

Abstract
Pathogenic bacteria modify the lipid A portion of their lipopolysaccharide to help evade the host innate immune response. Modification of the negatively charged phosphate groups of lipid A aids in resistance to cationic antimicrobial peptides targeting the bacterial cell surface. The lipid A of Helicobacter pylori contains a phosphoethanolamine (pEtN) unit directly linked to the 1-position of the disaccharide backbone. This is in contrast to the pEtN units found in other pathogenic Gram-negative bacteria, which are attached to the lipid A phosphate group to form a pyrophosphate linkage. This study describes two enzymes involved in the periplasmic modification of the 1-phosphate group of H. pylori lipid A. By using an in vitro assay system, we demonstrate the presence of lipid A 1-phosphatase activity in membranes of H. pylori. In an attempt to identify genes encoding possible lipid A phosphatases, we cloned four putative orthologs of Escherichia coli pgpB, the phosphatidylglycerol-phosphate phosphatase, from H. pylori 26695. One of these orthologs, Hp0021, is the structural gene for the lipid A 1-phosphatase and is required for removal of the 1-phosphate group from mature lipid A in an in vitro assay system. Heterologous expression of Hp0021 in E. coli resulted in the highly selective removal of the 1-phosphate group from E. coli lipid A, as demonstrated by mass spectrometry. We also identified the structural gene for the H. pylori lipid A pEtN transferase (Hp0022). Mass spectrometric analysis of the lipid A isolated from E. coli expressing Hp0021 and Hp0022 shows the addition of a single pEtN group at the 1-position, confirming that Hp0022 is responsible for the addition of a pEtN unit at the 1-position in H. pylori lipid A. In summary, we demonstrate that modification of the 1-phosphate group of H. pylori lipid A requires two enzymatic steps.
Unlike Gram-positive organisms, Gram-negative bacteria are further guarded from their environment by an asymmetric outer membrane that encapsulates their peptidoglycan. The outer leaflet of the outer membrane is composed mainly of lipopolysaccharide (LPS),1 the major surface molecule of Gram-negative bacteria. LPS is held in the outer membrane by its hydrophobic anchor lipid A (1, 2). Attached to lipid A are the 3-deoxy-D-manno-octulosonic acid (Kdo) sugars (Fig. 1) that act as a bridge connecting the lipid A anchor to the core and O-antigen (1–3). The lipid A domain is required to maintain the integrity of the outer membrane barrier (4) and is the bioactive component of LPS that is associated with Gram-negative septic shock (5–7). During Gram-negative bacterial infections, lipid A serves as one of the pathogen-associated molecular patterns activating the innate immune system (8, 9) through the pattern recognition receptor TLR-4 (10–12).
Fig. 1. Comparison of the chemical structures of the Kdo-lipid A domain of E. coli and the major and minor structures of H. pylori.

The covalent modifications of lipid A are indicated with dashed bonds, and lengths of the acyl chains are designated with numbers in circles. A, the major lipid A species of E. coli is a hexa-acylated disaccharide of glucosamine that is substituted at the 1- and 4′-positions with phosphate and glycosylated at the 6′-position with two Kdo (3-deoxy-D-manno-octulosonic acid) moieties (1, 5). In wild type K12 strains, a portion of the lipid A contains a pyrophosphate group at the 1-position (73). The modification of phosphate moieties of E. coli lipid A are regulated by PmrA (74) and may be substituted with L-Ara4N (red substituent) or pEtN (purple substituent) groups. In addition, the protein product of the PhoP-activated gene, pagP, further modifies E. coli lipid A by the addition of a palmitoyl chain (blue substituent) to the hydroxyl group of the N-linked (R)-3-hydroxymyristate chain on the proximal glucosamine unit of lipid A (40, 44). B, the minor lipid A species of H. pylori is similar to that seen in E. coli but has slightly longer acyl chains and only contains one Kdo moiety (13, 14, 19). C, the major lipid A species of H. pylori is a tri- to tetra-acylated structure that is lacking the 4′-phosphate group and is substituted at the C-1 position with a pEtN residue (13–15).
The typical lipid A backbone is a β,1′-6-linked disaccharide of glucosamine that is bisphosphorylated and multiply acylated. In Escherichia coli, the disaccharide backbone is polyacylated with (R)-3-hydroxymyristate at the 2-, 3-, 2′-, and 3′-positions. The hydroxyl groups of the 2′- and 3′-linked fatty acyl chains are further esterified with laurate (C12) and myristate (C14), respectively (Fig. 1A) (1, 2, 5). Although a negatively charged hexa-acylated lipid A structure is characteristic in a number of Gram-negative animal pathogens, the lipid A structure can be further modified.
The lipid A of Helicobacter pylori is quite unusual in that it lacks a 4′-phosphate group and is tri- or tetra-acylated with either (R)-3-hydroxystearate (C18) or (R)-3-hydroxypalmitate (C16) (Fig. 1C) (13–15). Another unusual feature is that the C-1 hydroxyl group of the proximal glucosamine is derivatized with a phosphoethanolamine (pEtN) residue (13–15). This is in contrast with the pEtN units of E. coli (16) (Fig. 1), Salmonella typhimurium (17), and Neisseria meningitidis (18), which are attached to the lipid A phosphate group to form a pyrophosphate linkage. Differences in the H. pylori lipid A structure are reflected in its low endotoxicity compared with lipid A produced by Gram-negative bacteria of the Enterobacteriaceae family (14, 19, 20). Pharmacological studies have demonstrated the importance of the phosphate groups and number of fatty acyl chains for cytokine induction via TLR-4 (21, 22). In addition to the low potency of H. pylori lipid A, the structure of the oligosaccharide portion of its LPS shows structural similarities to Lewis blood group antigens in the gastric mucosa, giving rise to a form of molecular mimicry that helps to camouflage the bacterium (23–25). Therefore, it has been theorized that H. pylori has evolved to have a less inflammatory LPS helping to prolong the chronic infections seen with this organism.
A minor lipid A species found in H. pylori is both bisphosphorylated and hexa-acylated (Fig. 1B) resembling enterobacterial lipid As. We reasoned that the lipid A structure of H. pylori is modified following the constitutive lipid A pathway and that these modifications are not regulated given the structure of the major lipid A species shown in Fig. 1A. We report here two proteins, Hp0021 and Hp0022, involved in the variation of the H. pylori lipid A structure.
EXPERIMENTAL PROCEDURES
Chemicals and Other Materials
[γ-32P]ATP and 32Pi were obtained from Amersham Biosciences. Silica Gel 60 (0.25-mm) thin layer plates were purchased from EM Separation Technology (Merck). Yeast extract and tryptone were from Difco. Triton X-100 and bicinchoninic acid were from Pierce. All other chemicals were reagent-grade and were purchased from either Sigma or Mallinckrodt.
Bacterial Strains and Growth Conditions
The bacterial strains and plasmids used in this study are summarized in Table I. H. pylori strains J99 and 26695 were obtained from the American Type Culture Collection. Primary plate cultures of H. pylori were grown from glycerol stock on blood agar medium at 37 °C for 36–60 h in a microaerobic atmosphere (5% O2, 10% CO2, and 85% N2). The resultant colonies were inoculated in Brucella broth (Invitrogen) supplemented with 10% fetal bovine serum (Hyclone) and vancomycin (10 μg/ml). Cells were grown to an A600 of 1.0 at 37 °C under microaerobic conditions for 24–36 h. Prior to every experiment, confirmation of H. pylori was performed by both Gram’s stain and urease test. E. coli was typically grown at 37 °C in Luria-Bertani (LB) broth, which consisted of 10 g of NaCl, 10 g of tryptone, and 5 g of yeast extract per liter (26). When required for selection of plasmids, cells were grown in the presence of ampicillin (100 μg/ml) or chloramphenicol (25 μg/ml).
Table I.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or description | Source or Reference |
|---|---|---|
| Strains | ||
| H. pylori | ||
| J99 | Wild type | ATCC 700824 |
| 26695 | Wild type | ATCC 700392 |
| E. coli | ||
| W3110 | Wild type, F−, λ− | E. coli Genetic Stock Center (Yale) |
| W3110A | Wild type, F−, λ−, aroA::Tn10 | 71 |
| WD2 | W3110, aroA::Tn10 msbA (A270T) | 72 |
| XL-1 Blue | recA1 endA1 gyrA96thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacIqZΔ M15::Tn10 (TetR)] | Stratagene |
| Nova F− | F−, endA1 hsdR17 (rK12− mK12+)supE44 thi-1 recA1 gyrA96 relA1 lac | Novagen |
| NovaBlue(DE3) | endA1 hsdR17 (rK12− mK12+)supE44 thi-1 recA1 gyrA96 relA1 lac (DE3) [F′ proAB lacIqZΔM15::Tn10 (TetR)] | Novagen |
| HSM174(DE3) | F− recA1 hsdR (rK12− mK12+) (DE3) Rif R | Novagen |
| BLR(DE3)pLysS | F− ompT hsdSB (rB− mB− gal dcm (DE3) Δ(srl-recA)306:: Tn10 pLysS, (TetR, CamR) | Novagen |
| Plasmids | ||
| pET21a(+) | Expression vector containing a T7 promoter, AmpR | Novagen |
| pETCoco-2 | Expression vector containing a T7 promoter, and two origins of replication control the single-copy (oriS) and medium-copy (oriV), AmpR | Novagen |
| pETDuet-1 | Expression vector containing two multiple cloning sites (MCS), each of which is preceded by a T7lac promoter, lacI, AmpR | Novagen |
| pHp0021 | pET21a (+) containing hp0021 | This work |
| pHp0350 | pET21a (+) containing hp0350 | This work |
| pHp0851 | pET21a (+) containing hp0851 | This work |
| pHp1580 | pET21a (+) containing hp1580 | This work |
| pHp0022 | pET21a (+) containing hp0022 | This work |
| pHp0021coco | pETCoco-2 containing hp0021 | This work |
| pHp0350coco | pETCoco-2 containing hp0350 | This work |
| pHp0851coco | pETCoco-2 containing hp0851 | This work |
| pHp1580coco | pETCoco-2 containing hp1580 | This work |
| pHp0021duet | pETDuet-1 containing hp0021 | This work |
| pHp0022duet | pETDuet-1 containing hp0022 | This work |
| pHp0021/Hp0022 | pETDuet-1 containing both hp0021 and hp0022 | This work |
| pWSK29 | Low-copy expression vector, Ampr | 77 |
| pWSHp0021 | pWSK29 containing hp0021 | This work |
| pMsbB | pET21a (+) containing msbB (lpxL) | 37 |
| pHtrB | pET21a (+) containing htrB (lpxM) | This work |
Recombinant DNA Technique
Plasmids were isolated by using the Qiagen Spin Prep Kit. Custom primers were obtained from Integrated DNA Technologies. PCR reagents were purchased from Stratagene. PCR clean up was performed using the QIAquick PCR Purification Kit (Qiagen). DNA fragments were isolated from gels using the QIAquick Gel Extraction Kit (Qiagen). Restriction endonucleases, T4 DNA ligase, and shrimp alkaline phosphatase were purchased from New England Biolabs. All modifying enzymes were used according to the manufacturer’s instructions.
