

Summary
T lymphocytes respond to foreign antigen by forming specialized junctions with antigen-presenting cells (APC) or target cells. A hypothesis is presented, illustrating the similarity between the T-cell recognition-activation process and the cell communication processes found in other organ systems, especially the nervous system. Based on data showing that a major neuronal protein, Thy-1, is also a mitogenic site on T cells, and based on predictions for the structures of the T-cell receptor (TcR) and Ia proteins, an activation model is presented as follows. 1) The T-cell receptor initiates cell-cell contact with the APC by interacting with Ia and antigen, forming an antigen-binding site. 2) Sets of adhesion molecules then bind, focusing the interacting proteins to the junctional site. One binding protein, L3/T4, binds Ia and concentrates the Ia molecules to the contact site. 3) The two-chain TcR then links together the TcR-Ia-antigen complexes, forming a linear chain of receptors which will self-associate once reaching a critical length, forming a cluster. This cluster juxtaposes associated channel subunits, the T3 membrane molecules, creating an ion channel, stimulating the T cell. 4) The MHC molecule is structurally a part of this activation complex, and therefore also forms a cluster on the APC surface, possibly activating the presenting cell. 5) Secretory products are then released into the synaptic site allowing for efficient and directed cell-cell communication. Cytolytic class-I-restricted cells use a similar pathway to focus the effect of cytolytic proteins.
This analogy views neuronal communication and lymphoid recognition as evolutionary descendents of a primordial lymphocytic type of cell interaction.
Keywords: T lymphocyte, MHC, Synapse, T-cell receptor, Antigen-presenting cell, Thy-1, Binding protein, Neuron, Position paper
Introduction
The activation of T cells involves a multistep process which begins with the recognition of antigen in association with major histocompatibility complex (MHC) proteins and results in the expression of specific immunological functions such as the secretion of regulatory factors or the expression of cytolytic activity. The initiation of this pathway is mediated by cell surface structures which regulate membrane contact and cell surface interactions as well as transduce signals to the cell interior. Although a number of structures have been described which participate in the early phases of T-cell recognition and activation, the actual physical mechanisms of how these proteins collectively interact and function is not fully understood. For several years, I have been involved in the study of T-cell activation using antibodies to the Thy-1 glycoprotein as an activation model. These studies describe mechanisms of receptor-mediated cell interaction which regulate T-cell activation and are now common to other T-cell activation models. In view of the high concentration of Thy-1 in the brain, especially in the neuronal cell-cell contact regions, the synapses, the interesting possibility can be raised that this molecule has a similar function in both the nervous and immune systems. In fact, as more functional cell surface structures become known, a striking resemblance in the forms of cell communication and interaction between these systems emerges. The model to be presented in this paper views the cell interaction complexes formed between T cells and antigen-presenting cells (APC) or target cells as evolutionary relatives of synaptic junctions in the nervous system and gap junctions in other systems. The lymphoid synaptic regions are formed by multiple adhesion proteins which stabilize the contact site and provide a focus for the formation of ion channels from independent protein subunits associated with the T-cell antigen receptor (TcR). Because the MHC proteins are structurally a part of the T-cell activation complex, they also simultaneously form a cluster which can transduce signals into the presenting cell. The junctional apparatus thus formed permits the focused secretion of labile mediators into synaptic spaces between the interacting cells, thereby creating an efficient method for cell-cell interaction and directed factor secretion. Although this relationship between immune recognition and neuronal communication is only a functional analogy, it is likely that many of the proteins which make up the interacting structures are structurally homologous and relatives of the immunoglobulin supergene family. This view of T-cell activation is derived from the Thy-1 model and from recent predictions for the three-dimensional structures and mechanisms of interaction of the MHC class II molecules and the T-cell antigen receptor. The Thy-1 system will first be briefly reviewed to illustrate the basic principles of receptor coupling and activation now consistent with recent findings in other systems. With these principles in mind, the structures implicated in the T-cell recognition of antigen will be discussed and then a hypothesis presented for the sequence of events which form the lymphocytic synapse, thus permitting cell coupling and signal transmission.
The Thy-1 molecule as an activation site on T cells
One of the first findings to suggest that T-cell activation involved synaptic structures was the demonstration that a major synaptic protein, Thy-1 [1], was not only a predominant T-cell surface structure, but also had the functional property of mediating T-cell activation. In fact, the first mitogenic antibodies [2] to Thy-1 were generated against mouse brain and, once absorbed to remove anti-mouse activity, bound to T cells and immunoprecipitated the classical 25,000-dalton species of Thy-1 [3]. This antibody was a potent mitogen for heterogeneous populations of cells from either spleen or lymph node, but failed to stimulate purified T cells at concentrations of antibody optimal for spleen cells. If B cells [2] or, as later found, a variety of Fc receptor (FcR)-bearing cells [3] were added to the T cells with antibody, the T-cell proliferative response was restored. Proliferation was dependent on an intact Fc portion of the antibody, suggesting the requirement for Fc-FcR binding in cell-cell interaction. This was also confirmed by blocking of the response with monoclonal antibodies against the FcR. Cell cooperation between the T cell and the FcR-positive cell was not MHC-restricted or dependent on the presence of Ia on the accessory cell. A clue to the role of FcR-bearing cells was the ability to substitute for their activity by adding to the culture a second antibody against the first antibody [3]. The second antibody cross-linked the Thy-1-bound antibody into stable large patches and caps on the T-cell surface which then initiated cell activation. This suggested that the FcR on the accessory cell was modifying or inducing the cross-linking of the T-cell-bound anti-Thy-1.
The major function of the accessory cell was to aid in the production of IL-2 [4]. Adding IL-2 to the T cells with antibody could replace the accessory cell requirement, indicating that antibody alone, even at low concentrations, induced the expression of IL-2 receptors, but was insufficient for IL-2 production. In the absence of exogenous IL-2, further cross-linking either by cell surface contact with the FcR accessory cell or by anti-Ig then initiated the production of IL-2, providing the growth stimulus for cell proliferation. These studies were extended with clones of EL-4 IL-2-producing T-cells, which responded to anti-Thy-1 with similar activation requirements [5]. In this system, antibody was found to bind to the surface of the cell and be partially endocytosed and shed without capping [6], and without stimulating IL-2 production. When anti-Ig was added to these anti-Thy-1-coated cells, surface stabilization of the primary antibody was seen, followed rapidly by membrane potential changes [6] and then, in 4-6 h, by the secretion of IL-2. Many of the above phenomena have now been confirmed in several laboratories using both polyvalent and monoclonal antibodies against Thy-1 [7, 8].
The Thy-1 model provides a simple model for receptor-mediated cell-cell coupling and surface focusing, and demonstrates a number of requirements for T-cell activation.
1) Cross-linking of specific sites on the T-cell surface initiates T-cell activity. This has now been found for antibodies directed against the T3 proteins [9] and for the putative anti-T-cell antigen receptor antibodies [9, 10]. Both the anticlonotypie antibodies and the anti-T3 antibodies trigger T-cell activation when presented in a multivalent form. Usually, this is accomplished by immobilizing the antibody on the surface of sepha-rose beads, allowing it to activate IL-2 receptor expression, IL-2 secretion and proliferation in T-cells and T-cell clones without accessory cells. Because of the biochemical and functional similarity between Thy-1 and T3 molecules, it was proposed that Thy-1 may actually be one of the T3 proteins [5, 6, 8], as will be discussed later. Therefore, it appears that of all the T-cell surface structures known to which antibodies have been made and tested, only a few can directly induce activation. Included in this set are the T-cell antigen receptor, the T3 proteins and the Thy-1 glycoprotein.
