

Abstract
Microcystins, a family of cyclic heptapeptides produced by the cyanobacteria, Microcystis aeruginosa, have documented hepatotoxic and tumor promoting activities. The purpose of this study was to evaluate the toxicity of inhaled microcystin LR (microcystin). Male BALB/c mice were exposed by nose-only inhalation to 260-265 μg microcystin/m3 for 7 days. The low-, mid- and high-dose groups were exposed for 0.5, 1, and 2 h, respectively. Control animals were sham exposed to aerosolized vehicle. Treatment-related microscopic lesions were observed only in the nasal cavity of the mid- and high-dose groups. These lesions consisted of minimal to moderate multifocal degeneration and necrosis of the respiratory epithelium, with variable neutrophilic inflammation and minimal to marked degeneration, necrosis, and atrophy of the olfactory epithelium. The no-adverse-effect dose for the nasal lesions was approximately 3 μg/kg body weight, or 20 ng/cm2 of nasal epithelium. In serum, only two protein peaks, occurring at m/zs of 11,688 and 11,829 Da, exhibited decreases in intensity that were microcystin dose-dependent. While these proteins have not been positively identified, they may be useful in the future as biomarkers of microcystin exposure in humans.
Keywords: Microcystin, Inhalation, Nasal epithelium, Protein expression in serum
1. Introduction
Cyanobacterial blooms in fresh surface waters occur world wide with ever increasing incidence (Duy et al., 2000; Falconer et al., 1994). Cyanobacteria produce hepatotoxins, neurotoxins, and lipopolysaccharide endotoxins that have caused adverse reactions or death in animals and humans (reviewed by Duy et al., 2000). Microcystins are a family of cyclic heptapeptides produced by the cyanobacteria, Microcystis aeruginosa (M. aeruginosa; Carmichael, 1992; Watanabe et al., 1996). Microcystins have been implicated in the death of both wild and domestic animals that consumed water containing dense blooms of the blue-green algae (Duy et al., 2000). A consistent pathological finding after lethal exposure to microcystin LR (microcystin) is diffuse hepatic centrilobular necrosis (Jackson et al., 1984; Theiss et al., 1988). In humans, a heavy bloom of M. aeruginosa in a reservoir at Malpus Dam, Australia, was correlated with a rise in plasma gamma-glutamyl transpeptidase suggestive of hepatic injury (Falconer et al., 1983). More recently, 100 of 131 hemodialysis patients in Caruaru, Brazil, developed acute liver failure and 76 of those patients died. Fifty-two of those deaths were directly attributed to the presence of microcystins (approximately 19 μg/l) in the reservoir water used for the dialysis treatments (Jochimsen et al., 1998; Carmichael et al., 2001; Azevedo et al., 2002). Microcystins are also potent tumor promoters in laboratory animals (Falconer and Buckley, 1989; Falconer, 1991; Nishiwaki-Matsushima et al., 1992; Ito et al., 1997). In humans, microcystins have been implicated in contributing to the high incidence of primary liver cancers in certain areas of China where the primary sources of drinking water are ponds, lakes, and shallow wells (Fleming et al., 2000; Ueno et al., 1996; Falconer and Humpage, 1996; Yu, 1995).
Ingestion of contaminated water is the most likely source of human and animal exposure to microcystin. Repeated inhalation exposure may also occur occupationally among groundskeepers or park managers in areas where surface water supplies are used to irrigate parks or golf courses. Inhalation exposure in homes whose domestic water supplies contain microcystin contamination can occur during showering because shower spray contains a high concentration of respirable water particles (W. Ollison and Y. Zhou, personal communication). Since many lakes and reservoirs are also used for recreational purposes, boaters, jet skiers, and water skiers may be exposed via inhalation to microcystin-contaminated spray. Although humans may be exposed to aerosolized microcystin in the workplace, home, or recreation areas, data are not available on the toxic effects associated with short-term, repeated inhalation of these toxins.
In laboratory animals, the acute toxicity of microcystin is significantly lower by ingestion than by other routes. For example, an oral dose of 500 μg microcystin/kg in mice produced only 24% mortality (Ito et al., 2000). An oral LD50 of 3000 μg/kg has been reported (Fitzgeorge et al., 1994). By comparison, LD50s of 36-250 μg/kg have been reported for i.p. or i.v. administration (Dawson, 1998; Fitzgeorge et al., 1994), 250 μg/kg for intranasal administration (Fitzgeorge et al., 1994), between 50 and 100 μg/kg for intratracheal administration (Ito et al., 2001), and 43 μg/kg by inhalation (Creasia, 1990).
Repeated intranasal administration of a nonlethal dose (31 μg microcystin/kg) once each day for 7 days resulted in extensive necrosis of the respiratory and olfactory epithelium (Fitzgeorge et al., 1994). Liver lesions consisted of centrilobular necrosis with hemorrhage and accumulation of large amounts of blood within the liver lobules. Adrenal lesions consisted of vacuolation and necrosis of the inner cortex accompanied by intense congestion of the medullary blood vessels. No lesions were observed in the trachea, lungs, esophagus, pancreas, spleen, lymph nodes, kidneys, or brain. These data suggest that toxic effects following exposure to microcystin may occur at lower doses following inhalation than ingestion, with different potential target organs for toxicity.
