

Abstract
The dietary polyphenolic compound resveratrol, by activating the protein deacetylase enzyme silent information regulator 2/sirtuin 1 (SIRT1), prolongs life span in evolutionarily distant organisms and may mimic the cytoprotective effects of dietary restriction. The present study was designed to elucidate the effects of resveratrol on cigarette smoke-induced vascular oxidative stress and inflammation, which is a clinically highly relevant model of accelerated vascular aging. Cigarette smoke exposure of rats impaired the acetylcholine-induced relaxation of carotid arteries, which could be prevented by resveratrol treatment. Smoking and in vitro treatment with cigarette smoke extract (CSE) increased reactive oxygen species production in rat arteries and cultured coronary arterial endothelial cells (CAECs), respectively, which was attenuated by resveratrol treatment. The smoking-induced upregulation of inflammatory markers (ICAM-1, inducible nitric oxide synthase, IL-6, and TNF-α) in rat arteries was also abrogated by resveratrol treatment. Resveratrol also inhibited CSE-induced NF-κB activation and inflammatory gene expression in CAECs. In CAECs, the aforementioned protective effects of resveratrol were abolished by knockdown of SIRT1, whereas the overexpression of SIRT1 mimicked the effects of resveratrol. Resveratrol treatment of rats protected aortic endothelial cells against cigarette smoking-induced apoptotic cell death. Resveratrol also exerted antiapoptotic effects in CSE-treated CAECs, which could be abrogated by knockdown of SIRT1. Resveratrol treatment also attenuated CSE-induced DNA damage in CAECs (comet assay). Thus resveratrol and SIRT1 exert antioxidant, anti-inflammatory, and antiapoptotic effects, which protect the endothelial cells against the adverse effects of cigarette smoking-induced oxidative stress. The vasoprotective effects of resveratrol will likely contribute to its anti-aging action in mammals and may be especially beneficial in patho-physiological conditions associated with accelerated vascular aging.
Keywords: tobacco, polyphenol, stroke, inflammation, apoptosis, vascular aging, sirtuin 1
Studies in the last few years have revealed that the dietary polyphenolic compound resveratrol, by activating the protein deacetylase enzyme silent information regulator 2/sirtuin 1 (SIRT1), prolongs life span in evolutionarily distant organisms (e.g., yeast, Drosophila, and the short-lived fish Nothobranchius furzeri) (67, 68) and may mimic the effects of dietary restriction to extend healthy life span. Knowing whether the antiaging action of resveratrol is conserved in mammals is one of the most exciting and important questions in the aging field today [recently reviewed elsewhere (10, 38)]. Because in humans cardiovascular aging is responsible for the largest portion of age-related morbidity and mortality, it is particularly important to elucidate whether resveratrol exerts antiaging effects in the cardiovascular system (2). Cardiovascular aging is characterized by oxidative stress, inflammation [i.e., NF-κB activation, endothelial activation, inflammatory cytokine expression, and upregulation of inducible nitric oxide (NO) synthase (iNOS)], disruption of endogenous tissue-protective mechanisms (27), and an increased rate of apoptotic cell death, which lead to an age-dependent deterioration of cardiovascular functions [recently reviewed elsewhere (19, 61, 64)]. In this regard, recent in vitro studies from this and other laboratories have shown that resveratrol in vitro can attenuate cellular oxidative stress, inhibits endothelial activation and monocyte adhesion (20, 28, 42), protects endothelial cells from oxidative stress-induced apoptosis (63), and attenuates proinflammatory gene expression by the inhibition of NF-κB activation in coronary arterial endothelial cells (CAECs) (20). Importantly, resveratrol is being considered as a therapeutic for humans for a variety of indications (10). To gain a better insight into the antiaging effects of resveratrol, in a series of studies we are currently characterizing the in vivo cytoprotective, antioxidant, and anti-inflammatory vascular effects of resveratrol in models of accelerated vascular aging.
The present study was designed to elucidate the effects of resveratrol on cigarette smoke-induced vascular oxidative stress and inflammation, which is a clinically highly relevant model of accelerated vascular aging. Cigarette smoking is the leading cause of preventable morbidity and mortality in the United States and constitutes a major risk factor for atherosclerotic vascular disease, including stroke and coronary artery disease. There is growing evidence suggesting that the increased production of reactive oxygen species (ROS) plays a central role in cigarette smoking-induced vascular pathophysiological alterations. Cigarette smoke can be divided into two phases: tar and gas-phase smoke. Both phases contain high concentrations of ROS, NO, peroxynitrite, and free radicals of organic compounds (45, 49, 50, 71). In addition to these short-lived, highly reactive substances, previous studies have shown that aqueous cigarette tar extracts also contain prooxidant substances that have the potential to increase the cellular production of ROS (3, 11, 50, 52, 56, 57, 71). Water-soluble components of cigarette smoke are likely to reach the systemic circulation, and thus they can directly promote vascular oxidative stress in systemic vascular beds. Indeed, our laboratory has recently shown that water-soluble components of cigarette smoke (which are likely to be present in the bloodstream in vivo in smokers) elicit oxidative stress in the vascular endothelium, at least in part, by activating the vascular NAD(P)H oxidase (46). Accordingly, a number of clinical and animal studies show that cigarette smoke produces generalized endothelial dysfunction in virtually every vascular bed (1, 12, 13, 24, 25, 46, 51), which is usually an indicator of an increased oxidative stress. Our laboratory has recently demonstrated that cigarette smoke-induced endothelial oxidative stress (especially increased levels of H2O2) results in NF-κB activation and, consequently, proinflammatory alterations in vascular phenotype (46). The aforementioned cigarette smoke-induced phenotypic and functional alterations resemble those observed in aging (8, 16, 17, 19, 21–23, 58, 61, 66). Yet, the effect of resveratrol in this model of oxidative stress-related accelerated vascular aging has not been elucidated.
On the basis of the aforementioned studies, we posit that water-soluble components of cigarette smoke increase ROS generation in endothelial and smooth muscle cells, activate NF-κB eliciting the expression of proinflammatory mediators, and promote endothelial cell apoptosis, and we hypothesize that resveratrol will prevent/attenuate these deleterious effects. To test this hypothesis, we characterized the vasoprotective effects of in vivo resveratrol treatment in cigarette smoke-exposed rats. In addition, the effects of in vitro resveratrol treatment on CSE-induced alterations in endothelial ROS production, NF-κB activation, expression of proinflammatory cytokines, and apoptosis induction were investigated.
MATERIALS AND METHODS
Animals and vessel isolation
Fourteen-to sixteen-week-old male Wistar rats (n = 20) were used. The protocols were approved by the Institutional Animal Care and Use Committee of New York Medical College and conformed to the current guidelines of the National Institutes of Health (NIH) and the American Physiological Society for the use and care of laboratory animals. Animals were euthanized by a lethal injection of pentobarbital sodium, and the carotid arteries, aortas, and coronary arteries were isolated for subsequent studies as described (46).
Cigarette smoke exposure and resveratrol treatment
The experimental group (n = 12) was exposed to the smoke of five commercial cigarettes (11 mg tar and 0.8 mg nicotine/cigarette) each day for 1 wk as described (46), whereas the control group was not exposed to cigarette smoke. To assess the vasoprotective effects of resveratrol, another group of rats was pretreated with resveratrol [25 mg·kg−1 ·day−1 in drinking water (66); for 2 days] and then exposed to the above-described cigarette smoking protocol (resveratrol treatment continued throughout the experimental period). We have used a modified dietary regimen of resveratrol feeding established by Dr. Rafael de Cabo’s laboratory at the NIH (9). The daily resveratrol intake was adjusted to the water consumption of the animals.
Functional studies
Endothelial function was assessed as previously described (31, 46). In brief, the carotid arteries were cut into ring segments 2 mm in length and mounted on 40-µm stainless steel wires in the myographs chambers (Danish Myo Technology, Atlanta, GA) containing Krebs buffer solution containing (in mM) 118 NaCl, 4.7 KCl, 1.5 CaCl2, 25 NaHCO3, 1.1 MgSO4, 1.2 KH2PO4, and 5.6 glucose at 37°C and gassed with 95% air-5% CO2 for the measurement of isometric tension. After an equilibration period of 1 h during which an optimal passive tension of 0.5 g was applied to the rings (as determined from the vascular length-tension relationship), the vessels were contracted by phenylephrine (10−6 mol/l) and relaxations to acetylcholine (from 10−9 to 10−4 mol/l) and the NO donor S-nitroso-N-acetyl-penicillamine (from 10−9 to 3 × 10−5 mol/l) were obtained.
Measurement of vascular O2 •− levels: lucigenin chemiluminescence
O2 •− production was assessed from vascular samples by the lucigenin chemiluminescence (5 µmol/l) method as previously described by our laboratory (15, 21, 46, 62). O2 •− production in the myocardiocytes was also assessed using the same method.
Measurement of vascular O2 •− levels: ethidium bromide fluorescence
Hydroethidine, an oxidative fluorescent dye, was used to assess O2 •− production in carotid arteries as previously reported by our laboratory (15, 21, 60, 62). This method provides sensitive detection of O2 •− levels in situ. In brief, cells are permeable to hydroethidine, which in the presence of O2 •− is oxidized to fluorescent ethidium bromide (EB). Ethidium is trapped by intercalation with DNA. Isolated, living vessels were incubated with hydroethidine (10−6 mol/l; at 37°C for 60 min). The arteries were then washed three times, embedded in optimum cutting temperature medium, and cryosectioned. Vascular sections were imaged using a Zeiss AxioCam Mrm camera mounted on a Zeiss Axiovert 200 microscope (Carl Zeiss, Thornwood, NY). Images were captured at ×20 magnification and analyzed using the Zeiss Axiovision imaging software. Ten to fifteen entire fields per vessel were analyzed with one image per field. The mean fluorescence intensities of EB-stained nuclei in the endothelium and medial layer were calculated for each vessel (n = 6 for each group). Thereafter, these intensity values for each animal in the group were averaged. Vessels coincubated with Tiron were used as negative controls.
Cigarette smoke extract preparation
Cigarette smoke extract (CSE; dissolved in DMSO, 40 mg/ml total particular matter, and nicotine content, 6%; kept at −80°C) was purchased from Murty Pharmaceuticals (Lexington, KY). From this stock solution, working solutions (from 0.004 to 40 µg/ml final concentration) were prepared immediately before the experiments by dilution with physiological HEPES buffer. In previous experiments, our laboratory has established a dose-response curve for the effects of CSE (0.004 ng/ml to 40 µg/ml) (46). With the assumption that cigarette smoke is extracted in the blood and equilibration occurs with the total blood volume, it is likely that the plasma levels of water-soluble components of cigarette smoke in smokers overlap with the CSE concentrations used in our present and previous studies (46). There are reliable data for the plasma concentrations of the particulate matter constituent nicotine during smoking. Thus, to correlate in vitro CSE concentrations with in vivo plasma levels, one can compare the nicotine concentration ranges in CSE and in the plasma. A commercially available cigarette contains ~15 mg of nicotine and a comparable amount of tar. Smoking a single cigarette under standardized conditions can produce peak plasma nicotine levels exceeding 25 ng/ml (41). In individuals who smoke more than one cigarette per day, the plasma level of nicotine is >50 ng/ml. In addition, there are 200–800 ng/ml cotinine and 100–500 ng/ml 3-OH cotinine, both metabolites of nicotine, in the blood stream (72). In our present and previous in vitro experiments, we have used CSE, which contained nicotine in a concentration range from 0.24 to 2,400 ng/ml. Thus CSE exerts its prooxidant and proinflammatory endothelial effects in a concentration range, which is likely to be comparable with those in the blood stream of smokers (46).