2) Regulation of T-cell activation is controlled by the level of receptor cross-linking or aggregation. Clustering of receptors is a universal event associated with receptor-mediated cell activation in a variety of biological systems [11, 12]. The final physiological effect depends directly on the number of receptors which are bound or occupied and then subsequently cross-linked. Low levels of cross-linking, usually with antibody alone as shown with anti-Thy-1, anti-T3 and anti-clonotypic antibodies, activates only IL-2 receptor expression, while higher levels are required for IL-2 production [4, 13, 14]. Regulation of activation can therefore be accomplished by controlling the amount of cross-linking. This can be done directly, either by changing the T-cell surface antigen or receptor concentration, or by modifying the dose of activating antibody or ligand. In the Thy-1 system, the production of IL-2 in response to anti-Thy-1 antibody was proportional to the surface concentration of Thy-1 on clones of EL-4 cells. Cells with lower Thy-1 expression also required higher antibody concentrations to produce equivalent amounts of IL-2. In addition to the concentration of the reactants, the formation and stability of aggregates should also be dependent on the affinity of the receptor or stimulating antibody.
3) A major pathway for physiological regulation of cross-linking is through receptor-mediated cell surface focusing. With anti-Thy-1, the FcR-bearing accessory cell binds to the Fc portion of the antibody and induces the aggregation of the antibody. Thus, the aggregation of the FcR simultaneously co-aggregates the Thy-1 molecule via an antibody bridge. A similar FcR-bearing accessory cell requirement has also been shown for the mitogenic anti-T3 antibody [15, 16]. This demonstrates two important principles of interactions at membrane interfaces. First is that the clustering of a set of receptors on one cell can influence the aggregation of an attached set of receptors or molecules on a second cell. This represents a simple coupling mechanism which is later postulated to be similar to the coupling between the T-cell receptor and MHC proteins. Secondly molecular interactions occurring between membrane interfaces favour a flow of receptors into the contact areas, because the free energy of binding stabilizes the molecules in these regions, as discussed by Bell [17]. This provides a focusing mechanism, which concentrates interacting proteins to contact sites. This concept is critical to the multipoint-multiprotein interaction for T-cell activation in response to antigen.
4) The final effect of receptor clustering can be regulated by controlling the cell activation « threshold » or sensitivity. The activation threshold represents a critical level of message such as Ca++, cyclic nucleotides, or phospholipid metabolite, required to trigger the cell. Usually this activation barrier is overcome by occupying or engaging a certain number of cell surface receptors. Thresholds can be modified either by controlling the intracellular resting state concentration of the inducer, for example, the intracellular Ca++ concentration, or by changing the reactivity of the intracellular target of this inducer. Because cellular growth regulation is also a complicated multistep process, its control may occur at a number of sites within the cell. In excitable cells, « thresholds » represent electrical potentials which, when reached, lead to rapid ion fluxes generating membrane potential spikes. Now that it appears that lymphoid cells may use membrane potential changes for cellular regulation [6, 18, 19], it is possible that threshold regulation may be similar to mechanisms involved in neuronal threshold control. The effects of IL-1 may be to modify the cell threshold by some of the above mechanisms. Overall, the sensitivity of the cell to stimulation will be determined both by the internal threshold setting and by the total number and affinity of cell surface receptors.
The question remains as to why receptor cross-linking or clustering is vital to the activation of not only T cells hut of any cell. Possibilities include the formation of ion channels from separate components, the formation of binding sites for other linkage or transducing proteins, and the formation of an active enzyme such as a kinase. In this context, how can the cross-linking of Thy-1 transduce a signal to the cell interior? The Thy-1 protein has been sequenced [20] and corresponding cDNA cloned [21, 22]. The amino acid sequence has homology with both variable region sequences and β2 microglobulin (β2m). The two cDNA clones for Thy-1 suggest that two forms may exist on the T-cell surface. One has no membrane extension and is actually shorter than the previously sequenced brain Thy-1 molecule, possibly representing a peripheral membrane molecule similar to β2m. The other cDNA clone has a hydrophobic stretch of C-terminal amino acids with a central aspartic acid residue suggesting a membrane-associated region. This hydrophobic sequence has striking homology with the membrane spanning region of surface IgM, indicating a possible structural overlap in the mechanism of signal transduction. However, the Thy-1 hydrophobic region is devoid of extensive charge residues characteristic of other channel-forming proteins and is not totally transmembrane. Therefore, it may not directly mediate membrane transduction or form a channel structure. To stimulate activation it must be associated with another protein or proteins to generate an activation complex. Since Thy-1 is a V-region-like domain, it may interact with itself, forming a V- to V-like dimer and thereby create an active site for another transducing protein. Alternatively, Thy-1 could associate with another complex such as the T3 and T-cell receptor proteins to generate a transmembrane signal through this apparatus.
The Thy-1 model was presented because it illustrates a mechanism of cell interaction which is postulated to be similar to the coupling of T cells and APC in the presence of antigen. The components necessary to the antigen specific activation pathway will next be presented.
Lymphocyte synaptic proteins
The main distinction between lymphoid synapse formation and synaptic junctions in the nervous system is that lymphocytes and their targets are mobile and separate before contact. In contrast to the synapses between neurons, which are stable fixed structures that transmit rapid intercellular communications, the lymphoid counterparts use mobile independant cell surface proteins which are brought together and focused in the presence of antigen to form junctional complexes. The cell structures in this model may themselves be evolutionary relatives and are classified as forms of cell adhesion and interaction molecules.
The synaptic model begins with independent molecules on the cell surface of both the T-cell and the APC or target cell. These molecules form sets of complementary binding proteins with one of each set localized to either the T cell or the APC.
Cell Adhesion and Binding Molecules
The first set of binding proteins is found on lymphoid cells and monocytes without cell subset distinction. An example of these molecules is the LFA-1 glycoprotein [23]. The LFA-1 glycoproteins are large MW non-covalently-linked dimers of 95 kDa and 160 kDa chains. Antibodies against these proteins block cell adhesion in class-I and-class-II-restricted functions by blocking at an early Mg++-dependent binding step. These structures are functionally analogous to the cell adhesion molecules (N-CAM) [24] described in nervous system development. N-CAM are large molecular weight glycoproteins which function in neuronal cell adhesion, especially during the development of synaptic pathways. They have the property of forming self-aggregates depending on the amount of protein glycosylation. It is possible that LFA proteins also change in binding properties with changes in glycosylation, thereby providing a mechanism for adhesion regulation. Although the binding sites for LFA molecules have not been described, it is possible that other LFA-related molecules on the presenting cell surface serve this purpose. Functionally related to LFA-1 are the more recently described LFA-2 and LFA-3 molecules [25]. Antibodies against these structures also block cytolysis, possibly at the adhesion stage [26].