Cloning of Putative H. pylori Orthologs of E. coli pgpB and hp0022
H. pylori orthologs of E. coli pgpB (hp0021, hp0350, hp0851, and hp1580) and N. meningitidis lptA (60) (hp0022) were cloned into pET21a(+) behind the T7lac promoter. The genes were amplified by PCR using H. pylori strain 26695 genomic DNA as the template. The forward and reverse primer sequences of H. pylori pgpB and lptA orthologs are described in Table II. The forward primers contained a clamp region, an NdeI site (Table II, underlined), and a match to the coding strand starting at the translation initiation start site. The reverse primers were designed with a clamp region, a BamHI site (Table II, underlined), and a match to the anti-coding strand that included a stop codon. The PCR mixture contained 100 ng of genomic DNA template, 150 ng of each primer, 200 μM of each dNTP, 100 mM Tris-HCl, pH 8.8, 35 mM MgCl2, 250 mM KCl, and 2.5 units of Pfu DNA polymerase (Stratagene) in a reaction volume of 0.05 ml. The reaction mixtures were subjected to a 90-s denaturation at 95 °C followed by 30 cycles of 95 °C for 60 s, 56 °C for 60 s, 72 °C for 60 s, and a final extension for 10 min at 72 °C, using the Stratagene RoboCyler Gradient 40 PCR system. The PCR products and vectors were digested with NdeI and BamHI, gel-purified, and ligated together. Plasmid constructs derived from pET21a(+) designated pHp0021, pHp0350, pHp0851, pHp1580, and pHp0022 were transformed into XL-1 Blue cells (Stratagene) for propagation of the plasmid. The plasmids were then screened for positive inserts and confirmed by DNA sequencing.
Table II.
Oligonucleotides
| Name | Sequence |
|---|---|
| U-hp0022-NdeI | 5′-GCGCGCCATATGTTGGCATCATTATTCCAT-3′ |
| L-hp0022-BamHI | 5′-GCGCGCGGATCCTTACTCTTTTTTGTGTTT-3′ |
| U-hp0021-NdeI | 5′-GCGCGCCATATGAAAAAATTCTTATTTAAA-3′ |
| L-hp0221-BamHI | 5′-GCGCGCGGATCCTTAAGGCTTTTTGGGGCT-3′ |
| U-hp0350-NdeI | 5′-GCGCGCCATATGCTGAAAATTCTTTCAAA-3′ |
| L-hp0350-BamHI | 5′-GCGCGCGGATCCTTAGATCTTTTGAAACAC-3′ |
| U-hp0851-NdeI | 5′-GCGCGCCATATGTATTTGACAGAACAATC-3′ |
| L-hp0851-BamHI | 5′-GCGCGCGGATCCTTATTGATTATAAGGGCG-3′ |
| U-hp1580-NdeI | 5′-GCGCGCCATATGCTATTTAATGGGCTATGC-3′ |
| L-hp1580-BamHI | 5′-GCGCGCGGATCCTTACCATTGATAAGAAAA-3′ |
| U-hp0021coco-NsiI | 5′-GCGCGCCATGCATAAAAAATTCTTATTTAAA-3′ |
| L-hp0021coco-BstBI | 5′-GCGCGCCTTCGAATTAAGGCTTTTTGGGGCT-3′ |
| U-hp0350coco-NsiI | 5′-GCGCGCCATGCATGCTGAAAATTCTTTCAAA-3′ |
| L-hp0350coco-BstBI | 5′-GCGCGCCTTCGAATTAGATCTTTTGAAACAC-3′ |
| U-hp0851coco-NsiI | 5′-GCGCGCCATGCATGTATTTGACAGAACAATC-3′ |
| L-hp0851coco-BstBI | 5′-GCGCGCCTTCGAATTATTGATTATAAGGGCG-3′ |
| U-hp1580coco-NsiI | 5′-GCGCGCCATGCATCTATTTAATGGGCTATGC-3′ |
| L-hp1580coco-BstBI | 5′-GCGCGCCTTCGAATTACCATTGATAAGAAAA-3′ |
| htrBUpper | 5′-GCGCGCCATATGATGACGAATCTACCC-3′ |
| htrBLower | 5′-GCGGCGGGATCCTTAATAGCGTGAAGG-3′ |
Additionally, hp0021 was cloned into the expression vector pWSK29 (77). The plasmid pHp0021 was subjected to restriction enzyme digest with XbaI and XhoI. The resulting fragment containing hp0021 was ligated into pWSK29 previously digested with XbaI and XhoI resulting in the vector pWSHp0021.
High Copy Expression of Putative PgpB and LptA Orthologs behind a T7lac Promoter
The H. pylori PgpB and LptA ortholog constructs derived from pET21a(+) were transformed into NovaBlue(DE3) (Table I) for overexpression of the protein. First, a single colony of E. coli harboring either the empty vector control or the recombinant expression plasmid were inoculated into 50 ml of LB broth and grown overnight (~16 h) in a rotary shaker at 37 °C toA600 ~1. The appropriate amount of overnight culture was used to inoculate 200 ml of fresh LB broth with an initial A600 of 0.05. Prior to induction with 1 mM IPTG, cells were allowed to reach mid-log phase of growth (A600 ~0.6). After induction with IPTG, the cells were left shaking for an additional 4 h. To determine the level of protein expression, membrane proteins were isolated (see below), separated by SDS-PAGE, and stained with Coomassie gel code blue staining reagent (Pierce). Unfortunately, expression of Hp0021, Hp0350, Hp0851, Hp1580, or Hp0022 in E. coli NovaBlue(DE3) did not result in any distinct protein bands (data not shown).
Cloning of Putative PgpB Orthologs into a Low Copy Expression Vector
PCR-amplified pgpB orthologs (hp0021, hp0350, hp0851, and hp1580) from H. pylori strain 26695 were cloned into pETCoco-2 for low copy replication and protein expression. The forward and reverse primer sequences of pgpB orthologs cloned into pETCoco-2 are described in Table II. The forward primers for pgpB orthologs contained a clamp region, an NsiI site (Table II, underlined), and a match to the coding strand beginning at the translation initiation start site. The reverse primers were designed with a clamp region, a BstBI site (Table II, underlined), and a match to the anti-coding strand that included a stop codon. Amplification was carried out as mentioned previously. The PCR products and vector were digested with NsiI and BstBI, gel-purified, and ligated together. Plasmid constructs derived from pET-Coco-2 designated pHp0021coco, pHp0350coco, pHp0851coco, and pHp1580coco were transformed into NovaF− cells (Novagen) for plasmid propagation. All constructs derived from pETCoco-2 were maintained in the single copy state with LB broth containing ampicillin (50 μg/ml) and 0.2% D-glucose at all times to ensure maximum stability, except during culturing for plasmid DNA isolation. Plasmid propagation of pETCoco-2-derived constructs was typically grown by inoculating a single colony of E. coli containing an empty or recombinant plasmid into 5 ml of LB broth supplemented with ampicillin and 0.2% glucose and grown overnight (~16 h) in a rotary shaker (250 rpm) at 37 °C. The overnight cultures were diluted to 1:50 with fresh LB broth containing ampicillin without glucose because the sugar prevents amplification of the plasmid. Cells were induced with 0.01% L-arabinose for plasmid replication at an A600 of 0.4. The cells were allowed to grow for an additional 4 h prior to harvest for plasmid purification. Plasmid constructs were then screened for positive inserts, confirmed by DNA sequencing, and transformed into E. coli K12 strain HMS174(DE3) cells (Novagen) for low copy expression.
Low Copy Expression of Putative Orthologs of PgpB
Low copy expression of pETCoco-2 derivatives was performed according to the instructions described by Novagen. Typically, a single colony of E. coli harboring either the empty vector control or the recombinant expression plasmid was inoculated into 5 ml of LB broth supplemented with ampicillin and 0.2% glucose and grown overnight at 37 °C. Overnight cultures were then used to inoculate 20 ml of fresh LB broth containing ampicillin with or without glucose at an initial A600 of 0.05. At mid-log phase of growth, the cells were induced with 0.5 mM IPTG and allowed to grow for an additional 3 h before harvesting.
