

Abstract
The lipopolysaccharide (LPS) core domain of Gram-negative bacteria plays an important role in outer membrane stability and host interactions. Little is known about the biochemical properties of the glycosyltransferases that assemble the LPS core. We now report the purification and characterization of the Rhizobium leguminosarum mannosyl transferase LpcC, which adds a mannose unit to the inner 3-deoxy-D-manno-octulosonic acid (Kdo) moiety of the LPS precursor, Kdo2-lipid IVA. LpcC containing an N-terminal His6 tag was assayed using GDP-mannose as the donor and Kdo2-[4′-32P]lipid IVA as the acceptor and was purified to near homogeneity. Sequencing of the N terminus confirmed that the purified enzyme is the lpcC gene product. Mild acid hydrolysis of the glycolipid generated in vitro by pure LpcC showed that the mannosylation occurs on the inner Kdo residue of Kdo2-[4′-32P]lipid IVA. A lipid acceptor substrate containing two Kdo moieties is required by LpcC, since no activity is seen with lipid IVA or Kdo-lipid IVA. The purified enzyme can use GDP-mannose or, to a lesser extent, ADP-mannose (both of which have the α-anomeric configuration) for the glycosylation of Kdo2-[4′-32P]lipid IVA. Little or no activity is seen with ADP-glucose, UDP-glucose, UDP-GlcNAc, or UDP-galactose. A Salmonella typhimurium waaC mutant, which lacks the enzyme for incorporating the inner L-glycero-D-manno-heptose moiety of LPS, regains LPS with O-antigen when complemented with lpcC. An Escherichia coli heptose-less waaC-waaF deletion mutant expressing the R. leguminosarum lpcC gene likewise generates a hybrid LPS species consisting of Kdo2-lipid A plus a single mannose residue. Our results demonstrate that heterologous lpcC expression can be used to modify the structure of the Salmonella and E. coli LPS cores in living cells.
Lipopolysaccharide (LPS)1 is a prominent structural component of the outer membranes of virtually all Gram-negative bacteria (1, 2). LPS may be viewed as a tripartite macromolecule. Its principal components include 1) the lipid A moiety (2–5), which is the hydrophobic outer membrane anchor; 2) the nonrepeating core oligosaccharide (1, 2); and 3) the distal repeating O-antigen polysaccharide (1, 2, 6). The lipid A moiety and its attached 3-deoxy-D-manno-octulosonic acid (Kdo) units represent the minimal LPS substructure required for bacterial viability (1, 2). All three domains of LPS are essential for full virulence during infection of animals and plants (1).
There are striking differences in the structures of diverse LPS molecules, such as those of the plant endosymbionts Rhizobium leguminosarum and Rhizobium etli compared with the enteric bacterium Escherichia coli (2, 7–9). R. leguminosarum and R. etli lipid A molecules lack the phosphate groups found in E. coli lipid A, are modified with a galacturonic acid moiety at position 4′, and contain an unusual 28-carbon secondary acyl chain at position 2′ (Fig. 1) (7–10). The inner core domains of R. leguminosarum and R. etli lack the L-glycero-D-manno-heptose residues found in E. coli, Salmonella, and most other Gram-negative bacteria and instead contain a mannose residue directly linked to position 5 of the inner Kdo sugar (Fig. 1) (11–14).
Fig. 1. Structures of R. leguminosarum and Escherichia coli lipid A and of the inner core oligosaccharides.
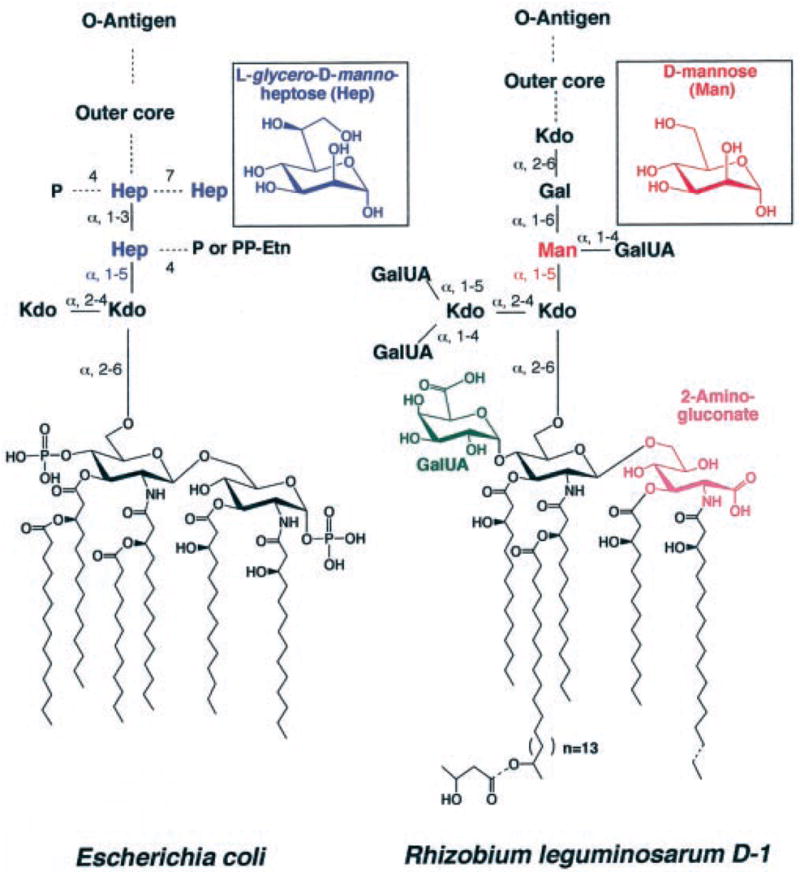
The closely related structures of L-glycero-D-manno-heptose and D-mannose are highlighted for comparison. There is, however, no sequence similarity between the enzyme that attaches the innermost heptose in E. coli (WaaC) and the enzyme that attaches the mannose residue in R. leguminosarium (LpcC) (14, 19). Only the major molecular species are indicated, which, in the case of R. leguminosarum, is designated component D-1 (2, 8, 9, 12).
Despite these and other differences, the first seven enzymes that catalyze the formation of the key intermediate Kdo2-lipid IVA (Fig. 2) are the same in both organisms (15). These bacteria diverge in their subsequent processing of Kdo2-lipid IVA. For instance, R. leguminosarum membranes contain phosphatases that remove the 1- and 4′-phosphate moieties late in the pathway (16, 17). The mannose residue of the inner core of R. leguminosarum is incorporated by a GDP-mannose-dependent enzyme, which is encoded by the lpcC gene (13, 14). In E. coli and Salmonella, WaaC(RfaC) adds L-glycero-D-manno-heptose to the inner Kdo unit at the same place where LpcC adds the mannose moiety in R. leguminosarum or R. etli (Fig. 1) (18, 19). There is, however, no sequence homology between WaaC and LpcC (14, 18, 19), despite the formal structural similarity of heptose and mannose (Fig. 1). WaaC is thought to utilize ADP-L-glycero-D-manno-heptose as its physiological sugar nucleotide donor (2, 18, 20), whereas LpcC employs GDP-mannose (14, 19). Both LpcC and WaaC can employ the analogue ADP-mannose (a natural product from corn) (21, 22) as an alternative substrate in vitro (13, 14, 19). However, bacteria do not normally synthesize ADP-mannose.
Fig. 2. Proposed reaction catalyzed by the R. leguminosarum mannosyl transferase LpcC.

The actual linkage generated in vitro by the cloned mannosyl transferase has not yet been confirmed but is presumed to be the same as that seen in the core domain of the LPS isolated from cells (11, 12).
Significant orthologs of R. leguminosarum lpcC are present in the genomes of several other important proteobacteria, including S. meliloti (see accompanying manuscript (58)) (23, 24), Agrobacterium tumefaciens (25, 26), Brucella melitenesis (27) and Francisella tularensis,2 all of which lack WaaC. The presence of a mannose residue in α-1,5 linkage to the inner Kdo moiety of F. tularensis LPS was recently established by chemical analysis (28). A. tumefaciens and B. melitenesis also contain the 28-carbon secondary acyl chain seen in R. leguminosarum (29, 30), whereas F. tularensis lipid A resembles that of R. leguminosarum in lacking both the 1- and 4′-phosphate residues (28). The significance of these correlations remains to be determined, but all three organisms can live inside of eucaryotic cells. R. leguminosarum lpcC mutants, which lack most core and all O-antigen sugars, are able to infect their hosts and form small nodules, but the nodules do not fix nitrogen (14).