2. Materials and methods
2.1. Chemicals
Microcystis aeruginosa was obtained from the University of Toronto Culture Collection (UTCC299). Microcystin LR was purified from cultures by established methods (Lawton and Edwards, 2001) involving a combination of methanol extraction, solid phase (C18) extraction and a final purification by HPLC (70:30 MeOH: 20 mM NH4OAc, Agilent Technologies, hypersil-ODS column), at the NIEHS Toxic Algae Culture Center at Florida International University, Miami, FL. Identity and purity were confirmed by mass spectrometry, 1H NMR (Bruker Avance, 400 MHz, in MeOH-d4; Harada et al., 1990) and reverse phase HPLC comparison to an authentic sample (Sigma-Aldrich, St. Louis, MO).
2.2. Animals
Thirty male BALB/c mice, 6-8 weeks old, were obtained from Charles River Laboratories, Wilmington, MA. The mice were housed in polycarbonate cages with hardwood chip bedding. The animal rooms were maintained at 20-22 °C with relative humidity at 20-50% and a 12-h light cycle beginning at 06:00. Rodent diet (Harlan Teklad, Madison, WI) and water were provided ad libitum, except during inhalation exposures. The mice were randomized by weight into exposure groups and identified by tail tattoo. All mice were conditioned to the nose-only inhalation restraint tubes before exposures began on 3 separate days for 0.5, 1 and 2 h on days 1, 2, and 3, respectively. The study protocol was approved by the Lovelace Respiratory Research Institute (LRRI) Institutional Animal Care and Use Committee.
2.3. Inhalation exposure system
The exposure system consisted of two (one microcystin, and one vehicle control), 36-port cylindrical nose-only inhalation chambers (InTox Products, Edgewood, NM). Aerosols generated by nebulization (Hospitak, Inc., Farmingdale, NY) were dried and diluted with supply air to achieve the desired chamber aerosol concentrations. Total airflow through the chambers was 11 l/min. Temperatures were monitored continuously with an acceptable range of 18-22 °C. Chamber oxygen concentration was monitored with an action level at ≤18%.
Stock solutions of microcystin were prepared in 100% ethanol (0.23 mg/ml). Generator solutions of microcystin contained 0.05 mg microcystin/ml in 20% ethanol. The vehicle control was 20% ethanol in water. Aerosol concentration was determined gravimetrically. Because of the low aerosol concentrations used in this study, all of the chamber exhaust was directed onto 25-mm filters (Zeflour, SKC Gulf Coast, Houston, TX). Twenty-minute filter samples were collected throughout the exposure periods. The stability of the aerosol concentration during the exposure was monitored using a real-time aerosol monitor (DustTrak, TSI Industries, Shoreview, MN) sampling from the breathing zone of the mice at a rate of 1 l/min. The particle size distribution of the microcystin aerosol was determined using a scanning mobility particle sizer (TSI Industries).
2.4. Experimental design
Groups of 6 mice each were exposed for 7 consecutive days to 260-265 μg microcystin/m3 for 30 min (low-dose group), 60 min (mid-dose group), and 120 min (high-dose group). Under these conditions, approximately 1 μg microcystin (or 50 μg microcystin/kg body weight) was expected to be deposited in the respiratory tract of the high-dose group, based on a minute volume of 0.04 l/min in mice and a total respiratory tract deposition fraction of 0.7 for an aerosol of 2 μm mass median aerodynamic diameter (Schlesinger, 1985, 1989). This dose, deposited by intratracheal instillation was not lethal in mice (Ito et al., 2001), whereas a slightly higher dose (1.5 μg) was. Therefore, a daily deposition of 50 μg microcystin/kg was considered the maximum dose that could be administered repeatedly in this experiment without mortality. The control group was exposed for 120 min to aerosol generated by nebulizing the aqueous ethanol vehicle. Following each exposure, animals were returned to their cages and were observed for clinical signs of toxicity.
2.5. Necropsy
On the day following the last exposure, all mice were sacrificed by intraperitoneal injection of an overdose of Euthasol®, and body weights were recorded. Blood was collected via cardiac puncture for evaluation of clinical chemistry and protein profiles in serum. Each animal received a complete necropsy. Adrenal glands, lung, liver, kidney, spleen, and thymus weights were recorded. A weighed portion of liver from each mouse was saved in RNA later. The remaining portion of liver, the respiratory tract tissues (including nasal cavity, larynx, trachea, and lung), adrenals, kidney, spleen, thymus, gastrointestinal tract, and testes were fixed in 10% neutral buffered formalin for histopathological evaluation.
2.6. Histopathology
Tissues were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin for evaluation. To evaluate the effect of microcystin on the epithelium lining the bony structure of the nasal passages (turbinates), several cross sections of the nose were taken. Locations of the sections of the nasal turbinates taken were as follows: Turbinate 1, immediately caudal to the upper incisors; Turbinate 2, at the dose of the incisive papilla; and Turbinate 3, at the dose of the first upper molar. Microscopic lesions, when present, were graded for severity.