Isolation and Analysis of Lipid A Species from 32 Pi-Labeled Cells
Cells containing pETCoco-2 derivatives were grown as described above. Upon induction at mid-log phase with IPTG, the cells were labeled uniformly with 2.5 μCi/ml 32 Pi for an additional 3 h at 37 °C. Bacteria were collected using a clinical centrifuge and washed with 5 ml of phosphate-buffered saline, pH 7.4. To extract the 32P-labeled phospholipids, the cell pellet was resuspended in 3 ml of a single-phase Bligh/Dyer mixture (27), consisting of chloroform/methanol/water (1:2:0.8, v/v). After mixing and incubating for 30 min at room temperature, the insoluble residue, containing the 32P-labeled lipid A still covalently bound to LPS, was recovered by centrifugation. The residue was subjected to hydrolysis at 100 °C in 12.5 mM sodium acetate buffer, pH 4.5, in the presence of 1% SDS to cleave the Kdo-lipid A linkage (16, 28). The 32P-labeled lipid A species were recovered by two-phase Bligh/Dyer extraction (16, 28) and spotted onto a Silica Gel 60 TLC plate (10,000 cpm/lane). The plate was developed in the solvent chloroform, pyridine, 88% formic acid, water (50:50:16:5, v/v). The plate was dried and exposed to a PhosphorImager Screen overnight to visualize the resolved 32P-lipid A species.
Cloning and Expression of H. pylori Hp0021 and Hp0022 in a Dual Expression System
The plasmid constructs, pHp0021 and pETDuet-1, were subjected to restriction enzyme digest with NcoI and BamHI. The resulting Hp0021 DNA fragment was isolated and ligated into the multiple cloning site 1 of pETDuet-1 to derive the plasmid construct pHp0021duet. In addition, the plasmid constructs pHp0022, pET-Duet-1, and pHp0021duet were subjected to restriction enzyme digested with NdeI and XhoI. The resulting Hp0022 DNA fragment was ligated into the MSC2 of both pETDuet-1 and pHp0021duet vectors to generate the plasmid constructs pHp0022duet and pHp0021/Hp0022, respectively. All of the plasmids derived from pETDuet-1 were transformed into XL-1 Blue cells for propagation of the plasmids, screened for positive inserts, and confirmed by DNA sequencing. The plasmids pHp0021duet, pHp0022duet, or pHp0021/Hp0022 were then transformed into the E. coli expression strain NovaBlue(DE3) and overexpressed as described previously.
Cloning and Expression of the E. coli Late Acyltransferases, LpxL and LpxM, behind a T7lac Promoter
The lpxL structural gene was obtained by PCR using W3110 genomic DNA as the template. The lpxM structural gene was cloned and the protein overexpressed as described previously (29). Primer sequences for lpxL are described in Table II. The lpxL PCR product was digested with NdeI and BamHI and ligated into the similarly digested vector pET21a(+). The resulting plasmid, pHtrB, was transformed into BLR(DE3)/pLysS cells (Novagen) for protein expression. Cells were grown in LB broth at 37 °C toA600 of ~0.6, followed by induction with 1 mM IPTG and further incubation at 37 °C for an additional 4 h.
Preparation of Cell-free Extracts, Double-spun Cytosol, and Washed Membrane
Typically, 500 ml of H. pylori or 200 ml of E. coli cultures were grown to an A600 of 1.0 at 37 °C and harvested by centrifugation at 6,000 × g for 30 min. All samples were prepared at 4 °C. Cell-free extract, double-spun cytosol, and washed membranes were prepared as described previously (30) and were stored in aliquots at −20 °C. Protein concentration was determined by the bicinchoninic acid method (31), using bovine serum albumin as the standard.
Preparation of Radiolabeled Substrate
The substrate 4′-32P-lipid IVA was generated from 100 μCi of [γ-32P]ATP and the tetraacyl-disaccharide 1-phosphate lipid acceptor, using the overexpressed 4′-kinase present in membranes of E. coli BLR(DE3)/pLysS/pJK2 (30, 32, 33), as described previously (30, 33). Kdo2-4′-32P-lipid IVA was prepared by adding purified E. coli Kdo transferase (WaaA) immediately after the 4′-kinase, as described previously (33, 34). To prepare Kdo2-4′-32P-lipid A (dilauroyl-Kdo2-4′-32P-lipid IVA), lauroyl-acyl carrier protein and E. coli overexpressed LpxL and LpxM (29) membranes were used in tandem with the Kdo transferase reaction under the following conditions: 50 mM Hepes, pH 7.5, 0.1% Triton X-100, 50 mM NaCl, 50 mM MgCl2, 0.1 mg/ml bovine serum albumin, 13.3 μM lauroyl-acyl carrier protein, and 0.05 mg/ml BLR(DE3)/pLysS/pMsbB and BLR(DE3)/pLysS/pHtrB membranes. Once the reaction was complete, the substrate was isolated as described previously (33).
Assay of the 1-Phosphatase
The 1-phosphatase activity from Hp0021 was assayed under optimized conditions in a 10- μl reaction mixture containing 50 mM MES, pH 6.0, 0.2% Triton X-100, and either 5μM 4′-32P-lipid IVA, Kdo2-4′-32P-lipid IVA, or Kdo2-4′-32P-lipid A (each at ~3000–5000 cpm/nmol) as the substrate. Washed membranes (0.005–1.0 mg/ml) were employed as the enzyme source, as indicated. Dephosphorylation reactions were incubated at 30 °C for the indicated times. Enzymatic reactions were terminated by spotting 4.5- μl portions of the mixtures onto Silica Gel 60 TLC plates, and the plate was dried under a cool air stream for 20 min.
When 4′-32P-lipid IVA was employed as the substrate, reaction products were separated using the solvent chloroform, pyridine, 88% formic acid, water (50:50:16:5, v/v). For reactions containing either Kdo2-4′32P-lipid IVA or Kdo2-4′-32P-lipid A as the substrate, the TLC plates were developed in the solvent chloroform, pyridine, 88% formic acid, water (30:70:16:10, v/v). After drying and overnight exposure of the plate to a PhosphorImager Screen, the product formation was detected and analyzed using a Bio-Rad Molecular Imager PhosphorImager equipped with Quantity One Software. The enzyme activity was calculated by determining the percentage of the substrate converted to product.
Separation of Inner and Outer Membranes
Membranes isolated from E. coli strain NovaBlue(DE3)/pHp0021 were separated by isopycnic sucrose gradient centrifugation. First, washed membranes were prepared as described above and were resuspended in 10 mM Hepes, pH 7.0, containing 0.05 mM EDTA at a protein concentration of 1–3 mg/ml. Membranes (2.2 ml) were layered onto a seven-step gradient, as described previously (35, 36), and subjected to ultracentrifugation at 35,000 rpm in a Beckman SW41 rotor for 19 h at 4 °C. The gradient was collected in ~0.5-ml fractions. Each fraction was then assayed for NADH oxidase, as the inner membrane marker, and for phospholipase A, as the outer membrane marker, as described previously (28). The amount of protein in each fraction was determined using the bicinchoninic acid assay (31). Each fraction was also assayed for 1-phosphatase activity using the standard conditions described above.
Large Scale Isolation of Lipid A from E. coli Cells Expressing H. pylori Hp0021 and Hp0022
Cultures of E. coli strain Nova-Blue(DE3) were grown in 1 liter of LB medium at 37 °C containing either pDuet-1, pHp0021duet, pHp0022duet, or pHp0021/Hp0022. The cultures were allowed to reach A600 ~0.6 prior to induction with 1 mM IPTG and further incubated for an additional 4 h at 37 °C. Cells were harvested by centrifugation at 6000 × g for 15 min and washed once with phosphate-buffered saline (37). The final cell pellets were resuspended in 40 ml of phosphate-buffered saline. Lipid A was released from cells and purified as described previously (16, 38) and stored frozen at −20 °C.
Purification of the 1-Phosphatase Reaction Product Generated in Vitro from Kdo2-Lipid A
A large scale 1-phosphatase reaction mixture (20 ml) was prepared by assembling the following components: 50 mM MES, pH 6.0, 0.25% Triton X-100, 50 μM of Kdo2-lipid A (39), and 0.3 mg/ml membrane proteins from NovaBlue(DE3)/pHp0021 (39). The reaction was initiated by the addition of the enzyme and incubated at 30 °C for 3 h. The reaction mixture was then converted into a two-phase Bligh-Dyer system consisting of chloroform, methanol, 0.05 M HCl (2: 2:1.8, v/v). The phases were separated by centrifugation at 4000 × g for 10 min. The lower phase was removed, and the resulting upper phase was extracted a second time by the addition of lower phase from a fresh two-phase acidic Bligh-Dyer system. The lower phases were pooled, and pyridine (1 drop/4 ml) was added to neutralize the minor amount of HCl carried over during the extraction process. The sample was dried by rotary evaporation, dissolved in 5 ml of chloroform/methanol/water (2:3:1, v/v), and applied to a 1-ml DE52 cellulose column equilibrated with the same solvent mixture. Following sample loading, the column was washed with 5 column volumes of chloroform/methanol/water (2: 3:1, v/v). The bound phospholipids and lipid A were eluted with 1-ml steps of chloroform/methanol/ammonium acetate (2:3:1, v/v), with increasing ammonium acetate concentrations of 60, 120, 240, and 480 mM successively in the aqueous component. For each step elution, 1-ml fractions were collected. The presence of lipids was determined by spotting 10 μl of each fraction onto a TLC plate followed by developing in chloroform, pyridine, 88% formic acid, water (30:70:16:10, v/v). After drying, lipids were visualized by spraying the plate with 10% sulfuric acid in ethanol and then charring. All fractions containing 1-dephosphorylated-Kdo2-lipid A were pooled and converted to a two-phase Bligh/Dyer mixture by addition of appropriate amounts of chloroform and water. The lower phase was dried under a stream of N2 and stored at −20 °C.
