

Abstract
Redox regulation of inducible nitric oxide synthase (iNOS) expression was investigated in lipopolysaccharide and interferon-γ (LPS+IFNγ)-stimulated microvascular endothelial cells from mouse skeletal muscles. Unstimulated endothelial cells produced reactive oxygen species (ROS) sensitive to inhibition of NADPH oxidase (apocynin and DPI), mitochondrial respiration (rotenone) and NOS (L-NAME). LPS+IFNγ caused a marked increase in ROS production; this increase was abolished by inhibition of NADPH oxidase (apocynin, DPI and p47phox deficiency). LPS+IFNγ induced substantial expression of iNOS protein. iNOS expression was prevented by the antioxidant ascorbate, NADPH oxidase inhibition (apocynin, DPI and p47phox deficiency), but not by inhibition of mitochondrial respiration (rotenone) and xanthine oxidase (allopurinol). iNOS expression also was prevented by selective antagonists of ERK, JNK, Jak2, and NFκB activation. LPS+IFNγ stimulated activation/phosphorylation of ERK, JNK, and Jak2 and activation/degradation of IκB, but only the activation of JNK and Jak2 was sensitive to ascorbate, apocynin and p47phox deficiency. Ascorbate, apocynin and p47phox deficiency also inhibited the LPS+IFNγ-induced DNA binding activity of transcription factors IRF1 and AP1 but not NFκB. In conclusion, LPS+IFNγ-induced NFκB activation is necessary for iNOS induction but is not dependent on ROS signaling. LPS+IFNγ-stimulated NADPH oxidase activity produces ROS that activate the JNK-AP1 and Jak2-IRF1 signaling pathways required for iNOS induction. Since blocking either NFκB activation or NADPH oxidase activity is sufficient to prevent iNOS expression, they are separate targets for therapeutic interventions that aim to modulate iNOS expression in sepsis.
Keywords: NADPH oxidase, inducible nitric oxide synthase, redox signaling, sepsis, endothelial cells
INTRODUCTION
In sepsis, endotoxin and cytokines stimulate the expression of inducible nitric oxide synthase (iNOS) and overproduction of nitric oxide (NO) in various tissues (Titheradge, 1999). While the high concentration of NO synthesized by iNOS is thought to be beneficial because it is bactericidal (Fierro et al., 1999), excessive NO may be detrimental because it causes endothelial dysfunction, hypotension and multiple organ failure (Chauhan et al., 2003; Su et al., 2007; Titheradge, 1999). These detrimental effects of NO may be mediated by peroxynitrite, the product of the reaction between NO and superoxide (Cuzzocrea et al., 2006).
Modulation of iNOS gene transcription is the major mechanism regulating iNOS activity (Kleinert et al., 2004; Lowenstein and Padalko, 2004). Stimulation with endotoxin (Escherichia coli lipopolysaccharide, LPS) and interferon-γ (IFNγ) has been used to induce iNOS in various cell types (Lowenstein and Padalko, 2004). In some cells, LPS+IFNγ stimulates signaling pathways that activate the transcription factors nuclear factor-κB (NFκB), interferon regulatory factor 1 (IRF1) and activator protein-1 (AP1), which are required for iNOS gene transcription (Kleinert et al., 2004; Lowenstein and Padalko, 2004). Importantly, the activation of some of these transcription factors may also be influenced by reactive oxygen species (ROS) (Liu et al., 2005; Macdonald et al., 2003).
NADPH oxidases are the major sources of ROS in endothelial cells under inflammatory conditions (Li et al., 2004; Ray et al., 2005). NADPH oxidase activation occurs over the short term through phosphorylation of the enzyme subunits and over the long term by increased subunit expression (Li et al., 2004; Ray et al., 2005). Like iNOS activity, excessive NADPH oxidase activity causes endothelial dysfunction. For instance, the endothelial barrier failure induced by an inflammatory cytokine is mediated by NADPH oxidase-derived ROS (Gertzberg et al., 2004). Additionally, ROS produced by NADPH oxidases have been implicated in the regulation of transcription of genes associated with inflammation (Griendling et al., 2000; Terada et al., 2002).
While NADPH oxidase has been shown to mediate iNOS expression in mouse macrophages (Lanone et al., 2005), the role of this oxidase in iNOS expression in endothelial cells has not been elucidated. Previous studies identified endothelial NADPH oxidases as potential targets for the antioxidant ascorbate, which has been shown in experimental sepsis to decrease iNOS expression, protect endothelial function and improve outcome (Armour et al., 2001; Feng et al., 2004; Kim et al., 2004; Tyml et al., 2005; Wu et al., 2004, 2007). Therefore, the aim of the present study was to test the hypothesis that NADPH oxidase-derived ROS signaling is required for the induction of iNOS protein by septic insult in microvascular endothelial cells.
Materials and Methods
Chemicals and reagents
Lipopolysaccharide (LPS, from Escherichia coli 055:B5), mouse IFNγ (recombinant), superoxide dismutase (SOD) and catalase were obtained from Sigma/Aldrich Chemical Co. (St. Louis, MO). Allopurinol, apocynin, ascorbic acid, diphenyleneiodonium chloride (DPI), genistein, rotenone, AG490, PD98059, SB203580, SP600125, MG132 and L-NAME were obtained from VWR (West Chester, PA). Anti-p-ERK, anti-IκB, anti-Nox1 and anti-Nox4 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-p-Jak2, anti-p-JNK and anti-JNK were obtained from Cell Signaling Technology (Danvers, MA). Anti-iNOS and anti-Nox2 antibodies were obtained from BD Biosciences (San Jose, CA). Anti-p47phox and anti-β-actin antibodies were obtained from Millipore Corporation (Temecula, CA) and EMD Chemicals (San Diego, CA), respectively.
Endothelial cells from skeletal muscle
Since the microvasculature in mouse skeletal muscle shows a robust iNOS-sensitive response to sepsis (Wu et al., 2004), we isolated microvascular endothelial cells from hind limb skeletal muscle of male C57BL/6 mice and p47phox-deficient mice with C57BL/6 background (obtained from Jackson Laboratory) using a cell-trapping technique as described by us (Wu et al., 2001). Briefly, muscle tissue was dissected out aseptically, minced into small pieces using dissecting scissors, and then digested in a collagenase solution. The tissue digests were filtered through a sterile 100 μm nylon mesh. The cells were pelleted by centrifugation, resuspended in DMEM/F12, and incubated with magnetic beads coated with Griffonia simplicifolia lectin I. After incubation at 4°C for 20 min, the magnetic beads and bound cells were trapped with a magnet and washed four times with serum-supplemented DMEM/F12. The beads and cells were then removed from the magnet and plated into 35 mm culture dishes.
Endothelial cells were grown in a culture medium consisting of DMEM/F12, 10% heat-inactivated fetal bovine serum, 0.2 mM L-glutamine, 5 U/ml heparin, 100 U/ml penicillin and 100 μg/ml streptomycin, in an incubator equilibrated with 5% CO2. Endothelial cell identification was carried out by immunocytochemical staining for von Willebrand factor, uptake of acetylated low-density lipoprotein labeled with 1,1-dioctadecyl-3,3,3-tetramethylindo- carbocyanine perchlorate, and binding of fluorescently labeled Griffonia simplicifolia lectin I (Wilson et al., 1996). Experiments were performed using confluent monolayers (passages 3 through 8) originating from at least three different mice for each experiment. Cells were treated with various agents as described in detail in the Figure legends.
Superoxide production by lucigenin assay
Measurement of superoxide in intact cells was carried out by lucigenin chemiluminescence assay. Microvascular endothelial cells isolated from wild type or p47phox deficient mice were cultured in 35 mm culture dishes. They were then incubated with culture medium (i.e., control condition) or the combination of LPS (25 ng/ml) and IFNγ (100 U/ml) for 24 h. The cells were scrape-harvested in phenol red-free DMEM (500 μl/dish) and dissociated by trituration with a pipette. Two aliquots (100 μl/aliquot) were saved for protein determination by Lowry protein assay. Other aliquots of cell suspension were added to wells of the 96-well scintillation plate. The cells were incubated for 1 h with vehicle [phosphate buffered saline (PBS) or 0.1 % DMSO in PBS], SOD (500 U/ml), SOD (500 U/ml)+catalase (1000 U/ml), ascorbate (500 μM), apocynin (250 μM), DPI (10 μM), rotenone (10 μM), allopurinol (100 μM) or L-NAME (1 mM). Chemiluminescence in cells was initiated by adding 100 μM NADPH and 5 μM lucigenin to each well and was measured at 2 min intervals at room temperature in a Bio-Tek Multifunction Plate Reader. In some experiments, NADPH or cells were omitted from the reaction mixtures. The chemiluminescence counts recorded during a 10 min period were averaged and superoxide production was expressed as counts/mg protein/s.
