor components. Purified platelets were stimulated with 100 ng/ml LPS, 100 ng/ml KDO2-Lipid A, 100 μg/ml Pam3Cys-SK4, or 0.1 units of thrombin before RNA isolated 3 hours later. Some samples contained soluble CD14 and LBP, while the competitive TLR4 receptor antagonist Rhodobacter sphaeroides LPS (RS-LPS) was present in one sample as stated. Bars represent standard errors. N = 4.
The response to E. coli LPS was receptor mediated because it was enhanced by a source of soluble CD14, which is needed to present LPS to TLR4, that we supplied as recombinant CD14 along with LPS-binding protein (Fig. 1D). Additionally the response to LPS was blocked by the competitive TLR4 antagonist (37) Rhodobacter sphaeroides LPS (Fig. 1D). The invariant lipid core of LPS was all that was required to stimulate platelet splicing because the homogeneous lipid ligand Kdo2-lipid A that lacks the outer carbohydrate chain of LPS was a highly effective agonist. Platelets express(4) the TLR2 receptor for endotoxic bacterial lipoproteins(38), and we found (Fig. 1D) that the synthetic TLR2 agonist Pam3-Cys-SK4, which contains the bioactive lipid modification of Gram negative and Gram positive bacterial lipoproteins, stimulated IL-1β RNA splicing. This synthetic lipopeptide was less effective than LPS, being about as effective as thrombin.
LPS stimulates IL-1β protein accumulation in platelets
Newly synthesized IL-1β primarily remains cell-associated in purified preparations of platelets stimulated by thrombin(16). Since platelet preparations contain contaminating monocytes, we used flow cytometery to define the population(s) of cells that synthesized and accumulated IL-1β in response to LPS stimulation. We gated the platelet population by forward and side scatter and then for cells that stained for surface CD42b (not shown). Staining this population for IL-1β after permeabilization revealed no staining beyond background levels in unstimulated cells, with an increase in IL-1β over time after LPS stimulation (Fig. 2A). This occurred in a way that varied with the concentration of agonist, with as little as 10 ng of E. coli LPS increasing the content of immunoreactive IL-1β (Fig. 2B).
Figure 2. LPS treatment induces IL1β expression.

A. Platelet IL-1β accumulation is time dependent. Purified platelets were treated with 100 ng/ml of E. coli LPS, recombinant CD14 and recombinant LBP for the stated times as in Fig. 1, permeabilized and stained with either anti-IL-1β or an isotype matched control antibody and then analyzed by flow cytometry as described in “Materials and Methods”. Both non-immune and anti-IL-1β antibodies were used at time 0 to define background binding, the shaded area and the light gray trace, that were equivalent. The ordinate is the number of events normalized to the maximal number of counts, the abscissa is the fluorescent channel 1 bin. B. LPS concentration/response profile. Platelets were treated with the stated concentration of LPS, along with recombinant CD14 and LBP, for 1 hour, and then assessed for IL-1β expression as above. Representative of four independent experiments.
Monocytes are not required in the platelet response to LPS
Monocytes may directly, or indirectly through cytokine production, contribute to platelet IL-1β accumulation, and quantification of small numbers of contaminating monocytes is problematic. We took advantage of the abundant expression of CD14 by monocytes and the paucity of this component of LPS signaling on platelets to establish a quantitative definition of monocyte contamination by real time PCR for CD14 mRNA. This mRNA was abundant in unstimulated monocytes (Fig. 3A), but was present in platelet-rich plasma even after filtration through 5μ mesh. We immunodepleted monocytes with a combination of anti-CD14, anti-CD15 and anti-CD45 conjugated to magnetic beads, while a second immunodepletion after addition of additional beads further reduced the CD14 RNA content. Based on RNA content, we calculate that 106 filtered, immunodepleted, and doubly immunodepleted platelets could contain up to 17 ± 16, 7.4 ± 3.6, and 2.5 ± 0.9 monocytes, respectively.
Figure 3. Platelets are a direct target of LPS stimulation.

