

Abstract
The lpcC gene of Rhizobium leguminosarum and the lpsB gene of Sinorhizobium meliloti encode protein orthologs that are 58% identical over their entire lengths of about 350 amino acid residues. LpcC and LpsB are required for symbiosis with pea and Medicago plants, respectively. S. meliloti lpsB complements a mutant of R. leguminosarum defective in lpcC, but the converse does not occur. LpcC encodes a highly selective mannosyl transferase that utilizes GDP-mannose to glycosylate the inner 3-deoxy-D-manno-octulosonic acid (Kdo) residue of the lipopolysaccharide precursor Kdo2-lipid IVA. We now demonstrate that LpsB can also efficiently mannosylate the same acceptor substrate as does LpcC. Unexpectedly, however, the sugar nucleotide selectivity of LpsB is greatly relaxed compared with that of LpcC. Membranes of the wild-type S. meliloti strain 2011 catalyze the glycosylation of Kdo2-[4′-32P]lipid IVA at comparable rates using a diverse set of sugar nucleotides, including GDP-mannose, ADP-mannose, UDP-glucose, and ADP-glucose. This complex pattern of glycosylation is due entirely to LpsB, since membranes of the S. meliloti lpsB mutant 6963 do not glycosylate Kdo2-[4′-32P]lipid IVA in the presence of any of these sugar nucleotides. Expression of lpsB in E. coli using a T7lac promoter-driven construct results in the appearance of similar multiple glycosyl transferase activities seen in S. meliloti 2011 membranes. Constructs expressing lpcC display only mannosyl transferase activity. We conclude that LpsB, despite its high degree of similarity to LpcC, is a much more versatile glycosyltransferase, probably accounting for the inability of lpcC to complement S. meliloti lpsB mutants. Our findings have important implications for the regulation of core glycosylation in S. meliloti and other bacteria containing LpcC orthologs.
As shown in the preceding paper (1), LpcC of Rhizobium leguminosarum is a highly selective mannosyl transferase that utilizes the sugar nucleotide GDP-mannose for the enzymatic glycosylation of the acceptor Kdo2-lipid IVA. These findings are consistent with earlier structural studies by Carlson and coworkers (2, 3) demonstrating that mannose is the sole sugar attached to the inner Kdo moiety of the core oligosaccharide of R. leguminosarum or Rhizobium etli lipopolysaccharide (LPS)1 (see Fig. 1 of the preceding paper). Furthermore, mutants of R. leguminosarum lacking lpcC are deficient in mannosyl transferase activity, are unable to form functional root nodules on Pisum sativum, and synthesize a truncated LPS molecule lacking the usual core sugar residues beyond the Kdo disaccharide (4).
Detailed structural studies of the LPS core of Sinorhizobium meliloti have not yet been reported.2 The S. meliloti core is complex, and it may contain glucose and galactose residues (5).2 The recent sequencing of the S. meliloti genome (6) and independent genetic studies by Lagares et al. (7) have demonstrated that a gene encoding an ortholog of LpcC is present in S. meliloti. Mutant 6963, which harbors a Tn5 insertion in this gene (designated lpsB), is defective in symbiosis with Medicago spp. (8, 9). Initially, mutant 6963 derived from strain 2011 was shown to be delayed in nodulation and poorly competitive for nodule occupancy on alfalfa (Medicago sativa) (8). In later studies, the same mutant was found to be unable to fix nitrogen (Fix−) on Medicago truncatula (9). Clear signs of plant defense were also observed with an abnormal pattern of infection as judged by electron microscopy (9). More recently, independent lpsB mutants generated with the related strain S. meliloti 1021 were reported to be Fix−in alfalfa. However, since 2011 and 1021 are both streptomycin-resistant derivatives of the wild type strain SU47, it may be that the defective nitrogen fixation phenotype seen in alfalfa with the lpsB mutant of strain 1021 is the consequence of both the lpsB lesion and some other as yet unidentified mutation in 1021 that is absent in strain 2011. Whatever the explanation, it is apparent that mutants defective in lpsB provide an interesting model system with which to probe the participation of LPS in symbiosis.
As shown in Fig. 1, S. meliloti LpsB is 351 amino acid residues long and displays 58% identity and 72% similarity to LpcC over the full length of the entire protein. Accordingly, LpsB, like R. leguminosarum LpcC, would be expected to function as a mannosyl transferase. The lpsB gene is, in fact, able to complement the truncation of lipopolysaccharide assembly and the nodulation deficiency seen in strain RSKnH, an lpcC mutant of R. leguminosarum (7). However, lpcC is not able to restore normal lipopolysaccharide assembly to the S. meliloti lpsB mutant 6963 (7).
Fig. 1. Sequence similarity comparison of R. leguminosarum LpcC and S. meliloti LpsB.

The middle line indicates identities and similarities. The proteins show 58% identity and 72% similarity over 350 of the 352 amino acid residues in LpcC. The E value (28) determined against the nonredundant data base is ~10−103.
To understand the biochemical basis for this genetic anomaly, we have now characterized the glycosyl transferase activity of the parental strain S. meliloti 2011 and its LPS core biosynthesis/nodulation-deficient mutant 6963. We have also cloned the lpsB gene behind the T7lac promoter and expressed it in Escherichia coli. The results show conclusively that LpsB, like LpcC, can function as an effective mannosyl transferase using GDP-mannose as the donor and Kdo2-[4′-32P]lipid IVA as the acceptor substrate. However, unlike LpcC, LpsB shows comparable activity with several other sugar nucleotides, including ADP-glucose and UDP-glucose. These additional activities of LpsB may account for the observation that lpcC cannot substitute for lpsB in cells of S. meliloti (7). Our findings further suggest that the most important functions of LpsB for S. meliloti biology are those related to the utilization of the sugar nucleotides other than GDP-mannose. How the LpsB protein acquires its relaxed sugar nucleotide selectivity when compared with LpcC can now be studied at the level of enzymology and structure.
