

As part of a program directed at the development of palladium- and nickel-catalyzed couplings of alkyl electrophiles,1 we have been exploring the possibility of achieving a variant of the Heck reaction wherein an alkyl, rather than an aryl, electrophile is coupled with an olefin. In an initial study, we were pleased to discover that such a transformation can indeed be accomplished, albeit in very poor yield (1.2%; eq 1). Interestingly, however, control reactions established that carbon-carbon bond formation proceeds more efficiently in the absence of palladium (24%; eq 1)!

Our mechanistic hypothesis for this unanticipated carbene-catalyzed cyclization is outlined in Figure 1.2,3 Thus, the catalyst (Nu:) adds to the electrophilic β carbon of the α, β-unsaturated ester, generating an enolate (A). Tautomerization then affords B,4 in which the β carbon is now nucleophilic (umpolung5,6). An intramolecular SN2 reaction provides C, which undergoes elimination to furnish the observed product and to regenerate the catalyst (Nu:).
Figure 1.
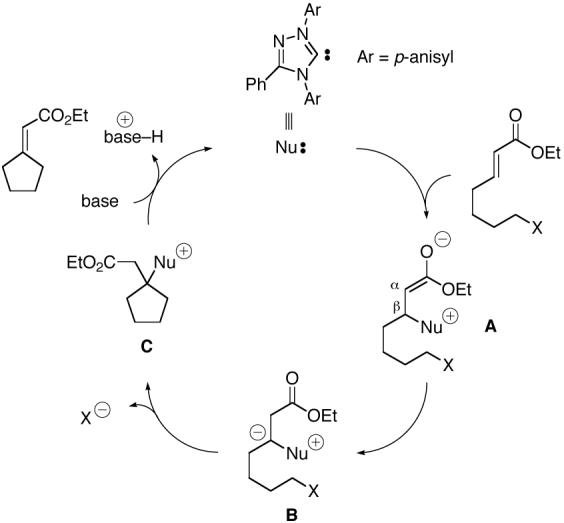
Possible mechanism for carbene-catalyzed β-alkylations of Michael acceptors.
For most of the previously described nucleophile-catalyzed reactions of Michael acceptors, the initial adduct (e.g., A) reacts with electrophiles in the α position (e.g., the Morita-Baylis-Hillman reaction7). Indeed, we have only been able to identify one previous report of a process wherein conjugate addition of a catalyst leads to the β carbon serving as a nucleophile.8 Furthermore, an interesting contrast is provided by the recent work of Krafft, who has established that enones/enals that bear pendant halides undergo cyclization in the α position when treated with stoichiometric PMe3 or PBu3 and then KOH (e.g., eq 2).9