A. Quantitative assessment of the effectiveness of platelet purification by immunodepletion of leukocytes. Platelet rich plasma (PRP) was purified using anti-CD14, -CD15, -CD45 and anti-glycophorin coated magnetic beads by passage once or twice over a Miltenyi AutoMACS column. RNA was extracted from the recovered cells and normalized for total RNA content before the content of CD14 RNA was quantitatively assessed in duplicate samples by real-time PCR as described in “Materials and methods”. B. ELISpot detection of IL-1β producing cells in a preparation of purified platelets. The top leftmost panel is a negative buffer control, 0.8 ng recombinant IL-1β (top middle), and 20,000 density purified monocytes stimulated by LPS (top right). The right panel shows 107 platelets stimulated by 100 ng LPS for 17 hours. C. Confocal microscopy of cells in a platelet preparation expressing IL-1β. Platelets suspended over gelatin-coated cover slips were treated with 100 ng/ml LPS for the stated times, permeabilized, and then stained with mouse anti-IL-1β and Alexa 488-conjugated anti-mouse antibody (green) and Alexa 594-conjugated wheat germ agglutinin (WGA) that stains plasma membrane sialated proteins and gangliosides (red). The fixative contained the nuclear stain DAPI to identify contaminating white blood cells (blue). Upper inset: Higher magnification (3.7×) shows IL-1β is localized as punctate intracytoplasmic inclusions at times shortly after LPS addition. Lower inset: 24 h of incubation in the absence of LPS reveals lower levels of IL-1β staining. Representative of three experiments.
Monocyte-depleted platelet preparations still produced IL-1β after LPS stimulation when analyzed by ELISpot (Fig. 3B), and the frequency of cells expressing IL-1β after LPS stimulation was significantly greater than the 25 ± 9 monocytes that could have been present in the well. We visualized the cells that contained IL-1β by confocal microscopy and used DAPI to stain any contaminating nucleated cells in the platelet preparation. Confocal microscopy showed that LPS increased the number and intensity of cells that stained for IL-1β protein (Fig, 3C), which was maximal by 6 h and then declined modestly after 18 h of LPS exposure. DAPI labeling of cell nuclei showed the platelet preparation was depleted of nucleated cells, and that all of the newly generated IL-1β protein was associated with anucleate platelets. We also found (not shown) that inhibition of transcription by actinomycin D suppressed monocyte production of IL-1β, but not that associated with anuclate platelets. These data show stimulation of monocyte cytokine production has no part in the platelet response to LPS.
LPS stimulates platelet cyclooxygenase-2 RNA splicing
Quiescent platelets are reported(33, 34) to contain no cyclooxygenase-2, but to test whether LPS-stimulated platelets might do so we designed primers that hybridized within exon 3 of cyclooxygenase-2 and at the junction formed by the splicing of exon 3 and 4. Little processed cyclooxygenase-2 RNA was present in unstimulated platelets, but we found that purified platelets stimulated by either LPS or the homogenous endotoxin Kdo2-Lipid A now contained spliced cyclooxygenase-2 RNA (Fig. 4A). Amplification across exons 9 and 10 confirmed that LPS initiated cyclooxygenase-2 splicing (not shown). The synthetic TLR2 agonist (Fig. 4A) and thrombin (not shown) also stimulated splicing, but again were not as effective as the TLR4 agonists. Two color flow cytometery (Fig. 4B) showed the platelets that accumulated IL-1β protein after LPS stimulation also accumulated cyclooxygenase-2 protein. Highly purified platelet preparations expressed immunoreactive cyclooxygenase-2 at the appropriate molecular weight after 17 h of LPS treatment (Fig. 4C). Staining of this band was abolished by the cognate peptide, and peptide sequencing of this band by mass spectrometry demonstrated unique cyclooxygenase-2 peptides (not shown).
Figure 4. Platelets express cyclooxygenase-2 after LPS stimulation.

A. Progression curve of real-time PCR for spliced cyclooxygenase-2 RNA. Purified platelets were stimulated with 100 ng E. coli LPS, 100 ng KDO2-Lipid A, 100 μg Pam3Cys-SK4 or left untreated. RNA was extracted and tested as in Figure 3 using primers anchored exon 3 and the junction formed by splicing exons 3 and 4. B. Platelet expression of IL-1β and cyclooxygenase-2. Purified platelets were stimulated with 100 ng/ml E. coli LPS for 3h or left untreated, permeabilized, fixed and stained with a combination of mouse anti-IL-1β and PerCP-conjugated goat anti-mouse antibody followed by a directly conjugated FITC-anti-cyclooxygenase-2 antibody. The cells were subjected to flow cytometry, gated by forward and side scatter, and FITC and PerCP fluorescence was assessed in channels FL1 and FL3, respectively. This experiment has been replicated more than fifteen times, although the level of cyclooxygenase-2 expression varied among donors. C. Cyclooxygenase-2 immunoblot in extracts of purified platelets. Platelets or density-purified monocytes were treated with LPS, recombinant soluble CD14 and LPS binding protein as in the preceding panel for 17 h, while monocytes were treated with 100 ng/ml LPS alone for 17 h. Upper panel, anti-cyclooxygenase-2 western blot. Middle panel, anti-cyclooxygenase-2 western blot using antibody pre-incubated with its cognate peptide. Lower panel, β-actin western blot.
