

Abstract
The orientation of the ELC region of myosin in skeletal muscle was determined by polarized fluorescence from ELC mutants in which pairs of introduced cysteines were cross-linked by BSR. The purified ELC-BSRs were exchanged for native ELC in demembranated fibers from rabbit psoas muscle using a trifluoperazine-based protocol that preserved fiber function. In the absence of MgATP (in rigor) the ELC orientation distribution was narrow; in terms of crystallographic structures of the myosin head, the LCD long axis linking heavy-chain residues 707 and 843 makes an angle (β) of 120–125° with the filament axis. This is ∼30° larger than the broader distribution determined previously from RLC probes, suggesting that, relative to crystallographic structures, the LCD is bent between its ELC and RLC regions in rigor muscle. The ELC orientation distribution in relaxed muscle had two broad peaks with β ∼70° and ∼110°, which may correspond to the two head regions of each myosin molecule, in contrast with the single broad distribution of the RLC region in relaxed muscle. During isometric contraction the ELC orientation distribution peaked at β ∼105°, similar to that determined previously for the RLC region.
INTRODUCTION
Generation of force and shortening by skeletal muscle is driven by a change in the orientation of the converter and LCD regions of the myosin head with respect to the actin-binding region (1–5). The LCD is thought to act as a lever arm, converting the structural change associated with the release of ATP hydrolysis products from the active site of the head into a 5–10 nm translation of the distal end of the LCD, which is connected to the myosin filament in muscle. The LCD is not rigid however, and its orientation in a muscle fiber is sensitive to elastic stress (3,6). LCD compliance is likely to dominate that of the actin-bound myosin head, and therefore has a fundamental role in its motor mechanism.
Most previous studies of LCD orientation in situ have used extrinsic fluorescence or spin probes attached to the myosin RLC in demembranated muscle fibers (3,5,7,8). The RLC is a convenient vector for introducing probes onto the LCD within the native myosin filament structure of the muscle sarcomere, because native RLCs can be replaced by exogenous RLCs under relatively mild conditions, with no significant modification of muscle fiber function (3,9,10). ELC exchange in muscle fibers is more demanding, but ELC probes have the potential to yield significant new information about the in situ orientation and flexibility of the LCD. The ELC is immediately adjacent to the converter region of the myosin head (Fig. 1), and its orientation might be expected to follow that of the converter region more closely than is the case for the RLC. Moreover, each myosin molecule has two head domains linked to a common coiled-coil heavy-chain tail at the C terminus of their RLC regions, so the two RLCs in each myosin molecule are unlikely to have the same orientation in situ. Finally, the combination of orientation data from RLC and ELC probes would allow the angle between the RLC and ELC regions of the LCD to be determined in situ; this angle differs significantly between published crystal structures of the LCD (1,11), and has not been measured in physiological conditions.
FIGURE 1.

Ribbon representation of the head region of chicken skeletal myosin in the nucleotide-free state (PDB 2mys; (1)) bound to an actin filament in the absence of ATP (2), showing the lever and hook axes, and the tilt (β) and twist (γ) angles. Pairs of cysteine residues introduced into the ELC and cross-linked using BSR-I2 (inset) are shown as spheres joined by rods.
We used polarized fluorescence from bifunctional rhodamine probes, an approach applied previously to the RLC (5,8) and troponin C (12) in muscle fibers, to determine the absolute orientation of the ELC with respect to the muscle fiber axis in relaxation, active contraction and rigor. We prepared three different ELC mutants, each containing a pair of cysteine residues that was cross-linked using BSR-I2 (Fig. 1, inset; see Asenjo et al. (13) and Julien et al. (14) for previous applications of this reagent). We chose BSR-I2 rather than the BR-I2 used in previous studies (5,12) because BSR-I2 gave substantially higher yields of labeled ELC in preliminary experiments, presumably as a result of its higher reactivity toward cysteine (14). We extracted the native ELC from demembranated muscle fibers using TFP (a small hydrophobic molecule that binds to calmodulin, troponin C, and the myosin light chains (15,16)), and replaced it by one of the purified ELC-BSRs. We measured the orientation of each BSR dipole with respect to the fiber axis by polarized fluorescence, and combined the orientation data from the three ELC-BSRs to determine the in situ orientation of the ELC.
METHODS
Preparation and mutagenesis of myosin ELC
The double-cysteine mutants E8C/L15C, K11C/Q73C, and P70C/N77C of the A2 isoform of the ELC of chicken skeletal myosin in the pGEX-2T expression vector (GE Healthcare, Chalfont St. Giles, UK) were obtained using the Stratagene (La Jolla, CA) QuikChange Kit and expressed in Escherichia coli Ca2+-competent BL21 (DE3) cells as GST fusion proteins. Native Cys136 was also replaced by Ala in each mutant. The entire mutant genes were sequenced. Wild-type ELC was expressed by the same methods for control experiments.
Transformed cells were grown overnight in 4 L of Terrific broth at 37°C, harvested by centrifugation and resuspended in ∼100 mL of ice-cold PBS (140 mM NaCl, 2.7 mM KCl, 10.1 mM Na2HPO4, pH 7.3). Pure ELC was obtained from these cells by the following procedures, all at 4°C. The cells were sonicated, and inclusion bodies containing GST-ELC were isolated by centrifugation at 31,500 × g for 25 min and subsequent resuspension in 50 mL of 1% sodium deoxycholate, 0.5% Nonidet P40, 200 mM NaCl, 25 mM Tris-HCl, 2 mM K2EDTA, pH 7.5. The suspension was centrifuged again and the pellet resuspended in 50 mL of 0.5% Triton, 1 mM K2EDTA, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM MnCl2, 10 μg/mL DNase, pH 8.0. This mixture was centrifuged as before, resuspended in 1 mM K2EDTA, 10 mM Tris-HCl, pH 8.0, and centrifuged again. This step was repeated four times, after which the supernatant had become clear. Sixty milliliters of 8 M urea, 20 mM Tris-HCl, 1 mM K2EGTA, 1 mM DTT, pH 8.0 was added to the final pellet and the mixture stirred slowly for 1.5 h to solubilize the pellet. The mixture was centrifuged at 31,500 x g for 1 h and the supernatant removed immediately. The supernatant was dialyzed into PBS and 1 mM DTT (2 × 5 L each for 2 h, then 1 × 5 L overnight).