2.7. Clinical chemistry
Blood urea nitrogen, creatinine, total bilirubin, alkaline phosphatase, aspartate transaminase, alanine transaminase, total protein, albumin, and globulin were analyzed in serum using a Hitachi 911 Automatic Analyzer (Roche Diagnostics Corporation, Indianapolis, IN).
2.8. Protein expression in serum
Serum protein profiling on serum from all mice was performed by retentate chromatography on ProteinChips® and analyzed by surface-enhanced laser desorption/ionization time-of-flight (SELDI-TOF) mass spectrometry on a PBSII reader (Ciphergen Biosystems, Fremont, CA). Serum samples (20 μl) were denatured in 9 M urea, 2% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate and 1% dithiothreitol at room temperature for 60 min. To increase the number of protein peaks visualized, a serum fractionation kit (Ciphergen Biosystems) was used to separate the denatured serum into six different fractions: (F1, flow through+pH 9; F2, pH 7; F3, pH 5; F4, pH 4; F5, pH3; and F6, organic). Protein profiles were obtained on three chemically distinct ProteinChip® array surfaces for each fraction (F1-F6): CM10 (weak cationic exchange), IMAC30 (immobilized metal affinity capture) and SAX2 (strong anionic exchange). Based upon the profiles on each chip (data not shown), fractions F4 and F5 on the IMAC30 chip afforded the greatest number of peak clusters. Therefore, the fractions F4 and F5 were combined from each animal and applied in duplicate, in random order, to the IMAC30 chip precharged with Cu2+. To elucidate the low-(1-20 kDa) and high-mass ranges (20-100 kDa), energy absorbing matrix 3-[3-dimethylammonio]-1-propanesulfonate and sinapinic acid, respectively, were applied to the chips after application of the sample. External calibrations with 3-5 standards were performed daily. Proteins peaks having intensities at least three times background noise and m/z ratios <2000 were grouped into clusters with similar m/z ratios using Protein Chip 3.1.1 software (Ciphergen Biosystems). Data were exported as a text file for further statistical analysis. A trend in cluster peak intensity across all dose groups was evaluated using GeneSpring software (Silicon Genetics, Redwood City, CA). When significant trends were found, pair-wise group comparisons were made using a Newman Keuls test. Preliminary identities of protein clusters showing microcystin dose-dependent changes were made, when possible, by searching online databases including TagIdent Tool (Swiss Institute of Bioinformatics, Geneva, Switzerland), Protein Prospector (University of California, San Francisco, CA), and PubMed (National Library of Medicine, Washington, DC). Positive identifications were not made due to the limited amounts of serum available.
2.9. Statistical analysis
Means, standard deviations, and standard errors for experimental parameters other than body and organ weights were calculated using Microsoft Excel® software. One-way analysis of variance was used to test if there was a statistically significant trend in the data (GraphPad Software, Inc., San Diego, CA). If group variances were not significantly different, the analysis of variance (ANOVA) was performed with a Dunnett’s post test to assess differences among exposure groups. If group variances were significantly different, the Kruskal-Wallis test was used with Dunn’s post test for multiple comparisons. Group mean body weight and organ weight data were tested for statistical significance using Path-Tox® software. Bartlett’s test was used to establish the homogeneity of the data. If the data were homogeneous, significance was evaluated using a modified Dunnett’s test. If data were nonhomogeneous, a modified t-test was used. For all parameters, the criterion for significance was set at p≤0.05.
3. Results
3.1. Aerosol characteristics and microcystin respiratory tract deposition
Low-, mid- and high-dose mice were exposed concurrently in the same chamber for 30, 60, and 120 min, respectively. The microcystin concentrations achieved during the 7 days of exposure for the low-, mid- and high-dose groups were (mean±SD) 260±89, 265±124, and 264±105 μg/m3, respectively. The aerosol had a mass median diameter of 0.53±0.01 μm and a geometric standard deviation σg of 1.69±0.04. This particle size was lower than the anticipated 2 μm, resulting in a lower daily microcystin deposition than originally anticipated. The daily deposition of microcystin was calculated using the following equation: μg deposited=aerosol concentration (μg/l air)×minute volume (l)×time (min)×deposition fraction. For mice, the minute volume is 0.04 l/min (Schlesinger, 1989), and the total respiratory tract deposition fraction of a 0.5 μm aerosol is approximately 0.2 (average of a presented range, Schlesinger, 1985). Using an average of 260 μg microcystin/m3 air (=260 ng/l air), approximately 0.06, 0.12, and 0.25 μg microcystin (3, 6, and 12.5 μg/kg) were deposited per day in the low-, mid- and high-dose microcystin animals, respectively.
3.2. In life observations and gross observations at necropsy
Mice exposed to microcystin exhibited no clinical signs of toxicity and there was no mortality associated with microcystin inhalation. Exposure had no effect on terminal body weights, or organ weights (data not shown). No gross lesions attributable to microcystin exposure were observed.