The second set of adhesion molecules are the cell class-specific binding proteins. These structures are usually correlated with the specific MHC restriction element recognized by the effector T cells, irrespective of effector function. The cytolvtic class-I-restricted cells express the Lyt2,3 (30 kDa, 33 kDa disulphide-linked dimers) [23, 27] or Leu2/T8 (33 kDa) [28] molecules, while class-II-restricted cells express the L3/T4(50 kDa) [29] molecule. Antibodies to these structures also block at an early cell binding step. However, not all cells expressing these structures can be blocked with antibody, and some Ia-restricted hybridomas totally lack the L3/T4 molecule [30], indicating that these proteins are not an absolute requirement for T-cell activation. This variation in the functional participation of these molecules in recognition has been proposed to be related to the affinity of the T-cell receptor for the MHC and antigen complex [30, 31]. This was shown by experiments in which in vivo primed CTL-cloned cells to virus plus self-class-I MHC were resistent to Lyt2 blocking when tested for anti-virus killing, but was sensitive to blocking when tested on a cross-reactive allogeneic target [31]. Blocking effector function with anti-LFA-1 has also revealed some heterogeneity, possibly for similar reasons of affinity. The target ligands for the Lyt2 and L3/T4 proteins may be a portion of the specific MHC protein because of the correlation with T-cell restriction [27, 31]. Binding therefore would appear to be independent of polymorphic residues, suggesting that monomorphic sites are used. If this is the case, then domain-domain interactions similar to C or V domain binding may occur between the N-terminal domains of, for example, the L3/T4 molecule and an external face of one of the Ia domains. The discovery of these adhesion proteins and their relationship to apparent affinity led to the view that T-cell binding and activation was a result of multi-protein binding collectively contributing to the final avidity required for cell stimulation.
The T-cell receptor for antigen and MHC proteins
The last set of binding proteins provides molecular specificity and is associated with the signal transducers for T-cell reactions. The set includes the T-cell antigen receptor and MHC glycoproteins, for our discussion, the class II molecule or Ia. In contrast to B cells, which use immunoglobulin as a membrane receptor and can bind antigen directly, the T cell classically appears not to take up or specifically bind native or degraded antigen, a phenomenon which has long puzzled immunologists. Instead, antigen recognition requires the simultaneous co-recognition of allelic determinants on MHC proteins. Although both single and dual receptor models have been previously proposed to explain this dual requirement, recently, because of a number of functional studies and with the published sequence of one chain of the T-cell receptor, the model of a single receptor composed of two chains expressing one functional site for antigen and MHC complexes is favoured. However, with one binding site, basically a monovalent receptor, the nature of the binding mechanism and the events which provide for receptor cross-linking, especially with univalent small peptides, are difficult to explain. An alternative to the single site model which provides for recognition and cross-linking has recently been proposed [32] and will briefly be described. This model relies upon the predictions for the three-dimensional structures of the Ia molecule and the T-cell receptor. The model states that the T-cell receptor has two chains with physically separated variable regions. One chain functions as only half of the antigen recognition unit, which, in the presence of antigen and a first domain of Ia, forms a stable antigen-binding site analogous to an antibody-combining site. The other chain of the T-cell receptor also has a V region and interfaces with the other Ia first domain on a separate Ia molecule, again similar to VH and VL. One T-cell receptor dimer therefore bridges two molecules of Ia only in the presence of antigen.
This proposed structure of the T-cell receptor was derived from a number of facts. Biochemically, the clonotypic receptor consists of two disulphide-linked chains, a basic β chain and an acidic α chain, both of which have a molecular weight between 40 and 50 kd [33-35]. By peptide mapping, these chains differ from each other and appear to have variable and constant sequence properties [34, 36]. In addition, the T-cell receptor complex has at least three « T3 » molecules [37]. Antibodies to the T3 proteins bind only to the 20 kd glycosylated form which, when precipitated, brings down the other two non-covalently associated molecules, the 25-28 kd glycosylated protein and the 20 kd non-glycosylated molecule. The non-glycosylated 20 kd protein may be mostly membrane-associated, since it is the only T3 molecule that is heavily labelled with lipid photo-affinity probes [38]. T3 appears to be required for the expression of the T-cell receptor [39] and, when quantitated, exists in numbers roughly equalling the clonotypic receptor number, 30-40,000 molecules. Antibodies to T3 co-modulate the receptor, and reciprocally, the anti-receptor antibodies modulate the T3 antibody specificities [39], suggesting that these molecules are tightly associated. The number of non-glycosylated 20 kDa or 25-28 kDa T3 molecules on the cell surface is not known because antibodies have not been reported which bind these proteins uniquely. The biochemical similarity and the functional property of mitogenicity led to the proposal that Thy-1 was analogous to one of the T3 molecules, possibly the 25-28 kd species [6]. Because there are 100-200,000 Thy-1 molecules on the mouse T-cell surface, it is unlikely that the T3 molecule recognized by anti-T3 and associated with the T-cell receptor is the Thy-1 equivalent. In addition, anti-Thy-1 antibody does not cap or consistently block T-cell effector function (unpublished results). For these reasons, I view the Thy-1 molecule and its possible human equivalent, the T3 25-28 kd species, as couplers between the T-cell receptor-T3 unit and the 20 kd T3 membrane (T3m) component.
Recently, two reports of the gene sequences of one chain of the T-cell receptor were published and demonstrated a striking resemblance between the TcR and immunoglobulin (Ig) by sequence homology [40, 41]. This chain has two domains, an N-terminal variable region domain and a constant region domain. A number of cysteines were found in addition to the four cysteines used for domain stabilization. One was at position 260 between the membrane spanning region and the constant region thought to be in a position homologous to the Ig light chain cysteine which binds the heavy chain. These sequences were analysed for amino acid and DNA homology with Ig and verified the reported homologies, except for a few critical findings [32]. First, the cysteine at position 260 in the T-cell receptor is in a position homologous to the hinge region in Ig heavy chains and not where heavy and light chains bind. Secondly, on the DNA level, the only significant homology found was between the T-cell constant region and the α3 region of class I molecules. Since this area in class I molecules binds to (β2m and the T-cell receptor is co-associated with and may require the T3 complex for expression, it was proposed that this region of the T-cell chain binds one of the T3 molecules. Knowing that the T-cell receptor has a hinge region and molecules which could function to stabilize isolated chains leads to the most likely conclusion that the T-cell receptor chains and at least the V-regions of the receptor are separated much like heavy chains in antibody. In order to form active binding sites, these T-cell V regions must interact with other V-region-like domains. The V-like domains which complement the T-cell receptor first domains are pro posed to be the N-terminal domains of Ia. Our model of the T-cell receptor is presented in figure 1. The two chains of the T-cell receptor are connected at the hinge and stabilized by the T3 glycoproteins. The V regions are facing in opposite directions and are separated. The Thy-1, T3 and TcR constant domains bind to each other by domain stacking similar to the mechanism by which constant regions and variable regions bind. The Thy-1 (T3 25-28 kd species) hydrophobic region with central negative charge would bind both to the positive charge in the T-cell receptor β chain transmembrane region by ionic bridging and to the T3 membrane component. The T3m is most likely the transducing component of this complex and is postulated to be one subunit of an ion channel.
Fig. 1. The T-cell antigen/MHC receptor complex and class II molecules.