Given the importance of LpcC in the biology of R. leguminosarum (14), its presence in certain human pathogens, and the general lack of biochemical information concerning glycosyl transferases involved in LPS core assembly (2, 31), we have now purified R. leguminosarum LpcC to homogeneity and characterized some of its properties. Pure LpcC is highly selective for GDP-mannose, consistent with the reported structure of the R. leguminosarum LPS core (11, 32). Interestingly, the lpcC gene can complement waaC mutants of Salmonella (18), as judged by restoration of O-antigen attachment. GDP-mannose is available to LpcC in Salmonella (2, 31, 33), and mannose is presumably similar enough to L-glycero-D-manno-heptose (Fig. 1) to permit the distal enzymes of Salmonella core assembly to function normally. When lpcC is expressed at 22 °C in a deletion mutant of E. coli lacking both waaC and waaF (34), a single hexose moiety (presumably mannose) is added to a portion of the Kdo2-lipid A made by this strain, further validating the utility of lpcC as a tool for LPS structure modification in living cells of diverse bacteria.
EXPERIMENTAL PROCEDURES
Chemicals and Materials
The [γ-32P]ATP was purchased from PerkinElmer Life Sciences. ADP-mannose, GDP-mannose, Kdo, and HEPES were obtained from Sigma. Bicinchoninic assay reagents and Triton X-100 were purchased from Pierce. Yeast extract and peptone-tryptone were from Difco. Silica Gel 60 thin layer chromatography plates were obtained from Merck. DNA primers and T4 DNA ligase were from Invitrogen, and PCR reagents were from Stratagene. Reagent grade pyridine, chloroform, and methanol were purchased from Mallinckrodt.
Bacterial Strains and Growth Conditions
The E. coli deep rough mutant WBB06 (ΔwaaC-waaF) was kindly provided by Werner Brabetz (34). Wild-type strain W3110 was obtained from the E. coli Genetic Stock Center at Yale University. Salmonella typhimurium strains SL1377 (waaC630) (18) and SL3770 (wild-type) were obtained from the Salmonella Genetic Stock Center (University of Calgary, Calgary, Canada). E. coli strain BLR(DE3)/pLysS was purchased from Novagen. All bacteria were grown in LB broth (10 g of NaCl, 10 g of peptone-tryptone, and 5 g of yeast extract per liter) (35). When necessary, the cultures were supplemented with tetracycline (12 μg/ml) or kanamycin (30 μg/ml).
Molecular Biology Techniques
Plasmids were prepared using the Qiagen Mini Prep Kit (Qiagen). Restriction endonucleases (New England Biolabs), shrimp alkaline phosphatase, and T4 ligase were used according to the manufacturer’s instructions (Invitrogen). Competent cells were prepared for transformation using the calcium chloride method (36, 37).
Construction of Plasmid pMKHN Expressing LpcC with a His6 Tag at the N terminus
The plasmid pJK6A (13) was used to retrieve the lpcC gene by digesting it with BamHI and NdeI. The desired fragment was ligated into the pET28b + vector (Novagen) behind the T7lac promoter that had been digested with the same enzymes, placing a His6 tag at the N terminus. This construct (pMKHN) contains a thrombin cleavage site immediately downstream of the His6 tag.
Construction of the High Copy Plasmid pMKCA Expressing lpcC
The plasmid pMKCA was used for complementation of the waaC-deficient S. typhimurium mutant SL1377 (18) or of the E. coli waaC-waaF deletion mutant WBB06 (34). pMKHN was digested with XhoI and BamHI to retrieve the lpcC gene, and the desired fragment was then cloned behind the lac promoter into the vector pBluescript KS+ (Stratagene), digested with the same enzymes, to generate pMKCA.
Mannosyl Transferase Assay
The mannosyl transferase LpcC was assayed using the LPS precursor Kdo2-[4′-32P]lipid IVA, which was isolated and stored as a frozen aqueous dispersion as described previously (38). Prior to each use, the radiolabeled substrate was subjected to ultrasonic irradiation in a water bath for 1 min. The mannosyl transferase was assayed as follows. The reaction mixture (10-μl final volume) contained 50 mM HEPES, pH 7.5, 0.1% Triton X-100, 1 mM GDP mannose, and 10 μM Kdo2-[4′-32P]lipid IVA (3000 – 6000 cpm/nmol). The reaction was started by the addition of an appropriate amount of enzyme (2.5–5 μg/ml of pure enzyme or about 50 μg/ml of membranes of cells overexpressing lpcC behind the T7lac promoter) and incubated for the indicated times at 30 °C. The reactions were stopped by spotting 4-μl samples directly onto a silica gel thin layer plate. After drying the spots at room temperature, the plate was developed in the solvent chloroform, pyridine, 88% formic acid, water (30:70:16:10, v/v/v/v). Following removal of the solvent with a hot air stream, the plate was analyzed using an Amersham Biosciences PhosphorImager (STORM 840), equipped with ImageQuant software.
Preparation of Membranes for Assay and Purification of LpcC
BLR(DE3)/pLysS/pMKHN was grown from a single colony in 1 liter of LB medium containing 100 μg/ml ampicillin and 30 μg/ml kanamycin at 37 °C until the optical density of 600 reached ~0.5. The culture was induced with 1 mM isopropyl-1-thio-β-D-galactopyranoside and incubated with shaking for an additional 3 h at 30 °C. Cells were harvested at 4 °C by centrifugation at 4,000 × g for 10 min, were resuspended in 30 ml of 50 mM HEPES, pH 7.5, and were broken by one passage through a French pressure cell at 18,000 p.s.i. Debris and inclusion bodies were removed by centrifugation at 4,000 × g for 15 min. Membranes were prepared by ultracentrifugation at 100,000 × g for 60 min at 4 °C. The membranes were resuspended in 20 ml of the same buffer, and the centrifugation was repeated a second time at 100,000 × g to remove any remaining cytosol. Protein concentration was determined by the bicinchoninic method (Pierce) using bovine serum albumin as the standard. The membrane pellet was resuspended at 10 –15 mg/ml in 50 mM HEPES, pH 7.5, and stored at −80 °C until further use.
Purification of His-tagged LpcC over a Nickel Column
The washed membranes were solubilized by adding an appropriate volume of a 10% Triton X-100 stock solution to give a final concentration of 1% Triton X-100 and incubated at 4 °C for 1 h with intermittent inversion on a rotating apparatus (19). The solubilization mixture was then centrifuged at 100,000 × g for 60 min at 4 °C to remove any remaining particulate proteins and inclusion bodies.
One ml of His-Bind resin (Novagen) was prepared for nickel affinity chromatography in a small disposable column by first washing the resin with 3 column volumes of water, 5 column volumes of 50 mM NiSO4, and 3 column volumes of the binding buffer (Novagen), which contains 5 mM imidazole, 0.5 M NaCl, and 20 mM Tris chloride, pH 7.9, with the further addition of 0.1% Triton X-100. A sample of the solubilized membrane proteins (11.2 mg), prepared as described above, was diluted 10-fold in binding buffer and was applied to the column at 4 °C at its natural flow rate. One-ml fractions were collected throughout. The column was washed and then eluted sequentially with 10 ml (5 mM), 6 ml (20 mM), and 6 ml (60 mM) of imidazole in 20 mM Tris-chloride buffer, pH 7.9, containing 0.5 M NaCl and 0.1% Triton X-100. The protein content of each fraction was determined. The peak of enzyme activity, which eluted in the 60 mM imidazole wash, was determined by assaying each fraction in the linear range under standard conditions. The proteins were visualized by polyacrylamide gel electrophoresis in the presence of 12% SDS using the Bio-Rad Mini Protean II electrophoresis system.