Mass Spectrometry of Lipid A Species
Mass spectra of the purified lipids were acquired in the negative and positive linear mode using a matrix-assisted laser desorption-ionization/time of flight (MALDI-TOF) mass spectrometer (AXIMA-CFR, Kratos Analytical, Manchester, UK) equipped with a nitrogen laser (337 nm). The instrument was operated using 20-kV extraction voltage and time-delayed extraction, providing a mass resolution of about ±1 atomic mass units for compounds with Mr ~2000. Each spectrum represented the average of 100 laser shots. Saturated 6-aza-2-thiothymine in 50% acetonitrile and 10% tribasic ammonium citrate (9:1, v/v) served as the matrix in negative and positive modes. The samples were dissolved in chloroform/methanol (4:1, v/v) and deposited on the sample plate followed by an equal portion of matrix solution (0.3 μl). The sample was dried at 25 °C prior to mass analysis.
RESULTS
1-Phosphatase Activity in Membranes of Wild Type Virulent Strains of H. pylori
Membranes from virulent strains of H. pylori were isolated and assayed with 4′-32P-lipid IVA, a key lipid A precursor substrate, for 1-phosphatase activity. As shown in Fig. 2, membranes from both H. pylori strains 26695 and J99 display 1-phosphatase activity, denoted as 1-dephospho-4′-32P-lipid IVA (Fig. 2, lanes 3, 4, 6, and 7). Removal of the 1-phosphate group from 4′-32P-lipid IVA increases the hydrophobicity of the lipid substrate, resulting in a faster migrating lipid species when analyzed by TLC in the solvent chloroform, pyridine, 88% formic acid, and water (50:50:16:5, v/v). The phosphatase activity seen in Fig. 2 is not a result of 4′-phosphatase activity, because 4′-phosphatase activity would catalyze the release of inorganic 32Pi from the 4′-position of lipid A when analyzed by TLC. However, a slower migrating species, lipid IVB, was detected from membranes of wild type E. coli strain W3110 after 3 h at 30 °C (Fig. 2, lane 5). Lipid IVB is generated by the addition of a palmitate chain to the amide-linked β-hydroxmyristate residue at position 2 of lipid IVA (40, 41) that is catalyzed by the E. coli outer membrane protein, PagP (40, 42–44). The lipid A of E. coli normally contains phosphate groups at both the 1- and 4′-positions (5) and is not dephosphorylated prior to the addition of a pEtN or L-Ara4N. As a result, membranes from wild type E. coli strain W3110 do not show 1-phosphatase activity (Fig. 2, lanes 2 and 5).
Fig. 2. Detection of 1-phosphatase activity in membranes of wild type strains of H. pylori.

Membranes from either E. coli wild type strain W3110 or H. pylori wild type strains (26695 and J99) were assayed for 1-phosphatase activity using assay conditions described under “Experimental Procedures.” The protein concentration was 1.0 mg/ml, and assays were carried out for both 30 min (A) and 3 h (B) at 30 °C. Each reaction contained 5 μM 4′-32P-lipid IVA substrate. Reaction products were separated by TLC and detected with PhosphorImager analysis. Removal of the 1-phosphate group was indicated by conversion of the 4′-32P-lipid IVA substrate to a faster migrating species (26).
Identification of the H. pylori 1-Phosphatase (Hp0021) from Putative Orthologs of E. coli PgpB
Recently, Karbarz et al. (46) identified LpxE, a phosphatase that selectively removes the 1-phosphate group of Rhizobium leguminosarum lipid A. Discovery of LpxE revealed that it is distantly related to the membrane-bound phosphatidylglycerol phosphatase in E. coli (PgpB) (45–48). Therefore, we searched the COG protein data base (49–52) using E. coli PgpB to identify possible H. pylori lipid phosphatases. COG0671 contains four putative orthologs of PgpB in H. pylori strain 26695, designated Hp0021, Hp0350, Hp0851, and Hp1580. The H. pylori PgpB orthologs were examined for 1-phosphatase activity by expressing the putative lipid phosphatases in the E. coli K12 strain NovaBlue(DE3). Membranes isolated from NovaBlue(DE3) containing either pET21a(+) (vector control), pHp0021, pHp1580, pHp0851, or pHp0350 were assayed with 4′-32P-lipid IVA for 1-phosphatase activity (Fig. 3). NovaBlue(DE3) membranes expressing Hp0021 displayed a considerable amount of 1-phosphatase activity (Fig. 3, lane 3) as compared with membranes from cells containing the vector control (Fig. 3, lane 2). In contrast, Nova-Blue(DE3) membranes expressing either Hp0350 or Hp1580 (Fig. 3, lanes 4 and 5) did not demonstrate any 1-phosphatase activity. NovaBlue(DE3) membranes expressing Hp0851 (Fig. 3, lane 6) did display minor amounts of 1-phosphatase activity; however, further examination revealed that the activity was nonspecific (see below).
Fig. 3. Assay for 1-phosphatase activity from E. coli membranes overexpressing putative H. pylori PgpB homologs (Hp0021, Hp1580, Hp0851, and Hp0350).

Membranes from E. coli K12 strain, NovaBlue(DE3), containing either the vector control or the indicated hybrid plasmids were assayed for 1-phosphatase activity using 4′-32P-lipid IVA. The protein concentration was 0.05 mg/ml, and assays were carried out for 1 h at 30 °C with 5 μM 4′-32P-lipid IVA substrate. The products were separated by TLC and detected with PhosphorImager analysis.
Assay Conditions and Catalytic Properties of Hp0021
The Hp0021 protein is predicted to be 190 amino acids long and contains three membrane-spanning regions (see sosui.proteome.bio.tuat.ac.jp/sosuiframe0.html). The 1-phosphatase activity of Hp0021 is absolutely dependent on the presence of the nonionic detergent, Triton X-100, with optimal activity at 0.2% detergent. The pH optimum is 6.0, but significant activity is observed from pH 5.5 to 8.0. Divalent cations are not required, and EDTA has no effect on activity. The substrate specificity of Hp0021 is relatively broad (Fig. 4A) and does not require the Kdo moiety or complete acylation for activity, as indicated by the higher amount of activity with 4′-32P-lipid IVA than with either Kdo2-4′-32P-lipid IVA or Kdo2-4′-32P-lipid A. Formation of the 1-dephosphorylated reaction products is linear with both protein concentration and time. As shown in Fig. 4B, the 1-de-phosphorylation of 5 μM 4′-32P-lipid IVA by 0.01 mg/ml Nova-Blue(DE3)/pHp0021 membranes is linear with time for ~40 min at 30 °C. Next, the complete 1-dephosphorylation of 5 μM 4′-32P-lipid IVA is seen after 1.5 h when using 0.05 mg/ml of membrane protein (Fig. 4A). Overall, the H. pylori lipid A 1-phosphatase is a very stable enzyme having a specific activity of ~170 nmol/min/mg with lipid IVA as the substrate and E. coli membranes overexpressing Hp0021 as the enzyme source.
Fig. 4. Substrate specificity and time dependence of the H. pylori lipid A 1-phosphatase.