Western blot analysis
Endothelial cells isolated from wild type or p47phox deficient mice were pretreated with vehicle, ascorbate (500 μM, 12 h), apocynin (250 μM, 1 h), DPI (10 μM, 1 h), rotenone (10 μM, 1 h), allopurinol (100 μM, 1 h), MG132 (10 μM, 1 h), AG490 (100 μM, 1 h), genistein (100 μM, 1 h), SB203580 (10 μM, 1 h), PD98059 (30 μM, 1 h) or SP600125 (20 μM, 1 h). They were then incubated with the same drugs and LPS+IFNγ for another 24 h for detection of iNOS protein expression. Endothelial cells also were pretreated with vehicle, ascorbate (500 μM, 12 h) or apocynin (250 μM, 1 h) and then incubated with the same drugs and LPS+IFNγ for 30 min for detection of total IκB protein levels and phosphorylation levels of Jak2, ERK and JNK. For each sample, 50 μg cell lysate protein was separated in 12% SDS-polyacrylamide gel by electrophoresis and then transferred to polyvinylidene fluoride membrane. For protein detection, blocked membranes were incubated with primary antibodies in Tris-buffered saline containing 5% nonfat milk (or 5% bovine serum albumin for anti-phosphor-protein antibodies) and 0.1% Tween 20. Subsequently, the membranes were washed and then further incubated with respective horseradish peroxidase-conjugated secondary antibodies in Tris-buffered saline containing 5% nonfat milk and 0.1% Tween 20. Immunoreactive proteins were detected using the ECL detection system (GE Health Life Sciences) and autoradiography film. Blots were further probed with anti-β-actin antibody to normalize for protein loading.
Nuclear extracts
Endothelial cells isolated from wild type or p47phox deficient mice were cultured in 35 mm culture dishes. They were pretreated with vehicle, ascorbate (500 μM, 12 h) or apocynin (250 μM, 1 h) and then incubated with the same drugs and LPS+IFNγ for 2 h. The cells in each 35 mm culture dish were washed in ice-cold phosphate-buffered saline, harvested by scraping, and pelleted by centrifugation. The cell pellet was suspended in 400 μl ice-cold buffer [containing 10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonylfluoride] by gentle trituration. The cells were allowed to swell on ice for 15 min, then 25 μl of 10% Nonidet NP-40 solution was added and the tube was vortexed vigorously for 10 s. The crude nuclei were collected by centrifugation for 30 s at 15000 × g. The nuclear pellet was resuspended in 50 μl of ice-cold buffer [containing 20 mM HEPES (pH 7.9), 0.4 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonylfluoride] and the tube was shaken vigorously at 4°C for 15 min on a platform. The nuclear extract was centrifuged for 5 min at 15000 × g at 4°C and the supernatant was frozen at −80°C.
Electrophoretic mobility shift assay (EMSA)
Double-stranded oligonucleotides were end-labeled with [γ-32P]ATP (Amersham) and T4 polynucleotide kinase (Fermentas). The sense sequences of double-stranded oligonucleotides used were NFκB (5′-AGT TGA GGG GAC TTT CCC AGG C-3′), IRF1 (5′-GGA AGC GAA AAT GAA ATT GAC T-3′) and AP1 (5′-CGC TTG ATG ACT CAG CCG GAA-3′) (Santa Cruz Biotechnology). For the binding reaction, 15 μg of nuclear protein was incubated with 0.06 pmol of γ-32P-labeled oligonucleotides (∼50000 cpm) and 1 μg Poly dI-dC (Amersham) in a binding buffer [containing 10 mM Tris-Cl (pH 7.5), 50 mM NaCl, 1 mM dithiothreitol, 1 mM EDTA and 5% glycerol] for 20 min at room temperature. Oligonucleotide-protein complexes were resolved in 4% non-denaturing polyacrylamide gel by electrophoresis in low ionic strength buffer (0.5×TBE, pH 8.0). NFκB, IRF1 and AP1 oligonucleotide specificity was verified by competition binding assay and supershift assay as described by us (Wu et al., 2001).
Data analysis
Data are presented as mean ± SEM values and were analyzed with the Prism statistical program (GraphPad Software, San Diego, CA). Comparisons between experimental groups were performed with one-way ANOVA followed by the Tukey′s multiple comparison test. P < 0.05 was considered significant.
Results
LPS+IFNγ increases ROS production in microvascular endothelial cells
Microvascular endothelial cells produced a baseline level of ROS, as determined by the chemiluminescence production. ROS production was markedly elevated in the cells exposed to LPS+IFNγ. The chemiluminescence was related to superoxide because SOD greatly decreased chemiluminescence levels (Fig. 1A). Compared to SOD, SOD+catalase did not further reduce the superoxide levels, suggesting that hydrogen peroxide did not interfere with superoxide measurement (Fig. 1A). Since omission of NADPH from the reaction mixture abolished superoxide production (i.e., decreased chemiluminescence to the levels determined without cells) (Fig. 1A), we infer that superoxide production by the cells is NADPH-dependent. The failure of SOD to completely inhibit the cell-dependent chemiluminescence (Fig. 1A) is likely because SOD is not cell-permeable and cannot dismute intracellular superoxide. Indeed, ascorbate, an antioxidant that can be taken up by the cells (Wilson et al., 1996), suppressed the superoxide production more effectively than did SOD (compare Fig. 1A and B).
Fig. 1.
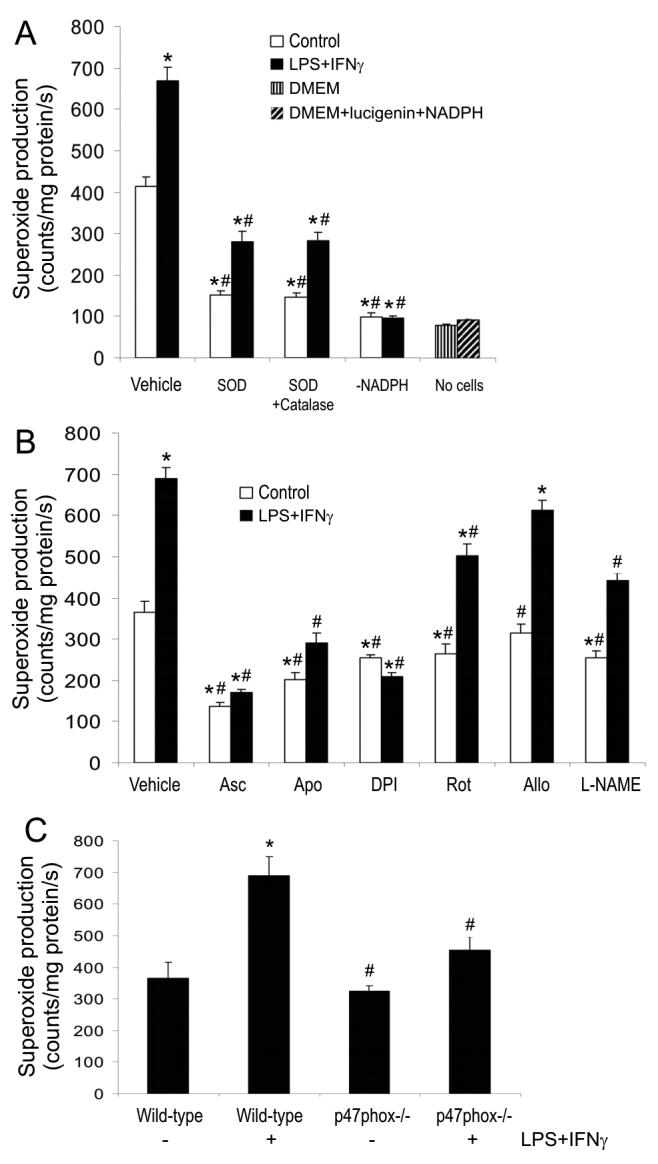
NADPH oxidase activity is responsible for LPS+IFNγ-stimulated superoxide production. Mouse skeletal muscle microvascular endothelial cells were incubated with culture medium (i.e., control condition) or the combination of LPS (25 ng/ml) and IFNγ (100 U/ml) for 24 h and subsequently the cells were suspended by scraping. Panel A shows lucigenin-based superoxide-sensitive chemiluminescence measured in wild type endothelial cells that were incubated for 1 h with vehicle (i.e., phosphate buffered saline, PBS), SOD (500 U/ml), or SOD (500 U/ml)+catalase (1000 U/ml) before addition of 100 μM NADPH and 5 μM lucigenin. The chemiluminescence also was measured in the absence of either NADPH or cells. Panel B shows the chemiluminescence measured in wild type endothelial cells that were incubated for 1 h with vehicle (PBS or 0.1 % DMSO in PBS), ascorbate (Asc, 500 μM), apocynin (Apo, 250 μM), DPI (10 μM), rotenone (Rot, 10 μM), allopurinol (Allo, 100 μM) or L-NAME (1 mM) before addition of 100 μM NADPH and 5 mM lucigenin. Panel C shows the chemiluminescence measured in wild type and p47phox knockout endothelial cells after incubation for 24 h with vehicle or LPS+IFNγ. Data are mean ± SEM of three separate experiments. *Significant difference from the baseline (i.e., vehicle control group) (P < 0.05). #Significant difference from vehicle LPS+IFNγ group (P < 0.05).