Measurement of O2 •− production in CSE-treated cultured arteries
To assess the direct effect of cigarette smoke constituents and resveratrol on vascular ROS generation, the ex vivo studies were complemented by in vitro experiments. Isolated carotid arteries were maintained in a stainless steel vessel culture chamber (Danish Myo Technology) under sterile conditions in F12 medium (GIBCO) containing antibiotics (100 UI/l penicillin and 100 mg/l streptomycin) and supplemented with 5% FCS (GIBCO/Invitrogen), as previously described (46) in the presence or absence of resveratrol (10 µmol/l for 24 h). In previous studies, our laboratory has demonstrated that resveratrol exerts antioxidant and anti-inflammatory action in a wide concentration range (0.3 to 30 µM), which overlaps with the resveratrol concentrations achievable in vivo by resveratrol feeding (20, 63). In the current study, we tested the effects of 10 µM resveratrol in vitro to compare the in vivo effects of resveratrol with its direct endothelial action in vitro and to dissect the molecular pathways responsible for the protective effects of resveratrol against cigarette smoke-induced endothelial alterations. We chose this concentration because it is close to the peak resveratrol concentration present in the plasma following oral resveratrol treatment. With the use of this concentration, the molecular pathways responsible for the effects of resveratrol can also be reliably assessed (20, 63). After the culture period, arteries were treated with CSE (0.4 µg/ml) or vehicle for 6 h. After the culture period, O2 •− production in the arterial segments was assessed by the hydroethidine staining method. En face preparations were imaged using a Zeiss Pascal laser-scanning confocal microscope. All fields were selected by random movement of the microscope stage to another area within an intact luminal surface of the artery. Images of the endothelial cell nuclei and nuclei of the underlying smooth muscle cells were captured at ×20 magnification and analyzed using the Zeiss Axionvision imaging software. Ten to fifteen entire fields per treatment group were analyzed with one image per field. The mean fluorescence intensities of EB-stained nuclei in the endothelium and medial layer were calculated for each vessel (n = 6 for each group). Thereafter, these intensity values for each animal in the group were averaged. Vessels coincubated with Tiron or polyethylene glycol (PEG)-SOD were used as negative controls.
Measurement of O2 •− and H2O2 production in CSE-treated endothelial cells: effects of SIRT1 overexpression and SIRT1 knockdown
Primary human CAECs (Cell Applications, San Diego, CA; after passage 4) were cultured in 96-well plates as described (15, 20, 63) in the presence or absence of resveratrol (10 µmol/l for 24 h). Thereafter, CAECs were treated with CSE (0.4 µg/ml) or vehicle for 24 h. After the culture period, the cells were washed three times and cellular O2 •− and H2O2 production was assessed using the dihydroethidine and C-H2DCFDA fluorescent methods, respectively (63). The time course of the buildup in EB and dichlorofluorescein (DCF) fluorescence was assessed by a Tecan Infinite M200 plate reader. The slope factors were calculated and normalized to Hoechst 33258 fluorescence representing DNA content (number of cells).
In separate experiments, the downregulation of SIRT1, the molecular target of resveratrol, in CAECs was achieved by RNA interference using the proprietary SIRT1 small-interfering RNA (siRNA) sequences (Origen) and the Amaxa Nucleofector technology (Amaxa, Gaithersburg, MD), as previously reported by our laboratory (15, 23, 65). Cell density at transfection was 30%. Specific gene silencing was verified with quantitative (q)RT-PCR and Western blot analysis (at the mRNA and protein level, respectively) as described (15, 23). On day 2 after the transfection, when gene silencing was optimal (>80%), the cells were treated with resveratrol (10 µmol/l for 24 h) or vehicle. Thereafter, CAECs were treated with CSE (0.4 µg/ml) or vehicle for 24 h. After the culture period, the cells were washed three times and cellular O2 •− production was assessed using the dihydroethidine fluorescent method.
In other experiments, SIRT1 was overexpressed (~7-fold) in CAECs using a proprietary cDNA construct (Strategene). The cells were treated with CSE (0.4 µg/ml) or vehicle for 24 h, and then cellular O2 •− production was assessed using the dihydroethidine fluorescent method. CAECs coincubated with Tiron or PEG-SOD served as negative controls.
Measurement of mitochondrial O2 •− production in CSE-treated endothelial cells
Mitochondrial O2 •− production was assessed in CSE-treated CAECs by flow cytometry (FAScalibur; BD Bioscience, San Jose, CA) using MitoSOX red as previously reported (43, 44). Cell debris (low forward and side scatter), dead cells (Sytox Green and annexin V positive), and apoptotic cells (annexin V positive) were gated out for analysis (43, 44). The data are presented by histogram of mean intensity of MitoSOX fluorescence or fold change when compared with those of the untreated control. The specificity of the method used was previously verified (43, 44).
Transient transfection and luciferase assays
CAECs were treated with resveratrol (24 h) and then treated with CSE. In some cells, SIRT1 was silenced (siRNA) or overexpressed before the resveratrol/CSE treatment protocols. The effect of CSE and resveratrol on NF-κB activity in CAECs was tested by a reporter gene assay. We used an NF-κB reporter composed of an NF-κB response element upstream of firefly luciferase (NF-κB-Luc; Stratagene) and a renilla luciferase plasmid under the control of the cytomegalovirus promoter (as an internal control). All transfections were performed with Novafector (Venn Nova, Pompano Beach, FL) following manufacturer protocols. Firefly and renilla luciferase activities were assessed after 42 h using the Dual Luciferase Reporter Assay Kit (Promega, Madison, WI) and a luminometer.
Real-time qPCR
qRT-PCR was used to elucidate the effect of resveratrol treatment on the smoking-induced expression of inflammatory master cytokines (TNF-α, IL-1β, and IL-6) and iNOS in coronary arteries. These factors were shown to be associated vascular inflammation induced by cigarette smoking (46) and are considered to be indicators of accelerated vascular aging. To elucidate the role of resveratrol on CSE-induced inflammatory gene expression in vitro, CAECs were treated with CSE with or without pretreatment with resveratrol (for 24 h). In some cells, SIRT1 was silenced (siRNA) or overexpressed before the resveratrol/CSE treatment protocols. Total RNA from the coronary arteries of the experimental animals and CAECs was isolated with a Mini RNA Isolation Kit (Zymo Research, Orange, CA) and was reverse transcribed using Superscript II RT (Invitrogen) as described previously (21, 22, 46). The real-time RT-PCR technique was used to analyze mRNA expression using the Strategene MX3000, as reported (15, 21–23, 46, 59). Samples were run in triplicates. The efficiency of the PCR reaction was determined using a dilution series of a standard vascular sample. Quantification was performed using the ΔΔCt method. The housekeeping gene β-actin was used for internal normalization. Oligonucleotides used for real-time qRT-PCR are listed in Table 1. Fidelity of the PCR reaction was determined by melting temperature analysis and visualization of product on a 2% agarose gel.
Table 1.
Oligonucleotides for real-time RT-PCR
| mRNA Targets | Sense | Antisense |
|---|---|---|
| Rat | ||
| TNF-α | TCGTAGCAAACCACCAAG | CTGACGGTGTGGGTGA |
| IL-1β | CAGCAATGGTCGGGAC | ATAGGTAAGTGGTTGCCT |
| IL-6 | TACCCCAACTTCCAATGC | GATACCCATCGACAGGAT |
| iNOS | TGCGGAGTGTCAGTGG | GGAGTAGCCTGTGTGC |
| ICAM-1 | CACAGCCTGGAGTCTC | CCCTTCTAAGTGGTTGGAA |
| HPRT | AAGACAGCGGCAAGTTGAATC | AAGGGACGCAGCAACAGAC |
| Human | ||
| TNF-α | GTGAGGAGGACGAACATC | AGCCAGAAGAGGTTGAGG |
| IL-6 | CCACCCCTGACCCAAC | AGTGTCCTAACGCTCATAC |
| iNOS | GGATTGATCGGAGCCT | ATGGGGAACAGACTGG |
| HPRT | AGATGGTCAAGGTCGCAAG | TTCATTATAGTCAAGGGCATATCC |
iNOS, inducible nitric oxide synthase; HPRT, hypoxanthine phosphoribosyltransferase.
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling assay
To quantitate the rate of endothelial cell apoptosis, terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) assay was performed as reported (18).
Caspase activity assay
The arterial and endothelial cell samples were homogenized in lyses buffer, and caspase activities were measured using Caspase-Glo 3/7 assay kit according to the manufacturer’s instruction (Promega) as described (18, 63). In 96-well plates, a 50-µl sample was mixed gently for 30 s with 50 µl Caspase-Glo 3/7 reagent and incubated for 2 h at room temperature. The lyses buffer with the reagent served as the blank. The luminescence of the samples was measured using an Infinite M200 plate reader (Tecan, Research Triangle Park, NC). Luminescent intensity values were normalized to the sample protein concentration.
Detection of apoptotic cell death by ELISA
Cytoplasmic histone-associated DNA fragments, which indicate apoptotic cell death, were quantified by the Cell Death Detection ELISAPlus kit (Roche Diagnostics, Indianapolis, IN) as described (18, 23, 63). Results are reported as normalized arbitrary optical density units.
DNA damage analysis by comet assay
Endothelial cells were pretreated with resveratrol (10−5 mol/l; for 24 h) and then treated with CSE (for 6 h) to induce DNA damage. After the culture period, the cells were harvested. Cells (105) in 100 µl PBS were mixed with 100 µl of 1.5% low-melting agarose, and 90 µl of this mixture were spotted on CometAssay slides (Trevigen, Gaithersburg, MD) between two layers of 1% low-melting agarose. The extent of DNA fragmentation was examined by single-cell electrophoresis (comet assay), as reported (63). Briefly, the comet assay is based on the alkaline lysis of labile DNA at sites of damage. The slides were treated with a lysis solution containing 2.5 M NaCl, 100 mM Na2EDTA, 1% Triton X-100, 10% DMSO, and 10 mM Trizma base (pH 10) for 1 h at 4°C in the dark, rinsed with a neutralization buffer 3 × 5 min in 0.4 M Tris (pH 7.5) to remove detergents and salts, and then submerged in a high pH buffer containing (in mM) 300 NaOH and 1 Na2EDTA (pH > 13) for 20 min to allow for unwinding of the DNA and the expression of alkali-labile damage. Electrophoresis was performed using the same buffer at 25 V (~0.74 V/cm) and 300 mA for 20 min. At the end of the run, the slides were neutralized in 0.4 M Tris •HCl (pH 7.5), submersed in absolute ethanol for 3 min, and air-dried and DNA was stained with SYBR green (Invitrogen). Fluorescent images of the nuclei were captured using a fluorescent microscope (at ×40 magnification). DNA damage was quantified by measuring the tail DNA content (as a percentage of total DNA) using the VisComet 2.0 software (Impulse).
Data analysis
Data were normalized to the respective control mean values. Data are means ± SE. Statistical analyses of data were performed by Student’s t-test or by two-way ANOVA followed by the Tukey’s post hoc test, as appropriate. A value of P < 0.05 was considered statistically significant.
RESULTS
Resveratrol prevents smoking-induced endothelial dysfunction
In vivo exposure of rats to cigarette smoke elicited impaired vascular relaxations to acetylcholine, which were prevented by resveratrol treatment (Fig. 1A).
Fig. 1.