We have optimized the carbene-catalyzed umpolung mode of β nucleophilicity for enoates (Figure 1) and determined that, through the appropriate choice of conditions, cyclization can be achieved with high efficiency (Table 1). Thus, p-methoxy-substituted variants of the Enders triazole-derived carbene10,11 have proved to be the best catalysts among those that we have examined to date, furnishing good yields of the target compounds (entries 1 and 2). The parent Enders carbene is somewhat less effective (entry 3), and IMes (entry 4), a thiazole derivative (entry 5), and a variety of amines (entries 6-8) and phosphines (entries 9-10), including PBu3, are essentially inactive as catalysts. With respect to the base, Cs2CO3 affords a somewhat lower yield than anhydrous K3PO4 (entry 11), whereas KOt-Bu is not suitable (entry 12). Regarding the solvent, 1,4-dioxane and EtOAc may be employed (entries 13 and 14), whereas N,N-dimethylacetamide may not (entry 15). The reaction is sluggish at room temperature (entry 16), and no cyclization is observed if the carbene precursor or the base is omitted (entries 17 and 18).
Table 1.
Effect of reaction parameters on the carbene-catalyzed β-alkylation of a Michael acceptor
 | |||
|---|---|---|---|
| entry | change from the standard conditions | yield (%)a | |
| n = 1 | n = 2 | ||
| 1 | no change | 92 | 85 |
| 2 | 10% 2 insted of 10% 1 | 88 | 85 |
| 3 | 10% 3 insted of 10% 1 | 91 | 37 |
| 4 | 10% IMes · HCl insted of 10% 1 | 5 | 5 |
| 5 | 10% 4 insted of 10% 1 | <1 | <1 |
| 6 | 10% DABCO insted of 10% 1 | <1 | <1 |
| 7 | 10% DMAP insted of 10% 1 | <1 | <1 |
| 8 | 10% quinidine instead of 10% 1 | <1 | <1 |
| 9 | 10% PBu3 instead of 10% 1 | <1 | <1 |
| 10 | 10% PPh3 instead of 10% 1 | <1 | <1 |
| 11 | 2.5 Cs2CO3 instead of 2.5 K3PO4 | 87 | 65 |
| 12 | 2.5 KOt-Bu instead of 2.5 K3PO4 | <1 | <1 |
| 13 | 1,4-dioxane instead of glyme | 91 | 70 |
| 14 | EtOAc instead of glyme | 89 | 60 |
| 15 | N,N-dimethylacetamide instead of glyme | 5 | 4 |
| 16 | r.t. instead of 80 °C | 16 | 2 |
| 17 | no 1 | <1 | <1 |
| 18 | no K3PO4 | <1 | <1 |
Ar = p-anisyl.
Yield according to GC analysis versus a calibrated internal standard (average of two experiments).
![]()
We were pleased to determine that cyclizations of a range of substrates proceed smoothly under our standard reaction conditions (Table 2). With respect to the leaving group, not only bromides (entry 1), but also tosylates (entry 2) and even chlorides (entry 3; cf. eq 2), can be employed. Furthermore, α,β-unsaturated allyl esters (entry 4), Weinreb amides (entry 5), and nitriles (entries 6 and 7) cyclize with good efficiency. The method may be applied to the synthesis of an oxygen heterocycle (entry 8), as well as six-membered rings (entry 9). Interestingly, four-membered rings can even be generated, albeit in modest yield (entry 10).12
Table 2.
Scope of the carbene-catalyzed β-alkylation of Michael acceptors
 | |||
|---|---|---|---|
| entry | starting material | reaction time (h) | yield (%)a |
| 1 (X = Br) |

|
8 | 94 |
| 2(X = OTs) | 16 | 94 | |
| 3(X = Cl) | 8 | 89 | |
| 4 |

|
8 | 88 |
| 5 |

|
16 | 68 |
| 6(X = Br) |

|
8 | 71 |
| 7(X = OTs) | 8 | 84 | |
| 8 |

|
6 | 77 |
| 9 |

|
16 | 81 |
| 10 |

|
33 | 48b |
Ar = p-anisyl.
Isolated yield (average of two experiments).
20% 1 was used, with dioxane as the solvent.
Clearly, tautomerization is a critical step in our proposed catalytic cycle, leading to nucleophilicity at the β carbon (Figure 1, A®B). In order to obtain insight into the origin of the difference in reactivity between the carbene derived from 1 and a phosphine (see above), we examined the deuterium-exchange process illustrated in eq 3. Scrambling occurs to a large extent with 10% 1, whereas little exchange is observed in the presence of PBu3.13