The ROS levels, detected either in unstimulated or in LPS+IFNγ-stimulated endothelial cells, were inhibited significantly by ascorbate, the NADPH oxidase inhibitors apocynin and DPI, the mitochondrial respiration blocker rotenone, and the NOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME), but not by the xanthine oxidase inhibitor allopurinol (Fig. 1B). These results indicate that the ROS are derived from NADPH oxidase, mitochondrial respiration and NOS. However, since inhibition of NADPH oxidase by apocynin, DPI and p47phox deficiency abolished the LPS+IFNγ-induced increase in superoxide production (Fig. 1B and C), we infer that NADPH oxidases are the major sources of ROS in LPS+IFNγ-stimulated endothelial cells.
NADPH oxidase-dependent redox signaling is required for iNOS induction
The NADPH oxidase catalytic subunits Nox1, Nox2 (i.e., gp91phox) and Nox4 have been reported present in endothelial cells (Bedard and Krause, 2007). Our results showed that only the Nox1 and Nox2 proteins (Fig. 2), but not Nox4 protein (data not shown), were detectable in microvascular endothelial cells. Both Nox1 and Nox2 were increased by LPS+IFNγ (Fig. 2). Their expression reached peak levels at 12 h and remained elevated at 24 h (Fig. 2). p47phox is a regulatory subunit of NADPH oxidases that controls superoxide production in endothelial cells under inflammatory conditions (Frey et al., 2002). Our results showed that p47phox protein was expressed in unstimulated endothelial cells. The expression of p47phox was elevated at 6 h post LPS+IFNγ and returned to baseline level by 24 h (Fig. 2).
Fig. 2.
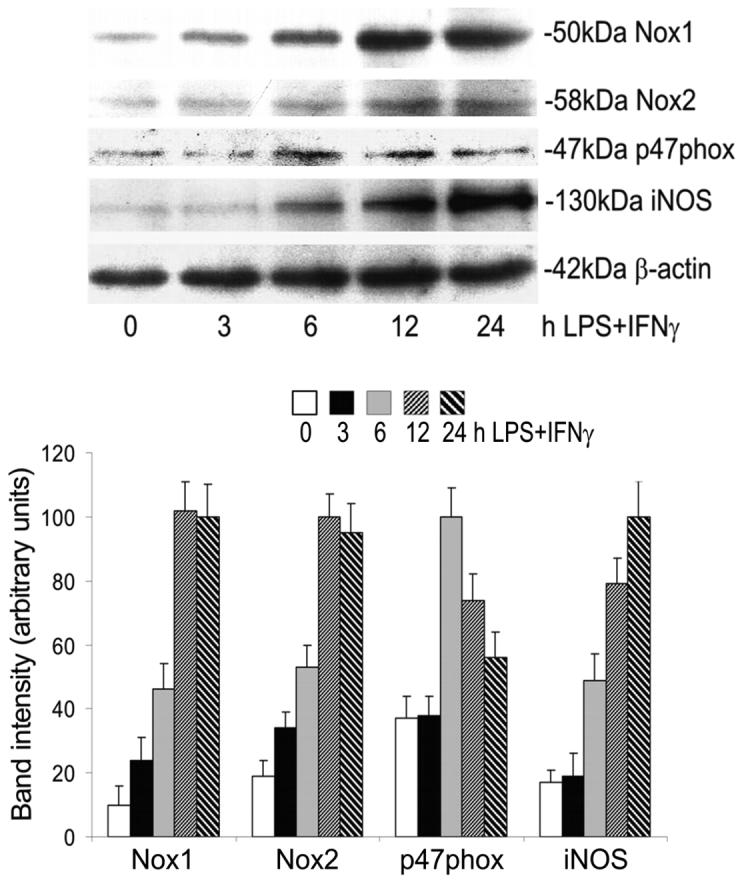
Time-dependent expression of Nox1, Nox2, p47phox and iNOS proteins in endothelial cells after exposure to LPS+IFNγ. Endothelial cells were incubated with LPS+IFNγ for 0, 3, 6, 12 and 24 h and then harvested for Western blot analysis. Shown are representative Western blot results and the summaries of band intensities (expressed in arbitrary units). β-actin was detected as a loading control.
iNOS can be induced by LPS+IFNγ in vascular endothelial cells (Koide et al., 2007; Wu et al., 2001). Consistently, our results showed that LPS+IFNγ induced a progressive increase in iNOS protein for up to 24 h (Fig. 2). Either LPS or IFNγ alone had negligible effect on iNOS protein expression in endothelial cells. However, their combination strongly induced iNOS protein (Fig. 3A). Ascorbate, apocynin and DPI attenuated iNOS protein induction by LPS+IFNγ (Fig. 3B). In contrast, rotenone and allopurinol did not inhibit iNOS protein expression (Fig. 3B). Further, iNOS induction by LPS+IFNγ was attenuated in endothelial cells that were deficient in p47phox (Fig. 3C). These results demonstrate that NADPH oxidase activity is necessary for iNOS induction.
Fig. 3.