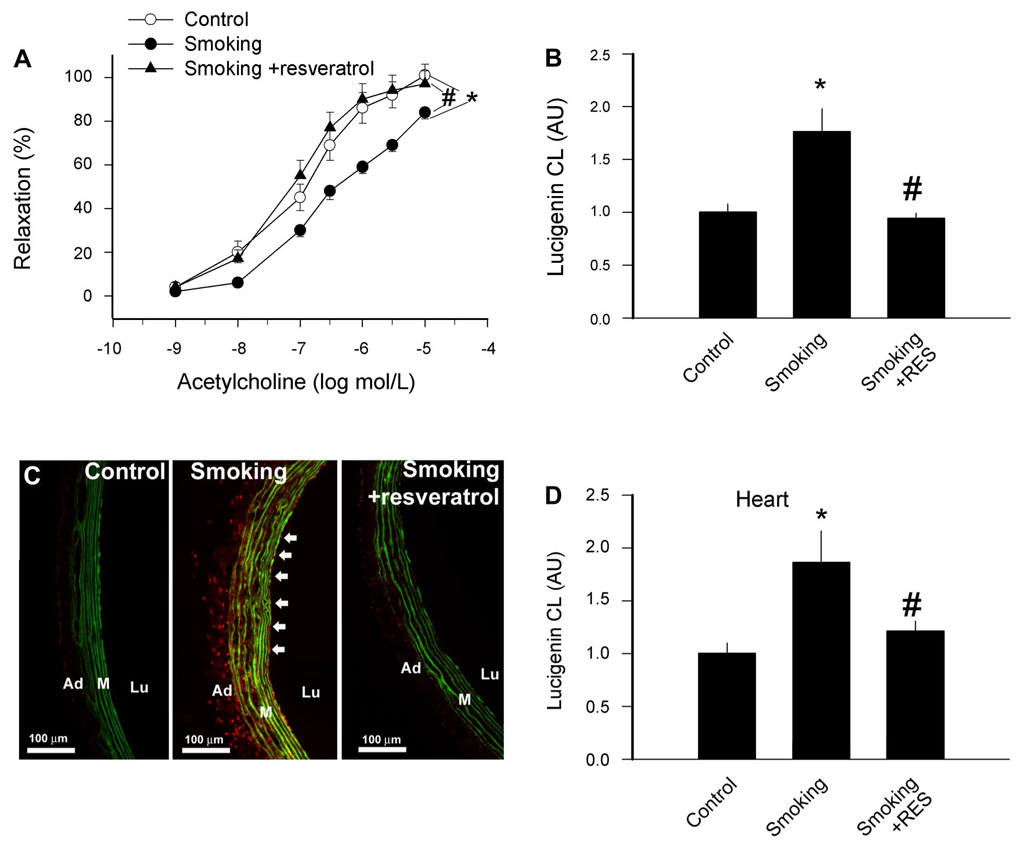
A: effects of resveratrol (Res) treatment on relaxation responses to acetylcholine in ring preparations of carotid arteries of control rats and rats exposed to cigarette smoke (smoking; see MATERIALS AND METHODS). Data are means ± SE; n = 6 animals for each group. B: effects of Res treatment on O2 •− generation in vessels of control rats and rats exposed to cigarette smoke. O2 •− generation was determined by the lucigenin (5 µmol/l) chemiluminescence (CL) method. Data are normalized to the mean value of the untreated control group. Data are means ± SE. C: representative fluorescent photomicrographs showing increased nuclear ethidium bromide (EB) fluorescence in endothelial cells (arrows) in sections of carotid arteries of rats exposed to cigarette smoke compared with vessels of control rats. Vessels were incubated with the dye dihydroethidium, which produces a red nuclear fluorescence when oxidized to EB by O2 •−. Res treatment prevented smoking-induced increases in vascular O2 •− production. Green autofluorescence of elastic laminae is shown for orientation purposes. Lu, lumen; M, media; Ad, adventitia. D: effects of Res treatment on O2 •− generation by myocardium of control rats and rats exposed to cigarette smoke (with or without Res treatment). O2 •− generation was determined by the lucigenin (5 µmol/l) CL method. Data are means ± SE. *P <0.05 vs. untreated; #P <0.05 vs. no Res. AU, arbitrary units.
Resveratrol prevents increases in vascular O2 •− production induced by smoking and in vitro CSE exposure
In carotid arteries of cigarette smoke-exposed rats, there was an increased O2 •− production as indicated by the increased lucigenin chemiluminescent signal (Fig. 1B). O2 •− generation was also increased in the myocardium (Fig. 1D). Using the EB staining method, we found that in cross sections of arteries of cigarette smoke-exposed rats, the mean fluorescence intensity of endothelial and smooth muscle cell nuclei was significantly greater than that of control rats (Fig. 1C). Resveratrol treatment prevented cigarette smoking-induced oxidative stress both in the vascular (Fig. 1, B and C) and cardiac (Fig. 1D) tissues.
Representative fluorescent photomicrographs of EB-stained untreated and CSE-treated carotid arteries (en face preparations) are shown in Fig. 2. When compared with that in untreated control vessels (Fig. 2A), in CSE-treated vessels there was an intensive nuclear EB staining, localized to the endothelial and smooth muscle cells, indicating that CSE directly promotes O2 •− generation in both cell types (Fig. 2B). In vitro resveratrol treatment (24 h) prevented CSE-induced increases in O2 •− production by the vascular endothelial and smooth muscle cells (Fig. 2C).
Fig. 2.
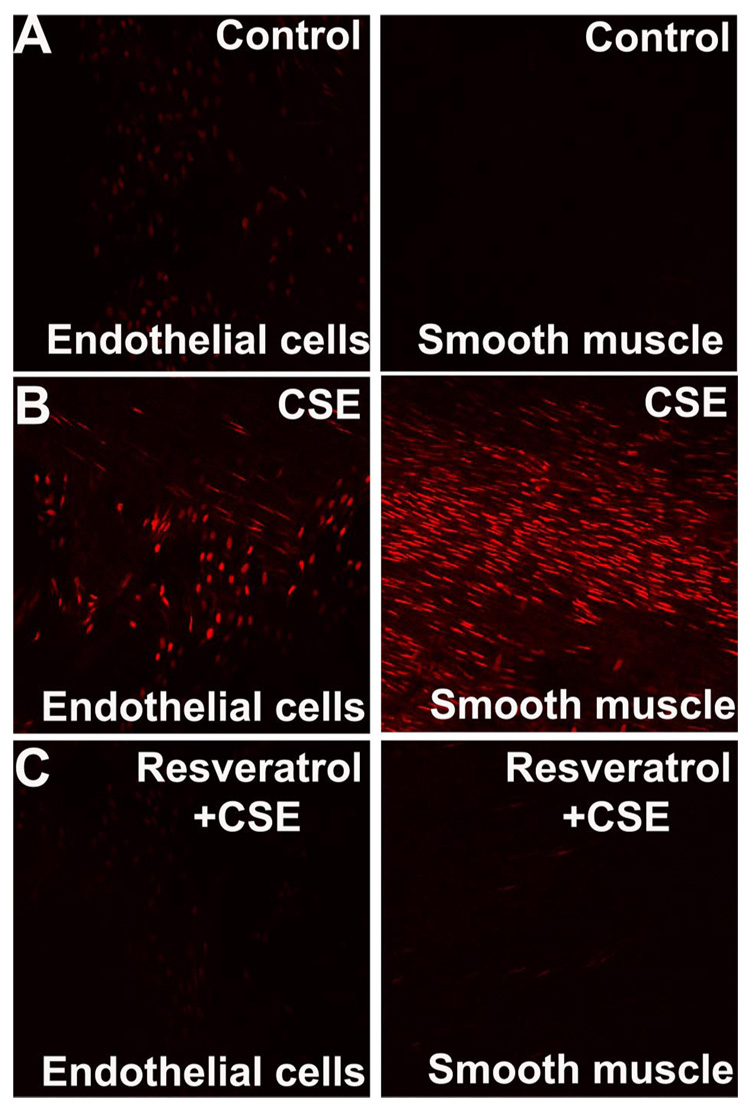
A: representative confocal images of en face preparations of rat carotid arteries incubated with cigarette smoke extract (CSE; 4 µg/ml; B) and untreated controls (A) after dihydroethidine staining. On these compressed Z stack images, the EB-stained elongated nuclei of vascular smooth muscle cells (A–C, right) and the round nuclei of endothelial cells (A–C, left) are visualized. The uneven distribution of the endothelial nuclei is due to the complex three-dimensional structure of the preparation. CSE resulted in a substantially increased nuclear EB staining in both endothelial and smooth muscle cells. Res pretreatment substantially reduced CSE-induced increases in EB staining in both cell types (C). Identical results were observed in 5 separate experiments (original magnification, ×20).
Resveratrol attenuates CSE-induced increases in O2 •− and H2O2 production in cultured endothelial cells: role of SIRT1
Treatment with CSE also significantly increased O2 •− (as indicated by the increased EB fluorescence; Fig. 3A) and H2O2 production (as indicated by the increased DCF fluorescence; Fig. 3F) in CAECs, which were prevented by pretreatment with resveratrol. To elucidate the cellular mechanism underlying the protective effects of resveratrol, the downregulation of SIRT1 in CAECs was achieved by RNA interference (Fig. 3, B and C). We found that the protective effects of resveratrol were abolished by knockdown of SIRT1 (Fig. 3D). In contrast, the overexpression of SIRT1 significantly attenuated CSE-induced oxidative stress in CAECs (Fig. 3E).
Fig. 3.
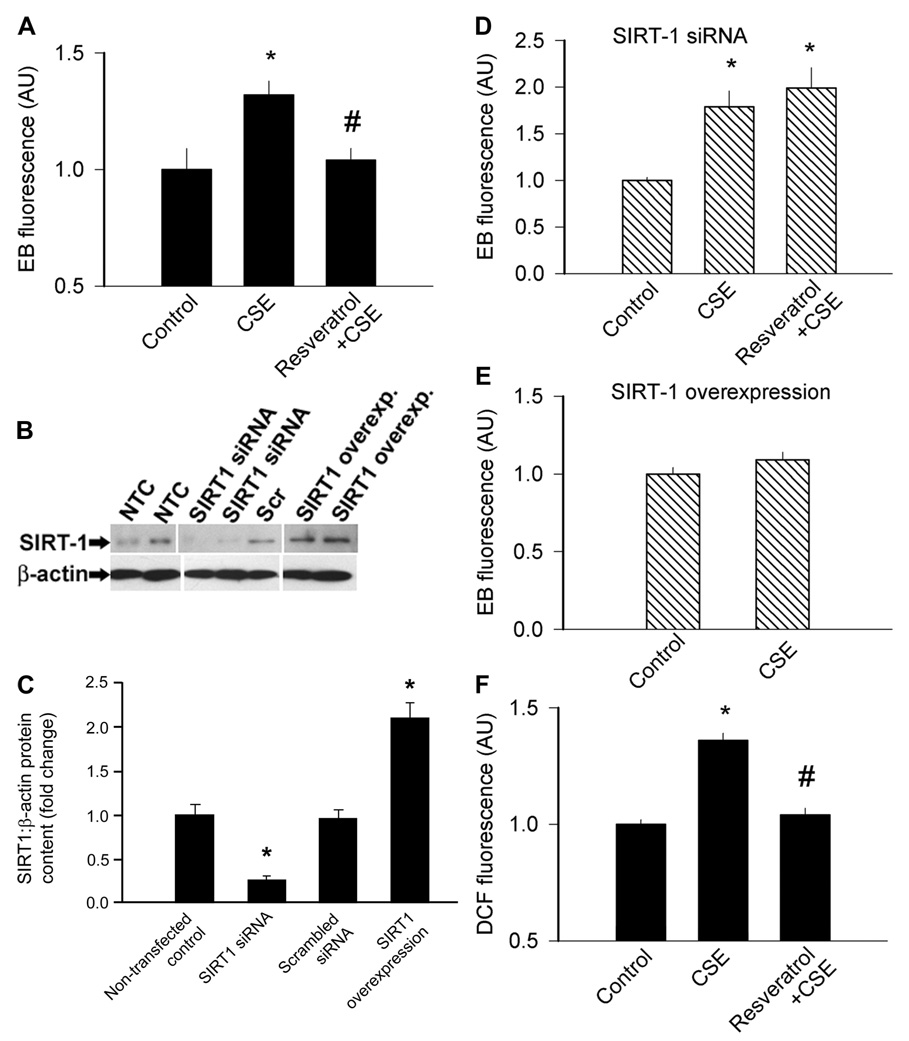
A: summary data from dihydroethidine staining experiments showing the time-dependent buildup of EB fluorescence in coronary arterial endothelial cells (CAECs). Bar graphs are summary data for slope factors normalized to cell number (i.e., nuclear DNA content, assessed by Hoechst fluorescence intensity), representing cellular O2 •− production. CSE (4 µg/ml) elicited significant increases in cellular O2 •− generation, which were prevented by Res (10 µmol/l) pretreatment (A). B: original Western blot showing downregulation of sirtuin 1 (SIRT1) in small-interfering RNA (siRNA)-treated CAECs. NTC, nontransfected control; Scr, scrambled siRNA. The effect of overexpression of SIRT1 is also shown. β-actin was used for normalization. Bar graphs (C) are summary densitometric data. Data are means ± SE. *P <0.05 vs. NTC. D: knockdown of SIRT1 (siRNA) in CAECs prevented the inhibitory effect of Res on CSE-induced oxidative stress. E: overexpression of SIRT1 in CAECs prevented CSE-induced oxidative stress, mimicking the effects of Res. F: CSE also significantly increased H2O2 production in CAECs [as shown by the dichlorofluorescein (DCF) fluorescence method], which was prevented by Res pretreatment. Data are means ± SE; n = 8 animals for each group. *P <0.05 vs. untreated; #P <0.05 vs. no Res. Overexp, overexpression.