EXPERIMENTAL PROCEDURES
Chemicals and Materials
Sugar nucleotides, Kdo, and HEPES were obtained from Sigma. Bicinchoninic assay reagents and Triton X-100 were purchased from Pierce. Yeast extract and peptone-tryptone were from Difco. Silica Gel 60 thin layer chromatography plates were obtained from Merck. Reagent grade pyridine, chloroform, and methanol were purchased from VWR. [γ-32P]ATP was purchased from PerkinElmer Life Sciences.
Bacterial Strains, Media, and Growth Conditions
The bacterial strains and plasmids used in this study are listed in Table I. S. meliloti strains have been described previously (7). S. meliloti strains were grown in TY medium (10 g of NaCl, 10 g of peptone-tryptone, and 5 g of yeast extract per liter, supplemented with 10 mM calcium chloride) at 30 °C. When necessary, the cultures were also supplemented with streptomycin (400 μg/ml) and/or neoymcin (30 μg/ml). E. coli strain BLR(DE3)/pLysS (Novagen) was grown in LB broth (10) at 37 °C.
Table I.
Sinorhizobium meliloti strains and plasmids used in this study
| Bacterial strains | Genotype | LPS properties | Source or reference |
|---|---|---|---|
| S. meliloti 1021 | Parental wild type | Full-length | Ref. 25 |
| S. meliloti 2011 | Parental wild type | Full-length | Ref. 26 |
| S. meliloti 6963 | lpsB::Tn5 | Truncated inner core | Ref. 8 |
| S. meliloti 6963-B | S. meliloti 6963 (pJBlpsSme) | Full-length | This work |
| S. meliloti 6963-C | S. meliloti 6963 (pPN120) | Truncated as in 6963 | This work |
| Plasmids | Relevant characteristics | ||
|
| |||
| pJB3Tc19 | Derivative of the minimal replicon of plasmid RK2, Apr, Tcr | Ref. 27 | |
| pAL100 | pACYC184-Gm-mob carrying in Dral a 5.5-kb SstI DNA fragment from S. meliloti 2011 harboring lpsB | Ref. 8 | |
| pJB1psSme | pJB3Tc19 carrying a 4.9-kb SstI-HindIII fragment with the region greA-lpsB-lpsE-lpsD from S. meliloti 2011 cloned into the polylinker of the vector between EcoRI-HindIII, Kmr | Ref. 7 | |
| pPN120 | pLAFR-1 with a 4.4 kb EcoRI fragment containing greA, lpcC, dctA, and the 53 region of dctB from R. leguminosarum bv. viciae, Tcr | Refs. 4 and 7 | |
Molecular Biology Techniques
Plasmids were prepared using the Qiagen Mini Prep Kit (Qiagen). Restriction endonucleases, Pfu polymerase (Stratagene), shrimp alkaline phosphatase, and T4 ligase (Invitrogen) were used according to the manufacturers’ instructions. Competent cells of E. coli were prepared for transformation using the calcium chloride method (11, 12). Introduction of plasmids into S. meliloti was carried out by conjugation as previously described (7).
Construction of a Plasmid Expressing LpsB behind the T7lac Promoter
PCR primers were designed to retrieve the S. meliloti lpsB gene from plasmid pAL100 (Table I) (7) and to introduce NdeI and BamHI restriction sites at the ends of the PCR product, as indicated. The primers used to obtain lpsB have the following sequences: forward, 5′-GAATTCCATATGGTCGATATCCGCGACGTC-3′; reverse, 5′-GAA-TTCGGATCCTCAACGCATCAGGCTTTCGTAAAC-3′. The conditions for the PCR were as follows: 45 s of denaturation at 98 °C and then 25 cycles of denaturation (45 s, 98 °C), annealing (45 s, 55 °C), and extension (1.5 min, 72 °C). After the last cycle, an additional 10-min extension at 72 °C was performed. The resulting 1056-base pair DNA product was digested with the NdeI and BamHI restriction enzymes and ligated into an expression vector, pET28b, that had been digested with the same enzymes. The resulting ligation mixture was used to transform E. coli XL1 Blue, selecting for kanamycin resistance (30 μg/ml). The presence of the proper inserts in plasmids from several colonies was verified by restriction mapping. The desired construct, designated pMKRM, was then transferred into E. coli BLR(DE3)/pLysS for high level heterologous expression of lpsB.
Preparation of Membranes for Assay of LpcC and LpsB
Each S. meliloti strain shown in Table I was grown from a single colony in 1 liter of TY medium containing the appropriate antibiotics to an optical density of 600 = 1.4 (late log phase). E. coli BLR(DE3)/pLysS/pMKRM was grown from a single colony in 1 liter of LB medium containing 30 μg/ml chloramphenicol and 30 μg/ml kanamycin at 37 °C until the A600 reached ~0.5. The culture was induced with 1 mM isopropyl-1-thio-β-D-galactopyranoside and incubated with shaking for an additional 3 h at 30 °C.
Cells from the above 1-liter cultures were harvested at 4 °C by centrifugation at 4,000 × g for 10 min, were each resuspended in 30 ml of 50 mM HEPES, pH 7.5, and were broken by one passage through a French pressure cell at 18,000 p.s.i. Debris and inclusion bodies were removed by centrifugation at 4,000 × g for 15 min. Membranes were prepared by ultracentrifugation at 100,000 × g for 60 min. The membranes were resuspended in 20 ml of the same buffer, and the centrifugation was repeated a second time at 100,000 × g to remove any remaining cytosol. Protein concentration was determined by the bicinchoninic method (13) (Pierce). The membrane pellet was resuspended at 10 –15 mg of protein/ml in 50 mM HEPES, pH 7.5, and stored at −80 °C until further use.