LPS alters platelet physiology over time
LPS was not a typical platelet agonist, and did not induce rapid platelet shape change or aggregation (not shown), as previously reported(10). However, both LPS and Kdo2-Lipid A augmented ADP-induced aggregation in platelet rich plasma (Fig. 5A). In contrast to splicing, the synthetic TLR2 agonist Pam3Cys was not an agonist for this response. Changes in platelet physiology in response to LPS included a rearrangement of their cytoskeleton with an increase in F-actin at the expense of monomeric G-actin (Fig. 5B). Direct quantification of actin nucleation in platelet lysates by incorporation of rhodamine-labeled G-actin showed 3h of LPS treatment increased actin polymerization 3.8 ± 0.8 fold. Changes in platelet physical structure after LPS exposure was also reflected in the change in both forward and side scatter during flow cytometry (Fig. 5C) reflecting changes in granularity and microparticle formation.
Figure 5. LPS primes platelet aggregation and alters platelet structure.
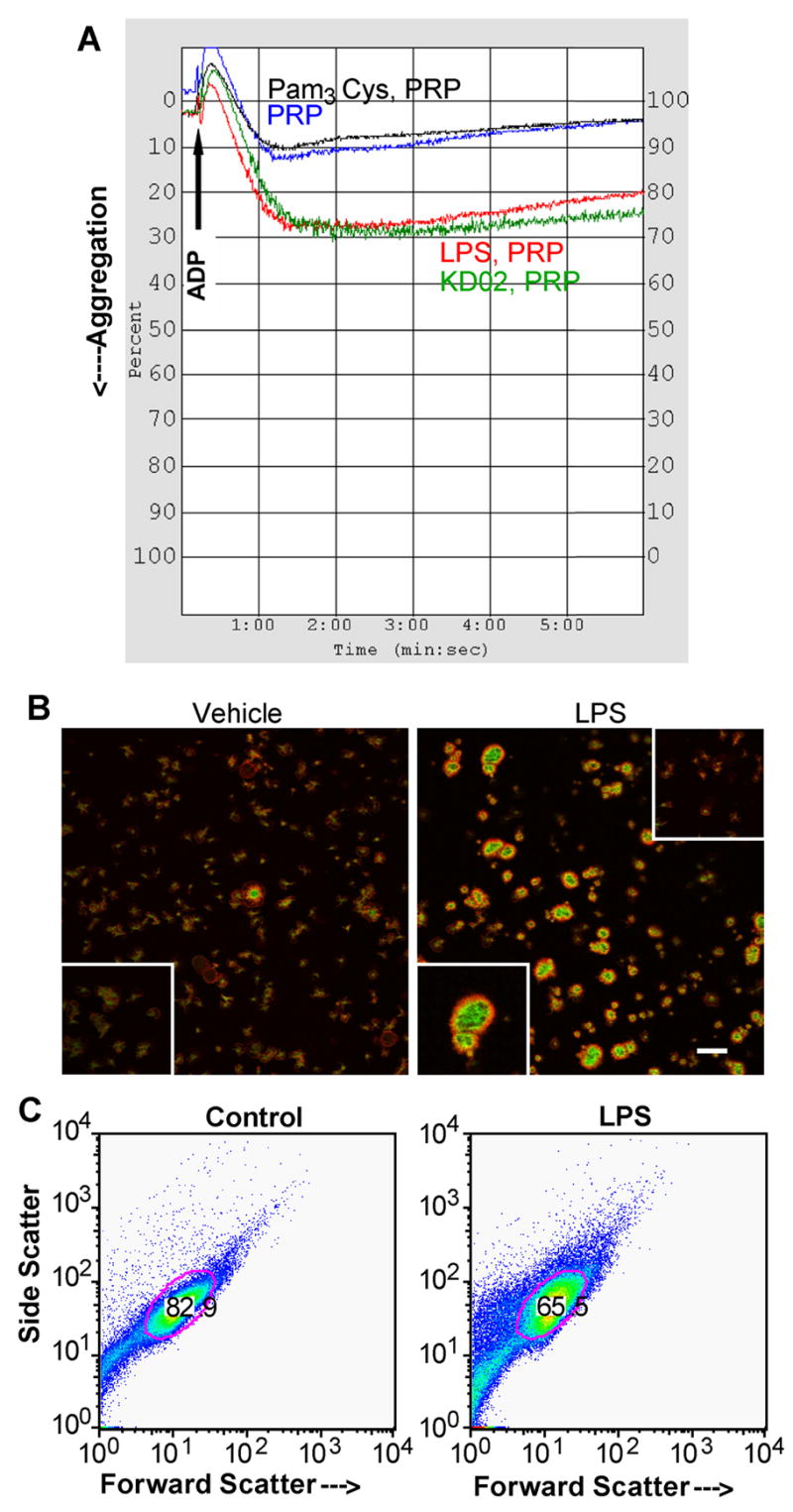
A. LPS primes ADP-induced aggregation in platelet-rich plasma (PRP). LPS (100 ng), KDO2-Lipid A (100 ng) or Pam3Cys-SK4 (100 μg) was added to 1 ml of PRP and incubated for 1h before addition to a stirred aggregometer cuvette. As shown by the arrow, 20 μM ADP was added and light transmittance was assessed over the subsequent 6 min. LPS did not induce aggregation without the addition of ADP (not shown). B. Polymerized actin staining of adherent platelets. Confocal microscopy images of control (left) and platelets stimulated with 100 ng/ml E. coli LPS (right) in the presence of autologous serum or soluble CD14 and LPS binding protein for 3h. Green staining is polymerized (F-actin) stained by FITC-phalloidin and red fluorescence is unpolymerized G-actin. The bar is 20 μm. Lower inserts: 2× magnified fields of respective images. Upper insert: pretreatment with cytochalasin B abrogated actin polymerization. C. LPS affects platelet structure. Platelets were treated or not with LPS for 3 h as in the preceding panel and then analyzed by flow cytometry for forward scatter (size) and side scatter (granularity). The area of the gate imposed on the dot blot stained for the platelet protein CD42b. This experiment has been repeated more than twenty times.