The GST-ELC was collected in two 50-mL screw-capped tubes, and 8 mL of 75% slurry of glutathione-Sepharose 4B (GE Healthcare) preequilibrated with PBS was added to each tube. The tubes were placed on a tube rotator for 1 h to allow the glutathione-Sepharose 4B to bind GST-ELC. The suspensions were centrifuged at 1940 x g for 5 min to pellet the beads, and the supernatant was removed. The beads were resuspended in 70 mL PBS, centrifuged and the supernatant removed as before. This was repeated three times and the beads were finally resuspended in 18 mL PBS. The column material was divided between three empty PD10 columns (GE Healthcare) and 500 units of thrombin (GE Healthcare; 1000 units/ml in PBS) was added to each column to cleave the ELC from GST, which remained bound to the column material. The columns were sealed and gently rotated for 16 h.
The columns were placed in an upright position and the column run-off collected in 15 mL screw-capped tubes. The columns were washed with 3 × 8 mL PBS. Most of the ELC was eluted from the columns in the run-off and first wash, and the second and third washes were generally discarded. A 50% slurry of benzamidine-Sepharose (GE Healthcare) in 0.05 M Tris-HCl, 0.5 M NaCl, pH 7.4 was added to these fractions to remove the thrombin. The tubes were gently rotated as before for 1 h. The benzamidine-Sepharose was removed by filtration, and 1 mM PMSF (final concentration) was added to each fraction to prevent further protease activity. ELC purity was determined by 15% acrylamide SDS-PAGE, reverse phase HPLC and electrospray mass spectrometry. Its concentration was estimated by comparing its absorbance at 215 nm with that of a troponin C standard (determined by Ferguson et al. (12)) that was included in reverse phase HPLC runs, assuming that both proteins had the same extinction coefficient. Pure fractions were pooled and concentrated to 128 μM by dialysis against solid sucrose.
Labeling double-cysteine ELC mutants with BSR
Of the mutant ELC preparation, ∼15 mL was incubated with 4 mM DTT at room temperature for 30 min to reduce any disulfides formed during storage, and gel-filtered (PD10 column; GE Healthcare) into labeling buffer (PBS containing 50 μM tris(carboxyethyl)phosphine to keep ELC sulfhydryls reduced). The ELC was diluted to 64 μM, and 30 μM BSR-I2 (Molecular Probes, Eugene, OR) was added from a 24 mM stock solution in dimethylformamide. The solution was incubated in the dark at room temperature and four further 10-μM aliquots of BSR-I2 were added at 10 min intervals, to a final concentration of 70 μM. The reaction was quenched with 2.4 mM sodium 2-mercaptoethanesulfonate (final concentration) 20 min after the final BSR-I2 addition, left at room temperature for a further 30 min, then gel-filtered through PD10 columns into FPLC buffer (10 mM potassium phosphate, 50 mM KPr, 1 mM MgCl2, pH 7.0) to remove unconjugated rhodamine.
To check the extent of BSR labeling, an aliquot (∼0.4 nmol ELC mutant) was taken after the sodium 2-mercaptoethanesulfonate quench and analyzed by reverse-phase HPLC (C18 Vydac 218TP54 column; Hichrom, Theale, UK) with a linear gradient from 65% solvent A (H2O/0.1% trifluoroacetic acid) and 35% solvent B (acetonitrile, 0.082% trifluoroacetic acid) to 45% solvent A and 55% solvent B over 20 min at a flow rate of 1 mL/min. Elution was monitored by absorbance at 215 nm and rhodamine fluorescence (λex 549 nm, λem > 580 nm). The elution profiles varied according to the mutant. In general, labeled and unlabeled ELC could not be resolved, but the extent of labeling could be monitored by the relative amplitudes of the absorbance and fluorescence peaks.
ELC-BSRs were purified in 500 nmol batches in FPLC buffer on a Mono-Q 10/10 FPLC anion-exchange column (GE Healthcare) using a 150–400 mM KCl gradient (total volume 150 ml) at 2 mL/min. Fractions eluting at ∼200 mM KCl were analyzed by reverse-phase HPLC (as described above) and electrospray mass spectrometry. Fractions containing >90% ELC-BSR were pooled, and concentrated in dialysis tubing against solid sucrose to a final concentration of 200–500 μM. The typical yield of purified labeled protein was 35%. Protein concentration was measured using an extinction coefficient for rhodamine of 52,000 M−1cm−1 at 528 nm, based on 1:1 stoichiometric labeling, which was confirmed for each of the ELC-BSRs by electrospray mass spectrometry. ELCs in which only one of the iodoacetamide groups of BSR-I2 has reacted with a cysteine can also be readily detected by this technique, because in this case the other iodoacetamide group reacts with the sodium 2-mercaptoethanesulfonate quencher. Intramolecular cross-linking of cysteines with bifunctional rhodamines produces two diastereoisomers (5,10,12); in the case of the ELC-BSRs these were not separated by either HPLC or FPLC.
For control experiments, wild-type ELC was expressed, purified, and characterized using the methods described above, and labeled (at 64 μM) on its native cysteine (Cys136) with 192 μM 6-iodoacetamidotetramethylrhodamine (17) in labeling buffer for 80 min in the dark at 20°C. The reaction was quenched with 6.3 mM sodium 2-mercaptoethanesulfonate, and the labeled protein was purified and analyzed by the methods described above for ELC-BSR.
Muscle fiber preparation and incorporation of ELC-BSR
Single fibers were prepared from psoas muscles of New Zealand White rabbits as described previously (18). Fiber bundles were stored at −20°C in a solution containing 70 mM KPr, 8 mM MgAc2, 5 mM K2EGTA, 7 mM Na2ATP, 6 mM imidazole, 0.1% (v/v) protease inhibitor cocktail P8340, (Sigma, St. Louis, MO) and 50% glycerol, pH 7.0. Fibers were used within 6 weeks. Single-fiber segments were dissected from a bundle, and aluminum T-clips crimped to each end of the segment were used to mount them between two hooks, one of which was attached to an AE801 force transducer (SensorOne, Sausalito, CA). The microscope stage contained five 60-μL glass troughs that could be rotated to change the solution bathing the fiber. Fibers were initially mounted in relaxing solution (25 mM imidazole, 10 mM K2EGTA, 56 mM KPr, 6.6 mM MgAc2, 5.5 mM Na2ATP, 5 mM Na2CrP, ionic strength 150 mM, pH 7.1). Fiber diameter and sarcomere length were measured using a 32× objective and an ocular graticule. Sarcomere length was set to 2.4 μm.