3.3. Histopathology
Significant, treatment-related microscopic lesions were observed only in the nasal cavity. These lesions consisted of minimal to moderate multifocal degeneration and necrosis of respiratory epithelium, with variable neutrophilic inflammation (Fig. 1), and minimal to marked degeneration, necrosis and atrophy of the olfactory epithelium (Fig. 2). Necrosis of the respiratory epithelial cells tended to involve one to a few cells, whereas necrosis of the olfactory epithelium tended to involve larger patches containing more cells. Nasal cavity lesions were, for the most part, bilaterally symmetric in distribution. The dorsal meatus, the medial free margins of the maxilloturbinates, and the ethmoid turbinates had the greatest concentration of lesions. The severity of these lesions increased with increasing dose of microcystin (Table 1). Other lesions, consisting of minimal to mild neutrophilic inflammation of the squamous epithelium lining the nasal vestibule and the floor of the ventral meatus, and lymphocytic inflammation associated with the nasolacrimal duct, are considered to be nontreatment related.
Fig. 1.

Respiratory epithelial necrosis (large arrow) with neutrophilic inflammation (small arrows). The level of magnification is 600×
Fig. 2.
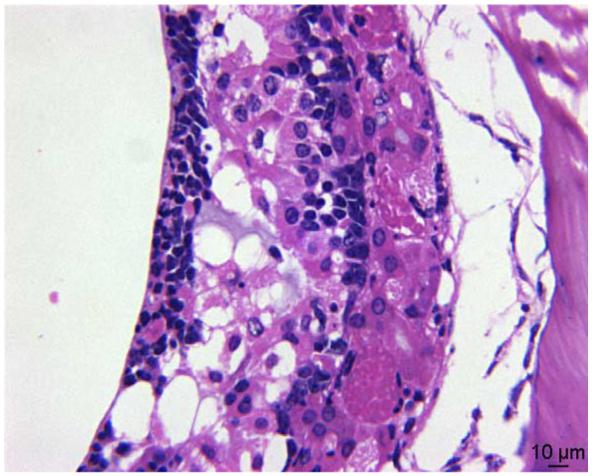
Olfactory epithelial degeneration and necrosis (600×).
Table 1.
Treatment related lesions of male BALB/c mice inhaling microcystin
| Microscopic finding | Severity grade | Exposure group |
|||
|---|---|---|---|---|---|
| Control | Low | Mid | High | ||
| Turbinate 1 | |||||
| Respiratory epithelial necrosis | Not noted | 6 | 5 | 0 | 4 |
| Minimal | 0 | 1 | 0 | 0 | |
| Mild | 0 | 0 | 6 | 0 | |
| Moderate | 0 | 0 | 0 | 2 | |
| Respiratory epithelial inflammation | Not noted | 6 | 5 | 6 | 5 |
| Mild | 0 | 1 | 0 | 1 | |
| Olfactory epithelial degeneration, necrosis and atrophy | Not noted | 6 | 6 | 6 | 1 |
| Mild | 0 | 0 | 0 | 4 | |
| Moderate | 0 | 0 | 0 | 1 | |
| Turbinate 2 | |||||
| Respiratory epithelial necrosis | Not noted | 6 | 6 | 0 | 0 |
| Mild | 0 | 0 | 6 | 3 | |
| Moderate | 0 | 0 | 0 | 3 | |
| Respiratory epithelial inflammation | Not noted | 6 | 5 | 6 | 6 |
| Mild | 0 | 1 | 0 | 0 | |
| Olfactory epithelial degeneration, necrosis and atrophy | Not noted | 6 | 6 | 0 | 0 |
| Mild | 0 | 0 | 6 | 0 | |
| Moderate | 0 | 0 | 0 | 6 | |
| Turbinate 3 | |||||
| Olfactory epithelial degeneration, necrosis and atrophy | Not noted | 6 | 6 | 0 | 0 |
| Mild | 0 | 0 | 6 | 0 | |
| Moderate | 0 | 0 | 0 | 4 | |
| Marked | 0 | 0 | 0 | 2 | |
There was a slight increase in the number of multi-nucleated giant cells in the testes of the treated mice; however, an exposure concentration-response relationship was not evident. Control mice had fewer than 5 giant cells per testicle. One low-dose mouse and one high-dose mouse had a slight increase in the number of giant cells in the testes (11 and 14 giant cells per testicle, respectively). Multi-nucleated giant cells generally represent spermatid degeneration and fusion, and may result from exposure to toxic agents or from increased testicular temperatures. The lack of dose response suggests that the changes were not related to the microcystin doses to which the mice were exposed.
3.4. Clinical chemistry
A statistically significant decrease in blood urea nitrogen (BUN) was observed with increasing dose of microcystin. Urea is produced by the liver from ammonia derived from protein catabolism and is excreted by the kidneys. Decreases in BUN may be seen with hepatic insufficiency, decreased protein consumption, or increased fluid intake. Neither the histologic appearance of the liver nor other biochemical parameters supports a diagnosis of liver failure. Increasing doses of microcystin exposure may have caused a subtle decrease in food consumption, sufficient to cause a decrease in BUN but not of sufficient duration to cause detectable weight loss. Alternatively, microcystin exposure may have induced an increase in water intake. Regardless of the etiology, the changes in BUN were slight and were not considered to be biologically relevant.