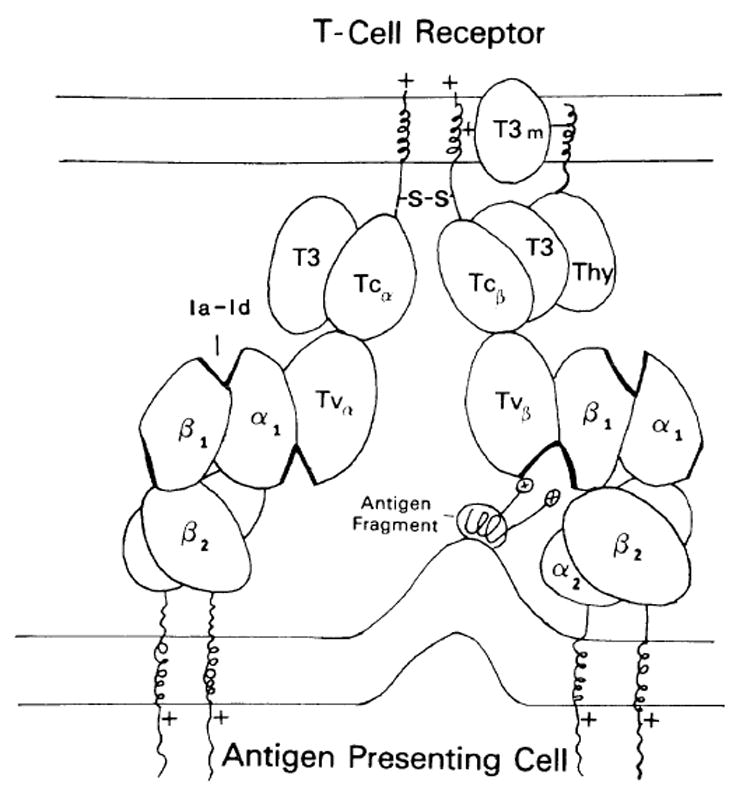
The oval structures represent Ig-like domains and the abbreviations are as follows. For the T-cell receptor shown on the T-cell membrane (above): Tvβ = β-chain variable region; Tcβ = β-chain constant region; Tvα = α-chain variable region; Tcα = α-chain constant region; T3 = 20 kDa glycosylated T3 peripheral protein: Thy – Thy-1 or T3 25-28 kDa molecule; T3m = T3 membrane component. For the Ia molecules: β1 = β-chain polymorphic first domain; β2 = β-chain membrane proximal domain; α1 = α-chain first domain; α2 = α-chain second domain; Ia-Id = Ia idiotype. The thickened areas represent polymorphic regions in the domain. The antigen fragment depicts the proposed α-helical cylochrome c fragment [50] with carboxy terminal positively charged lysine residues interacting with the recognition unit.
The structure of class II molecules
Ia glycoproteins are composed of non-covalently-linked dimers of β chains of 28 kDa and α chains of 33 kDa (see review [42]). Each chain has two external domains, a membrane-proximal Ig-homologous-constant-region-like domain (β2, α2) and an N-terminal polymorphic domain (β1, α1). The three-dimensional structure of Ia is not known. Recently, using a sensitive local sequence homology program, the β1 domains were found to show low but significant homology with Cκ and V κ domains, suggesting that they also are structurally members of the Ig-supergene family [32]. These sequence homologies were supported by a striking similarity in hydrophobicity profiles. These facts together with results of analysis using computer programs which predicted regions of β-pleated sheets in the molecule allowed the modeling of the β1 domain into an Ig-like folded sandwich of two anti-parallel β-pleated sheets. The polymorphic regions were then located and found to be present mostly, but not totally, on bends between strands, and juxtaposed in two areas on opposite ends of the domain. One region fell in a position which interfaced with the α1 domain at the N-terminal most peripheral edge of the molecule, while the other was on the opposite β-pleated sheet on the external face of the β1 domain pointing toward the membrane. This external face was postulated to have a structure which complements the T-cell receptor V-region. The polymorphic regions on this face therefore function similarly to hypervariable regions in Ig providing for both antigen and interdomain contacts. The α1 domain of the Ia heavy chain had lower homology with Ig, but because of the Ig relationship of the α chain membrane proximal region, it was assumed to have a structure similar to the β1 domain for the purposes of this model. A schematic model of the Ia molecule is shown in figure 1 on the APC membrane. The two membrane proximal domains are constant region-like and are tightly associated. The connecting peptides between the membrane and the second domains provide for flexibility at the base of the molecule to accommodate membrane-associated antigens. The Ia-β1 and −α1 domains bind to each other and provide surfaces for the TcR interaction. There are 3 main sites of Ia polymorphism on this molecule (indicated by the thick domain edge). One on the external face of β1, another site in an analogous position in the α1 domain and finally a site in a region where the β1 and αl domains interact. This last site is in a position similar to an Ig-combining site and is referred to as an Ia idiotype (Ia-Id). Functionally, the Ia-Id may not be involved in antigen binding, because it does not directly complement the TcR chains. It may play a role in MHC restriction, possibly by interacting with the TcR α chain or, alternatively, it may function as a site for suppressor cell or factor binding.
The interaction of the T-cell receptor with MHO is shown in figure 1. The V domain of one T-cell chain (TcR-Vβ) binds to the β1 domain external surface and is stabilized in this example by antigen bridging the gap between the TcR and Ia amino acids in « hypervariable » polymorphic regions. The site of antigen binding is analogous to the combining site of antibody, since it is formed by the interaction of a TcR V-region domain and a V-like Ia domain. The actual binding of TcR-Vβ and Ia-β1 is through multiple contacts between polymorphic and non-polymorphic framework regions of both domains similar to the binding between VII and VL in antibody. The TcR-Vα domain on the other chain is proposed to bind to the α1 domain on a separate Ia molecule, resulting in the bridging of two Ia molecules by one TcR complex. This TcR-V α and α1 interaction is, in principle, similar to the TcR-V β and β1binding, except that it binds without antigen. The TcR-Vα domain may already have a high affinity for the Ia-α1 domain or be induced to bind because of a conformational change in the α1 domain secondary to the stable TcR-Vβ and β1 interaction with antigen. With other antigens, the situation may be reversed and antigen will bridge the TcR-Vα with α1 instead of interacting with the β1 domain. In general, due to thymic selection, the TcR V regions have low but definite affinity for the Ia first domains in the absence of antigen.
Formation of T-cell activation complexes and ion channels
The model for TcR binding to Ia and antigen shown in figure 1 now provides a mechanism for generating receptor cross-linking and makes a clear prediction as to the, molecular events which lead to signal transduction. Following the binding of the TcR to the b1 domain with antigen, the TcR-α-chain then links these units by binding to the Ia-α1 domain (fig. 2). These complexes will not branch; instead, once reaching a critical length [43], will self-associate to form a ring. During the circularization step, the TcR-associated T3 and Thy-1 structures are forced to the center of this ring. These structures are then secondarily stabilized and juxtaposed, thereby creating a multi-subunit transducing complex. The T3m may exist as one subunit of an ion channel which, when stabilized as diagrammed, becomes a functional channel for either Ca++[44] or monovalent ions. The formation of an ion channel from multiple identical but separate subunits is not unprecedented, as a similar process may occur with the Fc receptor for IgG [45] and possibly with the IgE receptor. Purified FcR, when inserted into synthetic membranes, will mediate ion influx when aggregated by antibody-antigen complexes or monoclonal anti-FcR [46]. Once the TcR channels start functioning, the intracellular concentration of Ca++ increases until a threshold level is reached, activating the growth cascade.
Fig. 2. T-cell activation complexes; ion channel formation.