Each ELC-BSR was separately incorporated into single fibers using a protocol modified from Matthew et al. (16). Fibers were incubated in 20 mM PIPES, 10 mM K2EGTA, 150 mM KPr, and 0.75 mM TFP (Fluka, Poole, UK), pH 6.8 for 15 min at 27°C, washed for 3 × 4 min in relaxing solution at 22°C and then transferred into relaxing solution containing 10 mM DTT and 1–4 mg/mL ELC-BSR for 30 min at 22°C. RLC lost in the ELC extraction protocol was replaced by incubating for 15 min at 10°C in relaxing solution plus 1 mg/mL RLC (prepared as in Corrie et al. (5)) plus 10 mM DTT. Troponin and troponin C were similarly replaced in subsequent incubations of 10 min in 0.5 mg/mL chicken skeletal troponin (Sigma), then 5 min in 0.5 mg/mL troponin C (prepared as in Ferguson et al. (12)). The protocol described above was selected on the basis of preliminary experiments in which the concentration of TFP and ELC-BSR, temperature and timings were varied to select the most suitable compromise between extent of ELC-BSR exchange and preservation of fiber function. The concentration of ELC-BSR in the fibers was estimated from the relative fluorescence intensity of labeled fibers and a 100-μm microslide containing a known concentration of ELC-BSR (19).
Fibers were activated at 10°C by incubating for 2 min in a preactivating solution containing 25 mM imidazole, 0.1 mM K2EGTA, 89.5 mM KPr, 6.1 mM MgAc2, 5.6 mM Na2 ATP, 5 mM Na2CrP, 1 mg/mL rabbit muscle creatine phosphokinase (Sigma), 150 mM ionic strength, pH 7.1, before transferring to activating solution (pCa 4.6) containing 25 mM imidazole, 10 mM CaEGTA, 56 mM KPr, 6.1 mM MgAc2, 5.6 mM Na2ATP, 5 mM Na2CrP, 1 mg/mL creatine phosphokinase, ionic strength 150 mM, pH 7.1. Isometric force and polarized fluorescence intensities were measured after force had reached a plateau. Measurements were also made in rigor conditions in a solution containing 25 mM imidazole, 2.5 mM K2EGTA, 120 mM KPr, 2.2 mM MgAc2, ionic strength 150 mM, pH 7.1.
Polarized fluorescence and ELC-BSR orientation measurements
Steady-state polarized fluorescence intensities were measured using a modified epifluorescence microscope as described previously (10,18,19). Fluorescence was collected by a 0.25 N.A. objective above the fiber, and separated into components polarized parallel and perpendicular to the fiber axis, the intensities of which were measured with two photomultipliers. The fiber was illuminated either from above, through the objective, or from the side, with light propagating at 90° to both the vertical microscope axis and horizontal fiber axis (8). In each case the polarization was set alternately parallel and perpendicular to the fiber axis. Thus eight polarized intensities were measured, which can be reduced to three independent order parameters 〈P2d〉, 〈P2〉, and 〈P4〉 that contain all the available information about the orientation distribution of the BSR dipole with respect to the fiber axis (20). 〈P2d〉 describes the amplitude of independent motion of the dipoles with respect to the ELC backbone on a timescale that is fast compared with the fluorescence lifetime. 〈P2〉 and 〈P4〉 give information about the mean orientation ψ of the dipole with respect to the muscle fiber axis. The normalized distribution g(ψ) of such angles can be represented as a series expansion of Legendre polynomial functions PL(cosψ) with coefficients 〈PL〉, where
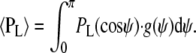 |
Given the symmetry of the distribution of probe dipoles in the muscle fiber, only the even 〈PL〉 are nonzero, and only the second- and fourth-rank order parameters, 〈P2〉 and 〈P4〉, can be obtained from the polarized fluorescence intensities measured here. P2(cosψ) and P4(cosψ) are the functions 0.5(3cos2ψ − 1) and 0.125(35cos4ψ − 30cos2ψ + 3) respectively. An estimate of the mean angle between each BSR dipole and the fiber axis, ψME, was obtained as the mean of the broadest orientation distribution consistent with the measured order parameters, the one-dimensional ME distribution (14,21).
The orientation of the ELC with respect to the fiber axis was estimated by combining 〈P2〉 and 〈P4〉 values from the three ELC-BSRs with the relative orientations of the three pairs of labeled cysteines in crystallographic structures of the myosin head domain of either chicken skeletal myosin (1) or scallop myosin (22). LCD orientations were described with the reference axes used previously for interpreting RLC probe data (5,8). For chicken skeletal myosin, the “lever” axis (Fig. 1) is defined as the line joining the α-carbons of Cys707 and Lys843 of the myosin heavy chain, and the hook axis is the line joining the mid-points of the α-carbons of the pairs Phe836, Ile838, and Met832, Leu834 of the myosin heavy chain. The angular coordinates of the ELC-BSR8–15, ELC-BSR11–73 and ELC-BSR70–77 dipoles in this reference frame are: θ = 96.6°, 15.0°, and 49.7°, φ = 82.9°, −106.0°, and −40.0° respectively, where θ is the angle between the BSR dipole and the lever axis, and φ is the angle between the plane containing the lever and BSR dipole axes and that containing the lever and hook axes. The corresponding angular coordinates for scallop myosin are θ = 120.0°, 11.9°, and 43.7°, φ = 75.1°, 129.7°, and −30.5°, with the lever axis defined by the α-carbons of scallop myosin residues Cys703 and Leu837 and the hook axis by the midpoint of those of Trp827 and Leu829 and the midpoint of Ser831 and Val833. The orientation of the ELC region with respect to the fiber axis is described by two angles: β, describing the tilt of the lever axis with respect to the fiber axis, and γ, describing the rotation of the hook axis around the lever axis (Fig. 1). Beta can vary between 0 and 180°. An increase in β is defined as producing a translation of Lys843 (chicken skeletal myosin) toward the Z-line of the half-sarcomere, as would occur in a myosin head attached to actin during muscle shortening. Gamma values are reported in the range −180° to +180°. When γ = 0°, the hook axis is coplanar with the lever and fiber axes and points toward the M-line of the sarcomere. An increase in γ denotes a counter-clockwise rotation of the hook around the lever axis when viewed from Lys843. The azimuthal orientation of the LCD around the filament axis cannot be determined from the present measurements. Three-dimensional alignment of myosin crystal structures used the protein structure comparison service SSM at European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/ssm), authored by E. Krissinel and K. Henrick.