In conclusion, we have demonstrated that, for a range of Michael acceptors, a nucleophilic catalyst can transiently transform the normally electrophilic β carbon into a nucleophilic site through an addition-tautomerization sequence. Specifically, we have established that an N-heterocyclic carbene can catalyze β-alkylations of a variety of α,β-unsaturated esters, amides, and nitriles that bear pendant leaving groups. We anticipate that it will be possible to exploit this intriguing umpolung reactivity in a variety of contexts.
Supplementary Material
Acknowledgment
Support has been provided by the NIH (NIGMS, R01-GM62871 and R01-GM57034), the German Academic Exchange Service (postdoctoral fellowship to C.F.), a Robert T. Haslam graduate fellowship (to S.W.S.), NSERC of Canada (postdoctoral fellowship to D.A.P.), Merck Research Laboratories, and Novartis. Funding for the MIT Department of Chemistry Instrumentation Facility has been furnished in part by the NSF (CHE-9808061 and DBI-9729592).
REFERENCES
- (1).For leading references, see: Powell DA, Maki T, Fu GC. J. Am. Chem. Soc. 2005;127:510–511. doi: 10.1021/ja0436300.
- (2)(a).For overviews of processes catalyzed by nucleophilic carbenes, see: Zeitler K. Angew. Chem., Int. Ed. 2005;44:7506–7510. doi: 10.1002/anie.200502617. Nair V, Bindu S, Sreekumar V. Angew. Chem., Int. Ed. 2004;43:5130–5135. doi: 10.1002/anie.200301714.
- (3).For a review of processes catalyzed by nucleophilic phosphines, see: Methot JL, Roush WR. Adv. Synth. Catal. 2004;346:1035–1050.
- (4).For precedent in a more highly activated substrate, see: Enders D, Breuer K, Teles JH, Ebel KJ. Prakt. Chem. 1997;339:397–399.
- (5)(a).Seebach D. Angew. Chem., Int. Ed. Engl. 1979;18:239–258. [Google Scholar]; (b) Hase TA, editor. Umpoled Synthons. Wiley; New York: 1987. [Google Scholar]
- (6)(a).For pioneering studies wherein addition of a catalyst to the carbonyl carbon of an α,β-unsaturated aldehyde, followed by deprotonation of the aldehyde proton, leads to nucleophilicity at the β position, see: Burstein C, Glorius F. Angew. Chem., Int. Ed. 2004;43:6205–6208. doi: 10.1002/anie.200461572. Sohn SS, Rosen EL, Bode JW. J. Am. Chem. Soc. 2004;126:14370–14371. doi: 10.1021/ja044714b. Of course, this approach can be applied to α,β-unsaturated aldehydes, but not to esters, amides, or nitriles.
- (7).For leading references, see: Basavaiah D, Rao AJ, Satyanarayana T. Chem. Rev. 2003;103:811–892. doi: 10.1021/cr010043d.
- (8).In a study of the Stetter reaction, an undesired adduct arising from reactivity at the β position of an α,β-unsaturated amide was produced in up to 54% yield in the presence of 1.0 equivalent of PBu3 : Gong JH, Im YJ, Lee KY, Kim JN. Tetrahedron Lett. 2002;43:1247–1251.
- (9)(a).Krafft ME, Haxell TFN. J. Am. Chem. Soc. 2005;127:10168–10169. doi: 10.1021/ja052146+. [DOI] [PubMed] [Google Scholar]; (b) Krafft ME, Seibert KA, Haxell TFN, Hirosawa C. Chem. Commun. 2005:5772–5774. doi: 10.1039/b512665g. [DOI] [PubMed] [Google Scholar]
- (10)(a).For reports of the parent carbene (Ar = Ph), see: Enders D, Breuer K, Raabe G, Runsink J, Teles JH, Melder J-P, Ebel K, Brode S. Angew. Chem., Int. Ed. 1995;34:1021–1023. Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534–541. doi: 10.1021/ar030050j. (c) See also: Walentowski R, Wanzlick H-WZ. Naturforsch., B: Chem. Sci. 1970;25:1421–1423.
- (11).For an early study of electronic effects on a process catalyzed by nucleophilic triazole-derived carbene, see: Teles JH, Melder J-P, Ebel K, Schneider R, Gehrer E, Harder W, Brode S, Enders D, Breuer K, Raabe G. Helv. Chim. Acta. 1996;79:61–83.,
- (12).Notes: (a) An α,β-unsaturated phenyl ketone can be cyclized, but a significant amount of the β,γ-unsaturated enone is produced. (b) We have not yet been able to achieve efficient seven-membered ring formation or intermolecular β-alkylation. (c) Substrates in which the olefin has a Z, rather than an E, configuration cyclize less rapidly. (d) Under our standard conditions, we have not been able to effectively cyclize α-substituted enoates.
- (13).In a competition experiment between the alkyl bromide and the alkyl chloride illustrated in entries 1 and 3 of Table 2, comparable reaction rates were observed, consistent with the hypothesis that cyclization may not be the turnover-limiting step for these substrates. In contrast, for the homologous substrates (six-membered ring formation), the chloride is essentially unreactive under the conditions in which the bromide cyclizes.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.