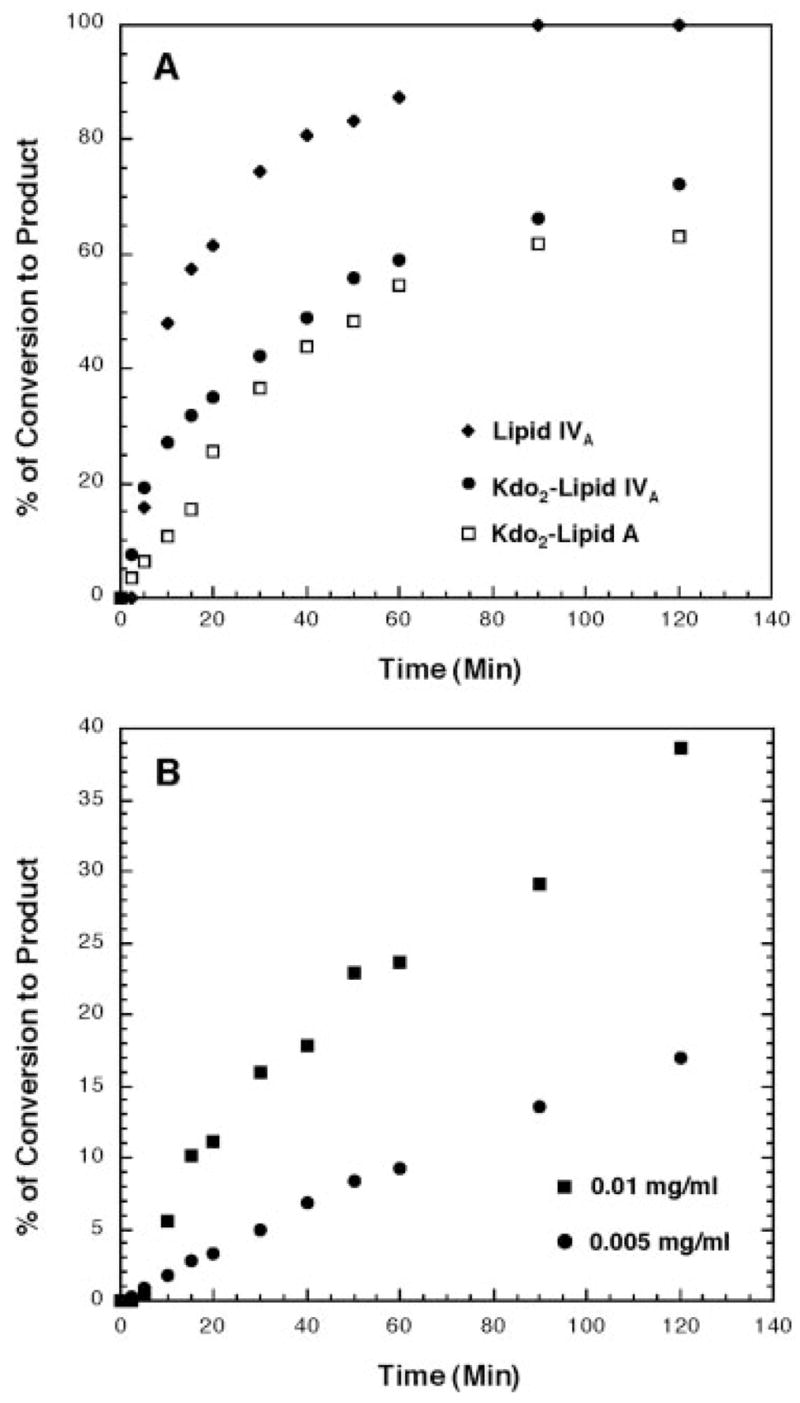
A, the 1-phosphatase activity of NovaBlue(DE3) membranes expressing Hp0021 was assayed under standard conditions described under “Experimental Procedures” with either 5 μM 4′-32P-lipid IVA, Kdo2-4′32P-lipid IVA, or Kdo2-4′32P-lipid A. Protein concentrations at 0.05 mg/ml from NovaBlue(DE3)/pHp0021 membranes served as the enzyme source. Reaction products were separated by TLC as described under “Experimental Procedures” and subjected to PhosphorImager analysis. B, the 1-phosphatase activity of Hp0021 was assayed under standard conditions with 5 μM 4′-32P-lipid IVA. Membranes from NovaBlue(DE3) expressing Hp0021 at the indicated concentrations served as the enzyme source. Reaction products were separated by TLC as described under “Experimental Procedures” and subjected to PhosphorImager analysis.
Low Copy Expression of H. pylori PgpB Orthologs in E. coli
Assay of E. coli membranes expressing Hp0851 showed minor amounts of 1-phosphatase activity (Fig. 3, lane 6). To confirm that Hp0021 functions as a lipid A phosphatase and to ensure that Hp0851 phosphatase activity was nonspecific, we isolated the lipid A from E. coli expressing the individual supposed H. pylori PgpB orthologs. The putative lipid phosphatases were cloned into the low copy expression vector pETCoco-2 to generate the following plasmids: pHp0021coco, pHp1580coco, pHp0851coco, and pHp0350coco. A low copy expression system was utilized to avoid nonspecific phosphatase activity generated from high levels of protein expression. Cultures of the E. coli K12 strain HMS174(DE3) containing one of the pETCoco-2 derived plasmids were labeled with 32Pi, and the lipid A fraction was isolated to determine possible modifications in vivo.
HMS174(DE3) containing the pETcoco-2 vector control produced the typical 32P-labeled lipid A species seen in wild type E. coli K12, 1,4′-bisphosphate lipid A, and 1-pyrophosphate lipid A. The latter contains a pyrophosphate at the 1-position of the disaccharide (Fig. 1) and represents approximately one-third of the lipid A found in E. coli grown on nutrient broth (16, 28). Low copy expression of Hp0021 resulted in the production of only 1-dephosphorylated lipid A (Fig. 5, lane 2) illustrating that Hp0021 functions as a lipid A phosphatase. 32P-Labeled lipid A fractions isolated from cells containing pHp0350coco, pHp0851coco, and pHp1580coco did not contain any significant amount of 1-dephosphorylated lipid A species (Fig. 5, lanes 3–5). The minor amount of 1-dephosphorylated lipid seen in Fig. 5, lanes 3–5, arises as a decomposition product during mild acid hydrolysis at 100 °C used to free lipid A from LPS (16, 17, 36). An increase in Hp0851 expression by increasing copy number of the pHp0851coco plasmid did not result in any production of dephosphorylated lipid A species (data not shown), confirming that only Hp0021 functions as a lipid A 1-phospha-tase in vivo. Bacteria showed normal growth during expression of either Hp0021 or Hp1580. However, induction of Hp0851 and Hp0350 resulted in a decrease in growth rate (data not shown). Although we were unable to isolate as much 32P-labeled lipid A from cells expressing Hp0851, it appeared that HMS174(DE3) containing pHp0851coco did not produce the 1-pyrophosphorylated lipid A species.
Fig. 5. Lipid A composition in E. coli K12 strains expressing putative H. pylori PgpB orthologs.

The 32P-labeled lipid A species from HMS174(DE3) containing the vector control (pETcoco-2) or the indicated hybrid plasmids were isolated as described under “Experimental Procedures.” The labeled lipid A species were separated by TLC using the solvent chloroform, pyridine, 88% formic acid, water (50:50: 16:5, v/v) and visualized by PhosphorImaging. The presence of 1-de-phosphorylated-lipid A from HMS174(DE3) expressing Hp0021 is indicated. A small amount of 1-dephosphorylated-lipid A may arise as a decomposition product during hydrolysis of the Kdo linkage at pH 4.5 at 100 °C (16, 17, 36). A reduced amount of 32P-labeled lipid A was isolated from bacteria expressing Hp0851 and Hp0350 because of toxic effects upon protein expression.
MALDI-TOF Mass Spectrometry of the 1-Phosphatase Reaction Product
To confirm that hp0021 is the structural gene for H. pylori lipid A 1-phosphatase, the reaction product utilizing mature Kdo2-lipid A as the substrate was analyzed by MALDI-TOF mass spectrometry in the negative mode. MALDI-TOF mass spectrometry of the initial substrate E. coli Kdo2-lipid A revealed a major ion peak at m/z 2238.7 atomic mass units with additional minor ions at m/z 2019.3 and 1798.7 atomic mass units (Fig. 6A). The signal at m/z 2238.7 atomic mass units is interpreted as [M – H] − of the predominant hexa-acylated Kdo2-lipid A 1,4′-bisphosphate molecule (Fig. 6A), characteristic of E. coli K12. The peaks at m/z 2019.3 and 1798.7 atomic mass units are interpreted as [M – H-Kdo] − and [M – H-Kdo2] −, respectively, and correspond to the loss of the labile Kdo moiety during mass spectrometry (Fig. 6A). MALDI-TOF mass spectrometry of the Hp0021 reaction product revealed a predominant peak at m/z 2158.4 atomic mass units confirming the enzymatic removal of the 1-phosphate group (Fig. 6B) from the Kdo2-lipid A substrate. Again, the minor peaks at m/z 1938.9 and 1718.5 arise from the loss of either one or two Kdo moieties from the 1-dephosphorylated reaction product, respectively (Fig. 6B).
Fig. 6. MALDI-TOF mass spectrometry of the 1-phosphatase reaction product generated from Kdo2-lipid A.

The reaction product generated from incubation of hexa-acylated Kdo2-lipid A with NovaBlue(DE3)/pHp0021 membranes was purified by DEAE-cellulose chromatography as described under “Experimental Procedures.” The spectra shown in A is that of the starting Kdo2-lipid A substrate, and the spectra shown in B shows the resulting 1-dephosphorylated reaction product, 1-dephosphorylated-Kdo2-lipid A. Both spectra were acquired in the negative ion mode.
Identification of the Structural Gene Encoding the H. pylori Lipid A pEtN Transferase (Hp0022)
We have demonstrated that Hp0021 is responsible for removing the 1-phosphate group from the proximal glucosamine of lipid A. However, the major lipid species of H. pylori has a pEtN unit directly linked to the 1-position of the disaccharide backbone (Fig. 1C). This suggests that the modification of H. pylori lipid A at the 1-position also requires a lipid A pEtN transferase. Recently, Cox et al. (53) identified NMB1638, renamed LptA, as a pEtN transferase specific for N. meningitidis lipid A based upon loss of pEtN from lipid A in an lptA mutant. Also, it has been shown in vitro that E. coli YjdB (EptA) transfers pEtN to E. coli lipid A precursors (54). Examination of the COG data base using either N. meningitidis NMB1638 or E. coli YjdB to search for orthologs in H. pylori strain 26695 revealed two possible orthologs designated Hp0022 and Hp1417m.
We decided to investigate Hp0022 as the possible H. pylori lipid A pEtN transferase primarily because of its homology to LptA (E value <10−48 using BLASTp) and also because its structural gene is located in a putative operon with hp0021 in the H. pylori genome. Hp0022 is estimated to be 521 amino acids long and is predicted to contain multiple membrane-spanning regions (see sosui.proteome.bio.tuat.ac.jp/sosuiframe0. html). Membranes isolated from NovaBlue(DE3) containing either pET21a(+) or pHp0022 were assayed for pEtN transferase activity using the substrates 4′-32P-lipid IVA, Kdo2-4′-32P-lipid IVA, and Kdo2-4′-32P-lipid A. Assaying Hp0022 with lipid A precursor substrates that were still phosphorylated at the 1-position did not demonstrate any 1-pEtN transferase activity when analyzed by TLC (data not shown). These data suggest that the activity of Hp0022 may require a lipid A substrate that is initially dephosphorylated at the 1-position prior to the addition of a pEtN.