The order parameters 〈P2〉 and 〈P4〉 yield a relatively low resolution view of the distribution of β and γ that is represented here as the distribution with the maximum informational entropy. The maximum entropy (ME) distribution fME is the broadest distribution that is consistent with the measured 〈P2〉 and 〈P4〉 values, calculated to maximize the informational entropy of the distribution, defined as 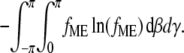 fME is proportional to exp Σ[λ2i.P2i(cosψi) + λ4i.P4i(cosψi)], where ψi is the angle between dipole i and the fiber axis and λ2i and λ4i are Lagrange multipliers for ELC-BSRi, chosen to fit the measured 〈P2i〉 and 〈P4i〉 values for that ELC-BSR. Psii is related to the angular coordinates (θi,φi) of the probe in the protein frame according to cosψi = cosβcosθi − sinβsinθicos(γ + φi). Note that a previous ME analysis (21) is modified here by omitting the sinβ term when calculating the entropy and displaying the (β,γ) distributions (14), so the ME distributions shown here represent orientation probability densities in the absence of the previous assumption (21) that the azimuthal distribution of probe angles in the cylindrically symmetrical coordinate system of the muscle fiber satisfies 3-D space-filling constraints. Each ME distribution gives an exact fit to the mean 〈P2〉 and 〈P4〉 for each condition, and therefore takes no account of measurement errors in these parameters. The effect of such errors was assessed by calculating ME distributions in which the 〈P2〉 and 〈P4〉 value for each ELC-BSR was chosen at random from normal distributions calculated from the experimental mean and SE for each parameter. Ten such ME distributions were calculated for each condition, and all 10 distributions were similar to that calculated from the corresponding mean 〈P2〉 and 〈P4〉 values and shown in Results. Thus the experimental uncertainties in the order parameters are sufficiently small to have no effect on the conclusions from ME analysis about the orientations and relative intensities of the peaks in the ME distributions at the level of precision with which these are reported.
fME is proportional to exp Σ[λ2i.P2i(cosψi) + λ4i.P4i(cosψi)], where ψi is the angle between dipole i and the fiber axis and λ2i and λ4i are Lagrange multipliers for ELC-BSRi, chosen to fit the measured 〈P2i〉 and 〈P4i〉 values for that ELC-BSR. Psii is related to the angular coordinates (θi,φi) of the probe in the protein frame according to cosψi = cosβcosθi − sinβsinθicos(γ + φi). Note that a previous ME analysis (21) is modified here by omitting the sinβ term when calculating the entropy and displaying the (β,γ) distributions (14), so the ME distributions shown here represent orientation probability densities in the absence of the previous assumption (21) that the azimuthal distribution of probe angles in the cylindrically symmetrical coordinate system of the muscle fiber satisfies 3-D space-filling constraints. Each ME distribution gives an exact fit to the mean 〈P2〉 and 〈P4〉 for each condition, and therefore takes no account of measurement errors in these parameters. The effect of such errors was assessed by calculating ME distributions in which the 〈P2〉 and 〈P4〉 value for each ELC-BSR was chosen at random from normal distributions calculated from the experimental mean and SE for each parameter. Ten such ME distributions were calculated for each condition, and all 10 distributions were similar to that calculated from the corresponding mean 〈P2〉 and 〈P4〉 values and shown in Results. Thus the experimental uncertainties in the order parameters are sufficiently small to have no effect on the conclusions from ME analysis about the orientations and relative intensities of the peaks in the ME distributions at the level of precision with which these are reported.
Confocal microscopy
Fibers into which ELC-BSR had been exchanged as described above were stretched to a sarcomere length of ∼3.2 μm in relaxing solution at 10°C and fixed in 4% (w/v) formaldehyde in PBS for 30 min at 20°C. The fibers were washed twice in PBS, and incubated in 5% normal goat serum, 1% BSA, 1 mM sodium azide in PBS for 30 min at 23°C. They were then rinsed in PBS, transferred to a petri dish and incubated overnight at 4°C in an anti-α-actinin primary antibody (Clone EA-53, Sigma) diluted in 10 mM Tris-HCl, 155 mM NaCl, 2 mM Na2EGTA, 2 mM MgCl2, 1% BSA, pH 7.5. The fibers were washed for 6 × 30 min in PBS and incubated overnight at 4°C in the same solution containing an anti-mouse Cy2-conjugated secondary antibody (Jackson ImmunoResearch, Newmarket, UK). Fibers were washed for 6 × 30 min in PBS, and mounted in 5% n-propyl gallate, 30 mM Tris-HCl, 70% glycerol, pH 9.5. Fluorescence images were obtained using a Zeiss LSM510 META confocal microscope with 63× (1.4 N.A.) oil-immersion objective, Cy2 excitation at 488 nm, BSR excitation at 543 nm, 580 nm beam-splitter, and 590 nm long-pass emission filter. The images were not optimized or recolored, so the relative fluorescence intensities are represented accurately.
RESULTS
Preparation and characterization of ELC-BSRs
Mutants of the A2 isoform of chicken fast skeletal muscle myosin ELC were prepared with pairs of surface-accessible cysteine residues at positions 8 and 15, 11 and 73, or 70 and 77 (Fig. 1). The cysteine pair in each mutant ELC was cross-linked using BSR-I2 (see Methods for details) to give a 1:1 ELC-BSR complex with >95% purity in each case. The measured masses of ELC-BSR8–15, ELC-BSR11–73 and ELC-BSR70–77 were 17,521, 17,491, and 17,536 Da, respectively. The corresponding calculated masses for the expressed ELCs, (including the N-terminal GSHM from the thrombin cleavage site) in which both cysteines have reacted with BSR are 17,521, 17,494, and 17,539 Da. The location of the cross-linking sites in ELC-BSR8–15 was confirmed by tryptic digestion (5), which produced a fluorescent product with measured mass 3282 Da, close to the calculated mass (3286 Da) for the product of tryptic cleavage at Lys11, i.e., the peptides GSHMSFSPDEICDFK and EAFCLFDR covalently linked by BSR by way of Cys8 and Cys15.