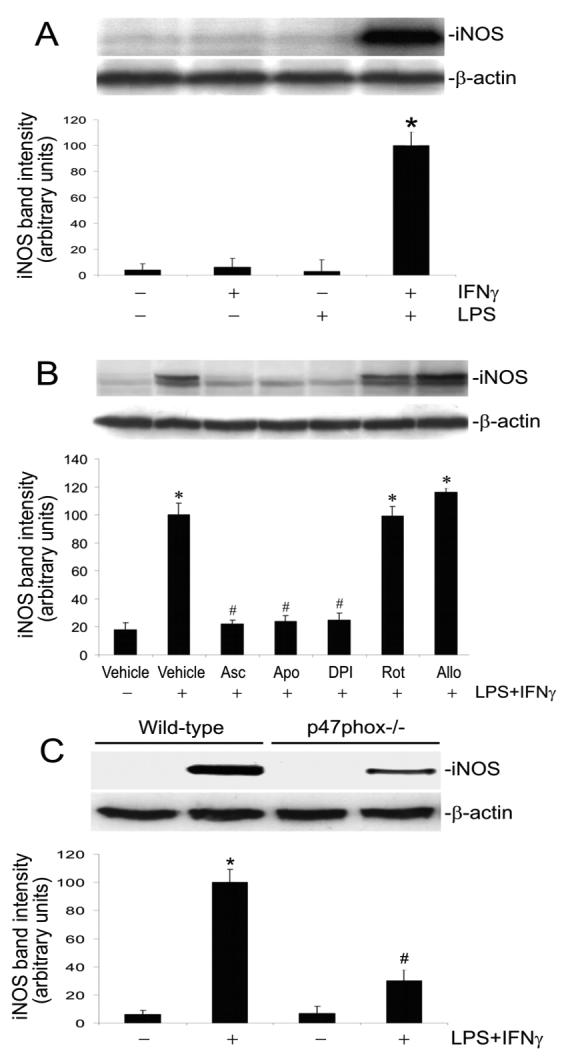
Suppression of NADPH oxidase activity attenuates iNOS induction. A: Endothelial cells were incubated for 24 h with vehicle, IFNγ, LPS, or LPS+IFNγ and then were harvested for Western blot analysis. Shown are iNOS and β-actin Western blot examples and the summary of iNOS protein band intensities (expressed in arbitrary units; mean ± SEM values for three experiments). *Significant difference from the vehicle, IFNγ alone, and LPS alone (P < 0.05). B: Endothelial cells were pretreated with vehicle, ascorbate (Asc, 500 μM, 12 h), apocynin (Apo, 250 μM, 1 h), DPI (10 μM, 1 h), rotenone (Rot, 10 μM, 1 h) or allopurinol (Allo, 100 μM, 1 h). Subsequently they were incubated with the same drugs plus LPS+IFNγ for another 24 h, and then harvested for Western blot analysis. Shown are iNOS and β-actin Western blot examples and the summary of iNOS protein band intensities (expressed in arbitrary units; mean ± SEM values for three experiments). *Significant difference from the vehicle group (P < 0.05). #Significant difference from the LPS+IFNγ group (P < 0.05). C: Wild type and p47phox phox knockout endothelial cells were incubated with vehicle or LPS+IFNγ for 24 h. Shown are iNOS and β-actin Western blot examples and the summary of iNOS protein band intensities (expressed in arbitrary units; mean ± SEM values for three experiments). *Significant difference from the vehicle group (P < 0.05). #Significant difference from the wild type LPS+IFNγ group (P < 0.05).
Effects of ascorbate and apocynin on signaling pathways in iNOS induction
Since iNOS induction requires the activation of NFκB, Jak2-IRF1 and ERK/JNK-AP1 pathways (Kleinert et al., 2004; Lowenstein and Padalko, 2004), we determined the effects of apocynin and ascorbate on the activation of these signaling molecules in microvascular endothelial cells exposed to LPS+IFNγ.
Incubation of endothelial cells with the proteasome inhibitor MG132, to prevent NFκB activation, abolished iNOS protein induction by LPS+IFNγ (Fig. 4A). LPS+IFNγ caused degradation of IκB but neither ascorbate nor apocynin prevented this action (Fig. 5). Consistently, LPS+IFNγ stimulated NFκB DNA binding activity and this effect, too, was not altered by ascorbate or apocynin (Fig. 6). These results show that NFκB activation is necessary for iNOS induction but is not affected by ascorbate and apocynin.
Fig. 4.
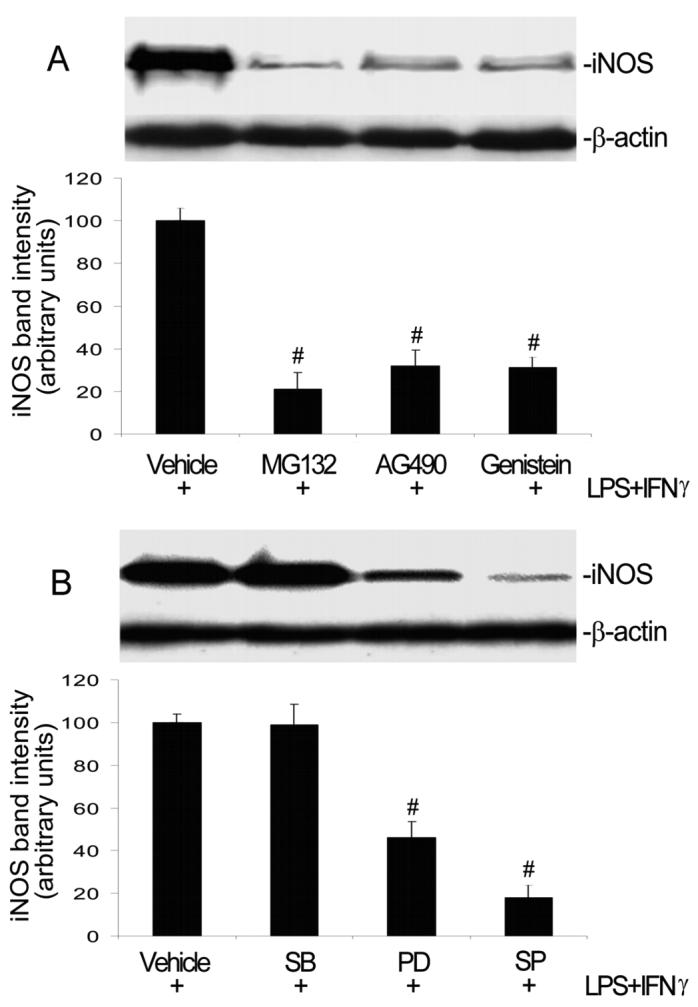
Inhibitors of NFκB, Jak2, ERK and JNK diminish iNOS induction. A: Endothelial cells were pretreated with vehicle, NFκB inhibitor MG132 (10 μM), Jak2 inhibitor AG490 (100 μM), or protein tyrosine kinase inhibitor genistein (100 μM) for 1 h. Subsequently they were incubated with the same drugs plus LPS+IFNγ for another 24 h, and then harvested for Western blot analysis. Shown are iNOS and β-actin Western blot examples and the summary of iNOS protein band intensities (e expressed in arbitrary units; mean ± SEM values for three experiments). #Significant difference from the LPS+IFNγ group (P < 0.05). B: Endothelial cells were pretreated with vehicle, p38 MAPK inhibitor SB203580 (10 μM), ERK inhibitor PD98059 (30 μM) or JNK inhibitor SP600125 (20 μM) for 1 h. Subsequently they were incubated with the same drugs plus LPS+IFNγ for another 24 h, and then harvested for Western blot analysis. Shown are iNOS and β-actin Western blot examples and the summary of iNOS protein band intensities (expressed in arbitrary units; mean ± SEM values for three independent experiments). #Significant difference from the LPS+IFNγ group (P < 0.05).
Fig. 5.
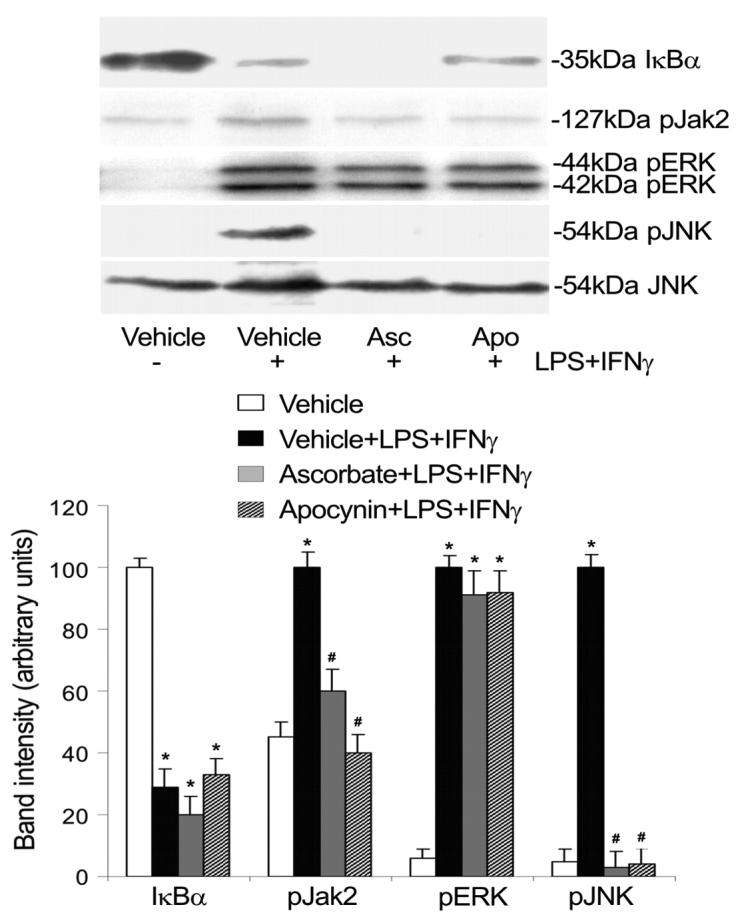
Ascorbate and apocynin inhibit Jak2 and JNK activation. Endothelial cells were pretreated with ascorbate (Asc, 500 μM, 12 h) or apocynin (Apo, 250 μM, 1 h). Subsequently they were incubated with the same drugs plus LPS+IFNγ for another 30 min, and then were harvested for detection of total IκB protein levels and phosphorylation levels of Jak2, ERK and JNK by Western blot analysis. Total JNK (54 kDa JNK) served as loading control. Shown are representative Western blot results and the summaries of band intensities (expressed in arbitrary units; mean ± SEM values for 3 independent experiments). *Significant difference from the vehicle group (P < 0.05). #Significant difference from the LPS+IFNγ group (P < 0.05).