Surface expression of granule-stored proteins was increased after 3h of LPS exposure. Flow cytometry showed that LPS increased expression of CD40L in some cells with the formation of a CD40L high population (Fig. 6A). LPS also initiated a time-dependent increase in surface P-selection expression that was evident by 1 h of exposure encompassing most platelets, with one population becoming particularly bright (Fig. 6B). Neutrophils constitutively express PSGL-1, which serves as a ligand for the P-selectin displayed by activated platelets. Platelets treated with LPS for 2.5 h to increase surface P-selectin readily associated with quiescent neutrophils (Fig. 6C). Blockade of this ligand-receptor interaction with anti-P-selectin monoclonal antibody suppressed platelet-induced neutrophil interaction (not shown).
Figure 6. LPS induces degranulation and P-selectin expression that promotes neutrophil binding.

A. CD40L surface expression. Platelets were stimulated by LPS, or not, for 3h, fixed and stained with either mouse anti-CD40L or an isotype matched control antibody before the cells were analyzed by flow cytometry. MFI, mean fluorescence intensity. B. P-selectin surface expression. Platelets were treated with buffer or 100 ng/ml LPS for 1 or 2 hours before fixation, antibody staining, and flow cytometry as before. The shadowed area shows unstained platelets, the thin line defines isotype control antibody staining, while the bold line shows P-selectin (CD62P) staining. C. LPS-stimulated platelets decorate quiescent neutrophils. Platelets were treated with buffer or LPS for 2.5 h as before, washed, and then suspended with quiescent autologous neutrophils for 10 min on gelatin-coated slides, fixed, and stained for P-selectin (green FITC fluorescence), plasma membrane by Alexa 594-conjugated wheat germ agglutinin (red), and nuclei with DAPI (blue). The images are representative of three experiments.
Discussion
These data show platelets are components of the innate immune system that recognize and then respond to endotoxin. Over time, LPS sensitized platelets to soluble agonists, altered the platelet cytoskeleton, altered platelet size and granularity, generated microparticles, promoted degranulation, induced surface expression of P-selectin and CD40L, and promoted leukocyte binding. LPS also proved to be a robust agonist of platelet RNA processing and translation that eclipsed thrombin as an agonist. This occurred in the virtual absence of contaminating monocytes, which we could document by quantifying CD14 mRNA. Therefore while LPS is a slowly acting secondary agonist for many of the responses typical of platelets, it now becomes the most robust agonist that alters the platelet transcriptome and proteome.