Incorporation of ELC-BSRs into muscle fibers
Native ELC was extracted from single demembranated muscle fibers by incubation in a rigor solution containing TFP (15,16). ELC-BSR (as well as RLC, troponin, and troponin C lost from the fiber during the TFP incubation) was then incorporated in sequential incubations in relaxing solution, as described in Methods. The extent of ELC-BSR incorporation could be increased by increasing either TFP concentration or temperature during the extraction step, but only at the expense of reduced isometric force generation after the ELC exchange protocol. Relatively mild extraction conditions (0.75 mM TFP for 15 min at 27°C) were chosen for the polarized fluorescence experiments, resulting in replacement of 33 ± 5% (mean ± SE, n = 17) of the native ELC by ELC-BSR and an isometric force of 72 ± 3% of that before the ELC exchange protocol. Isometric force after ELC exchange was the same within experimental variability for the three ELC-BSRs, unlabeled wild-type ELC (66 ± 8%; n = 4), and wild-type ELC labeled on the native Cys136 with 6-iodoacetamidotetramethylrhodamine (74 ± 5%; n = 5). Reduced isometric force after ELC exchange is therefore not due to ELC mutagenesis or labeling, and is likely to be a nonspecific effect of the exchange protocol.
The sarcomeric location of ELC-BSR70–77 incorporated into muscle fibers using this protocol was determined by confocal microscopy (Fig. 2). The green stripes show the location of an α-actinin antibody at the Z-discs. ELC-BSR70–77 (orange) is located in the A-band, with a narrow dark stripe at the midpoint of each A-band corresponding to the bare zone of the myosin filaments where there are no myosin heads. Thus, within the resolution of these measurements, ELC-BSR70–77 is located in the region of the sarcomere that contains myosin heads, as expected for specific replacement of the native ELC.
FIGURE 2.

Confocal microscope image of part of a rabbit psoas fiber after partial replacement of native ELC by ELC-BSR70–77 (orange). The Z-lines were labeled by an α-actinin antibody (green).
In situ orientation of BSR probe dipoles
Polarized fluorescence intensities from muscle fibers containing ELC-BSR8–15, ELC-BSR11–73, or ELC-BSR70–77 were used to calculate the order parameters 〈P2d〉, 〈P2〉, and 〈P4〉 that describe the orientation distribution of the BSR dipole with respect to the muscle fiber axis (20). 〈P2d〉 is a measure of the amplitude of rapid independent “wobble” of the dipole with respect to the ELC backbone, and was in the range 0.82–0.86 for each ELC-BSR in relaxation, active contraction and rigor (Fig. 3), corresponding to uniform wobble in a cone of semi-angle 25–29° on a timescale fast compared with the fluorescence lifetime. Similar values of 〈P2d〉 were reported previously for BR probes on the RLC (5,10), and slightly higher values for either BR or BSR on troponin C (14).
FIGURE 3.

Order parameters 〈P2d〉, 〈P2〉, and 〈P4〉 for the orientation of three ELC-BSR dipoles in relaxation, active contraction, and rigor. Mean ± SE of mean for n = 5–9 fibers. Sarcomere length, 2.4 μm.
〈P2〉 and 〈P4〉 are sensitive to dipole orientations averaged over timescales that are long compared with the fluorescence lifetime, with the rapid independent motion of the probes factored out. 〈P2〉 would be +1 if all the dipoles were parallel to the fiber axis (ψ = 0°), and −0.5 if they were all perpendicular to that axis (ψ = 90°). 〈P4〉 gives higher resolution orientation information; it is +1 for ψ = 0°, has a minimum of −0.43 near ψ = 50° and is 0.375 at ψ = 90°, passing through zero near ψ = 30° and 70°. 〈P2〉 and 〈P4〉 values for ELC-BSR8–15, ELC-BSR11–73, and ELC-BSR70–77 in relaxation and active contraction were generally closer to zero than the corresponding values for rigor (Fig. 3), reflecting a greater degree of orientational order in rigor. This trend is particularly marked for ELC-BSR70–77. 〈P2〉 and 〈P4〉 values for active contraction were generally intermediate between those for relaxation and rigor, but closer to the relaxed values. These general features were observed previously for BR probes on the RLC (5,8,10).
The angles between the ELC-BSR dipoles and the fiber axis were quantified using the mean (ψME) of the 1-D ME orientation distribution, the broadest such distribution consistent with the 〈P2〉 and 〈P4〉 data (14,21). ψME for the ELC-BSR8–15 dipole was 60.5 ± 0.2°, 61.0 ± 0.3°, and 64.7 ± 0.6° (mean ± SE, n = 5) in relaxation, active contraction, and rigor respectively. The corresponding values for ELC-BSR11–73 were 57.8 ± 0.1°, 59.7 ± 0.3°, and 61.6 ± 0.4° (n = 7), and those for ELC-BSR70–77 were 57.3 ± 0.3°, 57.0 ± 0.2°, and 42.8 ± 1.7° (n = 5). Although most of these changes in ψME are small, they were reproducible. The ELC-BSR8–15 and ELC-BSR11–73 dipoles are more perpendicular to the fiber axis in rigor than in the other two states, whereas the ELC-BSR70–77 dipole is more parallel.
ELC orientation distributions
More detailed information about the in situ orientation of the ELC region of myosin was obtained by combining 〈P2〉 and 〈P4〉 values from the three ELC-BSR probes with the relative orientations of the labeled cysteines in the protein coordinate frame (Fig. 1). Initially we assumed that the LCD is a rigid body, and defined its orientation in the fiber in terms of the “lever axis” joining myosin heavy chain residues Cys707 and Lys843 (Fig. 1). The angle between the lever axis and the fiber axis is the tilt angle, β. Rotation of the LCD around the lever axis is described by the twist angle (γ), and γ is zero when the short C-terminal helix of the myosin heavy chain (i.e., the “hook axis”) is in the same plane as the lever and filament axes, with its C terminus pointing toward the M-line (Fig. 1). For each condition we calculated the 2-D (β, γ) orientation distribution with the maximum informational entropy that is consistent with the measured 〈P2〉 and 〈P4〉 values for the three ELC-BSR probes (8,14,21). These ME distributions can be considered as low-resolution approximations of the actual orientation distribution.
The ME distribution of the ELC region in relaxed muscle, calculated using the crystal structure of chicken skeletal myosin, is displayed as a contour plot in Fig. 4 A, with the hotter colors denoting a higher probability of that (β, γ) orientation. It has two red peaks of similar intensity, centered on β = 105°, γ = −55°, and β = 110°, γ = 20°, suggesting the presence of two populations of ELC molecules, which may be associated with the two heads of each myosin molecule. Although only the range −90° < γ < 90° is plotted here, γ can take values from −180° to +180°, and each point in the range plotted here is equivalent to an orientation (180° − β, 180° + γ) to which it can be considered to be related by the bipolar symmetry of the sarcomere. Thus the left peak in Fig. 4 A cannot be distinguished from β = 75°, γ = 125°, and the right peak from β = 70°, γ = −160°. It follows that the difference in β between the two populations could be either ∼5° or ∼35°. Similarly, the corresponding difference in γ between the two populations could be either ∼75° or ∼105°.