Fig. 6.
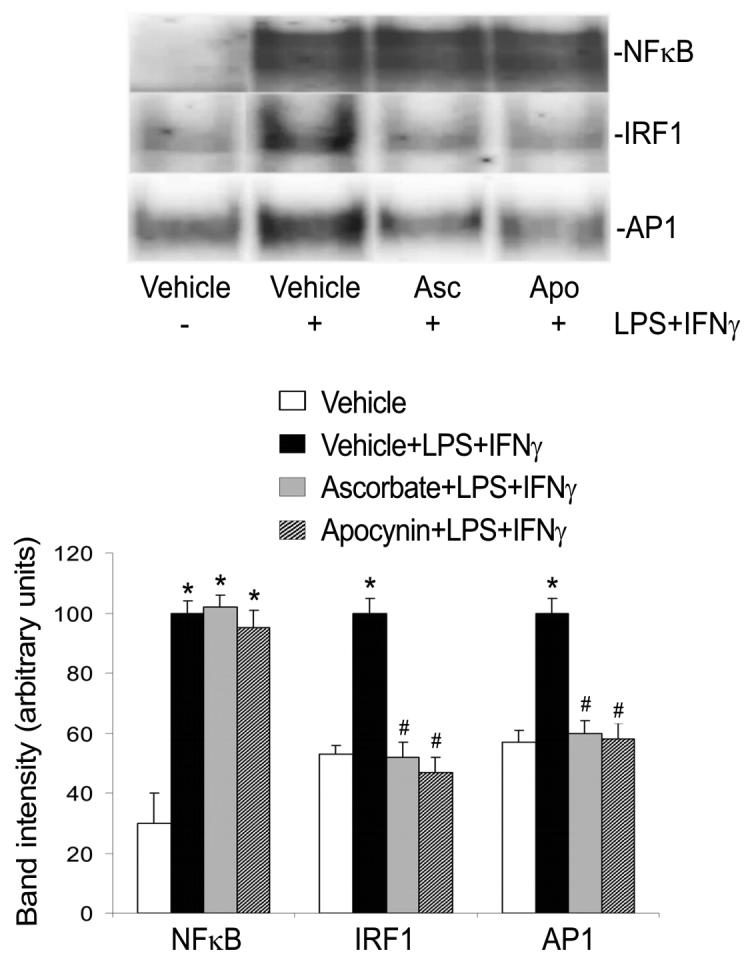
Ascorbate and apocynin inhibit IRF1 and AP1 activation. Endothelial cells were pretreated with ascorbate (Asc) for 12 h or apocynin (Apo) for 1 h. Subsequently they were incubated with the same drugs plus LPS+IFNγ for another 2 h, and then harvested. The nuclear proteins were extracted for determination of the DNA binding activities of NFκB, IRF1 and AP1 by EMSA. Shown are representative EMSA results and the summaries of band intensities (expressed in arbitrary units; mean ± SEM values for three independent experiments). *Significant difference from the vehicle group (P < 0.05). #Significant difference from the LPS+IFNγ group (P < 0.05).
Both the protein tyrosine kinase inhibitor genistein and the Jak2 selective inhibitor AG490 markedly suppressed iNOS protein induction by LPS+IFNγ (Fig. 4A). LPS+IFNγ stimulated phosphorylation of Jak2 and this action was prevented by ascorbate or apocynin (Fig. 5). Consistently, LPS+IFNγ stimulated IRF1 DNA binding activity and this effect was blocked by ascorbate or apocynin (Fig. 6). These results indicate that activation of the Jak2-IRF1 pathway is required for iNOS induction and is prevented by treatments that decrease intracellular ROS levels.
iNOS protein expression was partially inhibited by the ERK inhibitor PD98059, but was abolished by the JNK inhibitor SP600125 (Fig. 4B). In contrast, the p38 MAPK inhibitor SB203580 had no effect (Fig. 4B). Although both ERK and JNK were activated by LPS+IFNγ, only JNK activation was abolished by ascorbate and apocynin (Fig. 5). The activation of JNK and ERK can lead to the activation of transcription factor AP1 that is necessary for iNOS induction (Kleinert et al., 2004). Consistently, LPS+IFNγ stimulation increased AP1 DNA binding activity (Fig. 6). Further, this effect was blocked by ascorbate or apocynin (Fig. 6). These results indicate that, although both JNK and ERK are involved in iNOS induction, only JNK-AP1 activation is dependent on increases in intracellular ROS.
Effect of p47phox deficiency on signaling pathways in iNOS induction
To verify the role of NADPH oxidase in iNOS induction, we studied the effects of p47phox deficiency on the activation of NFκB, Jak2-IRF1 and ERK/JNK-AP1 pathways.
LPS+IFNγ caused IκB degradation and NFκB DNA binding activity in both wild type and p47phox-deficient endothelial cells (Fig. 7 and 8), indicating that NFκB activation is not dependent on NADPH oxidase activity. Nevertheless, LPS+IFNγ stimulated marked increases in Jak2 phosphorylation (Fig. 7) and IRF1 DNA binding activity (Fig. 8) in wild type but not in p47phox deficient endothelial cells, indicating that increased NADPH oxidase activity is essential for the activation of Jak2-IRF1 pathway. ERK activation was detected in both wild type and p47phox deficient endothelial cells (Fig. 7). In contrast, JNK activation and AP1 DNA binding activity were attenuated in p47phox deficient endothelial cells (Fig. 7 and 8). These results indicate that JNK is a target of NADPH oxidase-derived ROS and JNK inactivation alone is sufficient to attenuate AP1 DNA binding activity.
Fig. 7.