Mannosyl Transferase Assays
The mannosyl transferases LpcC and LpsB were assayed using the lipopolysaccharide precursor Kdo2-[4′-32P]lipid IVA, which was synthesized, isolated, and stored as a frozen aqueous dispersion, as described previously (14). Prior to each use, the radiolabeled substrate was subjected to ultrasonic irradiation in a water bath for 1 min. The mannosyl transferase was assayed as follows. The reaction mixture (10-μl final volume) contained 50 mM HEPES, pH 7.5, 0.1% Triton X-100, 1 mM GDP-mannose, and 10 μM Kdo2-[4′-32P]lipid IVA (3000 – 6000 cpm/nmol). The reaction was started by the addition of an appropriate amount of enzyme (usually at a final concentration of 1.0 mg/ml S. meliloti membranes or 0.02 mg/ml washed membranes of E. coli cells overexpressing lpsB behind the T7lac promoter) and incubated for the indicated times at 30 °C. The reactions were stopped by spotting 4-μl samples directly onto a silica gel thin layer plate. After drying the spots at room temperature, the plate was developed in the solvent chloroform, pyridine, 88% formic acid, water (30:70:16:10, v/v/v/v). Following removal of the solvent with a hot air stream, the plate was analyzed using an Amersham Biosciences PhosphorImager (Storm 840), equipped with ImageQuant software. Other glycosyl transferase activities of LpsB were assayed in the same way but in the presence of the indicated alternative sugar nucleotides present at 1 mM.
Purification of LPS from S. meliloti lpsB Mutant 6963
LPS (i.e. Kdo2-lipid A) was extracted from a 100-ml culture of late log phase cells (OD = 1.4) with a single phase Bligh-Dyer mixture, consisting of chloroform/methanol/water (1:2:0.8, v/v/v). Following phase partitioning, the LPS was purified by chromatography on a DEAE-cellulose column, prepared in chloroform/methanol/water (2:3:1, v/v/v) (1).
Mass Spectrometry
Spectra were acquired in the negative mode using a matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) Kompact 4 mass spectrometer (Kratos Analytical, Manchester, UK), equipped with a 337-nm nitrogen laser and set at a 20-kV extraction voltage. Conditions were the same as described in the preceding paper (1).
RESULTS
Complex Glycosylation of Kdo2-lipid IVA in Membranes of S. meliloti
As shown in Fig. 2A (lane 5), membranes of R. leguminosarum 3841 (15) glycosylate Kdo2-[4′-32P]lipid IVA in a highly selective manner using GDP-mannose as the preferred donor substrate. The nonphysiological analog ADP-mannose, a natural product found in corn (16, 17), can substitute for GDP-mannose (18), albeit not quite as effectively (Fig. 2A, lane 4). Other core sugars cannot be incorporated prior to the addition of the first mannose residue to Kdo2-lipid IVA (data not shown) (4, 19). The mannosyl transferase activity of R. leguminosarum is catalyzed by LpcC, which displays very little transferase activity with other sugar nucleotide donors, such as ADP-glucose or UDP-glucose (Fig. 2A, lanes 3 and 9, respectively) (1, 4).
Fig. 2. Distinct patterns of Kdo2-lipid IVA glycosylation with membranes of R. leguminosarum 3841 versus S. meliloti 1021.
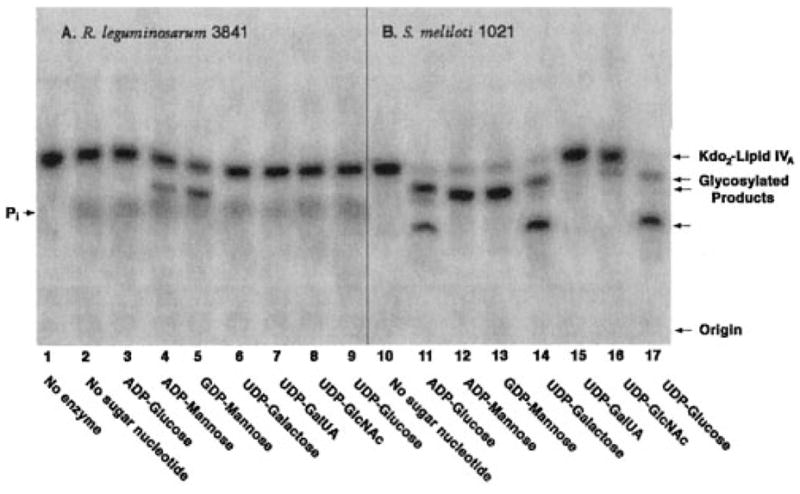
Reactions were carried out under standard conditions at pH 7.5 using a 1 mM concentration of each of the sugar nucleotides, as indicated. Membranes were used at 1 mg/ml. The incubation was carried out at 30 °C for 60 min.
In earlier studies with S. meliloti 1021 (18), little or no mannosyl transferase activity was detected in cell extracts or membranes prepared from cells grown on TY medium. S. meliloti 1021 cells were therefore used for the expression cloning and characterization of lpcC from R. leguminosarum (4). However, when grown in a more controlled manner to late log phase, as described above, membranes of S. meliloti 1021 do in fact catalyze rapid mannosylation of Kdo2-[4′-32P]lipid IVA (Fig. 2B, lanes 12 and 13). Interestingly, other glycosylations of Kdo2-[4′-32P]lipid IVA are observed with membranes of S. meliloti 1021 (Fig. 2B), which are absent in R. leguminosarum 3841 (Fig. 2A), as judged by the shift of the radioactive substrate band to more slowly migrating species with ADP-glucose, UDP-galactose, and UDP-glucose (Fig. 2B, lanes 11, 14, and 17, respectively). There is even the formation of some secondary glycosylation products of Kdo2-[4′-32P]lipid IVA with ADP-glucose, UDP-galactose, and UDP-glucose (Fig. 2B, lanes 11, 14, and 17), which are not seen when ADP-mannose or GDP-mannose are employed as the donor substrates (Fig. 2B, lanes 12 and 13). Under matched conditions, ADP-glucose, UDP-galactose, and UDP-glucose do not support any glycosylation of Kdo2-[4′-32P]lipid IVA in membranes of R. leguminosarum 3841 (Fig. 2A) or R. etli CE3 (data not shown). Neither R. leguminosarum nor S. meliloti 1021 membranes catalyze efficient glycosylation of Kdo2-[4′-32P]lipid IVA with UDP-GalUA or UDP-GlcNAc as sugar donors (Fig. 2, A (lanes 7 and 8) and B (lanes 15 and 16)). The formation of inorganic phosphate by membranes of R. leguminosarum is attributed to the 4′-phosphatase activity, which is absent in S. meliloti (Fig. 2, A versus B) (14, 20).