FIGURE 4.

Contour plots of the ME orientation distributions (β, γ) of the LCD in relaxation (A), active contraction (B), and rigor (C), calculated from the 〈P2〉 and 〈P4〉 data in Fig. 3 using the crystal structure of nucleotide-free chicken skeletal myosin (1). Hotter colors indicate a higher probability of that orientation. The pink square in C denotes the orientation of actin-attached chicken skeletal myosin in the absence of ATP (2).
The ME distribution of the ELC region in active contraction (Fig. 4 B) has a broad main peak at β = 105°, γ = 15°, close to one of the peaks observed in relaxation. Although the symmetry-related β = 75°, γ = −165° equivalent peak cannot be excluded, this would have the hook axis (Fig. 1) pointing away from the M-line, which seems unlikely in a load-bearing actin-attached myosin head. The ME distribution has a shoulder around β = 105°, γ = −55°, close to the second peak observed in relaxing conditions.
The ME distribution of the ELC region in rigor (Fig. 4 C) has a single peak centered on β = 120°, γ = 25°, and is much more compact than the relaxed or active distribution, with a full width at half-maximum of only ∼30° in β and 40° in γ. The symmetry-related solution with γ = −155° is highly unlikely in rigor, when all myosin heads are expected to be attached to actin with a conformation roughly similar to that shown in Fig. 1. The higher value of β in rigor than in active contraction corresponds to a forward motion through the power stroke, as expected for motion of the LCD-myosin rod junction away from the M-line (Fig. 1).
Broadly similar ME distributions for ELC orientation were obtained using a scallop myosin crystal structure (PDB 1sr6; (22)) to interpret the ELC-BSR polarized fluorescence data (see Supplementary Material Fig. S1, Data S1). Two populations of ELC orientations were apparent in relaxed muscle, at β = 80°, γ = −60° and β = 105°, γ = 10°. The ME distribution for active contraction is dominated by a main peak at β = 105°, γ = 10°, with a lower intensity peak at β = 85°, γ = −60°. The ME distribution for rigor had a narrow peak centered on β = 125°, γ = 15° (Fig. 5 A). The same general features were observed in ME distributions (not shown) calculated using a crystal structure of squid myosin in the “rigor-like” state (PDB 2ovk; (23)); in this case the rigor distribution, for example, had a similar narrow peak centered on β = 130°, γ = 5°.
FIGURE 5.

LCD orientation and conformation in rigor muscle. (A and B) ME orientation distributions calculated from probes on the ELC (A) and RLC (B) using the nucleotide-free scallop myosin structure (22). Inverted triangles denote the orientation calculated by fitting the catalytic domain of this structure into that of actin-attached chicken skeletal myosin in the absence of ATP (2). (C) Graphical representation of the orientation denoted by the inverted triangles with respect to a vertical actin filament. (D) Myosin head conformation with a bend between the ELC and RLC regions that is consistent with the probe data from both regions in rigor muscle (see text for details).
DISCUSSION
Preparation of ELC-BSRs
We determined the orientation of the ELC region of myosin in muscle fibers using the general approach applied previously to the RLC (5,8) and troponin C (12,14). Expressed proteins containing pairs of cysteine residues at suitable surface-accessible locations were cross-linked with a bifunctional rhodamine derivative, and the purified labeled proteins introduced into muscle fibers. In these experiments ELC mutants with cysteine pairs at positions 8 and 15, 11 and 73, or 70 and 77 were cross-linked using BSR-I2, which was chosen in preference to bifunctional rhodamine BR-I2 (5,24) because of its greater reactivity toward cysteine residues. A comparative study of BSR and BR on the C helix of troponin C indicated that both probes accurately report the orientation of the vector joining the two cysteines (14), and the solution structure of troponin C is not altered by attachment of either BR or BSR along its C helix (14,25).
Replacement of native ELC in muscle fibers by ELC-BSRs
RLC and troponin C can be exchanged into demembranated muscle fibers under relatively mild conditions by reducing the [Mg2+] and/or the ionic strength of the bathing solution (9,26). ELC is not extracted from muscle fibers under these conditions, but can be extracted or exchanged using TFP, a hydrophobic drug that binds to proteins of the calmodulin family (15,16,27–29). In these experiments, ∼⅓ of the native ELC was replaced by ELC-BSR. Isometric force generation was reduced by 28% on average after the ELC exchange protocol, but this decrease is not due to ELC mutagenesis or BSR labeling. Similar ELC exchange protocols were used previously to show the dependence of contraction speed and shortening velocity on ELC isoform in skeletal muscle (15) and smooth muscle (16), indicating that native ELC function is retained in these conditions. Confocal microscopy showed that the ELC-BSR introduced by this procedure is in the region of the sarcomere containing myosin heads (Fig. 2), as expected for specific replacement of endogenous ELC.
Orientation of the ELC region of myosin in muscle fibers
Relation to previous fiber studies using monofunctional ELC probes
ELCs with fluorescent probes attached to single cysteine residues were introduced previously into demembranated muscle fibers using a TFP-based protocol (28,29), and small differences (∼5°) in probe orientation between relaxing and rigor conditions were observed. However, two major differences between this study and previous studies preclude detailed comparison of the results. First, we introduced probes into the N-terminal lobe of the A2 isoform of the ELC at sites that do not interact with the myosin heavy chain (Fig. 1), whereas the previous studies used the A1 isoform labeled either at the C-terminus, near the catalytic domain of the heavy chain, or on the N-terminal extension that is not present in the A2 isoform (28,29). Second, the bifunctional rhodamine approach used here defines the orientation of the probe dipoles in the coordinate frame of the ELC, so the absolute orientation of the labeled region of the ELC with respect to the muscle fiber axis can be determined, in contrast to the previous studies with monofunctional probes in which the orientation of the probe dipole with respect to the protein was unknown.