N-terminal Sequencing of the Purified Protein
The thrombin cleavage capture kit (Novagen) was used to remove the N-terminal His6 tag and the adjacent thrombin cleavage site from the purified LpcC preparation. Approximately 5 μg of the cleaved purified protein was loaded onto a 12% polyacrylamide SDS gel along with two control lanes containing prestained standards to facilitate transfer to the blot and purified LpcC that had not been cleaved with thrombin. Electrophoresis was carried out at 200 V for 50 min using the Laemmli buffer system. The gel was then soaked in 10 mM CAPS buffer, pH 11, for about 5 min. A polyvinylidene difluoride membrane was prepared while the electrophoresis was in progress by brief soaking in methanol, rinsing with water, and then soaking in 10 mM CAPS, pH 11. Protein bands were transferred to the membrane with a Bio-Rad SD electroblotter, according to the manufacturer’s directions, at 4 °C. The transferred protein was visualized by Ponceau S staining, and the band of interest was excised. N-terminal sequencing was carried out by Dr. John Leszyk of the Worcester Foundation for Experimental Biology (Shrewsbury, MA). The sequence was determined as GSHMPDIRDV. The first two amino acid residues represent the residual thrombin cleavage site, and the single His residue is derived from the construction of the vector. The remaining sequence corresponds to the predicted N terminus of LpcC.
Mild Acid Hydrolysis of the Mannosyl Transferase Reaction Product
Two 10-μl reaction mixtures in plastic microcentrifuge tubes were prepared containing 50 mM Hepes, pH 7.5, 0.1% Triton X-100, and either Kdo2-[4′-32P]lipid IVA (8,000 cpm/reaction) or mannosyl-Kdo2-[4′-32P] lipid IVA (8,000 cpm/reaction). The mannosyl-Kdo2-[4′-32P] lipid IVA was first isolated from a larger scale mannosyl transferase assay system that had been allowed to go to completion. The purification of the mannosyl-Kdo2-[4′-32P] lipid IVA was achieved by thin layer chromatography, as for the preparation of Kdo2-[4′-32P]lipid IVA (38). Next, 4 μl of 10% SDS and 26 μl of 50 mM sodium acetate, pH 4.5, were added to each microcentrifuge tube, and the tubes were incubated in a boiling water bath. At various times, 4-μl samples were withdrawn and spotted onto a silica TLC plate. The plate was developed in the solvent chloroform, pyridine, 88% formic acid, water (30:70:16:10, v/v/v/v) and analyzed as described previously to determine the site of mannose attachment (19).
Purification of LPS from E. coli WBB06 Expressing lpcC
To isolate LPS, 100-ml cultures of the E. coli heptose deficient mutant WBB06 containing either pBluescript KS+ or pMKCA (expressing lpcC in pBluescript KS+) were grown to an optical density of 600 = 1.7 at 22 °C. A relatively low temperature was used for this work in order to induce the biosynthesis of colanic acid (39), and therefore presumably also GDP-mannose, in E. coli. Cells were harvested by centrifugation at 4,000 × g for 10 min and washed once with 62.5 ml of phosphate-buffered saline (40). The cell pellets were resuspended in 40 ml of phosphate-buffered saline. Glycerophospholipids and LPS (i.e. Kdo2-lipid A in the case of WBB06 or the putative mannosyl-Kdo2-lipid A in WBB06/pMKCA) were extracted for 1 h at room temperature using a single phase Bligh-Dyer mixture, consisting of chloroform/methanol/phosphate-buffered saline (1:2:0.8, v/v/v). The precipitated protein and nucleic acids were removed by centrifugation at 4,000 × g for 15 min. The supernatant was converted to a two-phase Bligh-Dyer mixture, consisting of chloroform/methanol/water (2:2:1.8, v/v/v) by the addition of appropriate amounts of chloroform and water. After thorough mixing, the phases were separated by centrifugation at 4,000 × g for 15 min. The lower phase was dried by rotary evaporation. The residue was redissolved in 5 ml of chloroform/methanol/water (2:3:1, v/v/v) and applied to a 1-ml DEAE-cellulose column (acetate form), equilibrated with chloroform/methanol/water (2:3:1, v/v/v). The column was washed with the same solvent mixture and then eluted with 5-ml portions of chloroform/methanol/aqueous ammonium acetate (2:3:1, v/v/v), in which the ammonium acetate concentration in the aqueous component was increased stepwise from 60 to 120 to 240 and finally to 480 mM. During the elution, 1-ml fractions were collected. The appearance of the Kdo2-lipid A and mannosyl-Kdo2-lipid A was determined by spotting 10-μl portions of each fraction onto the origin of a silica TLC plate, which was then developed in the solvent chloroform, pyridine, 88% formic acid, water (50:50:16:10, v/v/v/v). After drying the plate, the lipids were visualized by spraying with 10% sulfuric acid in ethanol followed by charring on a hot plate. Kdo2-lipid A or mannosyl-Kdo2-lipid A eluted with the 240 and 480 mM ammonium acetate components. To recover the intact lipids, the relevant fractions were pooled and converted to two-phase Bligh-Dyer mixtures. The lower phases were dried under a stream of N2 and stored at −20 °C.
Mass Spectrometry
Spectra were acquired in the negative mode using a matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) Kompact 4 mass spectrometer (Kratos Analytical, Manchester, UK), equipped with a 337-nm nitrogen laser and set at a 20-kV extraction voltage. Each spectrum was the average of 50 shots. The matrix used for this study was of a mixture of saturated 6-aza-2-thiothymine in 50% acetonitrile and 10% tribasic ammonium citrate (9:1, v/v). LPS samples were dissolved in a mixture of chloroform/methanol (4:1, v/v) before mixing with the matrix (1:1, v/v) on a slide. The sample was allowed to dry at room temperature prior to mass analysis.
NMR Spectroscopy
The substrates GDP-mannose and ADP-mannose (Sigma) were dissolved in 0.6 ml of 99% D2O in 5-mm NMR tubes. 1H and 13C NMR chemical shifts were referenced relative to 2,2-di-methylsilapentane-5-sulfonic acid at 0.00 ppm. 31P NMR chemical shifts were referenced to neat phosphoric acid. Spectra were recorded on Varian Inova 600 and 500 spectrometers equipped with Sun Ultra 5 computers and 5-mm Varian probes. NMR studies (COSY, NOESY, HMQC, and selective 31P-decouple/1H-observe experiments) were performed as described previously (9, 41).
Antibiotic Sensitivity Analysis and LPS Gels
Antibiotic sensitivity of selected strains was determined by a disc diffusion procedure, described previously (42). LPS samples were analyzed by 12% SDS gel electrophoresis and were prepared from membranes that were first subjected to complete digestion with proteinase K. Gels were stained with silver nitrate reagent (18, 43).
RESULTS
Purification of His6-tagged LpcC
As shown in Table I, a derivative of LpcC bearing a His6 tag followed by a thrombin cleavage site at its N terminus was expressed behind the T7lac promoter on pMKHN in E. coli BLR(DE3)/pLysS, resulting in overproduction of a protein of the expected molecular weight (Fig. 3, lane 1) and robust expression of membrane-associated (GDP-mannose-dependent) mannosyl transferase activity (Table I). LpcC activity is not detectable in wild type membranes or vector controls. These findings are consistent with previous studies of native lpcC expressed in E. coli (14).
Table I.
Purification of LpcC from E. coli BLR(DE3)/pLysS/pMKHN
| Step | Total activity | Total protein | Specific activity | Yield | Purification |
|---|---|---|---|---|---|
| units | mg | nmol/min/mg | % | -fold | |
| Membranes | 9017 | 39.9 | 226 | 100 | 1.0 |
| Solubilized membranes | 4716 | 11.2 | 421 | 52 | 1.9 |
| Nickel column | 540 | 0.15 | 3600 | 6 | 16a |
The purification relative to membranes of wild-type R. leguminosarum is estimated to be about 3600-fold, given a typical specific activity of about 1 nmol/min/mg in such preparations (13). One unit is defined as 1 nmol/min at 30 °C.
Fig. 3. SDS Gel electrophoresis of fractions generated during the purification of LpcC.