The completed genome of S. meliloti 1021 (6) and intriguing recent studies of the isogenic S. meliloti 2011 (7) have revealed the presence of a significant ortholog of LpcC (58% identity and 72% similarity over the full length of the protein) in both of these bacterial strains (Fig. 1). In the case of strain S. meliloti 2011, this LpcC ortholog (which was termed LpsB) (7) is required both for core oligosaccharide assembly and for symbiosis with Medicago spp., as judged by the analysis of mutant 6963, which contains a Tn5 insertion in lpsB. We therefore decided to examine the glycosylation of Kdo2-[4′-32P]lipid IVA using membranes of S. meliloti 2011 and mutant 6963. When membranes of S. meliloti 2011 are assayed at 1 mg/ml for their ability to glycosylate Kdo2-[4′-32P]lipid IVA, essentially the same complex pattern of sugar additions is observed (Fig. 3) as with S. meliloti 1021 membranes (Fig. 2B).
Fig. 3. Glycosylation of Kdo2-lipid IVA catalyzed by membranes of S. meliloti 2011.
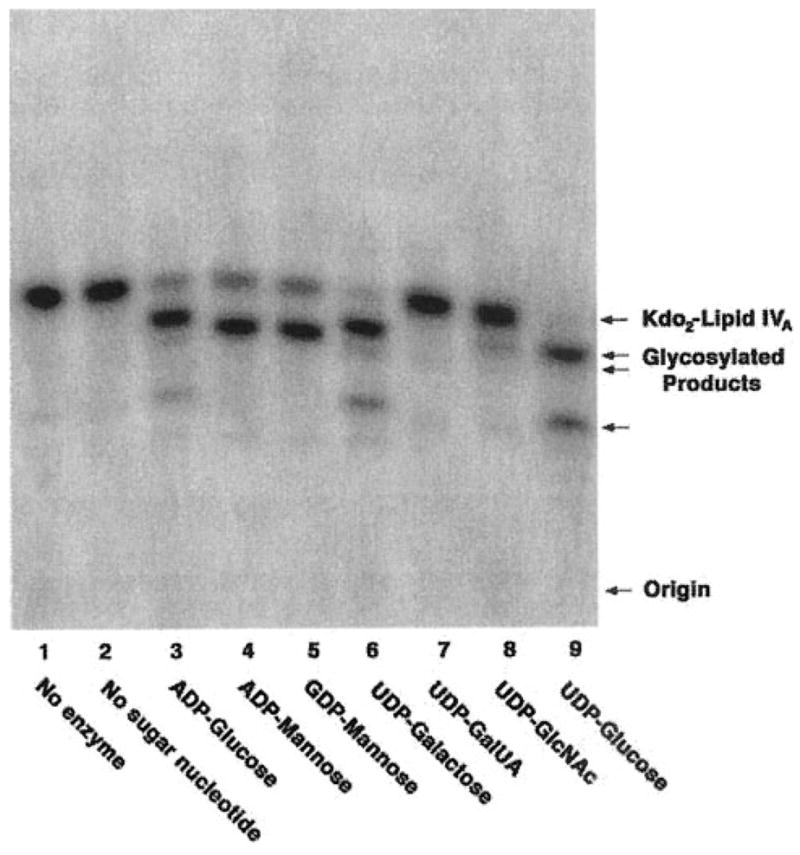
Reactions were carried out under standard conditions at pH 7.5 using a 1 mM concentration of each of the sugar nucleotides, as indicated. Membranes were used at 1 mg/ml. The incubation was carried out at 30 °C for 60 min.
Absence of Kdo2-Lipid IVA Glycosylation in Membranes of the lpsB Mutant 6963
When comparable washed membranes of S. meliloti mutant 6963 are assayed, no glycosylation of Kdo2-[4′-32P]lipid IVA of any kind is observed (data not shown). This striking result suggests that the diverse glycosylations seen in Figs. 2B and 3 are all the result of a single enzyme with relaxed sugar nucleotide substrate selectivity, when contrasted with LpcC. Based on this finding alone, however, some kind of pleiotropic regulatory effect secondary to the Tn5 insertion in the lpsB gene of mutant 6963 cannot be excluded.
Restoration of Mannosyl Transferase Activity in Mutant 6963 by DNA Fragments Harboring either lpsB or lpcC
We have previously described the plasmids pJBlpsSme, which contains lpsB on a 4.9-kb fragment of S. meliloti 2011 DNA, and pPN120, which harbors lpcC on a 4.4-kb fragment of R. leguminosarum DNA (7). As shown in Fig. 4, introduction of either of these plasmids into mutant 6963 (lanes 4 and 5) restores full mannosyl transferase activity, as judged by assay of cell membranes with Kdo2-[4′-32P]lipid IVA in the presence of GDP-mannose. In fact, all of the other glycosylations of Kdo2-[4′-32P]lipid IVA are also restored to mutant 6963 by plasmid pJBlpsSme (Fig. 5). In contrast, when membranes from mutant 6963 cells harboring the lpcC containing plasmid pPN120 are assayed with the other sugar nucleotide donors (Fig. 6), no activity is seen with ADP-glucose, UDP-galactose, or UDP-glucose, consistent with the substrate specificity of native R. leguminosarum membranes (Fig. 2A) and of purified LpcC (1). These results (Fig. 6) exclude the possibility that additional enzymes present in crude extracts of S. meliloti 2011 are interconverting all of the various sugar nucleotides added to the assay system to account for the diverse glycosylations seen in Figs. 2B, 3, and 5.
Fig. 4. Restoration of Kdo2-lipid IVA mannosylation in membranes of lpsB mutant 6963 expressing either R. leguminosarum lpcC or S. meliloti lpsB.