3.5. Serum protein profiles
Five protein clusters exhibited a trend toward decreasing peak intensity in association with microcystin exposure (Table 2). However, only two peaks, those occurring at m/z=11,688 and 11,829 Da exhibited decreases in intensity that were microcystin dose-dependent. The 11.8 kDa peak is consistent with β2-microglobulin, the light chain of the class J major histocompatibility antigens whose amino acid composition is similar among dogs, humans, mice, and rabbits, and is found free in biological fluids (Nakajima et al., 1999). The 11.8 kDa peak is also consistent with the molecular weight of murine serum amyloid A2 protein (SAA2; Yamamoto and Migita, 1985).
Table 2.
Microcystin-induced alterations in serum protein profiles
| Exposure group | Ion cluster (m/z) intensity (mean±SD; n=6) |
||||
|---|---|---|---|---|---|
| 5850 | 8120a | 11,688 | 11,829 | 13,990 | |
| Control | 30.1±8.0 | 6.06±1.8 | 2.89±1.5 | 11.1±2.7 | 2.13±0.8 |
| Low | 28.7±4.1 | 6.44±2.8 | 1.43±0.4 | 10.1±2.2b | 0.74±0.4b |
| Mid | 18.7±5.9b | 2.54±1.4b | 1.41±0.5b | 8.49±2.6b | 1.21±0.7b |
| High | 22.6±7.3b | 4.59±4.1 | 1.19±0.7b | 6.77±2.6b | 1.66±1.4b |
Ion cluster ranges mean m/z (range): 5850 (5845-5857), 8120 (8116-8126), 11,688 (11,678-11,699), 11,829 (11,816-11,843), 13,990 (13,990-13,990).
Mean significantly different from control.
The peak cluster at m/z=13,990 Da is consistent with a 14 kDa serum amyloid A protein (apoSAA) identified in both cow (Yamamoto et al., 1998) and human serum (Whitehead et al., 1992). In humans, the protein has been identified as a normal apolipoprotein component of nonacute phase high density lipoprotein that is constitutively expressed and not upregulated in response to tissue damage or inflammatory responses (Whitehead et al., 1992). Interestingly, the 14-kDA protein in cow serum is immunologically associated with two proteins having molecular masses of <6.5 and 7.5-9 kDa, possibly consistent with peak clusters found in mouse serum at 5850 and 8120 Da. The concentration of the 14 kDa protein in cow serum increases with inflammation, while the concentrations of the associated lower molecular weight proteins decrease. Since these SAAs have not been identified in mouse serum, the match is highly speculative. The cluster with m/z=8120 Da is also consistent with the molecular weight of human Rho GDP dissociation inhibitor protein (8118 and 8120 Da; [Leffers et al., 1993]). These proteins regulate the nucleotide (GDP) bound to Rho, thus determining the activity of Rho and its interaction with many of its downstream targets and are, therefore, important in cell signaling in relation to motility, shape and control of the actin cytoskeleton of cells. The Rho GDP inhibitor proteins have been found in neutrophil cytosol (Bowman et al., 1993; Bourmeyster et al., 1994), but no data were found regarding the presence of this protein in serum.
4. Discussion
In this study, mice were exposed by inhalation to monodisperse (σg<2), submicron aerosols of microcystin for 7 consecutive days, resulting in estimated daily deposition of 3, 6, and 12.5 μg microcystin/kg for the low-, mid- and high-dose groups, respectively.
Microcystin is a documented hepatotoxin, with some renal toxicity exhibited following administration of high doses. However, no hepatotoxicity or renal toxicity was observed either histologically or by clinical chemistry in mice inhaling 260-265 μg microcystin/m3 for 7 days. The lack of hepatotoxicity is probably because the deposited doses (2.5 μg microcystin/kg in the high-dose group) were substantially lower than the no adverse effect dose (NOAEL) of 200 μg/kg/day for induction of hepatotoxicity following repeated microcystin administration to mice by gavage (Fawell et al., 1999). The dose rate for the low-dose group was similar to the dose of 2.5 μg microcystin/kg/day resulting in no hepatotoxicity to mice administered the compound in drinking water for 18 months (Ueno et al., 1999).
Marked nasal lesions, including inflammation and necrosis of the respiratory epithelium and necrosis, degeneration, and atrophy of the olfactory epithelium were noted in turbinates 1, 2, and 3 of mid- and high-dose mice. These nasal lesions are consistent with those reported by Fitzgeorge et al. (1994) in mice following repeated administration of 34 μg microcystin/day. Since protein phosphatase 2A is located in olfactory epithelium (Kroner et al., 1996; Ishibashi et al., 2001) where it plays a role in odorant-induced second messenger response, it is likely that the lesions in the nose are mediated through microcystin adduction and inhibition of this enzyme, as occurs in liver (Ito et al., 2000; Duy et al., 2000). Lack of lesions in the larynx and lung may be a consequence of low or no microcystin deposition beyond the nose or to low doses of protein phosphatase 2A in these tissues. No lung lesions following repeated intranasal administration were reported by Fitzgeorge et al. (1994), despite the fact that a portion of an intranasally administered dose to anesthetized rodents reaches the lung (Balmelli et al., 1998). Protein phosphatase 2 plays a major role in lung development (Taylor et al., 2000; Xue et al., 1998), but no documentation of this enzyme in adult lung tissue could be found in the literature. The NOAEL for the nasal lesions occurred at an estimated deposited dose of 3 μg microcystin/kg/day, occurring after exposure to approximately 260 μg microcystin/m3 for 0.5 h/day. If a majority of the inhaled dose is deposited in the nose, the no-effect dose to the nasal epithelium is 20 ng/cm2, assuming a deposited dose of 60 ng and a nasal epithelium area of 2.8 cm2 for mouse (Gizurarson, 1990). No value can be predicted for the tumor promoting effect of microcystin in the nose.