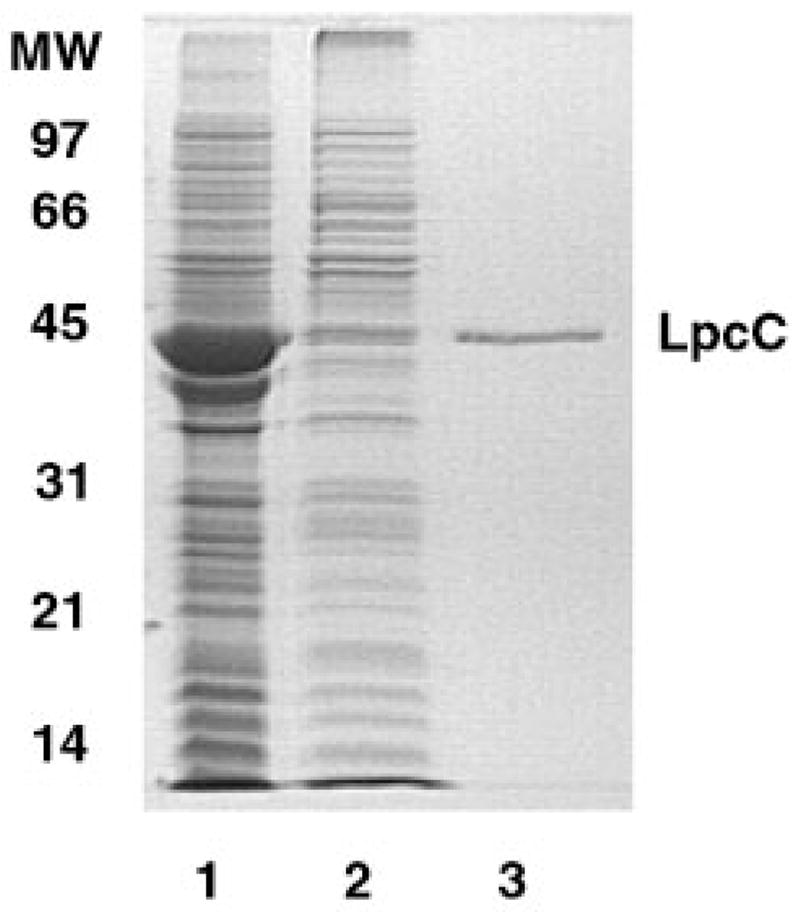
Lanes 1 and 2 contain 10 μg of protein from membranes and solubilized membranes, respectively, of BLR(DE3)/pLysS/pMKHN. Lane 3 contains 1.0 μg of LpcC after the nickel affinity column. Much of the overexpressed LpcC protein is not solubilized by Triton X-100, most likely because it is trapped in small inclusion bodies. However, about half of the mannosyl transferase activity present in whole membranes is recovered in the Triton X-100-solubilized fraction (see Table I).
Triton X-100 was used to solubilize the mannosyl transferase from the particulate fraction with a 52% recovery of activity (Table I). Relatively little of the overexpressed protein was actually solubilized (Fig. 3, lane 2), indicating that much of it is present in the form of small inclusion bodies. The active, solubilized material was applied to a nickel column prepared in 0.1% Triton X-100. Most of the activity was retained and was eluted by 60 mM imidazole together with a protein of the molecular weight expected for LpcC (Fig. 3, lane 3). The specific activity of the purified material was increased about 15-fold relative to the crude membrane fraction (Table I).
A portion of the purified preparation was cleaved with thrombin, displayed on a 12% SDS gel, and then transferred to a polyvinylidene difluoride membrane. N-terminal sequencing yielded the peptide GSHMPDIRDV …, in which the first two residues arise from the residual thrombin cleavage site and the third is a single His residue derived from the construction of the vector. The sequence MPDIRDV represents the true N terminus of LpcC (14).
Sugar Nucleotide Specificity of LpcC
As shown in Fig. 4 (lanes 2 and 3), 5 μg/ml purified LpcC catalyzes the quantitative conversion of Kdo2-[4′-32P]lipid IVA to mannosyl-Kdo2-[4′-32P]-lipid IVA in the presence of 1 mM GDP-mannose or 1 mM ADP-mannose in 30 min at 30 °C. In contrast, UDP-GlcNAc, UDP-galacturonic acid, or ADP-glucose (Fig. 4, lanes 4, 6, and 7, respectively) support little or no glycosylation of Kdo2-[4′-32P]lipid IVA under these conditions. A modest amount of glycosylated product is seen with UDP-glucose or UDP-galactose (Fig. 4, lanes 5 and 8). However, when these assays are conducted in the linear range with respect to the rate of product formation (data not shown), only GDP-mannose and to a lesser extent ADP-mannose function as substrates (also see accompanying manuscript (58)). We estimate that R. leguminosarum LpcC selectivity is at least 2 orders of magnitude greater for GDP-mannose than for UDP-galactose, UDP-glucose, UDP-Gl-cNAc, UDP-galacturonic acid, or ADP-glucose under our assay conditions. These findings are consistent with the reported structures of the R. leguminosarum and R. etli LPS core domains (11, 12).
Fig. 4. Sugar nucleotide specificity of purified LpcC.

Reactions were carried out under standard conditions at pH 7.5 using a 1 mM concentration of each of the sugar nucleotides, as indicated. Purified enzyme was used at 5 μg/ml. The incubation was carried out at 30 °C for 30 min. Under conditions where product evolution is linear with time (data not shown), the very slow reactions with UDP-glucose and UDP-galactose are not detectable.
Lipid Acceptor Specificity of LpcC
As shown in Fig. 5, only Kdo2-[4′-32P]lipid IVA functions as an acceptor substrate for the mannose residue. Kdo-[4′-32P]lipid IVA (prepared with KdtA from Hemophilus influenzae) (44) and [4′-32P]lipid IVA were completely inactive (Fig. 5). The failure of Kdo-[4′-32P]lipid IVA to function as an acceptor is somewhat surprising, given that LpcC incorporates the mannose unit on the inner Kdo moiety of Kdo2-[4′-32P]-lipid IVA (see below and Fig. 2). This unusual substrate specificity could be explained by the direct participation of the carboxylate moiety of the outer Kdo residue of Kdo2-[4′-32P]lipid IVA in LpcC catalysis.
Fig. 5. Lipid acceptor specificity of purified LpcC.

Reactions were carried out under standard conditions at pH 7.5 using either 10 μM Kdo2-[4′-32P]lipid IVA, Kdo-[4′-32P]lipid IVA, or [4′-32P]lipid IVA in the presence of 2.5 μg/ml protein.
Characterization of the Mannosyl Transferase Reaction Product
To determine the site of mannose attachment catalyzed by purified LpcC, the mannosyl-Kdo2-[4′-32P]lipid IVA product was subjected to hydrolysis at 100 °C in acetate buffer at pH 4.5 in the presence of SDS (19, 45). This procedure randomly cleaves glycosidic linkages of Kdo while leaving all other glycosidic linkages intact. As shown in Fig. 6, the products of mannosyl-Kdo2-[4′-32P]lipid IVA hydrolysis were subjected to thin layer chromatography and PhosphorImager analysis at various times of hydrolysis. No Kdo-[4′-32P]lipid IVA was detected as an intermediate at any time point. Instead, a compound migrating as would be expected for mannosyl-Kdo-[4′-32P]lipid IVA was observed (Fig. 6). These findings demonstrate that little or no mannose is added to the outer Kdo residue of Kdo2-[4′-32P]lipid IVA by LpcC, consistent with previous structural studies of mannosyl-Kdo containing core oligosaccharide fragments (11, 12) isolated from cells of R. etli.
Fig. 6. Time course of hydrolysis at 100 °C of Kdo2-[4′-32P]lipid IVA versus mannosyl-Kdo2-[4′-32P]lipid IVA.

A, the Kdo2-[4′-32P]lipid IVA control. B, mannosyl-Kdo2-[4′-32P]lipid IVA. The hydrolysis is carried out in sodium acetate buffer at pH 4.5 in the presence of SDS (19). The two Kdo glycosidic linkages are about equally susceptible to cleavage under these conditions, allowing discrimination between mannose addition to the outer versus the inner Kdo (19). LpcC modifies the inner Kdo as shown by the absence of unmodified Kdo-[4′-32P]lipid IVA during the time course of the hydrolysis of the mannosyl-Kdo2- [4′-32P]lipid IVA.