Whether LPS directly affects platelet function has been controversial(9, 10), but in vivo experiments are definitive and show the TLR4 receptor of platelets has a role at least in the systemic response to endotoxin(2, 4, 5). A critical issue to be answered in experiments examining LPS as an ex vivo platelet agonist is the potential role of monocytes remaining in the preparation because monocytes abundantly produce IL-1β that might then be incorrectly ascribed to activated platelets (24). Moreover, it is possible that LPS acts solely on monocytes to produce an agonist that then acts on platelets. Quantification of contaminating leukocytes at less than one per hundred thousand platelets(24) is problematic by flow cytometry(25), and platelet-monocyte aggregates will be excluded from the analysis. We approached this issue by first creating a protocol that produced highly purified platelet populations and then devising a method to quantitatively assess monocytic cell content in this preparation. We filtered platelet-rich plasma through 5 mircon mesh, and then negatively purified platelets by removing cells that expressed CD14, CD15 and CD45, along with anti-glycophorin to remove residual erythrocytes, by magnetic bead separation. While we found this reduced leukocyte contamination, we also found that a second pass through the AutoMax column after addition of additional antibodies and magnetic beads further reduced nucleated cell contamination, perhaps because the sample overloaded the capacity of the column.
Platelets are not known to express CD14 and accordingly we found it necessary to provide exogenous CD14, provided either as a recombinant protein or as the constitutively expressed protein in human serum (not shown), to aid their response to LPS. This enhancement varied among donors, but most often the response was greatly enhanced by exogenous CD14. Platelets, then, are analogous to endothelial cells(39, 40) that are aided by circulating soluble CD14 and LPS binding protein. In contrast, CD14 is a prominent mRNA and protein of monocytes, and so quantifying CD14 mRNA by real time PCR provides a quantifiable measure of the number of monocytes that could be present in any given platelet preparation. We used this value to show that the number of cells expressing IL-1β in an ELISpot assay was significantly larger than the maximum number of monocytes that could be present, and used this approach to show that an equivalent number of monocytes fail to produce detectable IL-1β (not shown). We found that reducing the number of contaminating monocytes to a few per million platelets did not diminish the responsiveness of the platelet population to LPS, and, as a complementary direct test, we found that blocking transcription with actinomycin D blocked IL-1β production by purified monocytes but not purified platelets. All these data show that LPS is a direct platelet agonist.
LPS stimulation of platelets initiated splicing of cyclooxygenase-2 RNA and ultimately, production of the corresponding protein. This occurred in highly purified platelet populations and in a population of cells defined as platelets during flow cytometry by forward, scatter, side scatter, and CD42b staining. Western blotting and mass spectrometry confirmed the presence of the protein in LPS-stimulated platelets. We emphasize, however, the extreme variability of this response, even when compared to IL-1β expression; the protein was present in some preparations without activation, while in others it could not be upregulated by LPS stimulation. The basis for this variability may lie in donor variability. More importantly, we could not demonstrate increased product formation nor sensitivity to the cyclooxygenase-2 inhibitor NS398 (not shown). Unstimulated platelets are not a source of cyclooxygenase-2 products(34), and it appears LPS stimulated platelets either generate protein that is not active, or it is present in insufficient quantities to affect arachidonate metabolism.
These data show that LPS synergizes with rapidly acting stimuli, but over time develops an independent capacity as a direct platelet agonist. Platelets also express TLR1, TLR2, TLR6 and TLR9 (4, 41, 42), and a synthetic TLR2 ligand—the only other TLR ligand we tested—proved to be an agonist, although a weak one, for splicing. Agonist stimulation of platelet splicing and activation-dependent translation of platelet mRNA includes IL-1β(18), Bcl-3 that aids clot retraction(43), tissue factor(19), now cyclooxygenase-2, and indeed many other currently unidentified proteins(11). LPS promoted platelet granule release accompanied by surface P-selectin expression that then promoted leukocyte interaction, so in vivo endotoxin exposure has the potential to directly alter platelet reactivity. Indeed, in vivo exposure to LPS shows platelet TLR4 to be a critical component of the response to endotoxemia (2).