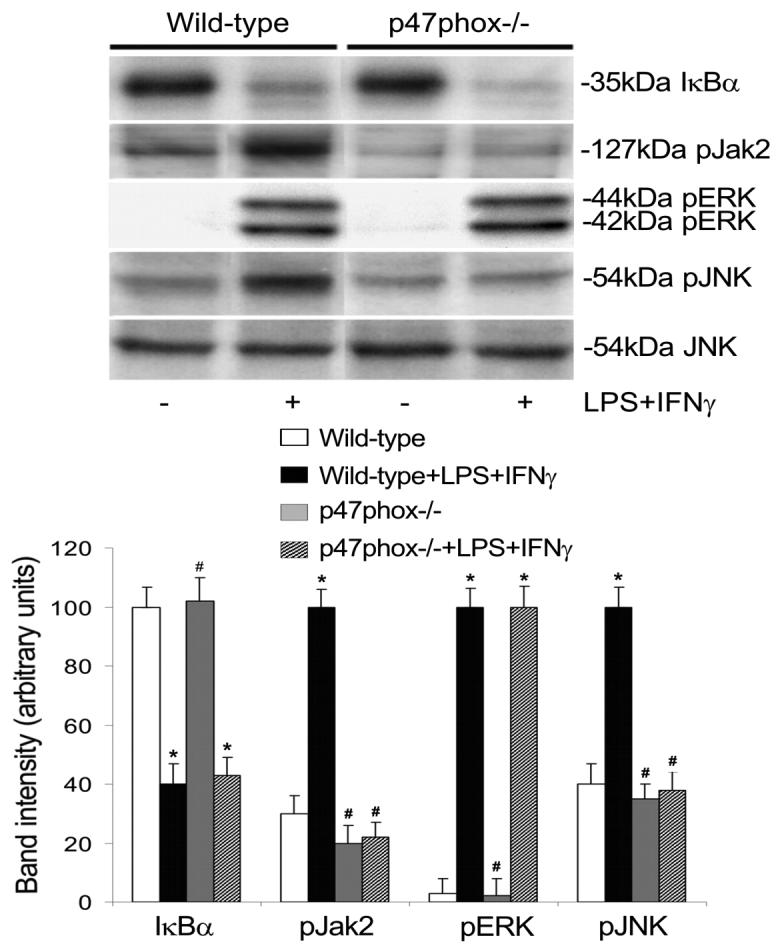
P47phox deficiency diminishes Jak2 and JNK phosphorylation. Wild type and p47phox deficient (p47phox−/−) endothelial cells were incubated with or without LPS+IFNγ for 30 min. Subsequently the cells were harvested for detection of total IκB protein levels and phosphorylation levels of Jak2, ERK, and JNK by Western blot analysis. Total JNK served as loading control. Shown are representative Western blot results and the summaries of band intensities (expressed in arbitrary units; mean ± SEM values for three independent experiments). *Significant difference from the vehicle treated wild type group (P < 0.05). #Significant difference from the LPS+IFNγ-stimulated wild type group (P < 0.05).
Fig. 8.
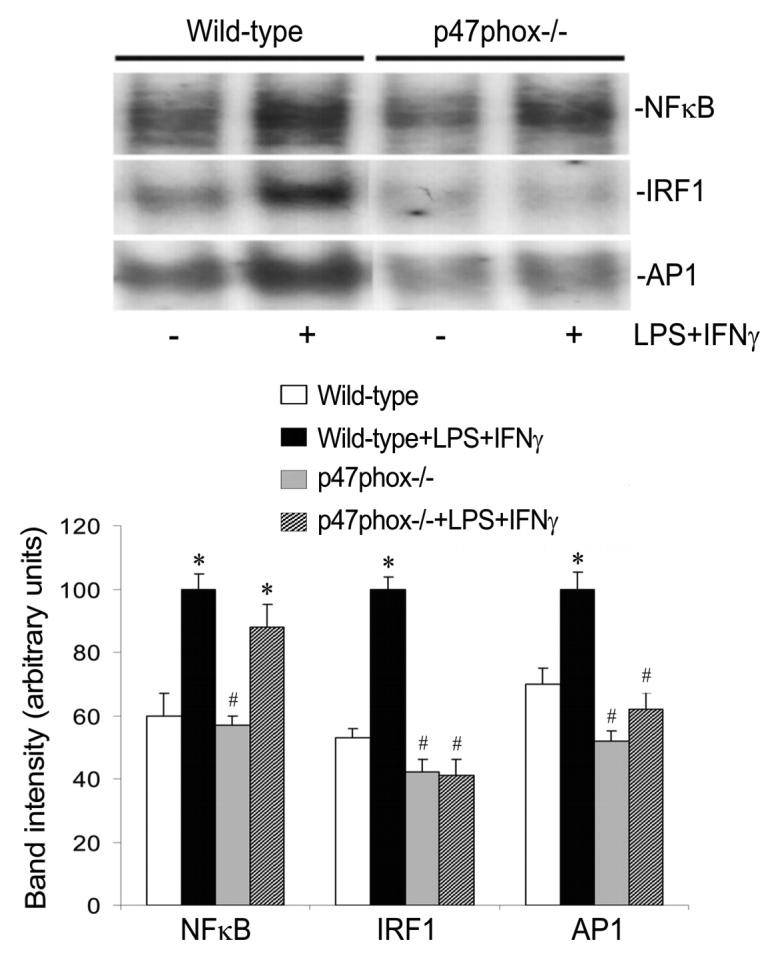
P47phox deficiency attenuates IRF1 and AP1 activation. Wild type and p47phox-deficient (p47phox−/−) endothelial cells were incubated with or without LPS+IFNγ for 2 h. Subsequently the nuclear proteins were extracted for determination of the DNA binding activities of NFκB, IRF1 and AP1 by EMSA. Shown are representative EMSA results and the summaries of band intensities (expressed in arbitrary units; mean ± SEM values for three independent experiments). *Significant difference from the vehicle treated wild type group (P < 0.05). #Significant difference from the LPS+IFNγ-stimulated wild type group (P < 0.05).
Discussion
The present study shows that increased NADPH oxidase activity is required for iNOS expression in LPS+IFNγ-stimulated microvascular endothelial cells. Suppression of NADPH oxidase activity by ascorbate, apocynin or NADPH oxidase subunit p47phox deficiency leads to attenuation of ROS-mediated signal transduction and iNOS expression.
NADPH oxidase and NFκB are independently required for iNOS induction
Our results show that microvascular endothelial cells produce ROS under basal conditions and this production is stimulated by LPS+IFNγ. The ROS produced under basal conditions are derived from NADPH oxidase, mitochondrial respiration and NOS. However, since inhibition of NADPH oxidase by apocynin, DPI or p47phox deficiency abolished the LPS+IFNγ-induced increase in superoxide production, we infer that NADPH oxidases are the major sources of ROS in LPS+IFNγ-stimulated endothelial cells. Furthermore, our results show that Nox1 and Nox2 proteins are detectable in microvascular endothelial cells and their expression is increased by LPS+IFNγ. Both of these Nox isoforms have been reported to contribute to ROS production in endothelial cells in response to various stimulations (Furst et al., 2005; Sorescu et al., 2004).
Although microvascular endothelial cells produced basal levels of ROS, only a low level of iNOS protein expression was detectable in these cells. We infer that oxidant signaling alone is not sufficient to trigger iNOS gene activation and that additional stimulation is required for iNOS induction. In support of this view, our previous study indicated that challenge of microvascular endothelial cells with ROS generated by xanthine oxidase/xanthine failed to induce iNOS protein (Wu et al., 2001). Exposure of the cells to LPS+IFNγ markedly increased NADPH oxidase activity and iNOS expression. The ROS derived from NADPH oxidases is required for iNOS induction, since NADPH oxidase inhibitors (apocynin and DPI) or p47phox gene deficiency attenuated iNOS expression in endothelial cells exposed to LPS+IFNγ.
One possible target of NADPH oxidase-derived ROS is transcription factor NFκB (Bowie and O'Neill, 2000; Gloire et al., 2006). NFκB activation is an essential requirement for the expression of inflammation-related genes, including cytokines, cyclooxygenase-2, iNOS and NADPH oxidase subunits (Zingarelli, 2005; Anrather et al., 2006). NFκB activation inducers include microbial products such as LPS and inflammatory cytokines, such as TNFα and IL-1β, that activate the ubiquitin-proteasome pathway (Perkins, 2007). Although it has been proposed that LPS and inflammatory cytokines trigger NFκB activation by increasing intracellular ROS levels (Gloire et al., 2006), in most cases the production of ROS is a consequence of NFκB activation rather than its cause (Bowie and O'Neill, 2000).
Our study found that the proteasome inhibitor MG132 blocked iNOS induction by LPS+IFNγ in microvascular endothelial cells. However, lowering of intracellular ROS levels by ascorbate, apocynin and p47phox deficiency had no effect on LPS+IFNγ-stimulated IκB degradation and NFκB activation. These results indicate that NFκB activation is necessary for iNOS induction but not dependent on ROS. This situation may also exist in other cell types. For instance, the cell permeant antioxidants pyrrolidine dithiocarbamate and dimethylthiourea did not influence TNFα-induced NFκB activation in human airway epithelial cells (Krunkosky et al., 2003). Similarly, the antioxidant octyl caffeate prevented LPS+IFNγ-stimulated iNOS induction but not IκB degradation in aortic smooth muscle cells (Hsiao et al., 2003). Thus, activation of NFκB by LPS and cytokines may be ROS-independent or only depend on cellular redox state in a cell- and stimulus-specific manner (Bowie and O'Neill, 2000; Gloire et al., 2006).