Partial Restoration of O-Antigen Synthesis and Antibiotic Resistance in a S. typhimurium waaC Mutant Expressing lpcC
Cells of S. typhimurium LT-2 synthesize O-antigen units containing mannose residues, and therefore possess the enzymatic machinery to make GDP-mannose when grown on nutrient broth (2, 31). As shown in Fig. 7 (lane 2), a mutant of S. typhimurium defective in waaC(rfaC) (18) (which encodes the heptosyl transferase that adds the inner heptose unit of the core illustrated in Fig. 1) generates a truncated LPS substructure lacking O-antigen. Expression of lpcC on a multicopy plasmid restores O-antigen production in a waaC mutant (Fig. 7, lane 4), although somewhat less efficiently than in wild type cells, as judged by the higher intensity of the most rapidly migrating bands (Fig. 7, lane 1 versus lane 4). These findings suggest that mannose can be incorporated into the core of S. typhimurium LPS in place of the inner heptose unit when R. leguminosarum lpcC is expressed in mutants lacking waaC.
Fig. 7. SDS gel electrophoresis of LPS from Salmonella waaC(rfaC) mutants complemented with R. leguminosarum lpcC.
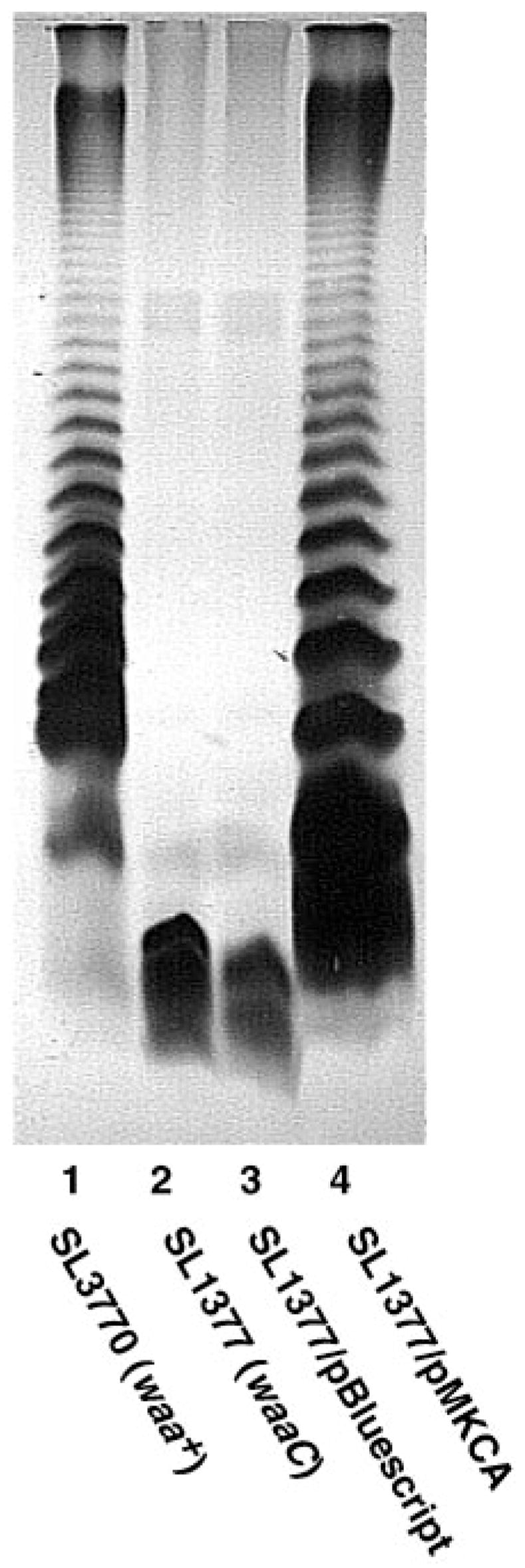
LPS preparations were separated on a 12% SDS gel and silver-stained by the method of Hitchcock and Brown (43). Lane 1, SL3770 (waa+); lane 2, SL1377 (waaC630); lane 3, SL1377 with pBluescript KS+; lane 4, SL1377 with pMKCA.
The antibiotic sensitivity data shown in Table II likewise support the idea that lpcC can partially rescue O-antigen production in an S. typhimurium mutant defective in waaC. Resistance to rifamycin and bacitracin is largely restored, but there is no effect on novobiocin hypersensitivity (Table II). The incomplete rescue of the antibiotic hypersensitivity of the waaC mutant by lpcC is consistent with the formation of an altered core domain containing mannose in place of the inner heptose residue and possibly also due to the reduced amount of O-antigen (Fig. 7).
Table II. Partial complementation of the antibiotic hypersensitivity of a S. typhimurium waaC mutant by lpcC.
A zone of inhibition of 6 mm corresponds to complete resistance and is equal to the diameter of the disc. All zones are in num
| Strain | Novobiocin | Rifamycin | Bacitracin |
|---|---|---|---|
| SL3770 (waa+) | <6 | 7 | <6 |
| SL1377 (waaC630) | 12 | 21 | 13 |
| SL1377/pBluescript KS+ | 14 | 21 | 13 |
| SL1377/pMKCA (lpcC*) | 12 | 12 | <6 |
Expression of lpcC in an E. coli Mutant Harboring a Deletion Spanning waaC and waaF
In the absence of analytical data demonstrating the incorporation of mannose into the inner core of waaC mutants expressing lpcC, the results shown in Fig. 7 and Table II could be interpreted to mean that LpcC is not incorporating mannose but might actually be utilizing endogenous ADP-L-glycero-D-manno-heptose at a slow rate (Fig. 1). The ability of recombinant LpcC to utilize ADP-L-glycero-D-manno-heptose has not been tested directly, since this nucleotide is not readily available (20). However, the ability of ADP-mannose (an analogue of ADP-L-glycero-D-manno-heptose) to substitute for GDP-mannose in vitro (Fig. 3) during LpcC catalysis glycosylation of Kdo2-[4′-32P]lipid IVA is well established (13). These considerations make it necessary to address the issue of whether LpcC is using GDP-mannose or ADP-L-glycero-D-manno-heptose when partially complementing the waaC mutation (Fig. 7).
To evaluate the LpcC donor substrate selectivity in living cells, the lpcC gene was expressed in an E. coli mutant (WBB06) (34) containing a deletion spanning both waaC and waaF. The latter gene encodes the second heptosyl transferase (Fig. 1) (2, 31). The LPS of WBB06 can be isolated directly by Bligh-Dyer extraction without prior mild acid hydrolysis, and it can be analyzed without further modification by MALDI-TOF mass spectrometry (Fig. 8A). It consists largely of Kdo2-lipid A, as judged by the signal in the negative mode spectrum attributed to [M − H]− at m/z 2237.8, consistent with hexa-acylated lipid A of E. coli K-12 (Mr 2238.7). When lpcC is expressed in living cells of WBB06, grown at 25 °C to stimulate GDP-mannose production, about one-third of the Kdo2-lipid A is derivatized with an additional hexose moiety (presumably a mannose, as indicated in Fig. 8C), as judged by the signal at m/z 2400.1. If LpcC had utilized endogenous ADP-L-glycero-D-manno-heptose (which is also present in WBB06) (20), the mass of the resulting LPS species would have been another 30 atomic mass units larger (equivalent to the extra CHOH unit in heptose versus mannose) than what was actually observed (Fig. 8B).
Fig. 8. MALDI-TOF mass spectra of LPS from E. coli WBB06 (ΔwaaC-waaF) containing pBluescript KS+ or pMKCA.

Spectra were acquired in the negative mode. A, LPS isolated from WBB06 cells containing the vector pBluescript+. B, LPS isolated from WBB06 cells containing pMKCA (lpcC+). C, the proposed structure of the mannosyl-Kdo2-lipid A.
Anomeric Carbon Configuration in GDP-mannose and ADP-mannose
Most common nucleotide diphosphate derivatives of D-sugars have the α-anomeric configuration. This point is well documented for GDP-mannose (33) but has not been evaluated for ADP-mannose, a natural product synthesized in corn (21, 22).
The 1H, 13C, and 31P NMR assignments for ADP-mannose in D2O are summarized in Table III. Fig. 9A shows the two-dimensional 1H-1H COSY analysis with the mannose through-bond cross-peaks and assignments indicated. As shown in Fig. 9B, a 1H-1H NOESY analysis of ADP-mannose provides the first indication of the expected α-anomeric stereochemistry in its mannose unit, as evidenced by the absence of significant cross-peaks between H-1″ and H-3″ or H-1″ and H-5″ of the mannose unit (Fig. 9B, arrows). The observed NOE between H-1″ and H-2″ (Fig. 9B) is identical to that seen in GDP-mannose (data not shown). However, this NOE is expected, regardless of whether H-1″ is in the axial position (α-anomer) or in the equatorial position (α-anomer), since H-2″ in mannose is in the equatorial position. Similarly, the coupling constant between H-1″ and H-2″ and also the C-1″ proton and carbon chemical shifts (Table III) are not diagnostic for the anomeric stereochemistry (46, 47).