A key distinction between the findings reported here and other experiments examining the effect of LPS on platelets that requires emphasis is that LPS is not a typical rapidly-acting, soluble agonist. Thus, the response of platelets to LPS developed over time and, for example, the binding of human neutrophils to LPS-stimulated platelets was apparent after several hours (Fig. 6C) and not after several minutes (not shown) of incubation. Platelet responses are typically analyzed as immediate responses to introduced agonists, and LPS modulation of platelet function does not fit the paradigm of established platelet agonists. The measured rate of change in platelet phenotype after LPS stimulation indicates that the effect of LPS on platelets will manifest in systemic changes, rather than local ones, as indeed has been observed in in vivo models of endotoxemia (2).
Acknowledgments
The technical aid of Mark Calabro, Manisha Sharma, Jessica Cemate and Stacy Macecevic is greatly appreciated, as was the helpful advice of Drs. Andrew Weyrich and Guy Zimmerman. The significant aid of Dr. Michael Kinter and the Mass Spectrometry core facility and the aid of Dr. Josephine Adams with the nucleation assay are gratefully acknowledged. We thank Dr. D. Zeldin (NIH/NIEHS, Research Triangle Park, NC) for the gift of recombinant cyclooxygenase-1. Figure preparation by Diana Lim is, again, greatly appreciated. We appreciate the support provided by the Flow Cytometry and Imaging core laboratories of the Lerner Research Institute. The authors have no commercial conflicts.
References
- 1.Palsson-McDermott EM, O’Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood. 2005;106:2417–2423. doi: 10.1182/blood-2005-03-0916. [DOI] [PubMed] [Google Scholar]
- 3.Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol. 2005;83:196–198. doi: 10.1111/j.1440-1711.2005.01314.x. [DOI] [PubMed] [Google Scholar]
- 4.Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, Ni H, Lazarus AH, Freedman J, Semple JW. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. 2006;107:637–641. doi: 10.1182/blood-2005-06-2202. [DOI] [PubMed] [Google Scholar]
- 5.Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, Watkins SL, Johnson R, Karpman D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. 2006;108:167–176. doi: 10.1182/blood-2005-08-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lodrup Carlsen KC, Granum B. Soluble CD14: role in atopic disease and recurrent infections, including otitis media. Curr Allergy Asthma Rep. 2007;7:436–443. doi: 10.1007/s11882-007-0067-2. [DOI] [PubMed] [Google Scholar]
- 7.Rumbaut RE, Bellera RV, Randhawa JK, Shrimpton CN, Dasgupta SK, Dong JF, Burns AR. Endotoxin enhances microvascular thrombosis in mouse cremaster venules via a TLR4-dependent, neutrophil-independent mechanism. Am J Physiol Heart Circ Physiol. 2006;290:H1671–1679. doi: 10.1152/ajpheart.00305.2005. [DOI] [PubMed] [Google Scholar]
- 8.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH, Kubes P. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 9.Montrucchio G, Bosco O, Del Sorbo L, Fascio Pecetto P, Lupia E, Goffi A, Omede P, Emanuelli G, Camussi G. Mechanisms of the priming effect of low doses of lipopolysaccharides on leukocyte-dependent platelet aggregation in whole blood. Thromb Haemost. 2003;90:872–881. doi: 10.1160/TH03-02-0085. [DOI] [PubMed] [Google Scholar]
- 10.Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, Dower SK, Buttle DJ, Sabroe I. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb Haemost. 2005;94:831–838. [PubMed] [Google Scholar]
- 11.Weyrich AS, Dixon DA, Pabla R, Elstad MR, McIntyre TM, Prescott SM, Zimmerman GA. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci U S A. 1998;95:5556–5561. doi: 10.1073/pnas.95.10.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bahou WF, Gnatenko DV. Platelet transcriptome: the application of microarray analysis to platelets. Semin Thromb Hemost. 2004;30:473–484. doi: 10.1055/s-2004-833482. [DOI] [PubMed] [Google Scholar]