NADPH oxidase-dependent signaling in iNOS induction
Our results indicate that activation of Jak2-IRF1 and JNK-AP1 pathways is dependent on NADPH oxidase-derived ROS signaling. LPS and inflammatory cytokines upregulate NADPH oxidase activity that produces excessive superoxide, which is then converted to hydrogen peroxide by superoxide dismutase (Tolando et al., 2000). Compared to superoxide, the longer half-life and radius of diffusion of hydrogen peroxide make it a more important autocrine agent (Ardanaz et al., 2006). NADPH oxidase-derived hydrogen peroxide likely mediates the activation of Jak2-IRF1 and JNK-AP1 pathways in LPS+IFNγ-stimulated endothelial cells, since activation of these signaling pathways can be blocked by catalase in microglia cells challenged with LPS+IFNγ (Pawate et al., 2004).
Our results show that ascorbate, apocynin and p47phox deficiency prevent Jak2 phosphorylation/activation, inhibit IRF1 DNA binding activity, and suppress iNOS expression. This evidence that the Jak2-IRF1 pathway mediates iNOS induction and is regulated by redox state in endothelial cells is consistent with the observation that other antioxidants (5,7-dihydroxyflavone; 3,4-dichloroisocoumarin; N-acetyl 5-hydroxytryptamine) blunt the DNA-binding activity of IRF1 that mediates iNOS induction by LPS+IFNγ in a macrophage cell line (Hecker et al., 1996). The mechanisms underlying ROS-induced activation of Jak2-IRF1 may involve NADPH oxidase-derived hydrogen peroxide that inhibits phosphotyrosine phosphatases SHP-1 and SHP-2, which are proposed to deliver negative feedback to Jak2 activation by dephosphorylating the critical tyrosines in Jak2 (Alblas et al., 2005).
We found that ERK and JNK are necessary for iNOS induction by septic insult in endothelialcells, but p38 MAPK is not. Similarly, LPS+IFNγ induced iNOS expression in macrophages through activation of ERK and JNK, but not through p38MAPK signaling pathways (Chan and Riches, 2001). However, our data indicate that ERK and JNK are activated by independent signal transduction pathways, since only JNK activation was sensitive to ascorbate, apocynin and p47phox deficiency. Indeed, to our knowledge, there is no previous report of a redox mechanism regulating ERK activation in endothelial cells exposed to septic insult. The observations that activation of JNK and AP1 is attenuated in apocynin-treated and p47phox deficient endothelial cells establish that NADPH oxidase mediates activation of the JNK-AP1 signaling pathway. This finding is in agreement with a report demonstrating that p47phox is involved in the TNFα-stimulated activation of JNK in human umbilical vein endothelial cells (Gu et al., 2002). A possible target of NADPH oxidase-derived ROS in JNK activation is thioredoxin. Oxidation of the cysteine residues of thioredoxin by ROS releases and activates apoptosis signal regulating kinase 1(ASK1) (Saitoh et al., 1998). ASK1 is an upstream kinase of JNK-AP1 pathways (Torres, 2003).
Septic insult stimulates excessive NADPH oxidase activity that facilitates iNOS expression to produce large amounts of NO. iNOS-derived NO and its metabolite peroxynitrite may contribute to the pathological alterations observed in sepsis such as endothelial dysfunction, hypotension and multiple organ failure (Chauhan et al., 2003; Su et al., 2007; Titheradge, 1999). The present study revealed that NADPH oxidase-derived ROS mediate the activation of Jak2-IRF1 and JNK-AP1 pathways necessary for iNOS induction. Attenuation of the signal ROS may be the mechanism by which antioxidants such as ascorbate inhibit iNOS expression and endothelial dysfunction in sepsis therapy (Armour et al., 2001; Feng et al., 2004; Kim et al., 2004; Tyml et al., 2005; Wu et al., 2004, 2007).
In conclusion, our study found that NFκB activation and NADPH oxidase-dependent redox signaling are independent requirements for iNOS induction by LPS+IFNγ in microvascular endothelial cells. Specifically, ROS derived from NADPH oxidases activate JNK-AP1 and Jak2-IRF1 pathways that mediate iNOS expression. Ascorbate, apocynin, DPI and p47phox deficiency suppress NADPH oxidase activity and consequently inhibit iNOS expression. Since blocking either NFκB activation or NADPH oxidase activity is sufficient to prevent iNOS expression,they are separate targets for therapeutic interventions that aim to modulate iNOS expression in sepsis.
Acknowledgments
This work was supported by grants from the University at Buffalo Foundation, the National Institutes of Health (J.X.W. and F.W.), the Canadian Institutes of Health Research, and the Heart and Stroke Foundation of Ontario (K.T. and J.X.W.), and by a Canada Graduate Scholarship Doctoral Award from the Canadian Institutes of Health Research to F.W.
Contract grant sponsor: Canadian Institutes of Health Research
Contract grant number: MOP 53342
Contract grant sponsor: Heart and Stroke Foundation of Ontario
Contract grant number: NA 5941
Contract grant sponsor: NIH/NCCAM
Contract grant number: 1RO1 AT003643-01A2
Literature Cited
- Alblas J, Honing H, de Lavalette CR, Brown MH, Dijkstra CD, van den Berg TK. Signal regulatory protein alpha ligation induces macrophage nitric oxide production through JAK/STAT- and phosphatidylinositol 3-kinase/Rac1/NAPDH oxidase/H2O2-dependent pathways. Mol Cell Biol. 2005;25:7181–7192. doi: 10.1128/MCB.25.16.7181-7192.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anrather J, Racchumi G, Iadecola C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- Ardanaz N, Pagano PJ. Hydrogen peroxide as a paracrine vascular mediator: regulation and signaling leading to dysfunction. Exp Biol Med (Maywood) 2006;231:237–251. doi: 10.1177/153537020623100302. [DOI] [PubMed] [Google Scholar]
- Armour J, Tyml K, Lidington D, Wilson JX. Ascorbate prevents microvascular dysfunction in the skeletal muscle of the septic rat. J Appl Physiol. 2001;90:795–803. doi: 10.1152/jappl.2001.90.3.795. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bowie A, O'Neill LA. Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol. 2000;59:13–23. doi: 10.1016/s0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- Chan ED, Riches DW. IFN-gamma + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38(mapk) in a mouse macrophage cell line. Am J Physiol Cell Physiol. 2001;280:C441–450. doi: 10.1152/ajpcell.2001.280.3.C441. [DOI] [PubMed] [Google Scholar]
- Chauhan SD, Seggara G, Vo PA, Macallister RJ, Hobbs AJ, Ahluwalia A. Protection against lipopolysaccharide-induced endothelial dysfunction in resistance and conduit vasculature of iNOS knockout mice. FASEB J. 2003;17:773–775. doi: 10.1096/fj.02-0668fje. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Mazzon E, Di Paola R, Esposito E, Macarthur H, Matuschak GM, Salvemini D. A role for nitric oxide-mediated peroxynitrite formation in a model of endotoxin-induced shock. J Pharmacol Exp Ther. 2006;319:73–81. doi: 10.1124/jpet.106.108100. [DOI] [PubMed] [Google Scholar]
- Feng NH, Chu SJ, Wang D, Hsu K, Lin CH, Lin HI. Effects of various antioxidants on endotoxin-induced lung injury and gene expression: mRNA expressions of MnSOD, interleukin-1beta and iNOS. Chin J Physiol. 2004;47:111–120. [PubMed] [Google Scholar]