The lack of hepatic lesions in our study with inhaled microcystin are inconsistent with the findings of lesions following repeated intranasal administration of 31 μg/kg microcystin for 7 days (Fitzgeorge et al., 1994). Since it is known that microcystin administered directly to lung by intratracheal administration is systemically absorbed and transported to liver (Ito et al., 2001), and that intranasally administered materials can reach lung, the lack of liver lesions following microcystin inhalation is likely because the dose to liver was insufficient to cause hepatotoxicity.
Some alterations in serum protein profiles were observed among the microcystin-exposed mice. It is not possible to say whether the effects observed were a direct result of interactions of microcystin in blood with these proteins, however it is likely that the microcystin-induced nasal lesions enhanced its absorption into the bloodstream. It is also possible that microcystin, once absorbed into the blood stream affected a target tissue, such as liver, causing alteration in blood protein types and amounts without causing frank lesions. The only microcystin dose-dependent changes in protein profiles were in the ion clusters occurring at m/zs of 11,688 and 11,829. The reason for lack of a consistent dose-response relationship for the three other ion clusters is not clear.
The alterations in serum protein profiles should be interpreted with a great deal of caution, since the analyses focused on a small fraction of the overall proteins present in serum. Very preliminary data suggest effects on SAA proteins, the components of high-density and low-density lipoproteins, and on β2-microglobulin, the light chain of the major histocompatability complex. A reduction in the concentration these proteins suggest a microcystin-modification in their production or possibly structure through adduction. No peaks were found in the combined F4 and F5 analyzed that corresponded to molecular weights of the 5 proteins listed in Table 2 plus the molecular weight of microcystin (995.2 Da). However, it is possible that such adducts may have separated outside of F4 and F5 in our serum fractionation scheme. Since protein phosphatase 2A is not found in serum, it is not surprising that effects on this protein were not detected. Further investigation will be required to determine the biological relevance of the alterations of protein patterns in microcystin-exposed mice or whether these changes will be useful biological indicators of microcystin exposure or effect.
Acknowledgements
This research was funded by NIEHS P30ES05705 10 P34 Pilot, NIEHS S11 ES11181, and matching funds from LRRI. We would like to thank B. Crowder, A. Dison, R. Garlick, A. Gomez, D.C. Santistevan and T. Zimmerman for their technical contributions. We thank James Aden for statistical support of the SELDI analyses.
References
- Azevedo SMFO, Carmichael WW, Jochimsen EM, Rinehart KL, Lau S, Shaw GR, Eaglesham GK. Human intoxication by microcystins during renal dialysis treatment in Caruaru-Brazil. Toxicology. 2002;181-182:441–446. doi: 10.1016/s0300-483x(02)00491-2. [DOI] [PubMed] [Google Scholar]
- Balmelli C, Roden R, Potts A, Schiller J, ge Grandi P, Nardelli-Haefliger D. Nasal immunization of mice with human papillomavirus type 16 virus-like particles elicits neutralizing antibodies in mucosal secretions. J. Virol. 1998;72:8220–8229. doi: 10.1128/jvi.72.10.8220-8229.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourmeyster N, Boquet P, Vignais PV. Role of bound GDP in the stability of the rho A-rho DGI complex purified from neutrophil cytosol. Biochim. Biophys. Res. Commun. 1994;205:174–179. doi: 10.1006/bbrc.1994.2646. [DOI] [PubMed] [Google Scholar]
- Bowman EP, Uhlinger DJ, Lambeth JD. Neutrophil phospholipase D is activated by a membrane-associated Rho family small molecular weight GTP-binding protein. J. Biol. Chem. 1993;268:21509–21512. [PubMed] [Google Scholar]
- Carmichael WW. Cyanobacteria secondary metabolites—the cyanotoxins. J. Appl. Bacteriol. 1992;7:445–459. doi: 10.1111/j.1365-2672.1992.tb01858.x. [DOI] [PubMed] [Google Scholar]