Table III. NMR spectroscopy of ADP-mannose.
1H and 13C chemical NMR data for ADP-mannose in D2O at 25 °C. shifts are relative to internal 2,2-dimethylsilapentane-5-sulfonic acid. 31P chemical shifts of ADP-mannose are −10.5 (α-P) and −12.9 ppm (β-P), respectively. 31P – 31P coupling = 20.8 Hz. Mult, multiplet
| Position | δH, J | δC, Ja |
|---|---|---|
| ppm (mult), Hz | ppm (mult), Hz | |
| Adenine | ||
| 2 | 8.486 (s) | 155.455 (d) |
| 4 | 151.706 (s) | |
| 5 | 121.233 (s) | |
| 6 | 158.243 (s) | |
| 8 | 8.237 (s) | 142.333 (d) |
| Ribose | ||
| 1′ | 6.118 (d), J1′,2′ 6.0 | 89.353 (d) |
| 2′ | ~4.73 (m) | 76.854 (d) |
| 3′ | 4.501 (m) | 72.960 (d) |
| 4′ | 4.372 (m) | 86.478 (dd), JC,P9.1b |
| 5a′ | ~4.19 (m) | 67.770 (dt), JC,P5.4b |
| 5b′ | ~4.19 (m) | |
| Mannose | ||
| 1″ | 5.485 (dd), J1″,2″ 1.3
JP″,P 7.8 |
99.027 (dd), JC,P 5.9b |
| 2″ | 4.021 (m), J2″,3″ 3.3 | 72.824 (dd), JC,P9.1b |
| 3″ | 3.893 (dd), J3″,4″ 9.8 | 72.370 (d) |
| 4″ | 3.645 (dd), J4″,5″ 9.8 | 76.235 (s) |
| 5″ | 3.818 (m) | 63.318 (t) |
| 6a″ | ~3.83 (m) | |
| 6b″ | 3.718 (dd), J5″6b″ 5.5
J6a″6b″ 12.4 |
|
Data obtained from natural abundance one-dimensional 13C spectrum at high digitization.
Carbon-phosphorus coupling constant.
Fig. 9. Two-dimensional 1H NMR spectra at 600 MHz in the sugar region of ADP-mannose.

A, COSY in D2O at 25 °C showing through-bond connectivities. Only the 1H resonances of the mannose residue are numbered. B, NOESY in D2O at 25 °C showing through-space connectivities. The mannose sugar 1H resonances and cross-peaks are as indicated. X indicates the residual HOD peak after reduction with presaturation and software processing. The NOESY was obtained with a 450-ms mixing time. No cross-peaks are seen between mannose H-1″ and H-3″ or between H-1″ and H-5″ (expected locations designated by the thin arrows), demonstrating that the anomeric configuration of the mannose residue must be α.
In addition to the lack of NOEs between H-1″ and H-3″ as well as between H-1″ and H-5″ (Fig. 9B, arrows), a very reliable distinguishing criterion for determining the mannose anomeric configuration is the one-bond 1H-13C coupling constant at the C-1″-position (48, 49). The mean one-bond 1H-13C couplings are 170 and 160 Hz for the α- and β-anomers, respectively (48, 49). This 10-Hz difference is attributed to the equatorial disposition of the H-1″ of the α-anomer, which has two gauche interactions with the lone pair orbitals of the ring oxygen, whereas the axial H-1″ of the β-anomer has one trans and one gauche interaction. The 1H-coupled 13C NMR spectrum of ADP-mannose revealed a C-1″ one-bond 1H-13C coupling constant of 173.4 Hz, confirming the α-anomeric stereochemistry.
The unequivocal demonstration of the α-anomeric stereochemistry of the mannose residue in ADP-mannose is of considerable interest in light of our previous finding that ADP-mannose can serve not only as an excellent surrogate donor substrate for LpcC (13) but also for purified WaaC (13, 19) in place of the physiologically relevant compound, ADP-L-glycero-D-manno-heptose (20). Based on these observations, one would expect that ADP-L-glycero-D-manno-heptose would likewise possess the α-anomeric configuration, but this point is controversial (20), as discussed below.
DISCUSSION
The structures of the lipid A and core domains of R. leguminosarum LPS differ significantly from those of E. coli LPS (Fig. 1). Nevertheless, except for subtle variations in acyl chain preferences, the first seven enzymes that generate the conserved intermediate Kdo2-lipid IVA (Fig. 2) are identical in both organisms (15). Kdo2-lipid IVA is further acylated both in E. coli and R. leguminosarum (Fig. 1) (50–53), but in R. leguminosarum, it is also dephosphorylated at the 1- and 4′-positions (16, 17) and oxidized at the 1 position (9) in the late stages of LPS assembly to generate the unusual phosphate-free lipid A species that are characteristic of this organism (Fig. 1, component D-1). In both systems, Kdo2-lipid IVA is also glycosylated with multiple core sugars (14, 18) prior to MsbA-mediated translocation to the periplasmic surface of the inner membrane (54).
The carbohydrate composition and linkages of the E. coli and R. leguminosarum core domains are quite different distal to the two Kdo residues (Fig. 1). In E. coli, S. typhimurium and most other Gram-negative bacteria that have been characterized to date, L-glycero-D-manno-heptose (Fig. 1) is attached at position 5 of the inner Kdo moiety (1, 2). In R. leguminosarum and R. etli, a mannose unit replaces this L-glycero-D-manno-heptose residue (Fig. 1) (11, 12). In previous studies with crude membrane preparations, we demonstrated that GDP-mannose is utilized as the mannose donor for R. leguminosarum and R. etli core glycosylation of Kdo2-lipid IVA (Fig. 2), and that R. leguminosarum lpcC is the structural gene encoding the mannosyl transferase (13, 14).
We have now purified the mannosyl transferase to homogeneity (Table I), using a His6-tagged lpcC construct expressed behind the T7lac promoter in E. coli. The properties of the pure enzyme are similar to those observed previously with crude membrane preparations (also see accompanying manuscript (58)). Of particular interest is the high degree of specificity of R. leguminosarum LpcC for GDP-mannose versus most other sugar nucleotides, which are utilized at least 2 orders of magnitude less rapidly under the assay conditions employed (Fig. 4). Only the analogue ADP-mannose can substitute for GDP-mannose effectively. As will be shown in the accompanying manuscript (58), a closely related LpcC ortholog found in S. meliloti (23, 24) can employ a much broader range of sugar nucleotide donors at comparable rates with the interesting consequence that the S. meliloti gene can complement a R. leguminosarum lpcC mutation but not vice versa (23).
Only a limited number of bacterial species possess orthologs of R. leguminosarum lpcC. These organisms include A. tumefaciens (25, 26), B. melitensis (27), S. meliloti (24), F. tularensis,2 and Rhodobacter sphaeroides. Mesorhizobium loti is unusual among these bacteria in that it lacks lpcC but instead contains an ortholog of WaaC(RfaC) as well as the enzymes that make L-glycero-D-manno-heptose (55). Interestingly, lpcC and waaC, which display no sequence similarity to each other, are never present simultaneously in the one organism, as judged by inspection of the available genomes.
Given the strong structural resemblance of L-glycero-D-manno-heptose to D-mannose (Fig. 1), we wished to determine whether or not lpcC could restore the assembly of a complete LPS molecule in a mutant of S. typhimurium lacking a functional waaC gene. As shown in Fig. 7, the O-antigen ladder that is characteristic of wild-type S. typhimurium is largely restored when lpcC is expressed on a low copy plasmid behind a lac promoter in a S. typhimurium waaC mutant. This result most likely reflects the utilization of GDP-mannose by LpcC in living cells of S. typhimurium and the incorporation of a mannose unit in place of the inner L-glycero-D-manno-heptose residue.