- 13.Macaulay IC, Carr P, Gusnanto A, Ouwehand WH, Fitzgerald D, Watkins NA. Platelet genomics and proteomics in human health and disease. J Clin Invest. 2005;115:3370–3377. doi: 10.1172/JCI26885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dittrich M, Birschmann I, Pfrang J, Herterich S, Smolenski A, Walter U, Dandekar T. Analysis of SAGE data in human platelets: features of the transcriptome in an anucleate cell. Thromb Haemost. 2006;95:643–651. [PubMed] [Google Scholar]
- 15.Pabla R, Weyrich AS, Dixon DA, Bray PF, McIntyre TM, Prescott SM, Zimmerman GA. Integrin-dependent control of translation: engagement of integrin alphaIIbbeta3 regulates synthesis of proteins in activated human platelets. J Cell Biol. 1999;144:175–184. doi: 10.1083/jcb.144.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, Weyrich AS. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001;154:485–490. doi: 10.1083/jcb.200105058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindemann S, Tolley ND, Eyre JR, Kraiss LW, Mahoney TM, Weyrich AS. Integrins regulate the intracellular distribution of eukaryotic initiation factor 4e in platelets. a checkpoint for translational control. J Biol Chem. 2001;276:33947–33951. doi: 10.1074/jbc.M104281200. [DOI] [PubMed] [Google Scholar]
- 18.Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, Fratto CM, Tolley E, Kraiss LW, McIntyre TM, Zimmerman GA, Weyrich AS. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005;122:379–391. doi: 10.1016/j.cell.2005.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwertz H, Tolley ND, Foulks JM, Denis MM, Risenmay BW, Buerke M, Tilley RE, Rondina MT, Harris EM, Kraiss LW, Mackman N, Zimmerman GA, Weyrich AS. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenicity of human platelets. J Exp Med. 2006;203:2433–2440. doi: 10.1084/jem.20061302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Panes O, Matus V, Saez CG, Quiroga T, Pereira J, Mezzano D. Human platelets synthesize and express functional tissue factor. Blood. 2007;109:5242–5250. doi: 10.1182/blood-2006-06-030619. [DOI] [PubMed] [Google Scholar]
- 21.Hawrylowicz CM, Howells GL, Feldmann M. Platelet-derived interleukin 1 induces human endothelial adhesion molecule expression and cytokine production. J Exp Med. 1991;174:785–790. doi: 10.1084/jem.174.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hawrylowicz CM, Santoro SA, Platt FM, Unanue ER. Activated platelets express IL-1 activity. J Immunol. 1989;143:4015–4018. [PubMed] [Google Scholar]
- 23.Loppnow H, Bil R, Hirt S, Schonbeck U, Herzberg M, Werdan K, Rietschel ET, Brandt E, Flad HD. Platelet-derived interleukin-1 induces cytokine production, but not proliferation of human vascular smooth muscle cells. Blood. 1998;91:134–141. [PubMed] [Google Scholar]
- 24.Pillitteri D, Bassus S, Boller K, Mahnel R, Scholz T, Westrup D, Wegert W, Kirchmaier CM. Thrombin-induced interleukin 1beta synthesis in platelet suspensions: impact of contaminating leukocytes. Platelets. 2007;18:119–127. doi: 10.1080/09537100600800792. [DOI] [PubMed] [Google Scholar]
- 25.van der Meer PF, Gratama JW, van Delden CJ, Laport RF, Levering WH, Schrijver JG, Tiekstra MJ, Keeney M, de Wildt-Eggen J. Comparison of five platforms for enumeration of residual leucocytes in leucoreduced blood components. Br J Haematol. 2001;115:953–962. doi: 10.1046/j.1365-2141.2001.03154.x. [DOI] [PubMed] [Google Scholar]
- 26.Semple JW, Speck ER, Cosgrave D, Lazarus AH, Blanchette VS, Freedman J. Extreme leukoreduction of major histocompatibility complex class II positive B cells enhances allogeneic platelet immunity. Blood. 1999;93:713–720. [PubMed] [Google Scholar]
- 27.Kinsella BT, O’Mahony D, Lawson JA, Pratico D, Fitzgerald GA. Cellular activation of thromboxane receptors. Ann N Y Acad Sci. 1994;714:270–278. doi: 10.1111/j.1749-6632.1994.tb12054.x. [DOI] [PubMed] [Google Scholar]
- 28.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 29.Patrono C, Rocca B. Drug insight: aspirin resistance--fact or fashion? Nat Clin Pract Cardiovasc Med. 2007;4:42–50. doi: 10.1038/ncpcardio0728. [DOI] [PubMed] [Google Scholar]