- Carmichael WW, Azevedo SM, An JS, Molica RJ, Jochimsen EM, Lau S, Rinehart KL, Shaw GR, Eaglesham GK. Human fatalities from cyanobacteria: chemical and biological evidence for cyanotoxins. Environ. Health Perspect. 2001;109:663–668. doi: 10.1289/ehp.01109663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creasia DA. Acute inhalation toxicity of microcystin-LR with mice. Toxicon. 1990;28:605. [Google Scholar]
- Dawson RM. Review article—the toxicology of microcystins. Toxicon. 1998;36:953–962. doi: 10.1016/s0041-0101(97)00102-5. [DOI] [PubMed] [Google Scholar]
- Duy TN, Lam PKS, Shaw GR, Connell DW. Toxicology and risk assessment of freshwater cyanobacterial (blue-green algal) toxins in water. Rev. Environ. Contam. Toxicol. 2000;163:113–185. doi: 10.1007/978-1-4757-6429-1_3. [DOI] [PubMed] [Google Scholar]
- Falconer IR. Tumor promotion and liver injury caused by oral consumption of cyanobacteria. Environ. Toxicol. Water Qual. 1991;6:177–184. [Google Scholar]
- Falconer IR, Buckley TH. Tumor promotion by Microcystis sp., a blue green alga occurring in water supplies. Med. J. Aust. 1989;150:351. doi: 10.5694/j.1326-5377.1989.tb136517.x. [DOI] [PubMed] [Google Scholar]
- Falconer IR, Humpage A. Tumor promotion by cyanobacterial toxins. Phycologia. 1996;35(Suppl 6):74–79. [Google Scholar]
- Falconer IR, Runnegar MTC, Jackson ARB, McInnes A. The occurrence and consequences of blooms of the toxic blue-green algae Microcystis aeruginosa in Eastern Australia. Toxicon. 1983;21(Suppl 3):119–121. [Google Scholar]
- Falconer IR, Burch MD, Steffensen AD, Choice M, Coverdale BR. Toxicity of the blue-green algae (cyanobacterium) Microcystis aeruginosa in drinking water to growing pigs, as an animal model for human injury and risk assessment. Environ. Toxicol. Water Qual. 1994;9:131–139. [Google Scholar]
- Fawell JK, Mitchell RE, Everet DJ, Hill RE. The toxicity of cyanobacterial toxins in the mouse: I microcystin-LR. Hum. Exp. Toxicol. 1999;18:162–167. doi: 10.1177/096032719901800305. [DOI] [PubMed] [Google Scholar]
- Fitzgeorge RB, Clark SA, Keevil CW. Routes of intoxication. In: Codd GA, Jefferies TM, Keevil CW, Potter C, editors. Detection Methods for Cyanobacterial Toxins. Royal Society of Chemistry; London: 1994. pp. 69–74. [Google Scholar]
- Fleming LE, Rivero C, Burns J. Report to Florida Harmful Algal Bloom (HAB) Taskforce, October 2000. NIEHS Marine and Freshwater Biomedical Sciences Center, University of Miami; Miami, FL: 2000. Blue green algal exposure, drinking water and primary liver cancer. [Google Scholar]
- Gizurarson S. Animal models for intranasal drug delivery studies. Acta Pharm. Nord. 1990;2:105–122. [PubMed] [Google Scholar]
- Harada K, Ogawa K, Matsuura K, Murata H, Suzuki M, Watanabe MF, Itezono Y, Nakayama N. Structural determination of geometrical isomers of microcystins LR and RR from cyanobacteria by two-dimensional NMR spectroscopic techniques. Chem. Res. Toxicol. 1990;3(5):473–481. doi: 10.1021/tx00017a014. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Shinogami M, Ishimoto S-I, Nibu K-I, Suzuki M, Kaga K. Identification of dual specificity phosphatases induced by olfactory bulbectomy in rat olfactory neuroeopthelium. Brain Res. 2001;902:205–211. doi: 10.1016/s0006-8993(01)02386-1. [DOI] [PubMed] [Google Scholar]
- Ito E, Kondo F, Terao K, Harada K-I. Neoplastic nodular formation in mouse liver induced by repeated intraperitoneal injections of microcystin LR. Toxicon. 1997;35(9):1453–1457. doi: 10.1016/s0041-0101(97)00026-3. [DOI] [PubMed] [Google Scholar]
- Ito E, Takai A, Kondo F, Hiroaki M, Susumu I, Harada K-I. Comparison of protein phosphatase inhibitory activity and apparent toxicity of microcystins and related compounds. Toxicon. 2000;40:1017–1025. doi: 10.1016/s0041-0101(02)00099-5. [DOI] [PubMed] [Google Scholar]
- Ito E, Kondo F, Harada K. Intratracheal administration of microcystin-LR and its distribution. Toxicon. 2001;39:265–271. doi: 10.1016/s0041-0101(00)00124-0. [DOI] [PubMed] [Google Scholar]
- Jackson ARB, McInnes A, Falconer IR, Runnegar MTC. Clinical and pathological changes in sheep experimentally poisoned by blue-green algae Microcystis aeruginosa. Vet. Pathol. 1984;21:102–113. doi: 10.1177/030098588402100117. [DOI] [PubMed] [Google Scholar]
- Jochimsen EM, Carmichael WW, An J, Cardo DM, Cookson ST, Holmes CE, Antunes M. Liver failure and death after exposure to microcystins at a hemodialysis center in Brazil. N. Engl. J. Med. 1998;338:873–878. doi: 10.1056/NEJM199803263381304. [DOI] [PubMed] [Google Scholar]