Resveratrol attenuates CSE-induced mitochondrial ROS production in endothelial cells
To test the direct effect of resveratrol on CSE-induced mitochondrial ROS generation, cultured cells were treated with resveratrol (10 µmol/l for 24 h) or vehicle. Subsequently, the cells were treated with CSE (from 0.04 to 4 µmol/l; for 24 h). Fluorescent microscopy (Fig. 4, A–C) and fluorescence-activated cell sorting analysis (Fig. 4, D–F) showed that when compared with that of the untreated controls, CSE significantly increased cellular MitoSox staining. Resveratrol pretreatment significantly attenuated CSE-induced increases in mitochondrial O2 •− production (Fig. 4, C, E, and F).
Fig. 4.
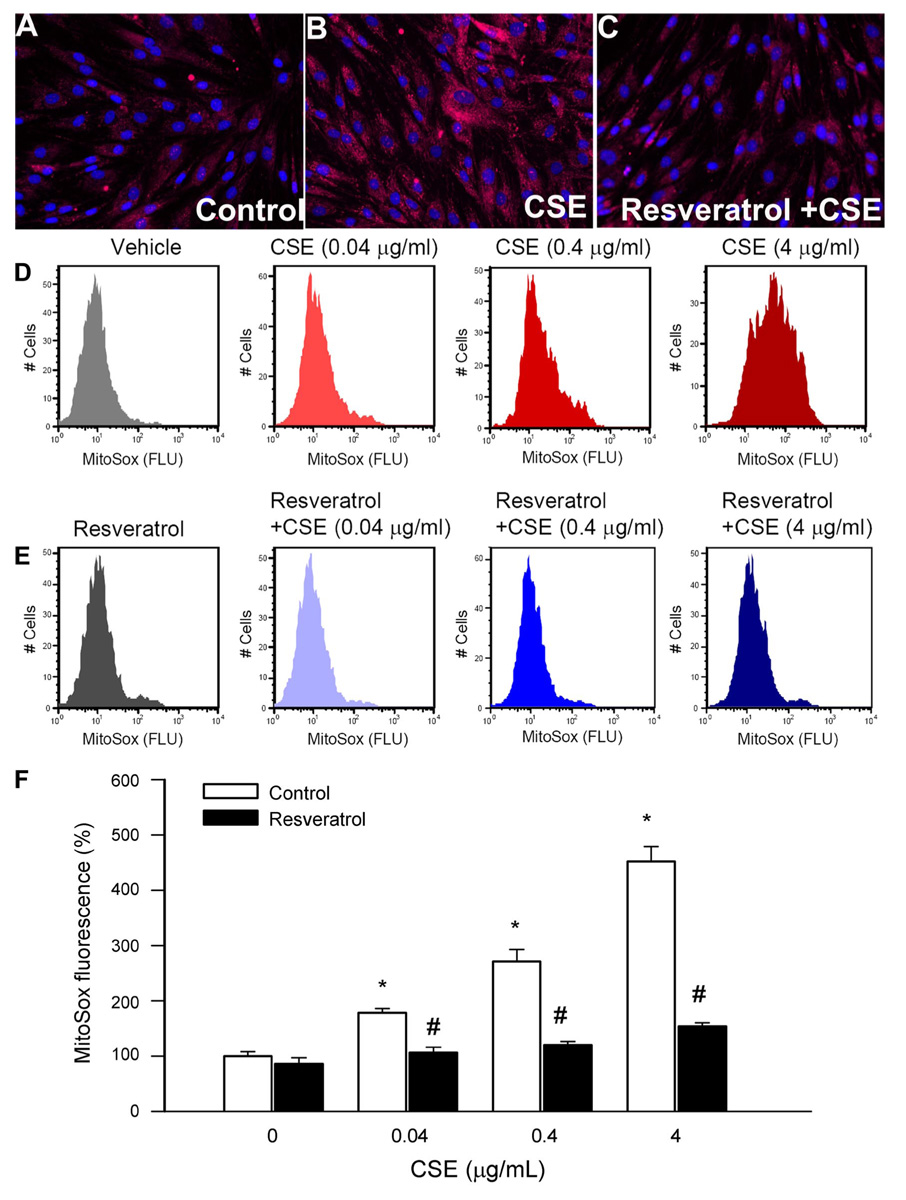
A–C: representative fluorescent images showing stronger perinuclear MitoSox staining [red fluorescence (Flu)] in CSE (4 µg/ml)-treated cells (B) compared with untreated controls (A). C: Res (10 µmol/l) pretreatment substantially attenuated CSE-induced mitochondrial O2 •− generation. Hoechst 33258 (blue fluorescence) was used for nuclear staining (original magnification, ×20). D–E: representative histograms of flow cytometry experiments demonstrating that CSE in a dose-dependent manner (D) elicits significant increases in mean fluorescent intensity of oxidized MitoSOX in CAECs, which were prevented by Res pretreatment (E). The experiments were performed in quadruplicates with identical results. F: summary data. Data are means ± SE. *P < 0.05 vs. untreated; #P < 0.05 vs. no Res.
Resveratrol attenuates CSE-induced activation of NF-κB in endothelial cells
We confirmed that CSE significantly enhanced the transcriptional activity of NF-κB in CAECs (as indicated by an increase in the luciferase activity; Fig. 5). Importantly, CSE-induced NF-κB activity was substantially attenuated by resveratrol pretreatment or the overexpression of SIRT1 (Fig. 5). The protective effects of resveratrol were significantly reduced by knockdown of SIRT1 (Fig. 5).
Fig. 5.
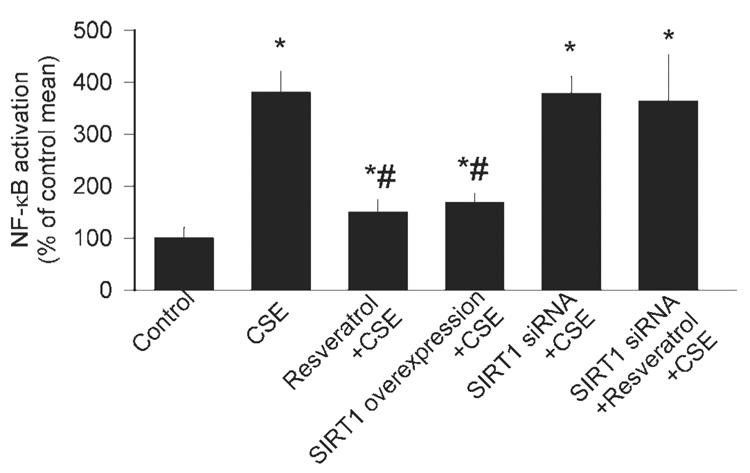
Reporter gene assay showing the effect of CSE (4 µg/ml) on NF-κB reporter activity in primary CAECs. Endothelial cells were transiently cotransfected with NF-κB-driven firefly luciferase and cytomegalovirus-driven ranilla luciferase constructs followed by CSE stimulation. Cells were then lysed and subjected to luciferase activity assay. After normalization, relative luciferase activity was obtained from 4 to 7 independent transfections. The effects of pretreatment with Res (10 µmol/l), SIRT1 overexpression, and knockdown of SIRT1 (siRNA) on CSE-induced NF-κB activation are also shown. Data are means ± SE. *P < 0.05. vs. untreated; #P < 0.05 vs. CSE only.
Resveratrol attenuates upregulation of inflammatory markers induced by smoking and in vitro CSE exposure in endothelial cells
In coronary arteries of cigarette smoke-exposed rats, the mRNA expression of iNOS, ICAM-1, IL-6, IL-1β, and TNF-α (Fig. 6, A–E) significantly increased. The expression of these inflammatory markers was significantly attenuated by in vivo resveratrol treatment (Fig. 6, A–E). The expression of SIRT1 was significantly increased by resveratrol treatment (Fig. 6F).
Fig. 6.
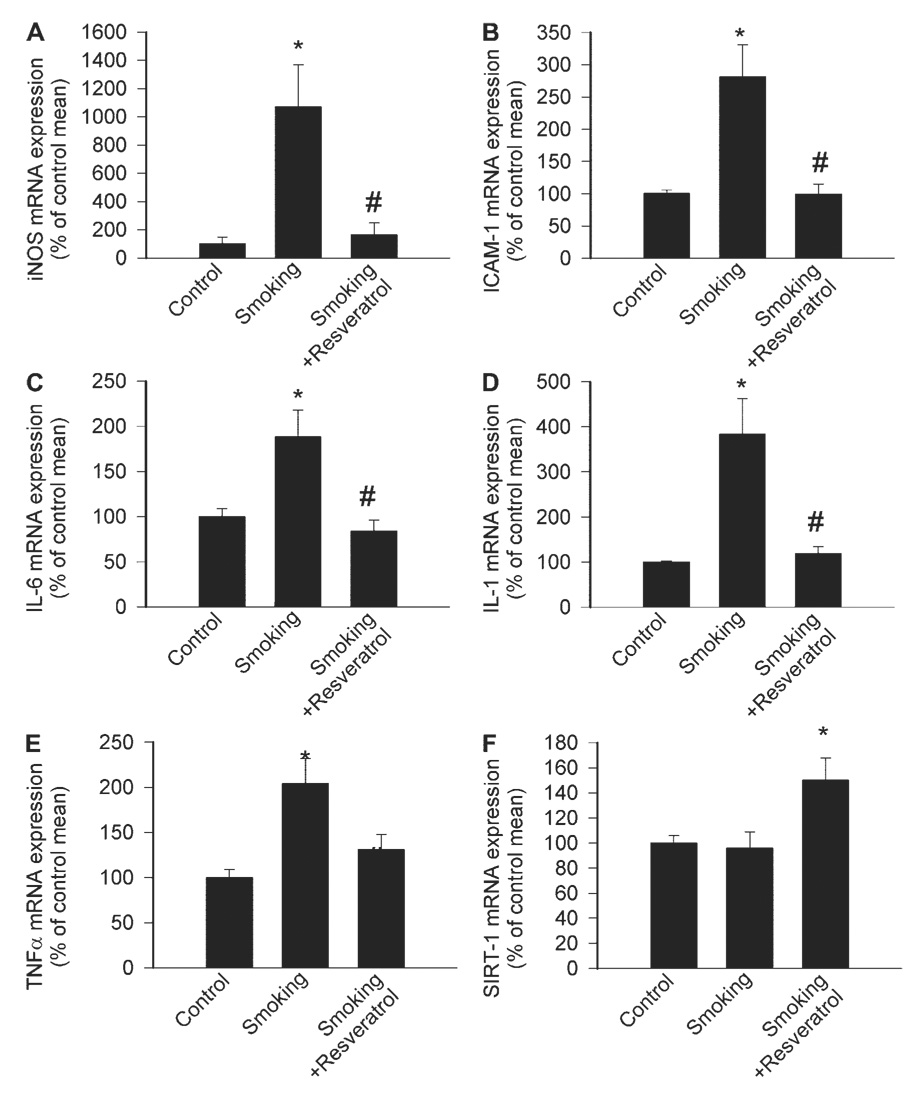
Expression of inducible nitric oxide synthase (iNOS; A), ICAM-1 (B), IL-6 (C), IL-1β (D), TNF-α (E), and SIRT1 (F) in coronary arteries of control rats, rats exposed to cigarette smoke (smoking; see MATERIALS AND METHODS), and rats exposed to cigarette smoke plus treated with Res. Analysis of mRNA expression was performed by real-time quantitative (q)RT-PCR. Hypoxanthine phosphoribosyltransferase (HPRT) was used for internal normalizations. Data are means ± SE; n = 5 to 6 animals for each group. *P <0.05 vs. untreated; #P <0.05 vs. no Res.