The orientation of the ELC region of myosin in rigor
In ATP-depleted (rigor) muscle all the myosin heads are attached to actin filaments, and the orientation of the ELC region of myosin calculated from the polarized fluorescence data using the crystal structure of chicken skeletal myosin in the nucleotide-free state (1) is centered on β = 120°, γ = 25° (Fig. 4 C, red region). These values of β and γ are larger than those calculated by fitting the same crystal structure into density maps determined by cryo-electron microscopy of chicken skeletal myosin head fragments bound to isolated actin filaments in the absence of ATP (2), which gave β = 102°, γ = −2° (Fig. 4 C, pink square; myosin head conformation shown graphically in Fig. 1). A subsequent higher resolution cryo-electron microscopy study of the same actomyosin rigor complex (30) found a similar LCD conformation, with β = 108°, γ = 8°.
The crystal structure of scallop myosin in the nucleotide-free state (31) has a markedly different conformation from that of chicken skeletal myosin (1), and the relative orientation of the LCD and actin-binding domains differs by ∼40°. The actin-binding regions of the two structures are similar, however, and it seems likely that they bind to actin in the same orientation. We therefore superimposed the actin-binding region of nucleotide-free scallop myosin on that of chicken myosin in the rigor complex (2). The resulting conformation of the scallop myosin (Fig. 5 C) has β = 132°, γ = 13° (Fig. 5 A, white inverted triangle), close to the β = 125°, γ = 15° orientation calculated from the polarized fluorescence data from the ELC probes using the scallop myosin structure (Fig. 5 A, red region). Thus, the in situ orientation of the ELC region of myosin bound to actin in the absence of ATP determined from the fluorescence data fits more closely with the crystallographic nucleotide-free conformation of scallop myosin (31) than with that of chicken skeletal myosin (1,2).
The crystallographic structures used in the above analysis were determined in the absence of both nucleotide and actin, but seem to mimic a state with low actin affinity that would be produced by nucleotide binding to the rigor complex, which has been called the “postrigor” state (23,30). However the myosin conformation in the rigor actomyosin complex might be closer to the “rigor-like” structure described recently for squid muscle myosin (PDB 2ovk; (23)), so we repeated the analysis using this structure. The results were similar to those obtained with the scallop myosin postrigor structure; fitting the actin-binding region of the squid myosin rigor-like structure to that of chicken skeletal myosin in the rigor complex (2) gave β = 128°, γ = 19°, close to the peak of the ME distribution calculated from the polarized fluorescence data from rigor muscle using the squid myosin rigor-like structure, β = 130°, γ = 5°.
A bend between the ELC and RLC regions of the LCD in rigor muscle
The ME distribution of the RLC region of the LCD in rigor muscle (Fig. 5 B), calculated from the 〈P2〉 and 〈P4〉 data from four BR probes on the RLC (10) using the scallop myosin postrigor structure peaks at β = 90°, γ = 40°. This distribution is broader in the β direction than that calculated from the ELC probes (Fig. 5 A). The difference between the peak values of β calculated from the RLC and ELC probe data sets (90°, Fig. 5 B, and 125°, Fig. 5 A, respectively) implies that the LCD is bent at the interface between the RLC and ELC in rigor muscle, by ∼35° with respect to the LCD conformation in the scallop myosin postrigor structure (31). The same 35° bend was obtained when either the chicken skeletal postrigor or squid myosin rigor-like structure was used for the analysis.
We used these results to estimate the average conformation of the myosin heads in rigor muscle. The actin-binding and ELC regions were assumed to retain the conformation determined by cryo-electron microscopy (Fig. 5 A, inverted triangle; Fig. 5 C), which for this structure is consistent with the fluorescence data from the ELC probes (Fig. 5 A, red region). Because azimuthal orientations cannot be measured by this technique, we made the simplest assumption that there is no azimuthal bending between the ELC and RLC regions. To fit the polarized fluorescence data from the RLC probes, it was necessary to both tilt the RLC region up by 45° and twist it by 25°, around a pivot point at heavy chain residue Ala797. The resulting structure (Fig. 5 D) is consistent with the polarized fluorescence data from both the ELC and RLC probes in rigor muscle. Note that the local reorientation of the RLC region is larger than the difference in the peak β,γ values determined using the ELC and RLC probe data sets separately (Fig. 5, A and B), because β and γ are defined using a coordinate frame that includes the whole of the LCD.
Crystal structures of the LCD (1,11,23,31) all have a significant bend in the long heavy-chain helix between the ELC and RLC regions, and this is presumably stabilized by the direct interaction between the ELC and RLC in those structures. This bend is largely abolished in the in situ LCD conformation in rigor muscle (Fig. 5 D), in which there is no direct interaction between the ELC and RLC. In scallop myosin this interaction is thought to be stabilized by a regulatory Ca2+ ion (11); the structure of this region in the absence of bound Ca2+ is unknown, but a change in LCD conformation associated with loss of the RLC:ELC interaction may be a key component of the mechanism of Ca2+-regulation in this myosin. There is no evidence for such a regulatory Ca2+ site in vertebrate myosin II, but the side-chain coordinates for the RLC and ELC are not defined in the only available structure of a vertebrate myosin II containing the RLC (PDB 2mys; (1)), so the structural details of the RLC/ELC interface in vertebrate skeletal myosins are unknown. The present results raise the intriguing possibility that this interface might be modulated during muscle contraction or its regulation in physiological conditions.
The in situ conformation of the LCD in rigor muscle shown in Fig. 5 D corresponds to the peaks of the ME distributions, but there is considerable orientational dispersion around these peaks, particularly in the axial orientation β of the RLC region (Fig. 5 B), which has a full width at half-maximum of 60°, about twice that of the ELC region (Fig. 5 A). The difference suggests that the ELC orientation is relatively constrained by the rigor orientation of the actin-binding region of myosin, and that a range of bends between the ELC and RLC regions is responsible for the greater axial dispersion of the RLC region. This range of RLC orientations could allow the two RLC regions of each myosin molecule to share a single junction with the coiled-coil myosin rod in situ without significant uncoiling of the rod.
Conformation of myosin heads in relaxed muscle
The ME distribution of the orientation of the ELC region in relaxed muscle (Fig. 4 A, chicken skeletal myosin coordinates; Fig. S1 A (Data S1), scallop myosin coordinates) suggests the presence of two populations of myosin heads with distinct orientations that may correspond to the two heads of each myosin molecule. Both populations have the long axis of the LCD (the lever axis, Fig. 1) roughly perpendicular to the filament axis. These LCD orientations are clearly different from those calculated by fitting myosin head structures into density maps determined from cryo-electron micrographs of myosin filaments isolated from tarantula muscle (32,33) or mouse cardiac muscle (filament region containing myosin-binding protein C (34)), in which one of the heads of each myosin has its LCD long axis almost parallel to the filament axis. There are many possible reasons for the different LCD orientations determined in those studies, related to the different species and muscle type, to filament isolation, or to other differences in methodology and analysis.