To demonstrate that the 1-phosphate group modification of H. pylori may require two enzymatic steps, we developed a dual protein expression system. The target proteins were expressed under the control of two separate T7lac promoters by cloning hp0021 and hp0022 into two separate multiple cloning sites of pETDuet-1 giving the plasmid pHp0021/Hp0022. In addition, Hp0021 and Hp0022 were individually cloned into pETDuet-1 giving the plasmids pHp0021duet and pHp0022duet. Separation of 32P-labeled lipid A species from NovaBlue(DE3) containing the vector pHp0021duet clearly demonstrates again that Hp0021 is responsible for the presence of 1-phosphatase-modified lipid A species (Fig. 7, lane 2). However, cells containing the pDuet-1 vector control contained the typical 1,4′-bisphos-phorylated and the 1-pyrophosphorylated E. coli lipid A species. As demonstrated in Fig. 7, lane 3, 32P-labeled lipid A species isolated from NovaBlue(DE3) containing pHp0022duet were similar to those found in cells containing the pDuet-1 vector control suggesting that Hp0022 alone does not modify E. coli lipid A. 32P-Labeled lipid A species isolated from Nova-Blue (DE3) containing the dual expression construct, pHp0021/Hp0022, revealed the following three lipid species: 1,4′-bisphosphorylated lipid A, 1-dephosphorylated lipid A, and lipid A containing a pEtN group at the C-1 position (see Fig. 8). Removal of the 1-phosphate group from 1,4′-bisphosphorylated lipid A followed by the addition of 1-pEtN unit generated a lipid species that migrates in a similar fashion to 1-pyrophosphate lipid A (Fig. 7, lane 4). Therefore, mass spectrometry was employed to differentiate between the two lipid species and to confirm that hp0022 encodes for a pEtN transferase acting after 1-dephosphorylation of H. pylori lipid A.
Fig. 7. Dual expression of Hp0021 and Hp0022 in E. coli results in the formation of 1-pEtN-lipid A species.

The 2P-labeled lipid A species from the indicated strains were isolated as described under “Experimental Procedures,” separated by TLC using the solvent chloroform, pyridine, 88% formic acid, water (50:50:16:5, v/v), and visualized by PhosphorImaging. A small amount of 1-dephosphorylated-lipid A may arise as a decomposition product during hydrolysis of the Kdo linkage at pH 4.5 at 100 °C (16, 17, 36). The structure of the lipid A species labeled as 1-pEtN-Lipid A was verified by mass spectrometry (Fig. 9D).
Fig. 8. MALDI-TOF mass spectrometry of lipid A from E. coli expressing the H. pylori lipid A 1-phosphatase and pEtN transferase genes.

The lipid A of E. coli NovaBlue(DE3) containing either pDuet-1 (A), pHp0021duet (B), pHp0022duet (C), or pHp0021/Hp0022 (D) was isolated and fractionated by DEAE-cellulose chromatography using published protocols (38). The lipid A samples were analyzed by MALDI-TOF mass spectrometry in the negative-ion mode. Although mass spectrometry data was acquired on lipids eluting in all salt fractions from the DEAE column, only the most representative data is shown for comparison. Data shown in A and C are from lipid A species found to elute in the 240 mM salt fraction, whereas data shown in B and D are from lipids eluting in the 60 mM salt fraction (see “Experimental Procedures”). The molecular weights of E. coli hexa-acylated bisphosphate lipid A, 1-dephosphorylated-lipid A, and 1-pEtN-lipid A are 1798.4, 1719.4, and 1841.4, respectively. E shows the positive ion spectrum of lipid A obtained from NovaBlue(DE3) containing pHp0021duet.
The lipid A of E. coli strain NovaBlue(DE3) containing either pETDuet-1, pHp0021duet, pHP0022duet, or pHp0021/Hp0022 was isolated and analyzed by MALDI-TOF mass spectrometry. The lipid A of NovaBlue(DE3) containing the control vector, pETDuet-1, consisted primarily of the hexa-acylated bisphosphate species with MALDI-TOF analysis resulting in a major peak at m/z 1798.4 (16, 38) (Fig. 8A). Mass spectrometry analysis of NovaBlue(DE3) containing the control vector also revealed the presence of hexa-acylated pyrophosphate (data not shown). Similar results were seen upon examination of the lipid A fraction of NovaBlue(DE3) expressing Hp0022 (Fig. 8C). As expected, expression of Hp0021 resulted in the loss of a single phosphate unit from lipid A (Fig. 8B), as indicated by [M – H]− at m/z 1719.0 in the negative mode. Furthermore, the positive ion spectrum showed the presence of B1+ ion at m/z 1087.3 indicating that the distal portion of the lipid A was not dephosphorylated (Fig. 8E). Mass spectrometry analysis of lipid A isolated from cells overexpressing both Hp0021 and Hp0022 demonstrated two ion peaks at m/z 1719.0 and 1840.9 atomic mass units in the negative mode (Fig. 8D). The ion peak at m/z 1719.0 corresponds to the loss of a phosphate group of lipid A. However, the ion peak at m/z 1840.9 correlates to the addition of a pEtN group that is interpreted as the addition of a pEtN unit to previously dephosphorylated lipid A. Therefore, based on these data, it can be concluded that the modification of the 1-phosphate group of H. pylori lipid A requires two enzymatic steps as follows: 1) removal of the 1-phosphate group by a specific phosphatase (Hp0021), and 2) addition of a pEtN unit directly to the glucosamine disaccharide backbone by a pEtN transferase (Hp0022).
Lipid A Transport across the Inner Membrane Is Required for Hp0021-catalyzed 1-Dephosphorylation
Kdo2-lipid A is synthesized on the cytoplasmic face of the inner membrane followed by the addition of the core oligosaccharide. The core-Kdo2-lipid A molecule is then transported across the inner membrane by the essential flippase, MsbA. As described previously by Doerrler et al. (67), MsbA-dependent transport of lipid A across the inner membrane is lost in the E. coli temperature-sensitive mutant WD2 by shifting cells from 30 to 44 °C for 30 min during the mid-log phase of growth. We have confirmed that Hp0021 1-phosphatase activity is localized to the inner membrane fraction based upon isopycnic density gradient separation of membranes from NovaBlue(DE3)-expressing Hp0021 (data not shown). By using WD2, we investigated whether or not the active site of Hp0021 was facing the periplasmic or cytosolic face of the inner membrane.
E. coli WD2 and its parent strain W3110A were transformed with either pWSK29 or pWSHp0021 (pWSK29 expressing Hp0021). Cells were grown at the permissive temperature of 30 °C until A600 of 0.6–0.7, following a temperature shift to 44 °C for 30 min, to inactivate MsbA-dependent lipid A transport. Cells were then labeled with 4 μCi/ml 32Pi for 20 min, and the newly synthesized 32P-labeled lipid A species was isolated and separated by TLC. At 30 °C, W3110A (temperature-sensitive control) and WD2 synthesized solely 1-dephosphorylated lipid A species (Fig. 9) when containing the plasmid pWSHp0021. Shifting W3110A expressing Hp0021 to 44 °C had no effect on production of 1-dephosphorylated lipid A. However, inactivation of MsbA at 44 °C in WD2/pWSHp0021 resulted in the loss of 1-dephospho-rylation (Fig. 9), indicating that the H. pylori 1-phosphatase active site is oriented toward the periplasm. Recently, Wang et al. (55) showed a similar result using a lipid A 1-phosphatase cloned from Franciscella novicida.
Fig. 9. Removal of the lipid A 1-phosphate group by Hp0021 is dependent upon the lipid A transporter, MsbA.

E. coli strains WD2 and W3110A containing either pWSK29 or pWSHp0021 were temperature-shifted to 44 °C for 30 min. Newly synthesized lipids were 2P-labeled for 20 min following temperature shift, and the lipid A was isolated as described under “Experimental Procedures.” Lipid A species from the indicated strains were separated by TLC using the solvent chloroform, pyridine, 88% formic acid, water (50:50:16:5, v/v) and visualized by PhosphorImaging.
DISCUSSION
The LPS of H. pylori is distinct from enterobacterial LPS in having a unique lipid A structure and many types of O-antigen polysaccharide, including Lewis antigen structures (23–25, 56, 57). H. pylori lipid A shows very low endotoxic properties (14, 19, 20) and has longer fatty acyl chains and fewer phosphate residues (Fig. 1C) (13, 14, 19). A number of Gram-negative bacteria modify the phosphate groups of their lipid A (58). Recently, Lee et al. (72) has shown that addition of pEtN to the 1-phosphate of S. enterica is important for resistance to the antimicrobial peptide polymyxin. In the case of H. pylori, the C-1 hydroxyl group of the proximal glucosamine sugar is derivatized with pEtN (13, 14, 19) (Fig. 1C). We report here that the pEtN moiety found at the C-1 position of H. pylori lipid A arises from the removal of the lipid A 1-phosphate group followed by the addition of pEtN to form a glycosidic phosphodi-ester linkage.
Recently, Karbarz et al. (46) reported the cloning of a membrane-bound enzyme, LpxE, which removes the 1-phosphate group of R. leguminosarum lipid A. In R. leguminosarum, the removal of the 1-phosphate group is followed by oxidation of the proximal sugar by the outer membrane enzyme LpxQ (59, 60). LpxE shares sequence homology to E. coli PgpB (45, 47, 48), primarily in the conserved active site motif (61) found in many lipid phosphatases. Examination of the COG data base shows that H. pylori strain 26695 contains four putative PgpB orthologs designated as Hp0021, Hp0350, Hp0851, and Hp1580. We had already detected the presence of a 1-phosphatase activity in membranes of H. pylori (Fig. 2). However, by assaying membranes of E. coli expressing the individual H. pylori PgpB orthologs using key 32P-labeled lipid A precursors as substrates, we showed that Hp0021 functions as a lipid A 1-phosphatase (Fig. 3).