The exposure of CAECs to CSE in vitro also elicited the upregulation of iNOS, IL-6, and TNF-α (Fig. 7, A–C). The expression of iNOS, IL-6, and TNF-α in CSE-treated vessels was significantly reduced by resveratrol pretreatment or SIRT1 overexpression (Fig. 7, A–C). The anti-inflammatory effects of resveratrol were significantly attenuated by knockdown of SIRT1 (Fig. 7, A–C).
Fig. 7.
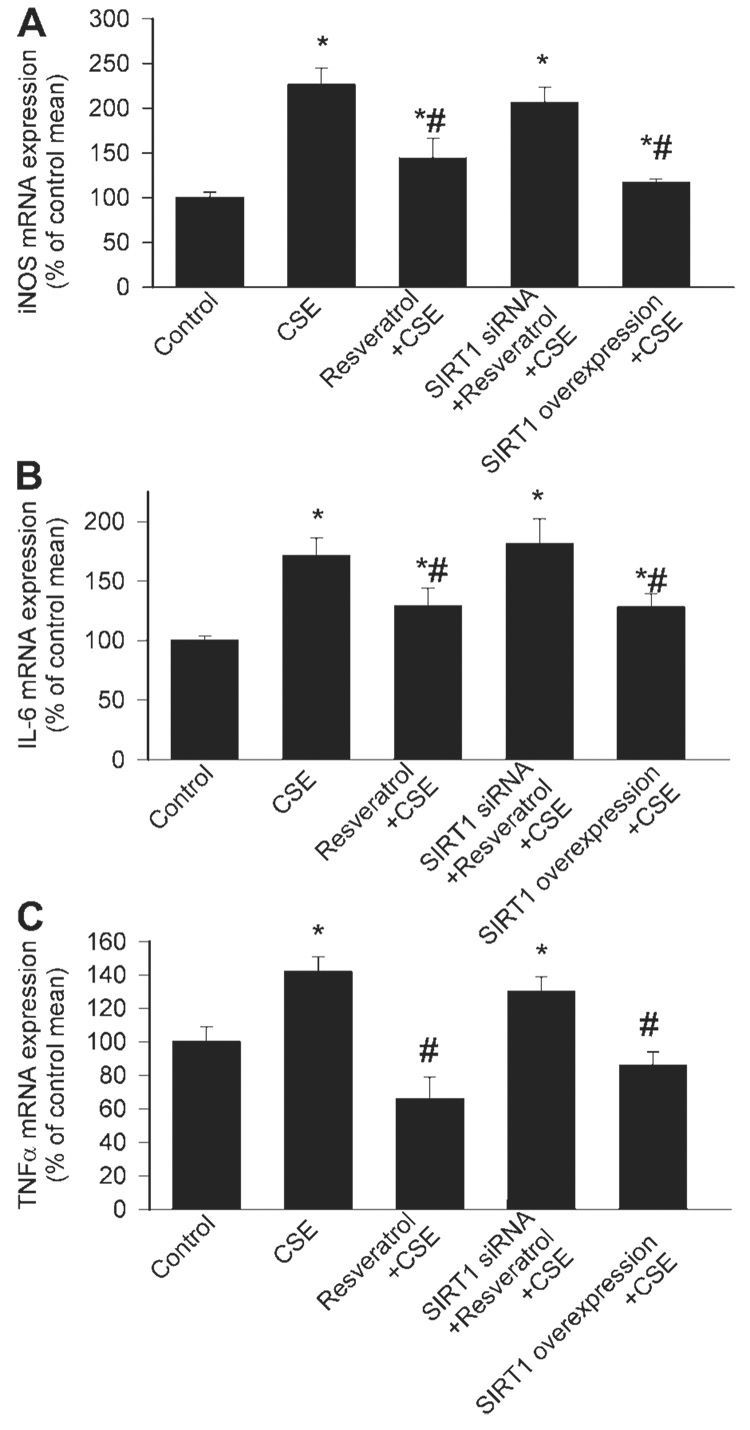
Effect of incubation with CSE (4 µg/ml) on expression of iNOS (A), IL-6 (B), and TNF-α (C) in CAECs. The effects of pretreatment with Res (10 µmol/l), SIRT1 overexpression, and knockdown of SIRT1 (siRNA) on CSE-induced inflammatory gene expression are also shown. Analysis of mRNA expression was performed by real-time qRT-PCR. HPRT was used for internal normalizations. Data are means ± SE; n = 4–6 animals for each group.*P < 0.05. vs. untreated; #P < 0.05 vs. CSE only.
Resveratrol attenuates cigarette smoke-induced endothelial apoptosis
The level of TUNEL-positive endothelial cells was low in vessels of untreated rats (Fig. 8A). Cigarette smoke exposure significantly increased the rate of endothelial cell apoptosis (Fig. 8B). In contrast, the number of TUNEL-positive endothelial cells remained low in vessels of resveratrol-treated rats after cigarette smoke exposure (Fig. 8C). Similar results were obtained when the DNA fragmentation rate was assessed in vascular homogenates (Fig. 8E) or when caspase 3/7 activities were compared as measures of apoptotic cell death (Fig. 8D). In vitro treatment of CAECs with CSE also induced apoptotic cell death, as indicated by the increased DNA fragmentation rate and caspase 3/7 activity (Fig. 8, F and G). Pretreatment of CAECs with resveratrol prevented CSE-induced increases in the rate of apoptotic cell death (Fig. 8, F and G). The antiapoptotic effects of resveratrol in CSE-treated CAECs were significantly attenuated by knockdown of SIRT1 (Fig. 8, F and G), whereas SIRT1 overexpression mimicked the effects of resveratrol (Fig. 8, F and G).
Fig. 8.
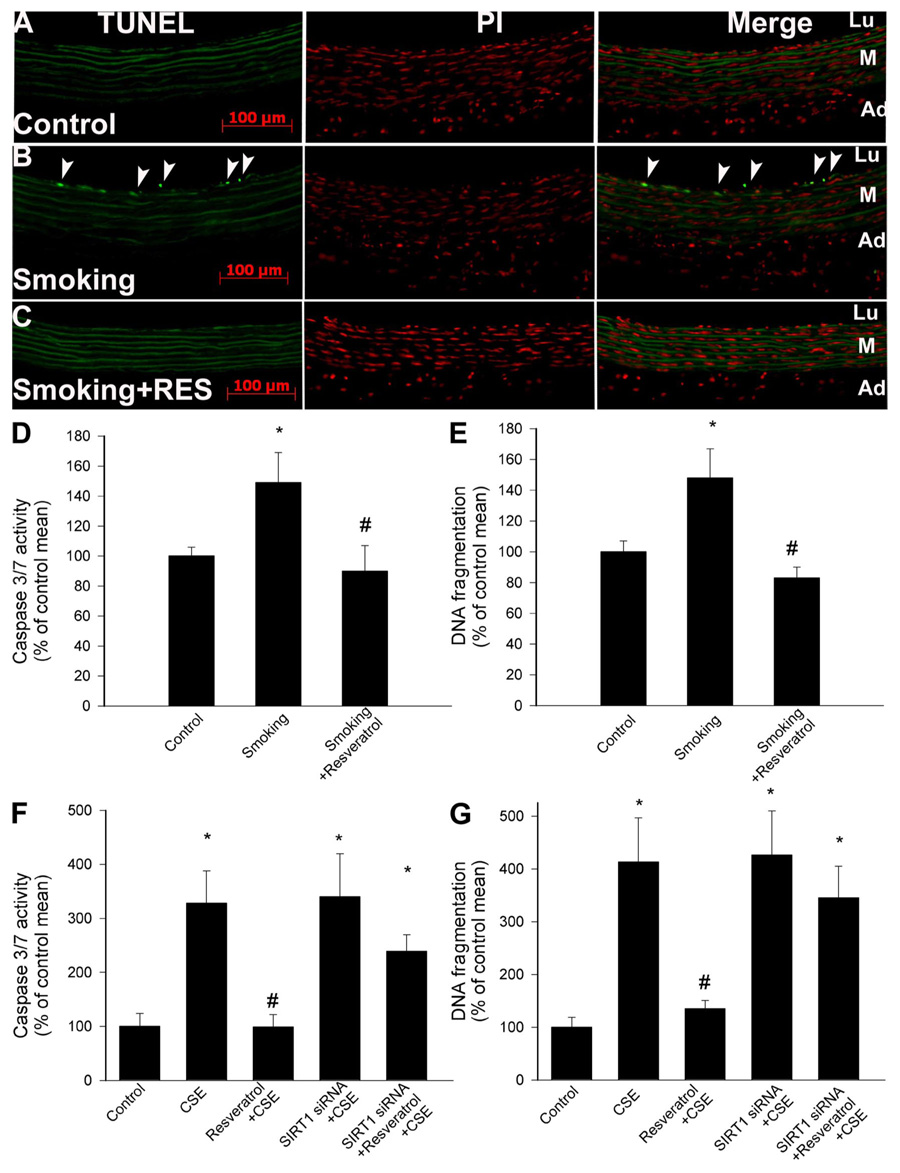
A: terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) assay results [left, TUNEL-positive cell nuclei are green and autofluorescence of elastic laminae are shown for orientation; middle, red fluorescence images of propidium iodide (PI)-stained nuclei; right, merged images]. When compared with that of untreated vessels (A), cigarette smoke exposure significantly increased the rate of endothelial cell apoptosis (B). In contrast, the number of TUNEL-positive endothelial cells remained low in vessels of Res-treated rats after cigarette smoke exposure (C). The green autofluorescence of elastic laminae is shown for orientation purposes. Caspase 3/7 activity (D) and DNA fragmentation (E) in carotid arteries of control rats and rats exposed to cigarette smoke (smoking; see MATERIALS AND METHODS) with or without Res treatment are shown. Data are means ± SE; n = 6 animals for each group (E). *P < 0.05 vs. untreated; #P < 0.05 vs. no Res. F and G: Res pretreatment inhibits the increases in caspase 3/7 activity (F) and DNA fragmentation (G) in CAECs induced by CSE (4 µg/ml). Knockdown of SIRT1 (siRNA) abolished the antiapoptotic effect of Res in CSE-treated CAECs. Data are means ± SE; n = 6 experiments for each group (G). Data are means ± SE. *P < 0.05. vs. untreated; #P < 0.05 vs. CSE only.