- Kroner C, Boekhoff I, Breer H. Phosphatase 2a regulates the responsiveness of olfactory cilia. Biochim. Biophys. Acta. 1996;1312:169–175. doi: 10.1016/0167-4889(96)00030-4. [DOI] [PubMed] [Google Scholar]
- Lawton LA, Edwards C. Purification of microcystins. J. Chromatogr. 2001;912:191–209. doi: 10.1016/s0021-9673(01)00592-1. [DOI] [PubMed] [Google Scholar]
- Leffers H, Nielsen MS, Andersen AH, Honore B, Madsen P, Vanderkerckhove J, Celis JE. Identification of two human Rho GDP dissociation inhibitor proteins whose over-expression leads to disruption of the actin cytoskeleton. Exp. Cell Res. 1993;209:165–174. doi: 10.1006/excr.1993.1298. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Hoshi F, Higuchi S, Kawamura AS. The complete amino acid sequence of dog beta2-microglobulin. J. Vet. Med. Sci. 1999;61:517–521. doi: 10.1292/jvms.61.517. [DOI] [PubMed] [Google Scholar]
- Nishiwaki-Matsushima R, Ohta T, Nishiwaki S, Suganuma M, Kohyama K, Ishikawa T, Carmichael WW, Fujiki H. Liver tumor promotion by the cyanobacterial cyclic peptide toxin microcystin-LR. J. Cancer Res. Clin. Oncol. 1992;118:420–424. doi: 10.1007/BF01629424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger RL. Comparative deposition of inhaled aerosols in experimental animals and humans: a review. J. Toxicol. Environ. Health. 1985;15:197–214. doi: 10.1080/15287398509530647. [DOI] [PubMed] [Google Scholar]
- Schlesinger RL. Deposition and clearance of inhaled particles. In: McClellan RO, Henderson RF, editors. Concepts in Inhalation Toxicology. Taylor and Francis; Washington, DC: 1989. pp. 191–217. [Google Scholar]
- Taylor BK, Stoops TD, Everett AD. Protein phosphatase inhibitors arrest cell cycle and reduce branching morphogenesis in fetal rat lung cultures. Am. J. Physiol. Lung Cell Mol. Physiol. 2000;278(L):1062–1070. doi: 10.1152/ajplung.2000.278.5.L1062. [DOI] [PubMed] [Google Scholar]
- Theiss WC, Carmichael WW, Wyman J, Bruner R. Blood pressure and hepatocellular effects of the cyclic heptapeptide toxin produced by Microcystis aeruginosa strain PCC-7820. Toxicon. 1988;26:603–613. doi: 10.1016/0041-0101(88)90243-7. [DOI] [PubMed] [Google Scholar]
- Ueno Y, Makita YI, Nagata S, Tsutumi T, Yosida F, Tamura S-I, Sekijima MK, Tashiro F, Harada T, Yoshida T. No chronic oral toxicity of a low dose of microcystin, a cyanobacterial hepatotoxin, in female BALB/c mice. Environ. Toxicol. 1999;14:45–55. [Google Scholar]
- Uneo Y, Nagata S, Tsutsumi T, Hasegawa A, Watanabe MF, Park HD, Chen GC, Yu SH. Detection of microcystins, a blue-green algal hepatotoxin in drinking water sampled in Haimen and Fusui, endemic areas of primary liver cancer in China, by highly sensitive immunoassay. Carcinogenesis (Oxf.) 1996;17:1317–1321. doi: 10.1093/carcin/17.6.1317. [DOI] [PubMed] [Google Scholar]
- Watanabe MF, Harada K-I, Carmichael WW, Fujiki H. Toxic Microcystis. CRC Press; Boca Raton, FL: 1996. [Google Scholar]
- Whitehead AS, de Beer MC, Steel DM, Rits M, Lelias JM, Lane WS, de Beer FC. Identification of novel members of the serum amyloid A protein super family as constitutive apolipoproteins of high density lipoprotein. Biol. Chem. 1992;267:3862–3867. [PubMed] [Google Scholar]
- Xue C, Heller F, Johns RA, Everett AD. Developmental expression and localization of the catalytic subunit of protein phosphatase 2A in rat lung. Dev. Dyn. 1998;211:1–10. doi: 10.1002/(SICI)1097-0177(199801)211:1<1::AID-AJA1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Yamamoto K-I, Migita S. Complete primary structures of two major murine serum amyloid A proteins deduced from cDNA sequences. Proc. Natl. Acad. Sci. USA. 1985;82:2915–2919. doi: 10.1073/pnas.82.9.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kathoh N, Adachi Y. The presence of two low molecular mass proteins immunologically related to 14 kilodalton serum amyloid A in the lipoprotein fraction and their decreased serum concentration in calves with experimentally induced pneumonia. Vet. Med. Sci. 1998;60:181–187. doi: 10.1292/jvms.60.181. [DOI] [PubMed] [Google Scholar]
- Yu S-J. Primary prevention of hepatocellular carcinoma. J. Gastroenterol. Hepatol. 1995;10:674–682. doi: 10.1111/j.1440-1746.1995.tb01370.x. [DOI] [PubMed] [Google Scholar]