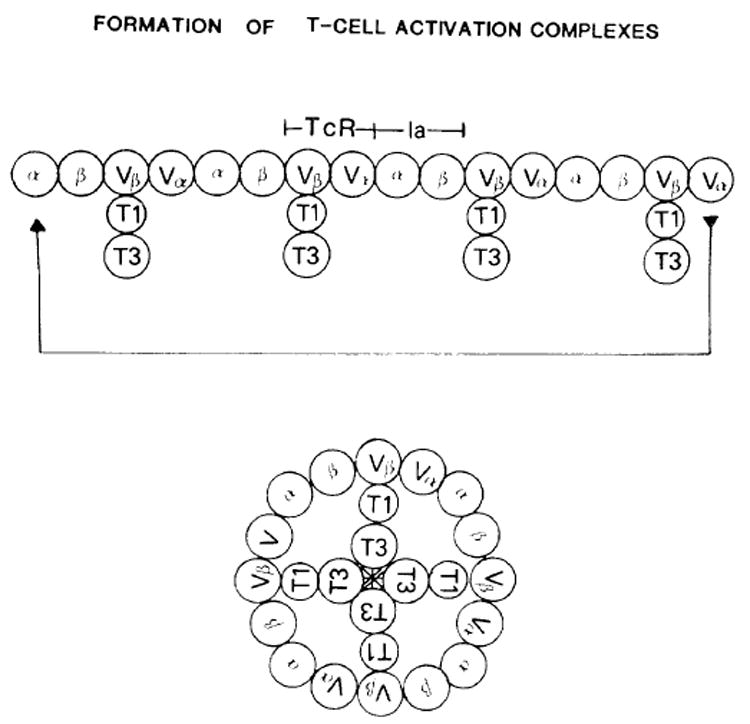
The complexes are viewed from above with the plane of the paper in the plane of the membrane. β and α represent the first domains of the Ia molecules. TcR.Vβ and Vα are the two variable regions. The Thy-1 molecule (T1) is attached to the TcR constant region via the T3(20 kDa) molecule (not shown) and to the T3 membrane component labelled as T3. The linear chain will self-associate to form a ring juxtaposing the T3 membrane proteins forming the ion channel.
As a consequence of TcR clustering and channel formation, a ring of Ia molecules will also form on the APC. If Ia has a transducing subunit associated with it, then it follows that a similar channel would form on this membrane permitting ion flux and thereby stimulating the macrophage or B cell. For instance, the macrophage could be triggered to release IL-1 through this mechanism while the B cell would undergo cell cycle progression and differentiation [48]. With this receptor coupling mechanism, activation of the presenting cell would be mediated directly by the TcR and Ia channel coupling and would not require other inducer factors from the T cell. Activation of both cells would be a simultaneous process driven by the formation of juxtaposed channels. In fact, the implicated close association of these channels raises the possibility that, in some interactions, actual direct cytoplasmic connection may occur, resembling a gap-type junction.
The synaptic T-cell activation casecade
Based on the above cell interaction molecules and the transducing cell coupling device, a sequence of events leading to cell activation for class-II-restricted recognition is postulated as schematically illustrated in figure 3.
Fig. 3. The T-cell synaptic-junctional complex.
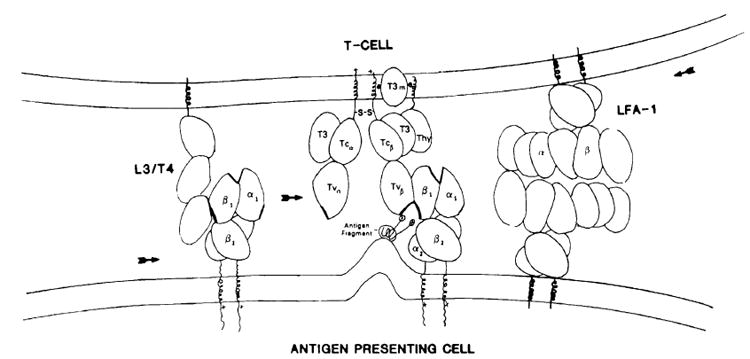
The sequence of binding events is described in the text. LFA-1 is shown binding to another LFA-like molecule on the APC which is not specifically LFA-1.
1) The pathway begins when the APC, either a macrophage, B cell or dendritic cell, takes up antigen and, either through lysosomal or cell surface processing [49], degrades the antigen to a small amphipathic peptide [50] of roughly 15-20 amino acids. This immunogenic peptide is then expressed on the cell surface with the hydrophobic groups associated with the plasma membrane and the hydrophilic groups exposed to solvent. These processed peptides are mobile on the cell surface and usually do not interact with class II MHC proteins.
2) The T cell bearing the MHC-restricted, antigen-specific T-cell receptor with two spatially separated V regions binds using one V domain, in this example from the β chain, to the N-terminal domain of the Ia β chain in the presence of antigen, thus forming a trimolecular complex. As individual receptors bind with Ia and antigen, the membranes from these cells are brought into close proximity.
3) The cell adhesion molecules, the LFA series and L3/T4, and possibly others then interact with their target ligands on the presenting cell and a flow of interaction molecules occurs into the contact (synaptic) region. The L3/T4 molecule binds to Ia and migrates to the contact site, actually increasing the local concentration of Ia.
4) This creates a focus for T-cell receptor migration resulting in the binding of additional TcR-Ia-antigen complexes. The TcR can compete for Ia binding with the L3/T4 molecule because of a higher binding affinity. L3/T4 may also remain attached to the Ia molecule if it doesn’t interfere with TcR and Ia binding. The T-cell receptor with one chain bound to the Ia-β1 domain and antigen then uses the other TcR α chain Lo bind to a second Ia molecule interacting with sites on the Ia-α1 domain. This becomes a nucleating focus for the sequential binding of TcR, Ia, and antigen, creating a linear chain of receptor-Ia-antigen complexes.
5) Once reaching a critical length, I his chain then binds to itself, forming a circular cluster (fig. 2). The T-cell-receptor-associated T3m-transducing subunits then become physically juxtaposed into a multi-subunit active complex, most likely a Ca++ ion channel. These channels then continually form until a critical level of Ca++ is reached, driving the metabolic processes of cell activation. Low levels of Ca++ or orther mediators induce the early expression of IL-2 receptors, possibly from a preformed pool.
6) As a consequence of TcR binding, Ia is simultaneously clustered, activating APC for the secretion of IL-1 or other APC factors. The accessory cell products could be secreted directly into the junctional space.
7) The intracellular Ca++ level then accumulates, secondary to further channel activation and possibly to the contribution of other hormones such as IL-1 stimulating the production of lymphokines, including IL-2. IL-2 then promotes the proliferation of the T cells with expressed IL-2 receptors.
8) The activation complex is then removed by endocytosis and shedding [6, 9], a down regulatory effect, turning off the receptor-generated signal and producing a refractory state to further antigen driven activation. If the antigen stimulus or Ia concentration is raised to high levels [51], responses can be generated from the few remaining or newly expressed T-cell receptors. Although these cells are depleted of antigen receptors, they may continue to express the IL-2 receptor and respond to IL-2 either generated by the same cell or from another source.