Resveratrol protects endothelial cells against CSE-induced DNA damage
Comet assay was performed to analyze CSE-induced DNA damage in CAECs. A random sample of 120 cells was analyzed from each slide. DNA strand breaks were quantified by assessing tail DNA content as described (18, 63). In cells without CSE treatment, all DNA was confined to the nuclei, as indicated by the percentage of DNA in the tail being <5%. In CSE, the treatment of CAECs resulted in a significant increase in DNA strand breaks (observed as a fluorescent tail along the electric field because small DNA fragments migrated out of the nuclei). Pretreatment with resveratrol led to a significant decrease in DNA damage (P < 0.05 vs. CSE treatment alone; Fig. 9).
Fig. 9.
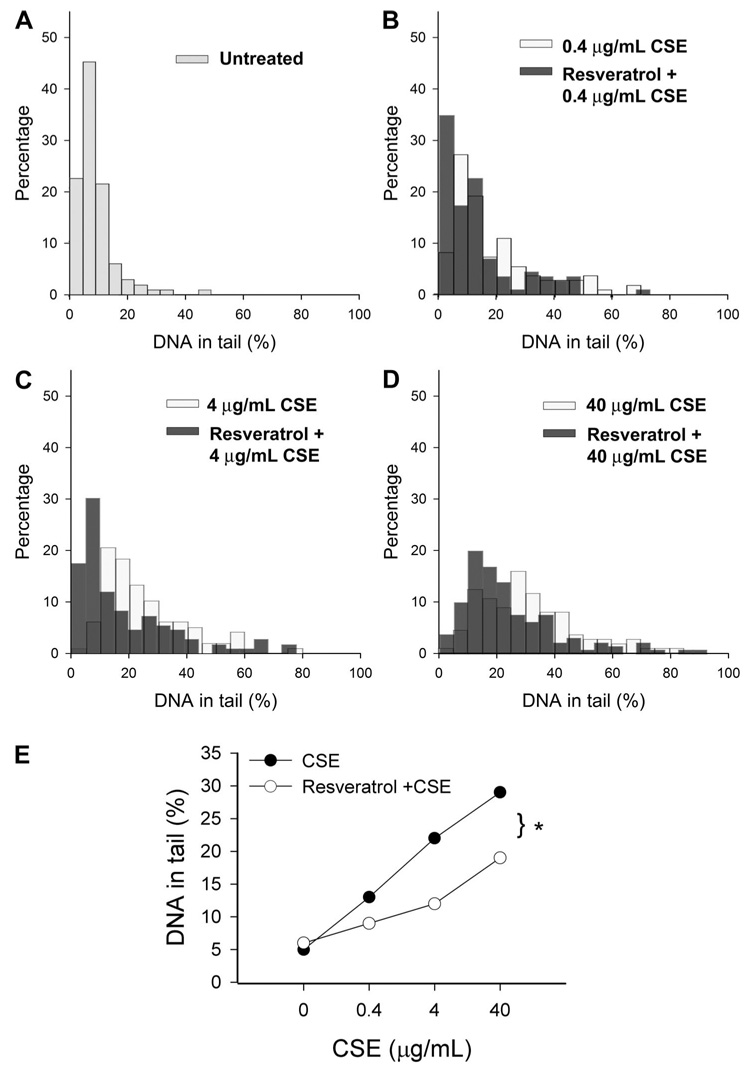
Results from comet assay experiments. CAECs were treated with Res (10−5 mol/l for 24 h) followed by exposure to CSE (from 0.4 to 40 µg/ml). A–D: frequency distribution of tail DNA content in untreated control cells (A) and CAECs treated with CSE or Res plus CSE [B–D; damaged DNA migrates during electrophoresis from the nucleus toward the anode, forming a shape of a comet with a head (cell nucleus with intact DNA) and a tail (relaxed and broken DNA)]. E: summary data. Res pretreatment significantly attenuated CSE-induced increases in tail DNA content. *P < 0.05.
DISCUSSION
There are five important findings in this study. First, in vivo cigarette smoke exposure elicits significant endothelial dysfunction in rat carotid arteries, which could be prevented by resveratrol (Fig. 1). This finding accords with the attenuation of increased O2 •− generation by resveratrol in these vessels (Fig. 1, B and C). These results are especially important because we demonstrated for the first time that per os resveratrol treatment exerts vasoprotective effects in a clinically highly relevant model of accelerated vascular aging. Our laboratory has previously shown that NAD(P)H oxidase is an important source of O2 •− in this model (46). It is likely that water-soluble components of cigarette smoke are directly responsible for the induction of the vascular ROS production, because exposure of isolated arteries to CSE in vitro, in the absence of activated leukocytes, elicits significant O2 •− production in a concentration-dependent manner (35, 46) (Fig. 2). Dihydroethidine staining revealed that both endothelial cells and vascular smooth muscle cells exhibit an upregulated O2 •− generation upon both in vivo and in vitro cigarette smoke exposure, and resveratrol was equally effective in attenuating oxidative stress in both cell types (Fig. 1C and Fig 2). Recent studies support the idea that CSE in vitro may induce NAD(P)H oxidase(s) and ROS production in other cell types as well (32, 35, 51), including the myocardium (Fig. 1D). Our laboratory has previously shown that increased NAD(P)H oxidase activity is responsible, at least in part, for enhanced endothelial O2 •− production in aging (21) and other pathophysiological conditions associated with accelerated aging, e.g., hyperhomocysteinemia (59) and hypertension (60, 62). O2 •− is known to react with NO-forming ONOO−, which is likely responsible for many of the adverse cellular effects of increased superoxide production (26, 47). Human studies suggest that smoking increases plasma 3-nitrotyrosine content (a biomarker of increased ONOO− generation) (48), thus in future studies it will be interesting to determine whether resveratrol treatment attenuates smoking-induced nitrosative stress as well.
At present, we do not know whether resveratrol directly inhibits the activation of the NAD(P)H oxidase in endothelial cells. We have strong evidence that resveratrol upregulates multiple antioxidant enzyme systems, which likely contribute to the attenuation of oxidative stress in endothelial cells (63). It is likely that the observed effects are not due to the antioxidant properties of resveratrol itself because pretreatment with resveratrol significantly attenuated CSE-induced ROS production (even when CSE treatment was applied in the absence of resveratrol) and the downregulation of SIRT1 prevented the antioxidant effects of resveratrol treatment (Fig. 3, B–D). The role of SIRT1 as a primary mediator of the effects of resveratrol is also suggested by the findings that the overexpression of SIRT1 mimicked many of the antioxidant effects of resveratrol (Fig. 3E).
In the present study, we have not measured the plasma concentration of resveratrol. Resveratrol is known to be easily absorbed from the digestive track, and per os resveratrol treatment was shown to induce substantial gene expression changes in a number of organs (9, 10). After per os administration, the plasma concentration of resveratrol increases to the micromolar range. In the circulation, in addition to the free form of resveratrol, trans-resveratrol-3-sulfate, trans-resveratrol-4′-sulfate, trans-resveratrol-3,5-disulfate, trans-resveratrol-3,4′-disulfate, trans-resveratrol-3,4′,5-trisulfate, trans-resveratrol-3-O-β-d-glucuronide, and resveratrol aglycone are also present, which likely retain much of the bioactivity of resveratrol. The findings that resveratrol consumption resulted in marked improvements in endothelial function and phenotype (similar to the observed direct in vitro effects of resveratrol) strongly suggest that resveratrol reaches the vascular cells in effective concentrations.
Mitochondria are also important sources of ROS in the vasculature, especially in aging (66). In the present study, we show that cigarette smoke exposure also increases mitochondrial O2 •− production in endothelial cells (Fig. 4). It has also been shown that cigarette smoke constituents impair mitochondrial function and elicit mitochondrial oxidative stress in other cell types as well (6, 29, 30, 33, 36, 37, 55). In this regard, a recent study demonstrated that acrolein, a major toxicant in cigarette smoke, causes oxidative mitochondrial damage (36). A higher level of oxidative mitochondrial DNA damage has been observed in smokers (7, 37, 39). These data support the hypothesis that cigarette smoke-induced mitochondrial damage and dysfunction may contribute an increased risk for cardiovascular disease in smokers. It is significant that in our study resveratrol substantially reduced mitochondrial oxidative stress in CSE-treated endothelial cells (Fig. 4), perhaps due to the previously demonstrated effects of resveratrol on cellular antioxidant enzymes (63). Because of efficient scavenging of O2 •− by high levels of SOD in mitochondria, it is likely that mitochondria-derived O2 •− is a minor factor in impairing endothelial vasomotor function in cigarette smokers. There are several lines of evidence supporting the view that O2 •− in the mitochondria is dismutated to H2O2, which easily penetrates the mitochondrial membranes. Accordingly, our laboratory has shown that cellular H2O2 levels are increased in CSE-treated endothelial cells (46), which are significantly reduced by resveratrol treatment (Fig. 3D). It is likely that increased cellular H2O2 levels play important proinflammatory signaling roles in endothelial cells. For example, increased H2O2 production was shown to be responsible for activating NF-κB in the cytoplasm of endothelial cells in aged rats (66).
The second important finding is that CSE can significantly increase NF-κB activation in endothelial cells, whereas resveratrol pretreatment was able to significantly attenuate CSE-induced activation of NF-κB (46, 53) (Fig. 5). In contrast, basal NF-κB activity in endothelial cells is low, thus resveratrol alone has only a limited effect on endothelial NF-κB activity and inflammatory gene expression in unstimulated vascular cells (20). The mechanism of action of resveratrol is not completely understood. Resveratrol is known to activate the protein deacetylase SIRT1 (34, 68), and knockdown of SIRT1 prevented the inhibitory effect of resveratrol on CSE-induced NF-κB activation (Fig. 5). SIRT1 was reported to physically interact with the RelA/p65 subunit of NF-κB and to inhibit transcription by deacetylating RelA/p65 (70). The role of SIRT1 in controlling NF-κB activation in endothelial cells is further supported by the finding that SIRT1 overexpression inhibited CSE-induced NF-κB activation in CAECs (Fig. 5). It is significant that SIRT1 also appears to regulate cigarette smoke-induced proinflammatory mediator release via the inhibition of NF-κB in macrophages in vitro and in rat lungs in vivo (69). It is also logical to hypothesize that the SIRT1-mediated antioxidant action of resveratrol (e.g., scavenging of H2O2) contributes to its inhibitory effects on NF-κB activation (38). This view is in line with our recent results showing that resveratrol effectively inhibits H2O2-induced NF-κB activation in endothelial cells (20).
In vivo exposure to cigarette smoke provoked an increase in the expression of proinflammatory cytokines (including IL-6, TNF-α, and IL-1β) and cytokine-sensitive inflammatory mediators (iNOS and ICAM-1) in the vascular wall (Fig. 6), extending the previous results of our laboratory (46). Importantly, many of these proinflammatory phenotypic alterations could also be mimicked by in vitro CSE challenge (46) (Fig. 7). The third significant finding in this study was that resveratrol both in vivo and in vitro inhibited cigarette smoke-induced vascular inflammatory gene expression (Fig. 6 and Fig. 7). Our previous studies demonstrated that CSE-induced NAD(P)H oxidase activation and oxidative stress play central roles in the induction of vascular inflammatory gene expression (46). Thus we attribute the anti-inflammatory effects of resveratrol, in part, to its antioxidant action (Fig. 1– Fig. 3). It is also likely that SIRT1 also exerts direct anti-inflammatory effects by interfering with specific cellular signaling pathways (e.g., iNOS, ICAM-1, IL-6, and TNF-α are known to be regulated by NF-κB, which is clearly inhibited by resveratrol/SIRT1 as shown in Fig. 5). The central role of SIRT1 in the anti-inflammatory effects of resveratrol is demonstrated by the findings that knockdown of SIRT1 attenuated the inhibitory effect of resveratrol on the CSE-induced upregulation of inflammatory genes (Fig. 7). Moreover, SIRT1 overexpression also mimicked the anti-inflammatory action of resveratrol (Fig. 7). Atherosclerosis is a chronic inflammatory disease, and pathological and epidemiological evidence suggest that proinflammatory cytokines play a central role orchestrating the pathological processes underlying the development of the atherosclerotic plaque. Thus our findings are of great significance, showing that resveratrol can abrogate the development of a proatherogenic microenvironment in the vascular wall induced by cigarette smoke-related oxidative stress. Relevant to the present discussion are the observations that resveratrol also attenuated oxidative stress-induced expression of iNOS and ICAM in arteries of aged F344 rats as well (66). We have good reason to believe that the aforementioned anti-inflammatory and antioxidant effects of resveratrol will contribute to the ability of chronic resveratrol treatment to delay vascular aging.