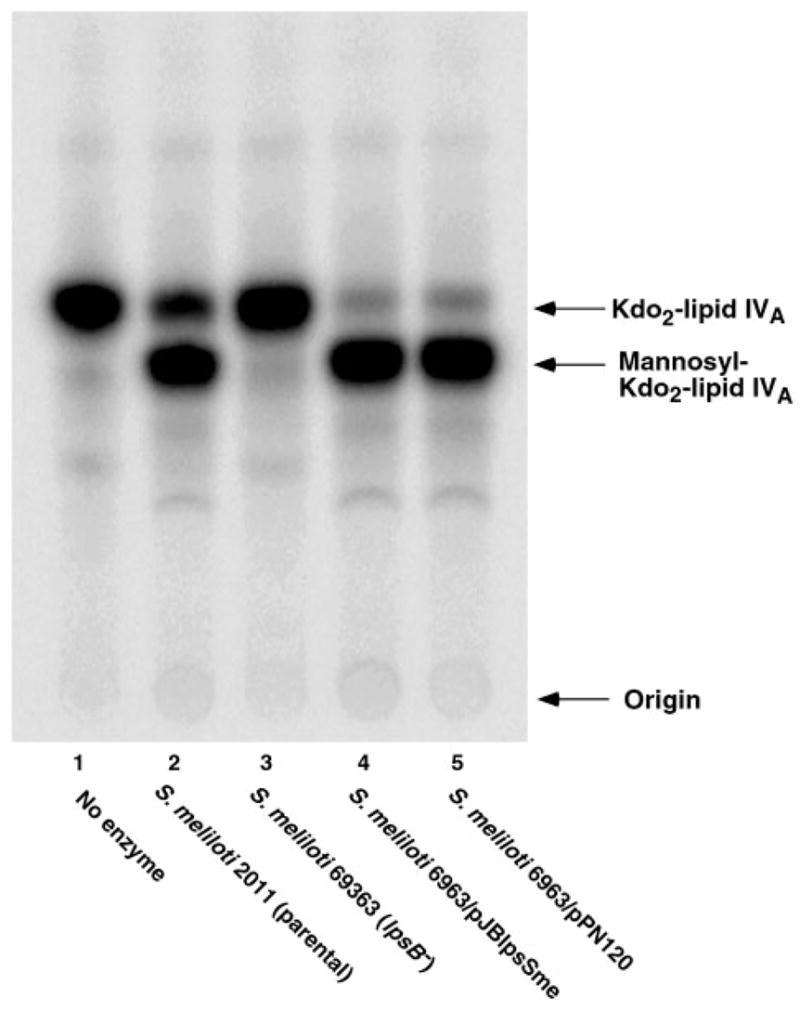
Reactions were carried out under standard conditions at pH 7.5 using 1 mM GDP-mannose as the sugar donor. Membranes were used at 1 mg/ml. The incubation was carried out at 30 °C for 60 min. Lane 1, no enzyme; lane 2, S. meliloti 2011; lane 3, S. meliloti 6963; lane 4, S. meliloti 6963/pJBlpsSme (lpsB); lane 5, S. meliloti 6963/pPN120 (lpcC).
Fig. 5. Restoration of other Kdo2-lipid IVA glycosylation reactions in membranes of lpsB mutant 6963 expressing S. meliloti lpsB.
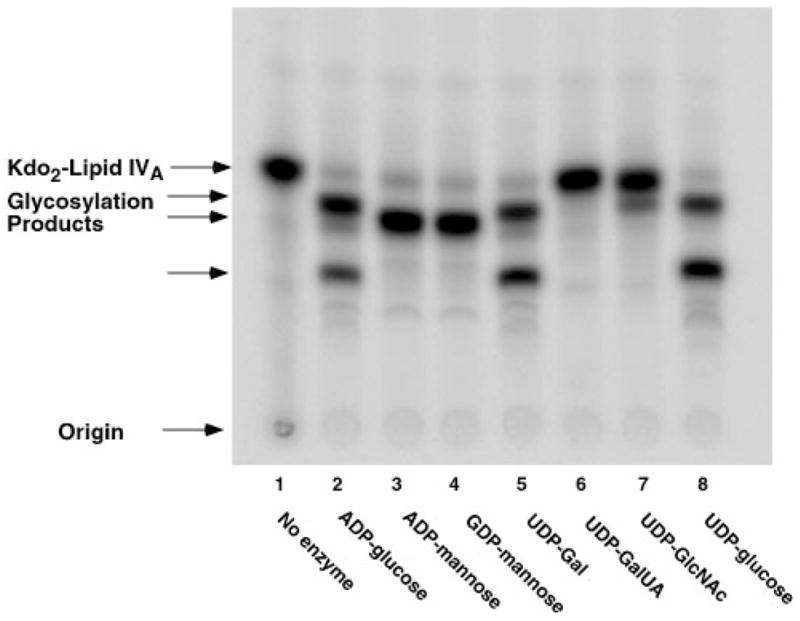
Reactions were carried out under standard conditions at pH 7.5 using a 1 mM concentration of each of the sugar nucleotides, as indicated. Membranes were used at 1 mg/ml. The incubation was carried out at 30 °C for 60 min.
Fig. 6. No Kdo2-lipid IVA glycosylation with sugars other than mannose in membranes of lpsB mutant 6963 expressing R. leguminosarum lpcC.
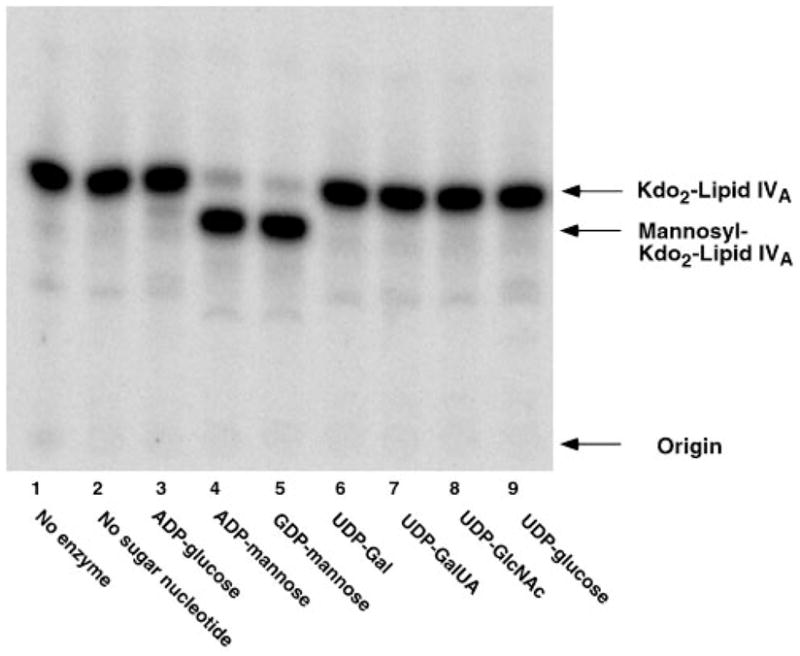
Reactions were carried out under standard conditions at pH 7.5 using a 1 mM concentration of each of the sugar nucleotides, as indicated. Membranes were used as the enzyme source at 1 mg/ml. The incubation was carried out at 30 °C for 60 min.