- Fierro IM, Nascimento-DaSilva V, Arruda MA, Freitas MS, Plotkowski MC, Cunha FQ, Barja-Fidalgo C. Induction of NOS in rat blood PMN in vivo and in vitro: modulation by tyrosine kinase and involvement in bactericidal activity. J Leukoc Biol. 1999;65:508–514. doi: 10.1002/jlb.65.4.508. [DOI] [PubMed] [Google Scholar]
- Frey RS, Rahman A, Kefer JC, Minshall RD, Malik AB. PKCzeta regulates TNF-alpha-induced activation of NADPH oxidase in endothelial cells. Circ Res. 2002;90:1012–1019. doi: 10.1161/01.res.0000017631.28815.8e. [DOI] [PubMed] [Google Scholar]
- Furst R, Brueckl C, Kuebler WM, Zahler S, Krotz F, Gorlach A, Vollmar AM, Kiemer AK. Atrial natriuretic peptide induces mitogen-activated protein kinase phosphatase-1 in human endothelial cells via Rac1 and NAD(P)H oxidase/Nox2-activation. Circ Res. 2005;96:43–53. doi: 10.1161/01.RES.0000151983.01148.06. [DOI] [PubMed] [Google Scholar]
- Gertzberg N, Neumann P, Rizzo V, Johnson A. NAD(P)H oxidase mediates the endothelial barrier dysfunction induced by TNF-alpha. Am J Physiol Lung Cell Mol Physiol. 2004;286:L37–L48. doi: 10.1152/ajplung.00116.2003. [DOI] [PubMed] [Google Scholar]
- Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- Gu Y, Xu YC, Wu RF, Souza RF, Nwariaku FE, Terada LS. TNFalpha activates c-Jun amino terminal kinase through p47(phox) Exp Cell Res. 2002;272:62–74. doi: 10.1006/excr.2001.5404. [DOI] [PubMed] [Google Scholar]
- Hecker M, Preiss C, Klemm P, Busse R. Inhibition by antioxidants of nitric oxide synthase expression in murine macrophages: role of nuclear factor kappa B and interferon regulatory factor 1. Br J Pharmacol. 1996;118:2178–2184. doi: 10.1111/j.1476-5381.1996.tb15660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao G, Shen MY, Chang WC, Cheng YW, Pan SL, Kuo YH, Chen TF, Sheu JR. A novel antioxidant, octyl caffeate, suppression of LPS/IFN-gamma-induced inducible nitric oxide synthase gene expression in rat aortic smooth muscle cells. Biochem. Pharmacol. 2003;65:1383–1392. doi: 10.1016/s0006-2952(03)00070-4. [DOI] [PubMed] [Google Scholar]
- Kim JY, Lee SM. Effect of ascorbic acid on hepatic vasoregulatory gene expression during polymicrobial sepsis. Life Sci. 2004;75:2015–2026. doi: 10.1016/j.lfs.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004;500:255–266. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Koide N, Mu MM, Hassan F, Islam S, Tumurkhuu G, Dagvadorj J, Naiki Y, Mori I, Yoshida T, Yokochi T. Lipopolysaccharide enhances interferon-gamma-induced nitric oxide (NO) production in murine vascular endothelial cells via augmentation of interferon regulatory factor-1 activation. J Endotoxin Res. 2007;13:167–175. doi: 10.1177/0968051907080894. [DOI] [PubMed] [Google Scholar]
- Krunkosky TM, Martin LD, Fischer BM, Voynow JA, Adler KB. Effects of TNFalpha on expression of ICAM-1 in human airway epithelial cells in vitro: oxidant-mediated pathways and transcription factors. Free Radic Biol Med. 2003;35:1158–1167. doi: 10.1016/s0891-5849(03)00498-2. [DOI] [PubMed] [Google Scholar]
- Lanone S, Bloc S, Foresti R, Almolki A, Taillé C, Callebert J, Conti M, Goven D, Aubier M, Dureuil B, El-Benna J, Motterlini R, Boczkowski J. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: implications for protection against endotoxic shock in rats. FASEB J. 2005;19:1890–1892. doi: 10.1096/fj.04-2368fje. [DOI] [PubMed] [Google Scholar]
- Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- Liu H, Colavitti R, Rovira II, Finkel T. Redox-dependent transcriptional regulation. Circ Res. 2005;97:967–974. doi: 10.1161/01.RES.0000188210.72062.10. [DOI] [PubMed] [Google Scholar]
- Lowenstein CJ, Padalko E. iNOS (NOS2) at a glance. J Cell Sci. 2004;117:2865–2867. doi: 10.1242/jcs.01166. [DOI] [PubMed] [Google Scholar]
- Macdonald J, Galley HF, Webster NR. Oxidative stress and gene expression in sepsis. Br J Anaesth. 2003;90:221–232. doi: 10.1093/bja/aeg034. [DOI] [PubMed] [Google Scholar]
- Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res. 2004;77:540–551. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NFκB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Ray R, Shah AM. NADPH oxidase and endothelial cell function. Clin Sci (London) 2005;109:217–226. doi: 10.1042/CS20050067. [DOI] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK)1. EMBO J. 1998;17:2596–2600. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorescu GP, Song H, Tressel SL, Hwang J, Dikalov S, Smith DA, Boyd NL, Platt MO, Lassegue B, Griendling KK, Jo H. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ Res. 2004;95:773–779. doi: 10.1161/01.RES.0000145728.22878.45. [DOI] [PubMed] [Google Scholar]
- Su CF, Yang FL, Chen HI. Inhibition of inducible nitric oxide synthase attenuates acute endotoxin-induced lung injury in rats. Clin Exp Pharmacol Physiol. 2007;34:339–346. doi: 10.1111/j.1440-1681.2007.04553.x. [DOI] [PubMed] [Google Scholar]
- Terada LS. Oxidative stress and endothelial activation. Crit Care Med. 2002;30(5 Suppl):S186–191. doi: 10.1097/00003246-200205001-00003. [DOI] [PubMed] [Google Scholar]
- Titheradge MA. Nitric oxide in septic shock. Biochim Biophys Acta. 1999;1411:437–455. doi: 10.1016/s0005-2728(99)00031-6. [DOI] [PubMed] [Google Scholar]
- Tolando R, Jovanovic A, Brigelius-Flohe R, Ursini F, Maiorino M. Reactive oxygen species and proinflammatory cytokine signaling in endothelial cells: effect of selenium supplementation. Free Radic Biol Med. 2000;28:979–986. doi: 10.1016/s0891-5849(00)00183-0. [DOI] [PubMed] [Google Scholar]
- Torres M. Mitogen-activated protein kinase pathways in redox signaling. Front Biosci. 2003;8:d369–391. doi: 10.2741/999. [DOI] [PubMed] [Google Scholar]
- Tyml K, Li F, Wilson JX. Delayed ascorbate bolus protects against maldistribution of microvascular blood flow in septic rat skeletal muscle. Crit Care Med. 2005;33:1823–1828. doi: 10.1097/01.ccm.0000172548.34622.de. [DOI] [PubMed] [Google Scholar]
- Wilson JX, Dixon SJ, Yu J, Nees S, Tyml K. Ascorbate uptake by microvascular endothelial cells of rat skeletal muscle. Microcirculation. 1996;3:211–221. doi: 10.3109/10739689609148290. [DOI] [PubMed] [Google Scholar]
- Wu F, Cepinskas G, Wilson JX, Tyml K. Nitric oxide attenuates but superoxide enhances iNOS expression in endotoxin- and IFNγ-stimulated skeletal muscle endothelial cells. Microcirculation. 2001;8:415–425. doi: 10.1038/sj/mn/7800113. [DOI] [PubMed] [Google Scholar]
- Wu F, Schuster DP, Tyml K, Wilson JX. Ascorbate inhibits NADPH oxidase subunit p47phox expression in microvascular endothelial cells. Free Radic Biol Med. 2007;42:124–131. doi: 10.1016/j.freeradbiomed.2006.10.033. [DOI] [PubMed] [Google Scholar]
- Wu F, Wilson JX, Tyml K. Ascorbate protects against impaired arteriolar constriction in sepsis by inhibiting inducible nitric oxide synthase expression. Free Radic Biol Med. 2004;37:1282–1289. doi: 10.1016/j.freeradbiomed.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Zingarelli B. Nuclear factor-kappaB. Crit Care Med. 2005;33(12 Suppl):S414–416. doi: 10.1097/01.ccm.0000186079.88909.94. [DOI] [PubMed] [Google Scholar]