ADP-mannose was originally described as a natural product isolated from corn (21, 22). Given its ability to substitute for GDP-mannose (Fig. 4) in the LpcC-catalyzed in vitro glycosylation of Kdo2-lipid IVA, we assume that its anomeric stereochemistry must beα. To validate this proposal, commercial ADP-mannose (Fig. 9 and Table III) and GDP-mannose (not shown) were evaluated by 1H-1H NOESY and one-bond 1H-13C coupling at C-1″. These criteria are especially useful for determining the anomeric stereochemistry of mannose residues, in which the coupling constants between H-1″ and H-2″ as well as the proton and carbon chemical shifts are not always diagnostic (46, 47). As shown in Fig. 9B, strong NOEs were observed between H-1″ and H-2″ but not between H-1″ and H-3″ or between H-1″ and H-5″ (arrows), which would be expected if the anomeric linkage had the β-configuration. The NOE results were identical with GDP-mannose (not shown). Furthermore, the 173.4-Hz one-bond 1H-13C coupling for C-1″ of ADP-mannose is diagnostic for the α-anomer. Accordingly, LpcC appears to utilize the α-anomer of both sugar nucleotides.
The demonstration that ADP-mannose has the α-anomeric configuration raises the question of whether or not the proposed endogenous ADP-L-glycero-D-manno-heptose (2) possesses the α- or the β-anomeric configuration. The few attempts to validate the structure of ADP-L-glycero-D-manno-heptose isolated from bacteria have not been altogether conclusive (20, 56, 57), since these preparations were not subjected to the 1H-1H NOESY analysis shown in Fig. 9. Based on chemical shifts and coupling constants, Gronow et al. (20) proposed that the naturally occurring ADP-L-glycero-D-manno-heptose has the β-anomeric configuration, but given the above observations, this issue should be reinvestigated. Purified WaaC can effectively utilize commercial ADP-mannose (but not GDP-mannose) (19) as a substrate for the glycosylation of Kdo2-lipid IVA, indicating that it too can utilize the α-anomer. However, other studies with synthetic ADP-L-glycero-D-manno-heptose preparations and crude membrane preparations containing WaaC (47) have suggested that only the β-anomer is active as the substrate for WaaC. Clearly, additional structural characterization of the natural product and enzymatic studies of purified WaaC in conjunction with well defined synthetic substrates are needed to resolve these issues.
One of the most interesting long term biological applications of the elucidation of the biochemistry of LPS core glycosylation is the possibility of creating hybrid strains of humans and animal pathogens, such as F. tularensis (28), with rationally reengineered cores. Such strains might show reduced virulence in infection models and therefore may prove useful in the development of novel vaccines. The functional subtleties of core glycosylation during membrane assembly may also be revealed by studies of the membrane composition and function of mutants with hybrid core structures.
Acknowledgments
We thank Kimberly White for assistance with the preparation of Kdo-lipid IVA with H. influenza KdtA.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants R37-GM-51796 (to C. R. H. R.) and GM54882 (to R. J. C.). The Duke NMR Center is supported in part by NCI, NIH, Grant P30-CA-14236. NMR instrumentation was funded by grants from the National Science Foundation, NIH, North Carolina Biotechnology Center, and Duke University.
The abbreviations used are: LPS, lipopolysaccharide(s); MALDI-TOF, matrix-assisted laser desorption-ionization/time of flight; Kdo, 3-deoxy-D-manno-octulosonic acid; NOE, nuclear Overhauser effect; CAPS, 3-(cyclohexylamino)propanesulfonic acid.
The F. tularensis genome is available on the World Wide Web at artedi.ebc.uu.se/Projects/Franciscella/.
References
- 1.Brade H, Opal SM, Vogel SN, Morrison DC, editors. Endotoxin in Health and Disease. Marcel Dekker, Inc; New York: 1999. [Google Scholar]
- 2.Raetz CRH, Whitfield C. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raetz CRH. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 4.Raetz CRH. J Bacteriol. 1993;175:5745–5753. doi: 10.1128/jb.175.18.5745-5753.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zähringer U, Seydel U, Di Padova F, Schreier M, Brade H. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 6.Reeves PR, Hobbs M, Valvano MA, Skurnik M, Whitfield C, Coplin D, Kido N, Klena J, Maskell D, Raetz CRH, Rick PD. Trends Microbiol. 1996;4:495–503. doi: 10.1016/s0966-842x(97)82912-5. [DOI] [PubMed] [Google Scholar]
- 7.Bhat UR, Forsberg LS, Carlson RW. J Biol Chem. 1994;269:14402–14410. [PubMed] [Google Scholar]
- 8.Que NLS, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2000;275:28006–28016. doi: 10.1074/jbc.M004008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Que NLS, Ribeiro AA, Raetz CRH. J Biol Chem. 2000;275:28017–28027. doi: 10.1074/jbc.M004009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhat UR, Mayer H, Yokota A, Hollingsworth RI, Carlson R. J Bacteriol. 1991;173:2155–2159. doi: 10.1128/jb.173.7.2155-2159.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhat UR, Krishnaiah BS, Carlson RW. Carbohydrate Res. 1991;220:219–227. doi: 10.1016/0008-6215(91)80020-n. [DOI] [PubMed] [Google Scholar]
- 12.Carlson RW, Reuhs B, Chen TB, Bhat UR, Noel KD. J Biol Chem. 1995;270:11783–11788. doi: 10.1074/jbc.270.20.11783. [DOI] [PubMed] [Google Scholar]
- 13.Kadrmas JL, Brozek KA, Raetz CRH. J Biol Chem. 1996;271:32119–32125. [PubMed] [Google Scholar]
- 14.Kadrmas JL, Allaway D, Studholme RE, Sullivan JT, Ronson CW, Poole PS, Raetz CRH. J Biol Chem. 1998;273:26432–26440. doi: 10.1074/jbc.273.41.26432. [DOI] [PubMed] [Google Scholar]
- 15.Price NPJ, Kelly TM, Raetz CRH, Carlson RW. J Bacteriol. 1994;176:4646–4655. doi: 10.1128/jb.176.15.4646-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Price NJP, Jeyaretnam B, Carlson RW, Kadrmas JL, Raetz CRH, Brozek KA. Proc Natl Acad Sci U S A. 1995;92:7352–7356. doi: 10.1073/pnas.92.16.7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brozek KA, Kadrmas JL, Raetz CRH. J Biol Chem. 1996;271:32112–32118. [PubMed] [Google Scholar]
- 18.Sirisena DM, Brozek KA, MacLachlan PR, Sanderson KE, Raetz CRH. J Biol Chem. 1992;267:18874–18884. [PubMed] [Google Scholar]
- 19.Kadrmas JL, Raetz CR. J Biol Chem. 1998;273:2799–2807. doi: 10.1074/jbc.273.5.2799. [DOI] [PubMed] [Google Scholar]
- 20.Gronow S, Oertelt C, Ervela E, Zamyatina A, Kosma P, Skurnik M, Holst O. J Endotoxin Res. 2001;7:263–270. [PubMed] [Google Scholar]
- 21.Passeron S, Recondo E, Dankert M. Biochim Biophys Acta. 1964;89:372–374. doi: 10.1016/0926-6569(64)90233-0. [DOI] [PubMed] [Google Scholar]