Heterologous Expression and Substrate Specificity of LpsB in E. coli
The lpsB gene of S. meliloti 2011 was cloned behind the T7lac promoter of the expression plasmid pET28b to generate pMKRM and transformed into the E. coli strain BLR(DE3)/pLysS. Washed membranes were then assayed from cells that had been induced in late log phase with isopropyl-1-thio-β-D-galactopyranoside for 3 h. As shown in Fig. 7A (upper panel), membranes of cells harboring the vector control (at 0.02 mg/ml) catalyze little or no glycosylation of Kdo2-[4′-32P]lipid IVA, with the possible exception of a small amount of activity seen with ADP-mannose, which might be attributed to the endogenous WaaC activity of the E. coli host (1, 21). In contrast, overexpression of lpsB behind the T7lac promoter results in robust glycosylation of Kdo2-[4′-32P]lipid IVA (Fig. 7B, lower panel) with a similar broad range of sugar nucleotide substrate utilization as observed in membranes of S. meliloti 1021 or 2011 (Figs. 2B and 3). Again, ADP-mannose, GDP-mannose, ADP-glucose, and UDP-glucose all function as efficient donor substrates and at comparable rates (Fig. 7B), whereas UDP-GalUA and UDP-GlcNAc are virtually inactive. No further secondary glycosylations are observed in this heterologous system (Fig. 7B versus Figs. 2B, 3, and 5), indicating that additional enzymes present only in S. meliloti must catalyze these reactions. Interestingly, UDP-galactose is not used very effectively as a sugar donor when LpsB is expressed in E. coli (Fig. 7B, lane 4). This finding suggests that the robust activity seen with UDP-galactose in S. meliloti membranes (Figs. 2B, 3, and 5) may be due to more effective 4′-epimerization of UDP-galactose to form UDP-glucose in S. meliloti membranes than in E. coli membranes.
Fig. 7. Glycosylations of Kdo2-lipid IVA catalyzed by membranes of E. coli expressing lpsB behind the T7lac promoter.

Reactions were carried out under standard conditions at pH 7.5 using a 1 mM concentration of each of the sugar nucleotides, as indicated. Enzyme was used at 0.02 mg/ml. The incubation was carried out at 30 °C for 60 min. A, membranes of BLR(DE3)/pLysS/pET28b; B, membranes of BLR(DE3)/pLysS/pMKRM.
The results of Fig. 7B establish unequivocally that S. meliloti LpsB can employ a much broader set of sugar nucleotides as donor substrates than can its ortholog LpcC of R. leguminosarum. These findings exclude pleiotropic regulatory effects as the explanation for the loss of all of the different glycosylation activities in membranes of mutant 6963. Supporting this result, we had previously suggested the absence of polar effects of the Tn5 insertion in 6963, since the open reading frames located immediately downstream of lpsB are transcribed in the opposite direction. Most importantly, our enzymatic data provide a straightforward biochemical rationale for the ability of S. meliloti lpsB to complement the R. leguminosarum lpcC mutation but not the reverse (7).
Mass Spectrometry of Truncated LPS Extracted from Mutant 6963
Chloroform/methanol extraction of the phospholipids and core-deficient LPS of mutant 6963, followed by chromatography on DEAE-cellulose, yields a purified preparation consisting of lipid A derivatized with two Kdo residues. As shown in the negative mode MALDI-TOF mass spectrum of this compound (Fig. 8A), the peak at m/z 2493.3 may be interpreted as the [M – H]− ion of a lipid A species that is acylated with two 3-OH-C14 chains, two 3-OH-C18 chains, and one secondary 29-OH-C30 moiety capped with a β-hydroxybutyrate residue (Fig. 8B). This pattern is consistent with the overall fatty acid composition of lipid A that was recently reported for lipid A of Sinorhizobium sp. NGR234 (22). Smaller peaks within the same cluster near m/z 2493.3 that differ by 14 atomic mass units might arise from the presence of shorter odd chain fatty acyl moieties and/or from shorter acyl chains in conjunction with partial O-methylation of the β-hydroxybutyrate residue in some molecular species (not shown) (22). The cluster of peaks around m/z 2392.3 might arise from one of the shorter chain lipid A species lacking the β-hydroxybutyrate group (or its methylated version) (22). The peaks around m/z 2052.6 are due to loss of the Kdo disaccharide from the parent compound. The peaks at m/z 2265.8, 1458.0, and 1003.2 could not be assigned and might be due to impurities.
Fig. 8. Negative mode MALDI-TOF mass spectrum of LPS purified from mutant 6963 by chloroform/methanol extraction.

The proposed LPS molecular species that could account for the major peaks observed in A is shown in B. This structure is consistent with the overall fatty acid composition recently reported for lipid A of Sinorhizobium sp. NGR234 (22).
The mass spectrum of the LPS recovered from mutant 6963 supports the idea that LpsB incorporates the first sugar of the inner core beyond Kdo in a major portion of the LPS molecules of S. meliloti and that further core glycosylation of these LPS molecules requires the presence of the sugar incorporated by LpsB (7).