In the model presented, the synapse, is initiated by the TcR-MHC-antigen binding and recruits other binding molecules to the contact site. The adhesion molecules probably have no transducing function alone but set the stage for the completion of the TcR and MHC antigen complexes which form the transducing apparatus. The formation of a synaptic structure by the focusing of interaction molecules may then provide for the local directed secretion of interleukins or other factors which would increase the effectiveness of the secretory mechanism. Effector molecules, many of which are extremely labile, would then be concentrated to synaptic spaces allowing for both efficient and specific intercellular communication. Some of these molecules have autostimulatory capacities and, once secreted into the junction, would feed back to the producer T cell. IL-2 may function by this pathway [14].
A more striking example of this synaptic process is clearly seen in the interactions between cytolytic T cells and target cells. CTL form conjugates with target cells in a manner similar to that described above for the Ia-restricted cells. However, the effect of this interaction is to specifically kill only the attached target cells and no others, suggesting a focused directed cytolytic process. Electron micrographs of cell conjugates demonstrate extensive « interdigitations » in the contact sites between the T cell and target cell reminiscent of synaptic foci [52, 53]. In fact, the lysosomal granules which contain the cytolytic mediators rearrange in the T-cell cytoplasm to directly underlie this region and granules are often seen releasing their contents into the junctional space. Following this release, channel formation and rings on the surface of target cells have been seen by a number of investigators and are thought to be created by the cytolytic secretory products [54, 55]. These channels are heterogeneous in size, with some similar to the channels created during complement-mediated cell lysis. The involvement of the T-cell receptor or MHC class I proteins in these channels has yet to be described. What is known is that some of the cytolytic proteins found in the CTL granules are unstable in Ca++ and serum-containing medium [55], suggesting that the transport of these substances to the target cells occurs either in an isolated environment or directly by membrane contact. In this case, the analogy of a presynaptic vesicle or granule releasing its contents which then insert into the postsynaptic membrane forming an ion channel, can easily be envisioned.
Conclusions
The functional similarity between neuronal synapses and lymphocyte junctions may represent more than just an analogy. Both systems involve extensive cell interaction and communication, many of which are mediated by small peptides. Recently, a number of neuropeptides were found to have effects on the immune system [56, 57]. Receptors once thought to be localized to the brain and nervous system have now been found on a variety of immunologically related cells, and some neuropeptides can modulate immune function both in vivo and in vitro. Even the predicted structures for many of these peptides [58] is remarkably similar to the amphipathic peptide antigens recognized by Ia-restricted T cells in some systems. Monovalent neuropeptides and other polypeptide hormones also induce receptor aggregation, by unknown mechanisms. To explain this cross-linking effect, a variety of mechanisms have been proposed, usually involving conformational changes induced in the receptor molecule secondary to binding, which then facilitates self-aggregation. Alternatively, I would suggest that the binding sites for these hormones may potentially only form when the peptide bridges receptor units, in a manner analogous to the T-cell receptor/Ia interaction with antigen. One receptor molecule would carry half of the binding site, and only in the presence of the peptide hormone would two subunits be brought together to form a recognition unit. This could be a simple mechanism for both recognition and the simultaneous cross-linking of receptors for monovalent peptides.
This view of receptor function could explain why many receptors and surface structures exist as subunit dimers [59]. In this scheme, the dimers would be back-to-back subunits with the potential binding (half) sites facing in opposite directions. Hormone binding would bridge the receptors into chains and then rings forming channels similar to the T-cell receptor complex. Alternatively, receptor clusters could either function as binding sites for coupling proteins or enzymes, or directly have enzymatic activity such as the kinase activity associated with growth factor receptors.
Stable ion channels like the acetylcholine receptor (AChR) may also function in this manner. Instead of separate independent channel sub-units, the AChR has five stably attached homologous chains [60]. The actual binding site for acetylcholine is thought to be in a region with β-pleated sheet characteristics [61] suggestive of a domain-like structure. Although the exact nature of this binding is unknown, it is possible that the binding site may be between two domains like the binding suggested above for T cells, and that this binding pulls together these domains resulting in a receptor conformational change, opening the channel. The conformational change would involve a shift in subunit alignment similar to that demonstrated for a Ca++ gap junction [62], rather than gross intramolecular conformational changes.
The structural building blocks for many of the proteins discussed in this paper may be related to the Ig domain. In the immune system, the Ig-supergene family provides structural mechanisms for creating fine specificity reactivity as well as allowing for the stable formation of large multidomain proteins. It is therefore likely that other molecules involved in cellular recognition, especially in the nervous system, will also have Ig domain-like characteristics. The apparent coincidental existence of the Thy-1 molecule on both lymphocytes and neurons may represent just the beginning of an extensive structural overlap possibly evolving from a primordial lymphocvtic type of interaction. With the advent of rapid molecular biological techniques for gene analysis and gene transfection, including the potential for generating large quantities of proteins for crystallographic analysis, together with biochemical and biophysical techniques for studying protein interactions, the above hypotheses for receptor structures and interactions should be testable and resolved in the near future.
Acknowledgments
I would like to thank Dr Minora Kanehisa for his collaborative efforts in protein modeling, and Drs Ronald Germain, Ronald Schwartz and Richard Weber for critical review of the manuscript and helpful comments.
- AChR
acetylcholine receptor
- APC
antigen-presenting cell
- CTL
cytolytic T lymphocyte
- FcR
Fc receptor
- IL-2
interleukin-2
- MHC
major histocompatibility complex
- N-CAM
neuronal-cell-adhesion molecule
- TcR
T-cell receptor
- T3m
T3 membrane
References
- 1.McClain LD, Tomana M, Acton RT. Purification and characterization of mouse brain Thy1.2 differentiation alloantigen. Brain Res. 1978;159:161–171. doi: 10.1016/0006-8993(78)90117-8. [DOI] [PubMed] [Google Scholar]