- 22.Dankert M, Passeron S, Recondo E, Leloir LF. Biochem Biophys Res Commun. 1964;14:358–362. doi: 10.1016/s0006-291x(64)80010-3. [DOI] [PubMed] [Google Scholar]
- 23.Lagares A, Hozbor DF, Niehaus K, Otero AJ, Lorenzen J, Arnold W, Puhler A. J Bacteriol. 2001;183:1248–1258. doi: 10.1128/JB.183.4.1248-1258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galibert F, Finan TM, Long SR, Puhler A, Abola P, Ampe F, Barloy-Hubler F, Barnett MJ, Becker A, Boistard P, Bothe G, Boutry M, Bowser L, Buhrmester J, Cadieu E, Capela D, Chain P, Cowie A, Davis RW, Dreano S, Federspiel NA, Fisher RF, Gloux S, Godrie T, Goffeau A, Golding B, Gouzy J, Gurjal M, Hernandez-Lucas I, Hong A, Huizar L, Hyman RW, Jones T, Kahn D, Kahn ML, Kalman S, Keating DH, Kiss E, Komp C, Lelaure V, Masuy D, Palm C, Peck MC, Pohl TM, Portetelle D, Purnelle B, Ramsperger U, Surzycki R, Thebault P, Vandenbol M, Vorholter FJ, Weidner S, Wells DH, Wong K, Yeh KC, Batut J. Science. 2001;293:668–672. doi: 10.1126/science.1060966. [DOI] [PubMed] [Google Scholar]
- 25.Goodner B, Hinkle G, Gattung S, Miller N, Blanchard M, Qurollo B, Goldman BS, Cao Y, Askenazi M, Halling C, Mullin L, Houmiel K, Gordon J, Vaudin M, Iartchouk O, Epp A, Liu F, Wollam C, Allinger M, Doughty D, Scott C, Lappas C, Markelz B, Flanagan C, Crowell C, Gurson J, Lomo C, Sear C, Strub G, Cielo C, Slater S. Science. 2001;294:2323–2328. doi: 10.1126/science.1066803. [DOI] [PubMed] [Google Scholar]
- 26.Wood DW, Setubal JC, Kaul R, Monks DE, Kitajima JP, Okura VK, Zhou Y, Chen L, Wood GE, Almeida NF, Jr, Woo L, Chen Y, Paulsen IT, Eisen JA, Karp PD, Bovee D, Sr, Chapman P, Clendenning J, Deatherage G, Gillet W, Grant C, Kutyavin T, Levy R, Li MJ, McClelland E, Palmieri A, Raymond C, Rouse G, Saenphimmachak C, Wu Z, Romero P, Gordon D, Zhang S, Yoo H, Tao Y, Biddle P, Jung M, Krespan W, Perry M, Gordon-Kamm B, Liao L, Kim S, Hendrick C, Zhao ZY, Dolan M, Chumley F, Tingey SV, Tomb JF, Gordon MP, Olson MV, Nester EW. Science. 2001;294:2317–2323. doi: 10.1126/science.1066804. [DOI] [PubMed] [Google Scholar]
- 27.DelVecchio VG, Kapatral V, Redkar RJ, Patra G, Mujer C, Los T, Ivanova N, Anderson I, Bhattacharyya A, Lykidis A, Reznik G, Jablonski L, Larsen N, D’Souza M, Bernal A, Mazur M, Goltsman E, Selkov E, Elzer PH, Hagius S, O’Callaghan D, Letesson JJ, Haselkorn R, Kyrpides N, Overbeek R. Proc Natl Acad Sci U S A. 2002;99:443–448. doi: 10.1073/pnas.221575398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vinogradov E, Perry MB, Conlan JW. Eur J Biochem. 2002;269:6112–6118. doi: 10.1046/j.1432-1033.2002.03321.x. [DOI] [PubMed] [Google Scholar]
- 29.Bhat UR, Carlson RW, Busch M, Mayer H. Int J Syst Bacteriol. 1991;41:213–217. doi: 10.1099/00207713-41-2-213. [DOI] [PubMed] [Google Scholar]
- 30.Basu SS, Karbarz MJ, Raetz CRH. J Biol Chem. 2002;277:28959–28971. doi: 10.1074/jbc.M204525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raetz CRH. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. 2. Neidhardt FC, editor. Vol. 1. American Society for Microbiology; Washington, D. C: 1996. pp. 1035–1063. [Google Scholar]
- 32.Carlson RW, Forsberg LS, Price NP, Bhat UR, Kelly TM, Raetz CRH. Prog Clin Biol Res. 1995;392:25–31. [PubMed] [Google Scholar]
- 33.Elling L, Ritter JE, Verseck S. Glycobiology. 1996;6:591–597. doi: 10.1093/glycob/6.6.591. [DOI] [PubMed] [Google Scholar]
- 34.Brabetz W, Muller-Loennies S, Holst O, Brade H. Eur J Biochem. 1997;247:716–724. doi: 10.1111/j.1432-1033.1997.00716.x. [DOI] [PubMed] [Google Scholar]
- 35.Miller JR. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 36.Bergmans HE, van Die IM, Hoekstra WP. J Bacteriol. 1981;146:564–570. doi: 10.1128/jb.146.2.564-570.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. John Wiley & Sons, Inc; New York: 1989. [Google Scholar]
- 38.Basu SS, York JD, Raetz CRH. J Biol Chem. 1999;274:11139–11149. doi: 10.1074/jbc.274.16.11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gottesman S, Stout V. Mol Microbiol. 1991;5:1599–1606. doi: 10.1111/j.1365-2958.1991.tb01906.x. [DOI] [PubMed] [Google Scholar]
- 40.Dulbecco R, Vogt M. J Exp Med. 1954;99:167–182. doi: 10.1084/jem.99.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Breazeale SD, Ribeiro AA, Raetz CRH. J Biol Chem. 2002;277:2886–2896. doi: 10.1074/jbc.M109377200. [DOI] [PubMed] [Google Scholar]
- 42.Odegaard TJ, Kaltashov IA, Cotter RJ, Steeghs L, van der Ley P, Khan S, Maskell DJ, Raetz CRH. J Biol Chem. 1997;272:19688–19696. doi: 10.1074/jbc.272.32.19688. [DOI] [PubMed] [Google Scholar]
- 43.Hitchcock PJ, Brown TM. J Bacteriol. 1983;154:269–277. doi: 10.1128/jb.154.1.269-277.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White KA, Kaltashov IA, Cotter RJ, Raetz CRH. J Biol Chem. 1997;272:16555–16563. doi: 10.1074/jbc.272.26.16555. [DOI] [PubMed] [Google Scholar]
- 45.Caroff M, Tacken A, Szabó L. Carbohydr Res. 1988;175:273–282. doi: 10.1016/0008-6215(88)84149-1. [DOI] [PubMed] [Google Scholar]
- 46.Agrawal PK. Phytochemistry. 1992;31:3307–3330. doi: 10.1016/0031-9422(92)83678-r. [DOI] [PubMed] [Google Scholar]
- 47.Zamyatina A, Gronow S, Oertelt C, Puchberger M, Brade H, Kosma P. Angew Chem Int Ed Engl. 2000;39:4150–4153. doi: 10.1002/1521-3773(20001117)39:22<4150::aid-anie4150>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 48.Ubrinova S, Ubrin D, Liptaj T, Bella J, Hirsch J. Magn Reson Chem. 1991;29:912–922. [Google Scholar]
- 49.Perlin AS, Casu B. Tetrahedron Lett. 1969:2921. [Google Scholar]
- 50.Brozek KA, Raetz CRH. J Biol Chem. 1990;265:15410–15417. [PubMed] [Google Scholar]
- 51.Clementz T, Bednarski JJ, Raetz CRH. J Biol Chem. 1996;271:12095–12102. doi: 10.1074/jbc.271.20.12095. [DOI] [PubMed] [Google Scholar]
- 52.Clementz T, Zhou Z, Raetz CRH. J Biol Chem. 1997;272:10353–10360. doi: 10.1074/jbc.272.16.10353. [DOI] [PubMed] [Google Scholar]
- 53.Brozek KA, Carlson RW, Raetz CRH. J Biol Chem. 1996;271:32126–32136. doi: 10.1074/jbc.271.50.32126. [DOI] [PubMed] [Google Scholar]
- 54.Doerrler WT, Reedy MC, Raetz CRH. J Biol Chem. 2001;276:11461–11464. doi: 10.1074/jbc.C100091200. [DOI] [PubMed] [Google Scholar]
- 55.Kaneko T, Nakamura Y, Sato S, Asamizu E, Kato T, Sasamoto S, Watanabe A, Idesawa K, Ishikawa A, Kawashima K, Kimura T, Kishida Y, Kiyokawa C, Kohara M, Matsumoto M, Matsuno A, Mochizuki Y, Nakayama S, Nakazaki N, Shimpo S, Sugimoto M, Takeuchi C, Yamada M, Tabata S. DNA Res. 2000;7:331–338. doi: 10.1093/dnares/7.6.331. [DOI] [PubMed] [Google Scholar]
- 56.Kontrohr T, Kocsis B. J Biol Chem. 1981;256:7715–7718. [PubMed] [Google Scholar]
- 57.Kocsis B, Kontrohr T. J Biol Chem. 1984;259:11858–11860. [PubMed] [Google Scholar]
- 58.Kanipes MI, Kalb SR, Cotter RJ, Hozbor DF, Lagares A, Raetz CRH. 2003;278:16365–16371. doi: 10.1074/jbc.M301256200. [DOI] [PMC free article] [PubMed] [Google Scholar]