Both LpxE and Hp0021 have multitransmembrane domains and, like other lipid phosphatases, require detergent for activity (45, 61, 62). Hp0021 was capable of removing the 1-phosphate group from various lipid A precursors, including mature Kdo2-lipid A (Figs. 4 and 6). Membranes from wild type H. pylori strains 26695 and J99 catalyzed the 1-dephosphorylation of lipid IVA at a specific activity of ~1.4 nmol/min/mg, whereas Rhizobium membranes showed a specific activity of only 0.014 nmol/min/mg. In comparison, membranes of E. coli overexpressing Hp0021 showed a specific activity of 170 nmol/min/mg resulting in more than a 100-fold increase over wild type levels. Most interestingly, expression of Hp0021 in E. coli K12 strains NovaBlue(DE3) (Fig. 7 and Fig. 8B) and HMS174(DE3) (Fig. 5) resulted in nearly 100% production of 1-dephosphorylated lipid A with no significant effects on bacterial growth, suggesting that removal of the 1-phosphate group of lipid A is not toxic to growth under normal laboratory conditions. The ability to generate E. coli strains lacking the 1-phosphate group of lipid A may prove to be useful for the development of specific vaccines. Monophosphorylated lipid A has been shown to be an effective adjuvant retaining the immunostimulatory properties of LPS but with greatly reduced toxic effects (63–65).
We also have presented data suggesting that Hp0022 functions as the H. pylori lipid A pEtN transferase. However, enzymatic data using defined substrates will be needed to confirm the function of Hp0022. The hp0021 and hp0022 genes appear to form a typical prokaryotic operon because the individual open reading frames are separated by only 10 bp, and the genes are unidirectionally transcribed beginning with hp0022. Hp0022 shares homology with the recently described LptA that is predicted to serve as a putative lipid A pEtN transferase in N. meningitidis (53). Based upon topology prediction programs (see sosui.proteome.bio.tuat.ac.jp/sosuiframe0.html), Hp0022 has six transmembrane regions with the soluble C-terminal region predicted to be located in the periplasmic region of the cell envelope. Expression of Hp0022 alone in an E. coli K12 strain did not result in modification to the bisphosphorylated E. coli lipid A structure. However, expression of both Hp0021 and Hp0022 together using a dual expression system resulted in the production of a lipid A species modified at the C-1 position with pEtN (Figs. 7 and 8). These findings confirmed our hypothesis that H. pylori 1-phosphate group modification is a two-step process. One would expect a similar mechanism to occur in the Gram-negative pathogen Porphyromonas gingivalis, the lipid A of which also contains a pEtN rather than a pyrophosphoethanolamine group at the 1-position. BLASTp analysis shows a distant homolog (E value <10−20) of Hp0022 in P. gingivalis. However, there are no significant homologs of Hp0021 (E value <10− 4 using BLASTp) in the sequenced genome of P. gingivalis W83. This is not too surprising considering Hp0021 is only distantly related to Rhizobium LpxE and is not detected as a H. pylori homolog using BLASTp. Therefore, further biochemical investigation will be required to determine whether P. gingivalis uses a mechanism similar to that of H. pylori during the modification of its lipid A.
With our evidence that Hp0021 phosphatase activity in E. coli is MsbA-dependent, we propose a periplasmic model for the modification of the H. pylori lipid A 1-phosphate group (Fig. 10). Following synthesis of bisphosphorylated Kdo2-lipid A via the constitutive pathway, the lipid is flipped across the inner membrane by the ABC transporter MsbA (28, 39, 66–68). Hp0021, an inner membrane phosphatase, then removes the proximal phosphate group from H. pylori lipid A. This is followed by addition of a pEtN moiety to the disaccharide backbone catalyzed by the lipid A pEtN transferase Hp0022. It is very likely that the glycerophospholipid PtdEtn serves as the donor substrate because E. coli that are unable to synthesize PtdEtn are unable to modify their lipid A with pEtN residues (69). On the whole, modification of the lipid A structure in the extracytoplasmic compartments has become a theme in the biosynthesis of lipid A given that a number of lipid A-modifying enzymes are outer membrane proteins (30, 40) or have active sites facing the periplasmic region (55, 69–72).
Fig. 10. Proposed topology of the cleavage and modification of the 1-phosphate group of H. pylori lipid A.

After assembly of Kdo2-lipid A bearing the core oligosaccharide (core-lipid A), the macromolecule is transported across the inner membrane by the ABC transporter, MsbA (28, 67). In H. pylori, the 1-phosphate group of the lipid A domain can then be removed by a specific phosphatase Hp0021. This is followed by the addition of a pEtN unit directly to the glucosamine disaccharide backbone catalyzed by the lipid A pEtN transferase, Hp0022. The predicted membrane topology of Hp0021 and Hp0022 was determined using the program SOSUI (sosui.proteome.bio.tuat.ac.jp/sosuiframe0.html) (75, 76).
Our work has corroborated the structure reported previously for H. pylori lipid A (13–15). We are currently in the process of constructing H. pylori mutants lacking functional copies of hp0021 and hp0022. These mutants will help us determine the importance of pEtN addition to the C-1 hydroxyl group of H. pylori lipid A during infection. Also, because none of the H. pylori PgpB orthologs showed lipid A 4′-phosphatase activity, we are actively trying to identify the structural gene encoding this enzyme. Changing the charge of the lipid A domain of H. pylori LPS most likely aids the bacterium in establishing the prolonged chronic infections seen with this organism by providing resistance to cationic antimicrobial peptides and reducing the endotoxic principle of H. pylori LPS.
Acknowledgments
A. X. Tran thanks C. M. Stead and A. L. Proffitt for help with preparation of this manuscript and P. B. Wyrick for continued support.
Footnotes
This work was supported by National Institutes of Health Grants K22-AI53645 (to M. S. T.), R37-GM51796 (to C. R. H. R.), and RO1-GM6440 (to R. J. C)
The abbreviations used are: LPS, lipopolysaccharide; pEtN, phosphoethanolamine; MES, 2-(N-morpholino) ethanesulfonic acid; IPTG, iso-propyl-1-thio- β-D-galactopyranoside; Kdo, 3-deoxy-D-manno-octulosonic acid; MALDI-TOF, matrix assisted laser-desorption/time of flight.
References
- 1.Raetz CR, Whitfield C. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raetz CR. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 3.Gronow S, Brade H. J Endotoxin Res. 2001;7:3–23. [PubMed] [Google Scholar]
- 4.Galloway SM, Raetz CR. J Biol Chem. 1990;265:6394–6402. [PubMed] [Google Scholar]
- 5.Raetz CRH. In: Cellular and Molecular Biology. Niedhardt F, editor. American Society for Microbiology; Washington, D. C: 1996. pp. 1035–1063. [Google Scholar]
- 6.Raetz CR. J Bacteriol. 1993;175:5745–5753. doi: 10.1128/jb.175.18.5745-5753.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bone RC. Clin Microbiol Rev. 1993;6:57–68. doi: 10.1128/cmr.6.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Medzhitov R, Janeway C., Jr Trends Microbiol. 2000;8:452–456. doi: 10.1016/s0966-842x(00)01845-x. [DOI] [PubMed] [Google Scholar]
- 9.Medzhitov R, Janeway C., Jr N Engl J Med. 2000;343:338–344. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- 10.Aderem A, Ulevitch RJ. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 11.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 12.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 13.Moran AP, Lindner B, Walsh EJ. J Bacteriol. 1997;179:6453–6463. doi: 10.1128/jb.179.20.6453-6463.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suda Y, Kim YM, Ogawa T, Yasui N, Hasegawa Y, Kashihara W, Shimoyama T, Aoyama K, Nagata K, Tamura T, Kusumoto S. J Endotoxin Res. 2001;7:95–104. [PubMed] [Google Scholar]
- 15.Suda Y, Ogawa T, Kashihara W, Oikawa M, Shimoyama T, Hayashi T, Tamura T, Kusumoto S. J Biochem (Tokyo) 1997;121:1129–1133. doi: 10.1093/oxfordjournals.jbchem.a021705. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Z, Lin S, Cotter RJ, Raetz CR. J Biol Chem. 1999;274:18503–18514. doi: 10.1074/jbc.274.26.18503. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Z, Ribeiro AA, Lin S, Cotter RJ, Miller SI, Raetz CR. J Biol Chem. 2001;276:43111–43121. doi: 10.1074/jbc.M106960200. [DOI] [PubMed] [Google Scholar]