DISCUSSION
R. leguminosarum LpcC (4) and S. meliloti LpsB (7) are protein orthologs that display 58% identity and 72% similarity over their entire lengths (Fig. 1). In other metabolic pathways, such as lipid A biosynthesis (23, 24), extensive similarity at this level is predictive of functional identity. Accordingly, LpsB of S. meliloti is classified as a mannosyl transferase in genomic databases (6), and it can indeed catalyze the mannosylation of Kdo2-lipid IVA in vitro (Fig. 7B). However, LpsB can also utilize several other sugar nucleotide substrates, namely ADP-glucose and UDP-glucose (Fig. 7B), which LpcC does not recognize effectively (Fig. 6) (1). We also know that whereas the lpsB gene can complement the lpcC mutation of R. leguminosarum, lpcC cannot complement lpsB-deficient strains of S. meliloti (7). These findings, taken together, demonstrate that LpsB donor substrates other than GDP-mannose (Fig. 7B) (i.e. most likely UDP-glucose or ADP-glucose) are critical for proper LPS core assembly in S. meliloti.
The results shown in Fig. 2B suggest that LpsB-catalyzed mannose incorporation (lanes 12 and 13) may not generate a core biosynthetic intermediate suitable for further glycosylation, as is observed when ADP-glucose or UDP-glucose is added as the sugar donor for LpsB (lower migrating bands in Fig. 2B, lanes 11 and 17). Additional evidence for the idea that mannosylation may prevent further core assembly in S. meliloti comes from the observation that lpcC expressed in the lpsB mutant 6963 does not significantly alter the migration of the truncated LPS species that is present in this strain (7), whereas lpcC and lpsB both restore full-length O-antigen to lpcC mutants of R. leguminosarum (7). LpcC presumably incorporates mannose into the nascent LPS of the S. meliloti mutant 6963, but the resulting product is not a substrate for further glycosylation.
The distinctly different sugar donor selectivity of LpcC versus LpsB demonstrates the need to conduct biochemical and enzymatic studies in conjunction with sequence comparisons when proposing in vivo functions for putative glycosyl transferases. Although LpsB does in fact catalyze the incorporation of mannose into Kdo2-lipid IVA in vitro, the role of LpsB within cells of S. meliloti is most likely the transfer of glucose units to the core of nascent LPS. The results of the present study reveal the difficulties associated with the correct functional assignment and data base annotation of glycosyl transferases. Thousands of diverse glycosyl transferase orthologs for which biochemical selectivity and function cannot be assigned are present in both procaryotic and eucaryotic genomes. LpcC and LpsB are members of a very large family of glycosyl transferases, most of which are probably membrane proteins (4). In E. coli, RfaG (the first glucosyl transferase of the core domain) is very distantly related to the LpsB/LpcC family (24). Although an x-ray structure has not been reported for any member of this enzyme family, it is likely that LpcC and LpsB will show similar protein folds. The unusual sugar nucleotide promiscuity of LpsB versus LpcC could be explained by subtle differences in the shapes of their donor substrate binding sites. Structural information might be helpful in redesigning the selectivity of glycosyl transferases such as LpsB with potential applications in the field of large scale carbohydrate synthesis. In that context, it remains to be determined whether or not LpsB actually glycosylates Kdo2-lipid IVA in the same position as does LpcC and generates the same anomeric configurations when using different sugar nucleotides as donor substrates.
A discrepancy between the results of Fig. 2B and our earlier studies (18) is the detection of significant mannosyl transferase activity in membranes of S. meliloti 1021. We previously reported that such preparations are inactive (18). It may be that the previously reported cultures were not harvested at the same time in late log phase. Furthermore, the cells were not grown in the presence of antibiotics in the previous studies (18, 19). Perhaps the mannosyl transferase is expressed in a regulated manner in response to growth phase or the composition of the medium. Whatever the explanation, the broad substrate selectivity of LpsB might allow for regulation of S. meliloti core sugar composition in response to changes in the relative intra-cellular concentrations of GDP-mannose versus UDP-glucose and/or ADP-glucose. The covalent structure of the S. meliloti core has not been reported. It appears to be complex2 compared with the well characterized core of R. leguminosarum (2, 3). Peculiar characteristics of the S. meliloti core were also suggested by earlier immunological studies. For instance, in contrast to most other Gram-negative bacteria, the immunodominant LPS epitopes of S. meliloti LPS are not located in the O-antigen domain but are in the core region (8). Interestingly, these core-associated epitopes are missing in the symbiotically defective lpsB mutant 6963 (8). Additional structural and physiological studies clearly will be necessary to explore the role of the S. meliloti core during symbiosis with plants.
Like the lpsB mutants of S. meliloti, the lpcC mutants of R. leguminosarum show aberrant nodulation (4). The LPS in the lpcC mutant RSKnH is entirely devoid of any residual species containing O-antigen (7), and both the lpcC and the lpsB genes can restore O-antigen to RSKnH (7). Similarly, the lpsB mutant 6963 contains an altered, rapidly migrating LPS species, not present in the wild type, which we believe consists of Kdo2-lipid A (7) (Fig. 8). This substance is soluble in chloroform/methanol mixtures, as described under “Experimental Procedures.” However, 6963 differs from RSKnH in that it retains some LPS molecules that are still derivatized with an O-antigen-like polymer or some other kind of polysaccharide (7). This more hydrophilic material would not be extracted with chloroform/methanol under our conditions. These complexities could explain why the total LPS, which was extracted with phenol from the lpsB mutant described by Campbell et al. (5), contains additional sugars besides glucosamine and Kdo. Xylose is reported by Campbell et al. (5) as a major component of the LPS in their lpsB mutant but is not present in our preparations (Fig. 8). As with the structure of the core domain of S. meliloti,2 the final resolution of these issues will require the development of better methods for LPS purification as well as direct structural analysis of intact material by mass spectrometry and NMR spectroscopy.
Many interesting questions remain regarding the relaxed substrate selectivity of LpsB versus LpcC. Is this phenomenon associated with other LpcC orthologs seen in organisms like Agrobacterium tumefaciens, Brucella meliltensis, or Franciscella tularensis?3 Is the mannosyl transferase activity of S. meliloti LpsB necessary under certain physiological circumstances when a truncated LPS core would be desirable? Could a mutant of LpsB be isolated that is unable to utilize GDP-mannose but that still functions normally with UDP- and ADP-glucose? Are the secondary glycosylation reactions that follow glucose incorporation (Fig. 2B, lanes 11 and 17) catalyzed by LpsC or LpsD (7)? The further biochemical and genetic analysis of this important class of glycosyl transferases should help to answer these questions and to advance our understanding of core assembly and function in the Rhizobiaceae and other Gram-negative bacteria. Molecular insights into the biochemistry of diverse LPS cores could prove useful in the development of new vaccines and in the improvement of agriculturally important crops.