The fourth important finding of our study is that cigarette smoking can induce apoptosis in endothelial cells, which was prevented by resveratrol treatment (Fig. 8). Resveratrol was also effective in preventing CSE-induced apoptosis in CAECs in vitro (Fig. 8). Moreover, our laboratory has recently shown that resveratrol, in a physiologically relevant concentration range, also prevents endothelial apoptosis induced by H2O2, TNF-α, and oxidized LDL (63). Present and previous findings of our laboratory (63) suggest that antiapoptotic effects of resveratrol are mediated by a SIRT1-dependent pathway (Fig. 8), which likely involves the induction of H2O2 scavenging mechanisms (e.g., glutathione peroxidase) (63). There is accumulating evidence that increased endothelial cell apoptosis may initiate atherosclerosis, whereas at later phases of atherogenesis, the increased rate of apoptosis of endothelial cells, vascular smooth muscle cells, and macrophages was observed in vulnerable lesions and at sites of plaque rupture (14). Because in humans cigarette smoke constituents, TNF-α, and oxLDL are physiologically relevant stimuli of apoptosis, which play an important role in the development of coronary artery disease, it is likely that the antiapoptotic action of resveratrol/SIRT1 will contribute to their cardioprotective effects in vivo.
Cigarette smoking was reported to increase oxidative DNA modification in humans (40) and laboratory animals. Earlier studies focused on cigarette smoking-induced DNA damage in the lung; however, it soon became obvious that systemic exposure to circulating cigarette smoke constituents results in an increased presence of elevated levels of DNA adducts in tissues not directly exposed to tobacco smoke. The fifth interesting finding of our present study is that soluble cigarette smoke constituents can induce significant oxidative DNA damage in endothelial cells (Fig. 9). Because endothelial cells in vivo represent the first line of defense against circulating toxic agents, it can be expected that significant oxidative DNA damage can occur in the vasculature of smokers as well. Indeed, previous studies demonstrated the presence of cigarette smoking-related oxidative DNA damage in human internal mammary artery specimens from smokers (73). The relationship between cigarette smoke-induced oxidative DNA damage in the lung and parenchymal tissues and carcinogenesis is widely appreciated, and there is good reason to believe that DNA damage also contributes to cardiovascular pathophysiological alterations. An important hypothesis put forward by Ames (4, 5) suggests a direct link between oxidative DNA modification and the aging process (54). In this regard, it is significant that resveratrol effectively protects endothelial cells against CSE-induced DNA damage (Fig. 9). Our laboratory has also recently demonstrated that resveratrol also attenuates UV254 nm-induced DNA damage (which is also mediated, at least in part, by ROS) in endothelial cells (63). Future studies need to elucidate whether the resveratrol-induced protection against oxidative DNA modifications and/or resveratrol-induced changes in DNA repair capacity contribute to the antiaging effects of resveratrol.
In conclusion, resveratrol, likely via a SIRT1-dependent mechanism, abrogated the adverse vascular effects of cigarette smoking by attenuating cigarette smoke-induced oxidative stress and preventing proinflammatory phenotypic alterations in vascular tissues. We hypothesize that the antioxidant, anti-inflammatory, and cytoprotective action of resveratrol will inhibit atherosclerotic plaque formation, decreasing the morbidity of stroke and myocardial infarction in pathophysiological conditions associated with accelerated vascular aging.
Acknowledgments
GRANTS
This study was supported by the National Heart, Lung, and Blood Institute Grants HL-077256 and HL-43023 (to Z. Ungvari and A. Csiszar) and the American Heart Association Grant 0435140-N (to A. Csiszar) and Philip Morris (to Z. Ungvari and A. Csiszar) and funded in part by the intramural program of the NIH.
REFERENCES
- 1.Adams MR, Jessup W, Celermajer DS. Cigarette smoking is associated with increased human monocyte adhesion to endothelial cells: reversibility with oral l-arginine but not vitamin C. J Am Coll Cardiol. 1997;29:491–497. doi: 10.1016/s0735-1097(96)00537-2. [DOI] [PubMed] [Google Scholar]
- 2.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 3.Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731–1737. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 4.Ames BN. Endogenous DNA damage as related to cancer and aging. Mutat Res. 1989;214:41–46. doi: 10.1016/0027-5107(89)90196-6. [DOI] [PubMed] [Google Scholar]
- 5.Ames BN. Endogenous oxidative DNA damage, aging, and cancer. Free Radic Res Commun. 1989;7:121–128. doi: 10.3109/10715768909087933. [DOI] [PubMed] [Google Scholar]
- 6.Anbarasi K, Vani G, Devi CS. Protective effect of bacoside A on cigarette smoking-induced brain mitochondrial dysfunction in rats. J Environ Pathol Toxicol Oncol. 2005;24:225–234. doi: 10.1615/jenvpathtoxoncol.v24.i3.80. [DOI] [PubMed] [Google Scholar]
- 7.Ballinger SW, Bouder TG, Davis GS, Judice SA, Nicklas JA, Albertini RJ. Mitochondrial genome damage associated with cigarette smoking. Cancer Res. 1996;56:5692–5697. [PubMed] [Google Scholar]
- 8.Batkai S, Rajesh M, Mukhopadhyay P, Hasko G, Liaudet L, Cravatt BF, Csiszar A, Ungvari ZI, Pacher P. Decreased age-related cardiac dysfunction, myocardial nitrative stress, inflammatory gene expression, and apoptosis in mice lacking fatty acid amide hydrolase. Am J Physiol Heart Circ Physiol. 2007;293:H909–H918. doi: 10.1152/ajpheart.00373.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 11.Bock FG, Swain AP, Stedman RL. Carcinogenesis assay of subfractions of cigarette smoke condensate prepared by solvent-solvent separation of the neutral fraction. J Natl Cancer Inst. 1972;49:477–483. [PubMed] [Google Scholar]
- 12.Celermajer DS, Sorensen KE, Bull C, Robinson J, Deanfield JE. Endothelium-dependent dilation in the systemic arteries of asymptomatic subjects relates to coronary risk factors and their interaction. J Am Coll Cardiol. 1994;24:1468–1474. doi: 10.1016/0735-1097(94)90141-4. [DOI] [PubMed] [Google Scholar]
- 13.Celermajer DS, Sorensen KE, Georgakopoulos D, Bull C, Thomas O, Robinson J, Deanfield JE. Cigarette smoking is associated with dose-related and potentially reversible impairment of endothelium-dependent dilation in healthy young adults. Circulation. 1993;88:2149–2155. doi: 10.1161/01.cir.88.5.2149. [DOI] [PubMed] [Google Scholar]
- 14.Choy JC, Granville DJ, Hunt DW, McManus BM. Endothelial cell apoptosis: biochemical characteristics and potential implications for atherosclerosis. J Mol Cell Cardiol. 2001;33:1673–1690. doi: 10.1006/jmcc.2001.1419. [DOI] [PubMed] [Google Scholar]
- 15.Csiszar A, Ahmad M, Smith KE, Labinskyy N, Gao Q, Kaley G, Edwards JG, Wolin MS, Ungvari Z. Bone morphogenetic protein-2 induces proinflammatory endothelial phenotype. Am J Pathol. 2006;168:629–638. doi: 10.2353/ajpath.2006.050284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Csiszar A, Labinskyy N, Orosz Z, Xiangmin Z, Buffenstein R, Ungvari Z. Vascular aging in the longest-living rodent, the naked mole rat. Am J Physiol Heart Circ Physiol. 2007;293:H919–H927. doi: 10.1152/ajpheart.01287.2006. [DOI] [PubMed] [Google Scholar]
- 17.Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z, Ungvari Z. Vasculoprotective effects of anti-tumor necrosis factor-α treatment in aging. Am J Pathol. 2007;170:388–698. doi: 10.2353/ajpath.2007.060708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Csiszar A, Labinskyy N, Zhao X, Hu F, Serpillon S, Huang Z, Ballabh P, Levy RJ, Hintze TH, Wolin MS, Austad SN, Podlutsky A, Ungvari Z. Vascular superoxide and hydrogen peroxide production and oxidative stress resistance in two closely related rodent species with disparate longevity. Aging Cell. 2007;6:783–797. doi: 10.1111/j.1474-9726.2007.00339.x. [DOI] [PubMed] [Google Scholar]
- 19.Csiszar A, Pacher P, Kaley G, Ungvari Z. Role of oxidative and nitrosative stress, longevity genes and poly(ADP-ribose) polymerase in cardiovascular dysfunction associated with aging. Curr Vasc Pharmacol. 2005;3:285–291. doi: 10.2174/1570161054368616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Csiszar A, Smith K, Labinskyy N, Orosz Z, Rivera A, Ungvari Z. Resveratrol attenuates TNF-α-induced activation of coronary arterial endothelial cells: role of NF-κB inhibition. Am J Physiol Heart Circ Physiol. 2006;291:H1694–H1699. doi: 10.1152/ajpheart.00340.2006. [DOI] [PubMed] [Google Scholar]
- 21.Csiszar A, Ungvari Z, Edwards JG, Kaminski PM, Wolin MS, Koller A, Kaley G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90:1159–1166. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- 22.Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in rat coronary arteries. FASEB J. 2003;17:1183–1185. doi: 10.1096/fj.02-1049fje. [DOI] [PubMed] [Google Scholar]
- 23.Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol Genomics. 2004;17:21–30. doi: 10.1152/physiolgenomics.00136.2003. [DOI] [PubMed] [Google Scholar]
- 24.Czernin J, Sun K, Brunken R, Bottcher M, Phelps M, Schelbert H. Effect of acute and long-term smoking on myocardial blood flow and flow reserve. Circulation. 1995;91:2891–2897. doi: 10.1161/01.cir.91.12.2891. [DOI] [PubMed] [Google Scholar]
- 25.Czernin J, Waldherr C. Cigarette smoking and coronary blood flow. Prog Cardiovasc Dis. 2003;45:395–404. doi: 10.1053/pcad.2003.00104. [DOI] [PubMed] [Google Scholar]
- 26.Ferdinandy P. Peroxynitrite: just an oxidative/nitrosative stressor or a physiological regulator as well? Br J Pharmacol. 2006;148:1–3. doi: 10.1038/sj.bjp.0706693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59:418–458. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- 28.Ferrero ME, Bertelli AE, Fulgenzi A, Pellegatta F, Corsi MM, Bonfrate M, Ferrara F, De Caterina R, Giovannini L, Bertelli A. Activity in vitro of resveratrol on granulocyte and monocyte adhesion to endothelium. Am J Clin Nutr. 1998;68:1208–1214. doi: 10.1093/ajcn/68.6.1208. [DOI] [PubMed] [Google Scholar]
- 29.Gairola C, Aleem HM. Cigarette smoke: in vitro effects of condensate fractions on mitochondrial respiration. Life Sci. 1974;14:2199–2207. doi: 10.1016/0024-3205(74)90102-7. [DOI] [PubMed] [Google Scholar]