- 18.Kulshin VA, Zahringer U, Lindner B, Frasch CE, Tsai CM, Dmitriev BA, Rietschel ET. J Bacteriol. 1992;174:1793–1800. doi: 10.1128/jb.174.6.1793-1800.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogawa T, Asai Y, Sakai Y, Oikawa M, Fukase K, Suda Y, Kusumoto S, Tamura T. FEMS Immunol Med Microbiol. 2003;36:1–7. doi: 10.1016/S0928-8244(03)00093-2. [DOI] [PubMed] [Google Scholar]
- 20.Ogawa T, Suda Y, Kashihara W, Hayashi T, Shimoyama T, Kusumoto S, Tamura T. Vaccine. 1997;15:1598–1605. doi: 10.1016/s0264-410x(97)00102-3. [DOI] [PubMed] [Google Scholar]
- 21.Schromm AB, Brandenburg K, Loppnow H, Moran AP, Koch MH, Rietschel ET, Seydel U. Eur J Biochem. 2000;267:2008–2013. doi: 10.1046/j.1432-1327.2000.01204.x. [DOI] [PubMed] [Google Scholar]
- 22.Schromm AB, Brandenburg K, Loppnow H, Zahringer U, Rietschel ET, Carroll SF, Koch MH, Kusumoto S, Seydel U. J Immunol. 1998;161:5464–5471. [PubMed] [Google Scholar]
- 23.Appelmelk BJ, Monteiro MA, Martin SL, Moran AP, Vandenbroucke-Grauls CM. Trends Microbiol. 2000;8:565–570. doi: 10.1016/s0966-842x(00)01875-8. [DOI] [PubMed] [Google Scholar]
- 24.Appelmelk BJ, Negrini R, Moran AP, Kuipers EJ. Trends Microbiol. 1997;5:70–73. doi: 10.1016/S0966-842X(96)10084-6. [DOI] [PubMed] [Google Scholar]
- 25.Appelmelk BJ, Vandenbroucke-Grauls CM. Gut. 2000;47:10–11. doi: 10.1136/gut.47.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller JR. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 27.Bligh EG, Dyer WJ. Can J Med Sci. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CR. J Biol Chem. 1998;273:12466–12475. doi: 10.1074/jbc.273.20.12466. [DOI] [PubMed] [Google Scholar]
- 29.Vorachek-Warren MK, Ramirez S, Cotter RJ, Raetz CR. J Biol Chem. 2002;277:14194–14205. doi: 10.1074/jbc.M200409200. [DOI] [PubMed] [Google Scholar]
- 30.Trent MS, Pabich W, Raetz CR, Miller SI. J Biol Chem. 2001;276:9083–9092. doi: 10.1074/jbc.M010730200. [DOI] [PubMed] [Google Scholar]
- 31.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 32.Garrett TA, Kadrmas JL, Raetz CR. J Biol Chem. 1997;272:21855–21864. doi: 10.1074/jbc.272.35.21855. [DOI] [PubMed] [Google Scholar]
- 33.Basu SS, York JD, Raetz CR. J Biol Chem. 1999;274:11139–11149. doi: 10.1074/jbc.274.16.11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belunis CJ, Raetz CR. J Biol Chem. 1992;267:9988–9997. [PubMed] [Google Scholar]
- 35.Guy-Caffey JK, Rapoza MP, Jolley KA, Webster RE. J Bacteriol. 1992;174:2460–2465. doi: 10.1128/jb.174.8.2460-2465.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osborn MJ, Gander JE, Parisi E, Carson J. J Biol Chem. 1972;247:3962–3972. [PubMed] [Google Scholar]
- 37.Dulbecco R, Vogt M. J Exp Med. 1954;99:167–182. doi: 10.1084/jem.99.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Odegaard TJ, Kaltashov IA, Cotter RJ, Steeghs L, van der Ley P, Khan S, Maskell DJ, Raetz CR. J Biol Chem. 1997;272:19688–19696. doi: 10.1074/jbc.272.32.19688. [DOI] [PubMed] [Google Scholar]
- 39.Doerrler WT, Raetz CR. J Biol Chem. 2002;277:36697–36705. doi: 10.1074/jbc.M205857200. [DOI] [PubMed] [Google Scholar]
- 40.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CR. EMBO J. 2000;19:5071–5080. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brozek KA, Bulawa CE, Raetz CR. J Biol Chem. 1987;262:5170–5179. [PubMed] [Google Scholar]
- 42.Belden WJ, Miller SI. Infect Immun. 1994;62:5095–5101. doi: 10.1128/iai.62.11.5095-5101.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gunn JS, Belden WJ, Miller SI. Microb Pathog. 1998;25:77–90. doi: 10.1006/mpat.1998.0217. [DOI] [PubMed] [Google Scholar]
- 44.Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, Hackett M, Miller SI. Cell. 1998;95:189–198. doi: 10.1016/s0092-8674(00)81750-x. [DOI] [PubMed] [Google Scholar]
- 45.Icho T. J Bacteriol. 1988;170:5117–5124. doi: 10.1128/jb.170.11.5117-5124.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karbarz MJ, Kalb SR, Cotter RJ, Raetz CR. J Biol Chem. 2003;278:39269–39279. doi: 10.1074/jbc.M305830200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dillon DA, Wu WI, Riedel B, Wissing JB, Dowhan W, Carman GM. J Biol Chem. 1996;271:30548–30553. doi: 10.1074/jbc.271.48.30548. [DOI] [PubMed] [Google Scholar]
- 48.Icho T, Raetz CR. J Bacteriol. 1983;153:722–730. doi: 10.1128/jb.153.2.722-730.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, Tatusova TA, Wagner L. Nucleic Acids Res. 2003;31:28–33. doi: 10.1093/nar/gkg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tatusov RL, Galperin MY, Natale DA, Koonin EV. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tatusov RL, Koonin EV, Lipman DJ. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 52.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox AD, Wright JC, Li J, Hood DW, Moxon ER, Richards JC. J Bacteriol. 2003;185:3270–3277. doi: 10.1128/JB.185.11.3270-3277.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trent MS, Raetz CRH. J Endotoxin Res. 2002;8:158. [Google Scholar]
- 55.Wang X, Karbarz MJ, McGrath SC, Cotter RJ, Raetz CR. J Biol Chem. 2004;279:49470–49478. doi: 10.1074/jbc.M409078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monteiro MA, Appelmelk BJ, Rasko DA, Moran AP, Hynes SO, MacLean LL, Chan KH, Michael FS, Logan SM, O’Rourke J, Lee A, Taylor DE, Perry MB. Eur J Biochem. 2000;267:305–320. doi: 10.1046/j.1432-1327.2000.01007.x. [DOI] [PubMed] [Google Scholar]
- 57.Moran AP, Prendergast MM, Appelmelk BJ. FEMS Immunol Med Microbiol. 1996;16:105–115. doi: 10.1111/j.1574-695X.1996.tb00127.x. [DOI] [PubMed] [Google Scholar]
- 58.Trent MS. Biochem Cell Biol. 2004;82:71–86. doi: 10.1139/o03-070. [DOI] [PubMed] [Google Scholar]
- 59.Que-Gewirth NL, Karbarz MJ, Kalb SR, Cotter RJ, Raetz CR. J Biol Chem. 2003;278:12120–12129. doi: 10.1074/jbc.M300379200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Que-Gewirth NL, Lin S, Cotter RJ, Raetz CR. J Biol Chem. 2003;278:12109–12119. doi: 10.1074/jbc.M300378200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stukey J, Carman GM. Protein Sci. 1997;6:469–472. doi: 10.1002/pro.5560060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carman GM. Biochim Biophys Acta. 1997;1348:45–55. doi: 10.1016/s0005-2760(97)00095-7. [DOI] [PubMed] [Google Scholar]
- 63.Johnson AG. Clin Microbiol Rev. 1994;7:277–289. doi: 10.1128/cmr.7.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson AG, Tomai M, Solem L, Beck L, Ribi E. Rev Infect Dis. 1987;9(Suppl 5):512–516. doi: 10.1093/clinids/9.supplement_5.s512. [DOI] [PubMed] [Google Scholar]
- 65.Tomai MA, Solem LE, Johnson AG, Ribi E. J Biol Response Modif. 1987;6:99–107. [PubMed] [Google Scholar]
- 66.Chang G, Roth CB. Science. 2001;293:1793–1800. doi: 10.1126/science.293.5536.1793. [DOI] [PubMed] [Google Scholar]
- 67.Doerrler WT, Reedy MC, Raetz CR. J Biol Chem. 2001;276:11461–11464. doi: 10.1074/jbc.C100091200. [DOI] [PubMed] [Google Scholar]
- 68.Schmitt L. Chembiochem. 2002;3:161–165. doi: 10.1002/1439-7633(20020301)3:2/3<161::AID-CBIC161>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 69.Trent MS, Ribeiro AA, Doerrler WT, Lin S, Cotter RJ, Raetz CR. J Biol Chem. 2001;276:43132–43144. doi: 10.1074/jbc.M106962200. [DOI] [PubMed] [Google Scholar]
- 70.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CR. J Biol Chem. 2001;276:43122–43131. doi: 10.1074/jbc.M106961200. [DOI] [PubMed] [Google Scholar]
- 71.Doerrler WT, Gibbons HS, Raetz CR. J Biol Chem. 2004;279:45102–45109. doi: 10.1074/jbc.M408106200. [DOI] [PubMed] [Google Scholar]
- 72.Lee H, Hsu FF, Turk J, Groisman EA. J Bacteriol. 2004;186:4124–4133. doi: 10.1128/JB.186.13.4124-4133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou Z, Ribeiro AA, Raetz CR. J Biol Chem. 2000;275:13542–13551. doi: 10.1074/jbc.275.18.13542. [DOI] [PubMed] [Google Scholar]
- 74.Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI. Mol Microbiol. 1998;27:1171–1182. doi: 10.1046/j.1365-2958.1998.00757.x. [DOI] [PubMed] [Google Scholar]
- 75.Mitaku S, Ono M, Hirokawa T, Boon-Chieng S, Sonoyama M. Biophys Chem. 1999;82:165–171. doi: 10.1016/s0301-4622(99)00116-7. [DOI] [PubMed] [Google Scholar]
- 76.Hirokawa T, Boon-Chieng S, Mitaku S. Bioinformatics. 1998;14:378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 77.Wang RF, Kushner SR. Gene (Amst) 1991;100:195–199. [PubMed] [Google Scholar]