- 2.Norcross MA, Smith RT. Regulation of T-cell mitogen activity of anti-lymphocyte serum by a B helper cell. J Immunol. 1979;122:1620–1628. [PubMed] [Google Scholar]
- 3.Maino VC, Norcross MA, Perkins MS, Smith RT. Mechanisms of Thy-1 mediated T-cell activation: roles of Fc receptors, T200, and H-2 glycoproteins in accessory cell function. J Immunol. 1981;126:1829–1836. [PubMed] [Google Scholar]
- 4.Konaka Y, Norcross MA, Maino VC, Smith RT. Anti-Thy-1 mediated T-cell activation. Role of soluble factors and expression of growth factor receptors on T cells. Europ J Immunol. 1981;11:445–450. doi: 10.1002/eji.1830110602. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu S, Smith RT, Norcross MA, Maino VC. Mechanisms controlling TCGF production by cloned sublines of EL-4 azgr in response to stimulation by anti-Thy-1 antibody. J Immunol. 1982;128:296–301. [PubMed] [Google Scholar]
- 6.Norcross MA, Smith RT, Shimizu S. Regulation of TCGF production in T-cells. — II. Early membrane events after anti-Thy-1 binding by the TCGF producing T lymphoma EL-4 G-12. J Immunol. 1984;132:833–838. [PubMed] [Google Scholar]
- 7.Jones B, Janeway C. Functional activities of antibodies against brain associated T-cell antigens. — I. Induction of T-cell proliferation. Europ J Immunol. 1981;11:584–592. doi: 10.1002/eji.1830110712. [DOI] [PubMed] [Google Scholar]
- 8.Gunter KC, Malek TR, Shevach EM. T-cell activating properties of an anti-Thy-1 monoclonal antibody: possible analogy to OKT3/Leu 4. J exp Med. 1984;159:716–730. doi: 10.1084/jem.159.3.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meurer SC, Hodgdon JC, Hussey RE, Protentis JP, Schlossman SF, Reinherz EL. Antigen-like effects of monoclonal antibodies directed at receptors on human T-cell clones. J exp Med. 1983;158:988–993. doi: 10.1084/jem.158.3.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kappler J, Kubo R, Haskins K, White J, Marrack P. The mouse T-cell receptor: comparison of MHC-restricted receptors on two T-cell hybridomas. Cell. 1983;34:727–737. doi: 10.1016/0092-8674(83)90529-9. [DOI] [PubMed] [Google Scholar]
- 11.King AC, Cuatrecasas P. Peptide hormone-induced receptor mobility, aggregation, internalization. New Engl J Med. 1981;305:77–88. doi: 10.1056/NEJM198107093050206. [DOI] [PubMed] [Google Scholar]
- 12.Metzger H, Ishizaka T. Transmembrane signaling by receptor aggregation: the mast cell receptor for IgE as a case study. Symposium Fed Proc. 1982;41:7–34. [Google Scholar]
- 13.Larrson EL, Coutinho A. Mechanism of T-cell activation. — I. Screening of « step one » ligands. Europ J Immunol. 1980;10:93–99. doi: 10.1002/eji.1830100205. [DOI] [PubMed] [Google Scholar]
- 14.Meurer SC, Hussey BE, Cantrell DA, Hodgdon JC, Schlossman SF, Smith KA, Reinherz EL. Triggering of the T3-Ti antigen receptor complex results in clonal T-cell proliferation through an interleukin-2-dependent autocrine pathway. Proc nat Acad Sci (Wash) 1984;81:1509–1513. doi: 10.1073/pnas.81.5.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tax WJM, Willems HW, Reekers PPM, Capel PJA, Koene RAP. Polymorphism in mitogenic effort of IgG1 monoclonal antibodies against T3 antigen on human T cells. Nature (Lond) 1983;304:445–447. doi: 10.1038/304445a0. [DOI] [PubMed] [Google Scholar]
- 16.Kaneoka HP, Sasasuki T, Benike CJ, Engleman EG. Human T lymphocyte proliferation induced by a pan-T monoclonal antibody (anti-Leu-4): Heterogeneity of response is a function of monocytes. J Immunol. 1983;131:158–164. [PubMed] [Google Scholar]
- 17.Bell GI. Theoretical models for cell-cell interactions in immune responses. In: Delisi C, Blumenthal R, editors. « Physical chemical aspects of cell surface events in cellular recognition ». Elsevier North Holland Publ. Co; Amsterdam: 1979. pp. 371–386. [Google Scholar]
- 18.Keifer HA, Blune AJ, Kaback HR. Membrane potential changes during mitogenic stimulation of mouse spleen lymphocytes. Proc nat Acad Sci (Wash) 1980;77:2200–2204. doi: 10.1073/pnas.77.4.2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Decoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature (Lond) 1984;307:465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- 20.Williams AF, Gagnon J. Neuronal cell Thy-1 glycoprotein: homology with immunoglobulin. Science. 1981;216:696–703. doi: 10.1126/science.6177036. [DOI] [PubMed] [Google Scholar]
- 21.Moriuchi T, Chang J, Denome R, Silver J. Thy-1 cDNA sequence suggests a novel regulatory mechanism. Nature (London) 1983;301:80–82. doi: 10.1038/301080a0. [DOI] [PubMed] [Google Scholar]
- 22.Moriuchi T, Silver J. Thy-1 has a hydrophobic transmembrane segment at the carboxy terminus. in press. [PubMed] [Google Scholar]
- 23.Springer TA, Davignon D, Ho MK, Kurzinger K, Martz E, Sanchez-Madrid F. LFA-1 and Lyt2,3 molecules associated with T-lymphocyte-mediated killing; and Mac-1, an LFA-1 homologue associated with complement receptor functions. Immunol Rev. 1982;68:171–195. doi: 10.1111/j.1600-065x.1982.tb01064.x. [DOI] [PubMed] [Google Scholar]
- 24.Edelman GM. Cell adhesion molecules. Science. 1983;219:450–457. doi: 10.1126/science.6823544. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez-Madrid F, Krensky AM, Ware CF, Robbins E, Strominger JL, Burakoff SJ, Springer TA. Three distinct antigens associated with human T-lymphocyte-mediated cytolysis: LFA-1, LFA-2, and LFV-3. Proc nat Acad Sci (Wash) 1982;79:7489–7493. doi: 10.1073/pnas.79.23.7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krensky AM, Robbins E, Springer TA, Burakoff SJ. LFA-1, LFA-2, and LFA-3 antigens are involved in CTL-target conjugation. J Immunol. 1984;132:2180–2182. [PubMed] [Google Scholar]
- 27.Swain SL. Significance of Lyt phenolypes: Lyt2 antibodies block activities of T-cells that recognize class I major histocompatibility complex antigens regardless of their function. Proc nat Acad Sci (Wash) 1981;78:7101–7107. doi: 10.1073/pnas.78.11.7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ledbetter JA, Evans RL, Lipinbski M, Cunningham-Rundes C, Good RA, Herzenberg LA. Evolutionary conservation of surface molecules that distinguish T-lymphocyte helper/inducer and cytoxic/suppressor subpopulations in mouse and man. J exp Med. 1981;153:310–323. doi: 10.1084/jem.153.2.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dialynas DP, Wilde DB, Marrack P. Characterization of the murine antigenic determinant designated L3-T4a, recognized by monoclonal antibody GK1.5: expression of L3/T4a by functional T-cell clones appears to correlate primarily with class II MHC antigen reactivity. Immunol Rev. 1983;74:29–56. doi: 10.1111/j.1600-065x.1983.tb01083.x. [DOI] [PubMed] [Google Scholar]
- 30.Marrack P, Endres R, Shimonkevitz R, Zlotnik A, Dialynas D, Fitch F, Kappler J. The major histocompatibility complex-restricted antigen receptor on T-cells. II. — Role of the L3/T4 product. J exp Med. 1983;158:1077–1091. doi: 10.1084/jem.158.4.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macdonald JR, Glasebrook AL, Bron C. Clonal heterogeneity in the functional requirement for Lyt-2/3 molecules on cytolytic T lymphocytes (CTL): Possible implications for the affinity of CTL antigen receptors. Immunol Rev. 1982;68:89–115. doi: 10.1111/j.1600-065x.1982.tb01061.x. [DOI] [PubMed] [Google Scholar]