- 30.Gairola C, Aleem MI. Cigarette smoke: effect of aqueous and nonaqueous fractions on mitochondrial function. Nature. 1973;241:287–288. doi: 10.1038/241287a0. [DOI] [PubMed] [Google Scholar]
- 31.Gupte SA, Arshad M, Viola S, Kaminski PM, Ungvari Z, Rabbani G, Koller A, Wolin MS. Pentose phosphate pathway coordinates multiple redox-controlled relaxing mechanisms in bovine coronary arteries. Am J Physiol Heart Circ Physiol. 2003;285:H2316–H2326. doi: 10.1152/ajpheart.00229.2003. [DOI] [PubMed] [Google Scholar]
- 32.Guthikonda S, Sinkey C, Barenz T, Haynes WG. Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circulation. 2003;107:416–421. doi: 10.1161/01.cir.0000046448.26751.58. [DOI] [PubMed] [Google Scholar]
- 33.Gvozdjakova A, Bada V, Sany L, Kucharska J, Kruty F, Bozek P, Trstansky L, Gvozdjak J. Smoke cardiomyopathy: disturbance of oxidative processes in myocardial mitochondria. Cardiovasc Res. 1984;18:229–232. doi: 10.1093/cvr/18.4.229. [DOI] [PubMed] [Google Scholar]
- 34.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 35.Jaimes EA, DeMaster EG, Tian RX, Raij L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler Thromb Vasc Biol. 2004;24:1031–1036. doi: 10.1161/01.ATV.0000127083.88549.58. [DOI] [PubMed] [Google Scholar]
- 36.Jia L, Liu Z, Sun L, Miller SS, Ames BN, Cotman CW, Liu J. Acrolein, a toxicant in cigarette smoke, causes oxidative damage and mitochondrial dysfunction in RPE cells: protection by (R)-alpha-lipoic acid. Invest Ophthalmol Vis Sci. 2007;48:339–348. doi: 10.1167/iovs.06-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knight-Lozano CA, Young CG, Burow DL, Hu ZY, Uyeminami D, Pinkerton KE, Ischiropoulos H, Ballinger SW. Cigarette smoke exposure and hypercholesterolemia increase mitochondrial damage in cardiovascular tissues. Circulation. 2002;105:849–854. doi: 10.1161/hc0702.103977. [DOI] [PubMed] [Google Scholar]
- 38.Labinskyy N, Csiszar A, Veress G, Stef G, Pacher P, Oroszi G, Wu J, Ungvari Z. Vascular dysfunction in aging: potential effects of resveratrol, an anti-inflammatory phytoestrogen. Curr Med Chem. 2006;13:989–996. doi: 10.2174/092986706776360987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lewis PD, Fradley SR, Griffiths AP, Baxter PW, Parry JM. Mitochondrial DNA mutations in the parotid gland of cigarette smokers and non-smokers. Mutat Res. 2002;518:47–54. doi: 10.1016/s1383-5718(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 40.Loft S, Vistisen K, Ewertz M, Tjonneland A, Overvad K, Poulsen HE. Oxidative DNA damage estimated by 8-hydroxydeoxyguanosine excretion in humans: influence of smoking, gender and body mass index. Carcinogenesis. 1992;13:2241–2247. doi: 10.1093/carcin/13.12.2241. [DOI] [PubMed] [Google Scholar]
- 41.Mendelson JH, Sholar MB, Mutschler NH, Jaszyna-Gasior M, Goletiani NV, Siegel AJ, Mello NK. Effects of intravenous cocaine and cigarette smoking on luteinizing hormone, testosterone, and prolactin in men. J Pharmacol Exp Ther. 2003;307:339–348. doi: 10.1124/jpet.103.052928. [DOI] [PubMed] [Google Scholar]
- 42.Moon SO, Kim W, Sung MJ, Lee S, Kang KP, Kim DH, Lee SY, So JN, Park SK. Resveratrol suppresses tumor necrosis factor-alpha-induced fractalkine expression in endothelial cells. Mol Pharmacol. 2006;70:112–119. doi: 10.1124/mol.106.022392. [DOI] [PubMed] [Google Scholar]
- 43.Mukhopadhyay P, Rajesh M, Hasko G, Hawkins BJ, Madesh M, Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc. 2007;2:2295–2301. doi: 10.1038/nprot.2007.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukhopadhyay P, Rajesh M, Yoshihiro K, Hasko G, Pacher P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem Biophys Res Commun. 2007;358:203–208. doi: 10.1016/j.bbrc.2007.04.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakayama T, Church DF, Pryor WA. Quantitative analysis of the hydrogen peroxide formed in aqueous cigarette tar extracts. Free Radic Biol Med. 1989;7:9–15. doi: 10.1016/0891-5849(89)90094-4. [DOI] [PubMed] [Google Scholar]
- 46.Orosz Z, Csiszar A, Labinskyy N, Smith K, Kaminski PM, Ferdinandy P, Wolin MS, Rivera A, Ungvari Z. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: role of NAD(P)H oxidase activation. Am J Physiol Heart Circ Physiol. 2007;292:H130–H139. doi: 10.1152/ajpheart.00599.2006. [DOI] [PubMed] [Google Scholar]
- 47.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petruzzelli S, Puntoni R, Mimotti P, Pulera N, Baliva F, Fornai E, Giuntini C. Plasma 3-nitrotyrosine in cigarette smokers. Am J Respir Crit Care Med. 1997;156:1902–1907. doi: 10.1164/ajrccm.156.6.9702075. [DOI] [PubMed] [Google Scholar]
- 49.Pryor WA, Prier DG, Church DF. Electron-spin resonance study of mainstream and sidestream cigarette smoke: nature of the free radicals in gas-phase smoke and in cigarette tar. Environ Health Perspect. 1983;47:345–355. doi: 10.1289/ehp.8347345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pryor WA, Stone K, Zang LY, Bermudez E. Fractionation of aqueous cigarette tar extracts: fractions that contain the tar radical cause DNA damage. Chem Res Toxicol. 1998;11:441–448. doi: 10.1021/tx970159y. [DOI] [PubMed] [Google Scholar]
- 51.Raij L, DeMaster EG, Jaimes EA. Cigarette smoke-induced endothelium dysfunction: role of superoxide anion. J Hypertens. 2001;19:891–897. doi: 10.1097/00004872-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 52.Schmeltz I, Stills CD, Chamberlain WJ, Stedman RL. Analysis of cigarette smoke fraction by combined gas chromatography-infrared spectrophotometry. Anal Chem. 1965;37:1614–1616. doi: 10.1021/ac60231a603. [DOI] [PubMed] [Google Scholar]
- 53.Shen Y, Rattan V, Sultana C, Kalra VK. Cigarette smoke condensate-induced adhesion molecule expression and transendothelial migration of monocytes. Am J Physiol Heart Circ Physiol. 1996;270:H1624–H1633. doi: 10.1152/ajpheart.1996.270.5.H1624. [DOI] [PubMed] [Google Scholar]
- 54.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slebos DJ, Ryter SW, van der Toorn M, Liu F, Guo F, Baty CJ, Karlsson JM, Watkins SC, Kim HP, Wang X, Lee JS, Postma DS, Kauffman HF, Choi AM. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am J Respir Cell Mol Biol. 2007;36:409–417. doi: 10.1165/rcmb.2006-0214OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Squadrito GL, Cueto R, Dellinger B, Pryor WA. Quinoid redox cycling as a mechanism for sustained free radical generation by inhaled airborne particulate matter. Free Radic Biol Med. 2001;31:1132–1138. doi: 10.1016/s0891-5849(01)00703-1. [DOI] [PubMed] [Google Scholar]
- 57.Swain AP, Cooper JE, Stedman RL. Large-scale fractionation of cigarette smoke condensate for chemical and biologic investigations. Cancer Res. 1969;29:579–583. [PubMed] [Google Scholar]
- 58.Ungvari Z, Buffenstein R, Austad SN, Podlutsky A, Kaley G, Csiszar A. Oxidative stress in vascular senescence: lessons from successfully aging species. Front Biosci. 2008;1:5056–5070. doi: 10.2741/3064. [DOI] [PubMed] [Google Scholar]
- 59.Ungvari Z, Csiszar A, Edwards JG, Kaminski PM, Wolin MS, Kaley G, Koller A. Increased superoxide production in coronary arteries in hyperhomocysteinemia: role of tumor necrosis factor-alpha, NAD(P)H oxidase, and inducible nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2003;23:418–424. doi: 10.1161/01.ATV.0000061735.85377.40. [DOI] [PubMed] [Google Scholar]
- 60.Ungvari Z, Csiszar A, Huang A, Kaminski PM, Wolin MS, Koller A. High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD(P)H oxidase. Circulation. 2003;108:1253–1258. doi: 10.1161/01.CIR.0000079165.84309.4D. [DOI] [PubMed] [Google Scholar]
- 61.Ungvari Z, Csiszar A, Kaley G. Vascular inflammation in aging. Herz. 2004;29:733–740. doi: 10.1007/s00059-004-2625-x. [DOI] [PubMed] [Google Scholar]
- 62.Ungvari Z, Csiszar A, Kaminski PM, Wolin MS, Koller A. Chronic high pressure-induced arterial oxidative stress: involvement of protein kinase C-dependent NAD(P)H oxidase and local renin-angiotensin system. Am J Pathol. 2004;165:219–226. doi: 10.1016/S0002-9440(10)63290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ungvari Z, Orosz Z, Rivera A, Labinskyy N, Xiangmin Z, Olson S, Podlutsky A, Csiszar A. Resveratrol increases vascular oxidative stress resistance. Am J Physiol Heart Circ Physiol. 2007;292:H2417–H2424. doi: 10.1152/ajpheart.01258.2006. [DOI] [PubMed] [Google Scholar]
- 64.Ungvari Z, Parrado-Fernandez C, Csiszar A, de Cabo R. Mechanisms underlying caloric restriction and lifespan regulation: implications for vascular aging. Circ Res. 2008;102:519–528. doi: 10.1161/CIRCRESAHA.107.168369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ungvari ZI, Labinskyy N, Gupte SA, Chander PN, Edwards JG, Csiszar A. Dysregulation of mitochondrial biogenesis in vascular endothelial and smooth muscle cells of aged rats. Am J Physiol Heart Circ Physiol. 2008;294:H2121–H2128. doi: 10.1152/ajpheart.00012.2008. [DOI] [PubMed] [Google Scholar]
- 66.Ungvari ZI, Orosz Z, Labinskyy N, Rivera A, Xiangmin Z, Smith KE, Csiszar A. Increased mitochondrial H2O2 production promotes endothelial NF-κB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007;293:H37–H47. doi: 10.1152/ajpheart.01346.2006. [DOI] [PubMed] [Google Scholar]
- 67.Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A. Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate. Curr Biol. 2006;16:296–300. doi: 10.1016/j.cub.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 68.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 69.Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-κB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol. 2007;292:L567–L576. doi: 10.1152/ajplung.00308.2006. [DOI] [PubMed] [Google Scholar]
- 70.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zang LY, Stone K, Pryor WA. Detection of free radicals in aqueous extracts of cigarette tar by electron spin resonance. Free Radic Biol Med. 1995;19:161–167. doi: 10.1016/0891-5849(94)00236-d. [DOI] [PubMed] [Google Scholar]
- 72.Zevin S, Jacob P, 3rd, Benowitz NL. Dose-related cardiovascular and endocrine effects of transdermal nicotine. Clin Pharmacol Ther. 1998;64:87–95. doi: 10.1016/S0009-9236(98)90026-1. [DOI] [PubMed] [Google Scholar]
- 73.Zhang YJ, Weksler BB, Wang L, Schwartz J, Santella RM. Immunohistochemical detection of polycyclic aromatic hydrocarbon-DNA damage in human blood vessels of smokers and non-smokers. Atherosclerosis. 1998;140:325–331. doi: 10.1016/s0021-9150(98)00136-1. [DOI] [PubMed] [Google Scholar]