Footnotes
This work was supported by National Institutes of Health Grants R37-GM-51796 (to C. R. H. R.) and GM54882 (to R. J. C.).
The abbreviations used are: LPS, lipopolysaccharide; Kdo, 3-deoxy-D-manno-octulosonic acid; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight.
R. Carlson, personal communication.
The F. tularensis genome is available on the World Wide Web at artedi.ebc.uu.se/Projects/Franciscella/.
References
- 1.Kanipes MI, Ribeiro AA, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2003;278:16356–16364. doi: 10.1074/jbc.M301255200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhat UR, Krishnaiah BS, Carlson RW. Carbohydr Res. 1991;220:219–227. doi: 10.1016/0008-6215(91)80020-n. [DOI] [PubMed] [Google Scholar]
- 3.Carlson RW, Reuhs B, Chen TB, Bhat UR, Noel KD. J Biol Chem. 1995;270:11783–11788. doi: 10.1074/jbc.270.20.11783. [DOI] [PubMed] [Google Scholar]
- 4.Kadrmas JL, Allaway D, Studholme RE, Sullivan JT, Ronson CW, Poole PS, Raetz CRH. J Biol Chem. 1998;273:26432–26440. doi: 10.1074/jbc.273.41.26432. [DOI] [PubMed] [Google Scholar]
- 5.Campbell GR, Reuhs BL, Walker GC. Proc Natl Acad Sci U S A. 2002;99:3938–3943. doi: 10.1073/pnas.062425699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galibert F, Finan TM, Long SR, Puhler A, Abola P, Ampe F, Barloy-Hubler F, Barnett MJ, Becker A, Boistard P, Bothe G, Boutry M, Bowser L, Buhrmester J, Cadieu E, Capela D, Chain P, Cowie A, Davis RW, Dreano S, Federspiel NA, Fisher RF, Gloux S, Godrie T, Goffeau A, Golding B, Gouzy J, Gurjal M, Hernandez-Lucas I, Hong A, Huizar L, Hyman RW, Jones T, Kahn D, Kahn ML, Kalman S, Keating DH, Kiss E, Komp C, Lelaure V, Masuy D, Palm C, Peck MC, Pohl TM, Portetelle D, Purnelle B, Ramsperger U, Surzycki R, Thebault P, Vandenbol M, Vorholter FJ, Weidner S, Wells DH, Wong K, Yeh KC, Batut J. Science. 2001;293:668–672. doi: 10.1126/science.1060966. [DOI] [PubMed] [Google Scholar]
- 7.Lagares A, Hozbor DF, Niehaus K, Otero AJ, Lorenzen J, Arnold W, Puhler A. J Bacteriol. 2001;183:1248–1258. doi: 10.1128/JB.183.4.1248-1258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lagares A, Caetano-Anolles G, Niehaus K, Lorenzen J, Ljunggren HD, Puhler A, Favelukes G. J Bacteriol. 1992;174:5941–5952. doi: 10.1128/jb.174.18.5941-5952.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niehaus K, Lagares A, Pühler A. Mol Plant Microbe Interact. 1998;11:906–914. [Google Scholar]
- 10.Miller JR. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 11.Bergmans HE, van Die IM, Hoekstra WP. J Bacteriol. 1981;146:564–570. doi: 10.1128/jb.146.2.564-570.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 13.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 14.Basu SS, York JD, Raetz CRH. J Biol Chem. 1999;274:11139–11149. doi: 10.1074/jbc.274.16.11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poole PS, Schofield NA, Reid CJ, Drew EM, Walshaw DL. Microbiology. 1994;140:2797–2809. doi: 10.1099/00221287-140-10-2797. [DOI] [PubMed] [Google Scholar]
- 16.Dankert M, Passeron S, Recondo E, Leloir LF. Biochem Biophys Res Commun. 1964;14:358–362. doi: 10.1016/s0006-291x(64)80010-3. [DOI] [PubMed] [Google Scholar]
- 17.Passeron S, Recondo E, Dankert M. Biochim Biophys Acta. 1964;89:372–374. doi: 10.1016/0926-6569(64)90233-0. [DOI] [PubMed] [Google Scholar]
- 18.Kadrmas JL, Brozek KA, Raetz CRH. J Biol Chem. 1996;271:32119–32125. [PubMed] [Google Scholar]
- 19.Brozek KA, Kadrmas JL, Raetz CRH. J Biol Chem. 1996;271:32112–32118. [PubMed] [Google Scholar]
- 20.Price NJP, Jeyaretnam B, Carlson RW, Kadrmas JL, Raetz CRH, Brozek KA. Proc Natl Acad Sci U S A. 1995;92:7352–7356. doi: 10.1073/pnas.92.16.7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kadrmas JL, Raetz CR. J Biol Chem. 1998;273:2799–2807. doi: 10.1074/jbc.273.5.2799. [DOI] [PubMed] [Google Scholar]
- 22.Gudlavalleti SK, Forsberg LS. J Biol Chem. 2002;278:3957–3968. doi: 10.1074/jbc.M210491200. [DOI] [PubMed] [Google Scholar]
- 23.Raetz CRH. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 24.Raetz CRH, Whitfield C. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meade HM, Long SR, Ruvkun GB, Brown SE, Ausubel FM. J Bacteriol. 1982;149:114–122. doi: 10.1128/jb.149.1.114-122.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Casse F, Boucher C, Julliot JS, Michell M, Dénarié J. J Gen Microbiol. 1979;113:229–242. [Google Scholar]
- 27.Blatny JM, Brautaset T, Winther-Larsen HC, Haugan K, Valla S. Appl Environ Microbiol. 1997;63:370–379. doi: 10.1128/aem.63.2.370-379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]