- 30.Censarek P, Freidel K, Udelhoven M, Ku SJ, Hohlfeld T, Meyer-Kirchrath J, Schror K, Weber AA. Cyclooxygenase COX-2a, a novel COX-2 mRNA variant, in platelets from patients after coronary artery bypass grafting. Thromb Haemost. 2004;92:925–928. doi: 10.1160/TH04-05-0302. [DOI] [PubMed] [Google Scholar]
- 31.Weber AA, Przytulski B, Schumacher M, Zimmermann N, Gams E, Hohlfeld T, Schror K. Flow cytometry analysis of platelet cyclooxygenase-2 expression: induction of platelet cyclooxygenase-2 in patients undergoing coronary artery bypass grafting. Br J Haematol. 2002;117:424–426. doi: 10.1046/j.1365-2141.2002.03423.x. [DOI] [PubMed] [Google Scholar]
- 32.Rocca B, Secchiero P, Ciabattoni G, Ranelletti FO, Catani L, Guidotti L, Melloni E, Maggiano N, Zauli G, Patrono C. Cyclooxygenase-2 expression is induced during human megakaryopoiesis and characterizes newly formed platelets. Proc Natl Acad Sci U S A. 2002;99:7634–7639. doi: 10.1073/pnas.112202999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patrignani P, Sciulli MG, Manarini S, Santini G, Cerletti C, Evangelista V. COX-2 is not involved in thromboxane biosynthesis by activated human platelets. J Physiol Pharmacol. 1999;50:661–667. [PubMed] [Google Scholar]
- 34.Reiter R, Resch U, Sinzinger H. Do human platelets express COX-2? Prostaglandins Leukot Essent Fatty Acids. 2001;64:299–305. doi: 10.1054/plef.2001.0276. [DOI] [PubMed] [Google Scholar]
- 35.Weyrich AS, Lindemann S, Tolley ND, Kraiss LW, Dixon DA, Mahoney TM, Prescott SP, McIntyre TM, Zimmerman GA. Change in protein phenotype without a nucleus: translational control in platelets. Semin Thromb Hemost. 2004;30:491–498. doi: 10.1055/s-2004-833484. [DOI] [PubMed] [Google Scholar]
- 36.Weyrich AS, Denis MM, Kuhlmann-Eyre JR, Spencer ED, Dixon DA, Marathe GK, McIntyre TM, Zimmerman GA, Prescott SM. Dipyridamole selectively inhibits inflammatory gene expression in platelet-monocyte aggregates. Circulation. 2005;111:633–642. doi: 10.1161/01.CIR.0000154607.90506.45. [DOI] [PubMed] [Google Scholar]
- 37.Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, Fenton MJ, Oikawa M, Qureshi N, Monks B, Finberg RW, Ingalls RR, Golenbock DT. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J Clin Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morr M, Takeuchi O, Akira S, Simon MM, Muhlradt PF. Differential recognition of structural details of bacterial lipopeptides by toll-like receptors. Eur J Immunol. 2002;32:3337–3347. doi: 10.1002/1521-4141(200212)32:12<3337::AID-IMMU3337>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 39.Frey EA, Miller DS, Jahr TG, Sundan A, Bazil V, Espevik T, Finlay BB, Wright SD. Soluble CD14 participates in the response of cells to lipopolysaccharide. J Exp Med. 1992;176:1665–1671. doi: 10.1084/jem.176.6.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pugin J, Schurer-Maly CC, Leturcq D, Moriarty A, Ulevitch RJ, Tobias PS. Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14. Proc Natl Acad Sci U S A. 1993;90:2744–2748. doi: 10.1073/pnas.90.7.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Semple JW, Coopamh M, Freedman J. Murine platelets express toll like receptor 2: A potential regulator of innate and adaptive immunity. Platelets. 2004;15:267. [Google Scholar]
- 42.Shiraki R, Inoue N, Kawasaki S, Takei A, Kadotani M, Ohnishi Y, Ejiri J, Kobayashi S, Hirata K, Kawashima S, Yokoyama M. Expression of Toll-like receptors on human platelets. Thrombosis Res. 2004;113:379–385. doi: 10.1016/j.thromres.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 43.Weyrich AS, Denis MM, Schwertz H, Tolley ND, Foulks J, Spencer E, Kraiss LW, Albertine KH, McIntyre TM, Zimmerman GA. mTOR-dependent synthesis of Bcl-3 controls the retraction of fibrin clots by activated human platelets. Blood. 2006;109:1975–1983. doi: 10.1182/blood-2006-08-042192. [DOI] [PMC free article] [PubMed] [Google Scholar]