- 32.Norcross MA, Kanehisa M. The predicted structure of the Ia β1 domain: a hypothesis for the structural basis of MHC-restricted T-cell recognition of antigen. in press. [PubMed] [Google Scholar]
- 33.Samelson LE, Germain RN, Schwartz RH. Monoclonal antibodies against the antigen receptor on a cloned T-cell hybrid. Proc nat Acad Sci (Wash) 1983;80:6972–6976. doi: 10.1073/pnas.80.22.6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McIntyre BW, Allison JP. The mouse T-cell receptor: structural heterogeneity of molecules of normal T-cells defined by xenoantisera. Cell. 1983;34:739–746. doi: 10.1016/0092-8674(83)90530-5. [DOI] [PubMed] [Google Scholar]
- 35.Meurer SC, Acuto O, Hussey RE, Hodgdon JC, Fitzgerald KA, Schlossman SF, Reinherz EC. Evidence for the T3-associated 90k heterodimer as the T-cell antigen receptor. Nature (Lond) 1983;303:808–810. doi: 10.1038/303808a0. [DOI] [PubMed] [Google Scholar]
- 36.Acuto O, Meurer SC, Hodgdon JC, Schlosman SF, Reinherz EL. Peptide variability exists within a and b subunits of the T-cell receptor for antigen. J exp Med. 1983;158:1368–1373. doi: 10.1084/jem.158.4.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borst J, Alexander S, Elder J, Terhorst C. The T3 complex on human T lymphocytes involves four structurally distinct glyco-proteins. J biol Chem. 1983;258:5135–5141. [PubMed] [Google Scholar]
- 38.Borst J, Prendiville MA, Terhorst C. The T3 complex on human thymus-derived lymphocytes contains two different subunits of 20 kDa. Europ J Immunol. 1983;13:576–580. doi: 10.1002/eji.1830130712. [DOI] [PubMed] [Google Scholar]
- 39.Meuer SC, Fitzgerald KA, Hussey RE, Hodgdon JC, Schlossman SF, Reinherz EL. Clonotypic structures involved in antigen-specific human T-cell function: relationship to the T3 molecular complex. J exp Med. 1983;157:705–719. doi: 10.1084/jem.157.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hedrick SM, Nielson EA, Kavalier J, Cohen DI, Davis MM. Sequence relationships between putative T-cell receptor polypeptides and immunoglobulins. Nature (Lond) 1984;308:153–158. doi: 10.1038/308153a0. [DOI] [PubMed] [Google Scholar]
- 41.Yanagi Y, Yoshikai Y, Legett K, Clark SP, Aleksander I, Mak T. A human T-cell-specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains. Nature (Lond) 1984;308:145–149. doi: 10.1038/308145a0. [DOI] [PubMed] [Google Scholar]
- 42.Kaufman JF, Auffray C, Korman AJ, Schackelford DA, Strominger J. The class II murine major histocompatibility complex. Cell. 1984;36:1–13. doi: 10.1016/0092-8674(84)90068-0. [DOI] [PubMed] [Google Scholar]
- 43.Dintzis HM, Dintzis RZ, Vogelstein B. Molecular determinants of immunogenicity: the immunon model of immune response. Proc nat Acad Sci (Wash) 1976;73:3671–3675. doi: 10.1073/pnas.73.10.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weiss A, Imboden J, Shoback P, Stobo J. Role of T3 surface molecules in human T-cell activation: T3-dependent activation results in arise in cytoplasmic free calcium. Proc nat Acad Sci (Wash) doi: 10.1073/pnas.81.13.4169. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young JDE, Unkeless JC, Kaback HR, Cohn ZA. Macrophage membrane potential changes associated with gamma2b gammal Fc receptor ligand binding. Proc nat Acad Sci (Wash) 1983;80:1357–1361. doi: 10.1073/pnas.80.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Young JDE, Unkeless JC, Kaback HR, Cohn ZA. Mouse macrophage Fc receptor for IgG2b/1 in artificial and plasma membrane vesicles function as a ligand-dependent ionophore. Proc nat Acad Sci (Wash) 1983;80:1636–1640. doi: 10.1073/pnas.80.6.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gilman SC, Rosenberg JS, Feldman JD. Inhibition of interleukin synthesis and T-cell proliferation by a monoclonal anti-Ia antibody. J Immunol. 1983;130:1236–1240. [PubMed] [Google Scholar]
- 48.Palacios R, Martinez-Maza D, Guy K. Monoclonal antibodies against HLA-DR antigen replace T helper cells in activation of B-Iym-phocytes. Proc nat Acad Sci (Wash) 1983;80:3456–3460. doi: 10.1073/pnas.80.11.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ziegler K, Unanue ER. Identification of a macrophage antigen processing event required for I-region-restricted antigen presentation to T lymphocytes. J Immunol. 1981;127:1869–1875. [PubMed] [Google Scholar]
- 50.Pincus MR, Gerewitz F, Schwartz RH, Scheraga HA. Correlation between the conformation of cylochrome c peptides and their stimulatory activity in a T-lymphocyte proliferation assay. Proc nat Acad Sci (Wash) 1983;80:3297–3300. doi: 10.1073/pnas.80.11.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matis LA, Glimcher LH, Paul WE, Schwartz RH. Magnitude of response of histocompatibility-restricled T-cell clones is a function of the product of the concentrations of antigen and Ia molecules. Proc nat Acad Sci (Wash) 1983;80:6019–6023. doi: 10.1073/pnas.80.19.6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zagury D. Direct analysis of individual killer T-cells: Susceptibility of target cells to lysis and secretion of hydrolytic enzymes by CTL. In: Clark WR, Golf-stein P, editors. « Mechanisms in cell mediated cyloloxicity ». Plenum Press; New York: 1981. [DOI] [PubMed] [Google Scholar]
- 53.Thiernesse N, Bernard DA, Jeannesson P, Zangury D. Activité phosphatasique acide de la cellule T cytolytique au cours du processus de cytolyse. C R Acad Sci (Paris) (Sér: D) 1977;285:713. [PubMed] [Google Scholar]
- 54.Dennert G, Podack ER. Cytolysis by H-2-specific T killer cells assembly of tubular complexes on target membranes. J exp Med. 1983;157:1483–1495. doi: 10.1084/jem.157.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henkart P, Millard PJ, Reynolds CW, Henkart M. Cytolytic activity of purified cytoplasmic granules from cytotoxic rat large granular lymphocyte tumors. J exp Med. 1984 doi: 10.1084/jem.160.1.75. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Payan DG, Levine JD, Goetzl EL. Modulation of immunity and hypersensitivity by sensory neuropeptides. J Immunol. 1984;132:1601–1604. [PubMed] [Google Scholar]
- 57.Weber R, Pert C. Opiatergic modulation of the immune system. In: Muller EE, Genazzani AR, editors. Central and peripheral endorphin receptors: basic and clinical, aspects. Raven Press; New York: 1984. in Press. [Google Scholar]
- 58.Kaiser ET, Kezdy FJ. Amphiphilic secondary structure design of peptide hormones. Science. 1984;223:249–256. doi: 10.1126/science.6322295. [DOI] [PubMed] [Google Scholar]
- 59.Schneider C, Sutherland R, Newman R, Greaves M. Structural features of the cell surface receptor for transferrin that is recognized by the monoclonal antibody OKT9. J biol Chem. 1982;257:8516–8522. [PubMed] [Google Scholar]
- 60.Moore HP, Hartig PR, Rafiery MA. Correlation of polypeptide composition with functional events in acetylcholine receptor-enriched membranes from Torpedo californica. Proc nat Acad Sci (Wash) 1979;76:6265–6269. doi: 10.1073/pnas.76.12.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fimer-Moore J, Stroud RM. Amphipathic analysis and possible formation of the ion channel in an acetylcholine receptor. Proc nal Acad Sci (Wash) 1984;81:155–159. doi: 10.1073/pnas.81.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Unwin PNT, Ennis PD. Two configurations of a channel forming membrane protein. Nature (Lond) 1984;307:609–613. doi: 10.1038/307609a0. [DOI] [PubMed] [Google Scholar]