The ME distribution for the RLC region in relaxed muscle, calculated from the polarized fluorescence data from the RLC probes (10) using either chicken skeletal myosin (Fig. S2 B, Data S1) or scallop myosin postrigor coordinates (Fig. S2 D, Data S1), is narrower than that for the ELC region in the same conditions, and shows only a single peak. This qualitative difference suggests that there might be different bend angles between the ELC and RLC regions of the two heads of each myosin molecule in relaxed muscle, but the more complex orientation distribution in relaxed muscle precludes the type of quantitative analysis of RLC:ELC bending presented above for rigor muscle.
Conformation of myosin heads during active contraction
The ME distribution of ELC orientations during active contraction peaked at β = 105°, γ = 10–15° (Fig. 4 B and Fig. S1 B, Data S1), but was much broader than that in rigor (Fig. 4 C, and Fig. S1 C, Data S1), with a minor population of molecules around γ = −60°, close to the second population observed in relaxed muscle (Fig. 4 A and Fig. S1 A, Data S1). In the conditions of these measurements, ∼30% of the myosin heads are attached to actin and bearing force. (35), and it seems likely that these force-bearing heads are within the γ = 10–15° population, with the hook helix of the myosin heavy chain pointing toward the M-line, as in rigor. The peak value of β, 105°, is similar to that estimated previously by fitting polarized fluorescence data from four RLC probes with a Gaussian orientation distribution, 96° (95% confidence limits 91–120°; (5)). A subsequent ME analysis of similar data from three of these RLC probes using the chicken skeletal myosin postrigor structure showed a more complex orientation distribution with peaks around β = 70°, γ = −44° and β = 130°, γ = 20° (8), which differs in detail from that calculated here from the ELC probes (Fig. 4 B and Fig. S1 B, Data S1). However, a quantitative comparison of the orientations of the RLC and ELC regions is again impeded by the presence of multiple orientation populations. Further experiments with mechanical and/or biochemical perturbations and additional probe sites will be required to determine the orientation of the RLC and ELC regions of the force-generating population of myosin heads in contracting muscle.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Dr. J. Kendrick-Jones for the gift of the wild-type ELC clone, Dr. J.E.T. Corrie for 6-iodoacetamidotetramethylrhodamine and BR-I2, Drs. E. Ehler and S. Lange for help with antibody labeling and confocal microscopy, and Drs. J.E.T. Corrie and R.E. Dale for helpful comments on the manuscript.
This work was supported by the Biotechnology and Biological Sciences and Medical Research Councils (UK).
Roisean E. Ferguson's present address is Cancer Research UK Clinical Centre, St. James University Hospital, Leeds L59 7TF, UK.
Abbreviations used: BR and BSR, bifunctional carborhodamine and bifunctional sulforhodamine moieties, respectively, attached to proteins after two-site labeling with BR-I2 or BSR-I2 (bis-((N- iodoacetyl)piperazinyl) sulforhodamine-I2) reagents; BSA, bovine serum albumin; CrP, creatine phosphate; DTT, dithiothreitol; ELC, myosin essential light chain; ELC-BSR, mutant ELC labeled with BSR-I2; FPLC, fast protein liquid chromatography; GST, glutathione S-transferase; HPLC, high performance liquid chromatography; KPr, potassium propionate; LCD, light-chain domain of myosin; MgAc2, magnesium acetate; ME, maximum entropy; PBS, phosphate-buffered saline; PMSF, phenylmethylsulfonyl fluoride; RLC, myosin regulatory light chain; TFP, trifluoperazine; PDB, Protein Data Bank.
Editor: Christopher Lewis Berger.
References
- 1.Rayment, I., W. R. Rypniewski, K. Schmidt-Base, R. Smith, D. R. Tomchick, M. M. Benning, D. A. Winkelman, G. Wesenberg, and H. M. Holden. 1993. Three dimensional structure of myosin subfragment-1: a molecular motor. Science. 261:50–58. [DOI] [PubMed] [Google Scholar]
- 2.Rayment, I., H. M. Holden, M. Whittaker, C. B. Yohn, M. Lorenz, K. C. Holmes, and R. A. Milligan. 1993. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 261:58–65. [DOI] [PubMed] [Google Scholar]
- 3.Irving, M., T. St C. Allen, C. Sabido-David, J. S. Craik, B. Brandmeier, J. Kendrick-Jones, J. E. T. Corrie, D. R. Trentham, and Y. E. Goldman. 1995. Tilting of the light chain region of myosin during step length changes and active force generation in skeletal muscle. Nature. 375:688–691. [DOI] [PubMed] [Google Scholar]
- 4.Dominguez, R., Y. Freyzon, K. M. Trybus, and C. Cohen. 1998. Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Cell. 94:559–571. [DOI] [PubMed] [Google Scholar]
- 5.Corrie, J. E. T., B. D. Brandmeier, R. E. Ferguson, D. R. Trentham, J. Kendrick-Jones, S. C. Hopkins, U. A. van der Heide, Y. E. Goldman, C. Sabido-David, R. E. Dale, S. Criddle, and M. Irving. 1999. Dynamic measurement of myosin light-chain domain tilt and twist in muscle contraction. Nature. 400:425–430. [DOI] [PubMed] [Google Scholar]
- 6.Dobbie, I., M. Linari, G. Piazzesi, M. Reconditi, N. Koubassova, M. A. Ferenczi, V. Lombardi, and M. Irving. 1998. Elastic bending and active tilting of myosin heads during muscle contraction. Nature. 396:383–387. [DOI] [PubMed] [Google Scholar]
- 7.Baker, J. E., I. Brust-Mascher, S. Ramachandran, L. E. W. LaConte, and D. D. Thomas. 1998. A large and distinct rotation of the myosin light chain domain occurs upon muscle contraction. Proc. Natl. Acad. Sci. USA. 95:2944–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hopkins, S. C., C. Sabido-David, U. A. van der Heide, R. E. Ferguson, B. D. Brandmeier, R. E. Dale, J. Kendrick-Jones, J. E. T. Corrie, D. R. Trentham, M. Irving, and Y. E. Goldman. 2002. Orientation changes of the myosin light-chain-domain during filament sliding in active and rigor muscle. J. Mol. Biol. 318:1275–1291. [DOI] [PubMed] [Google Scholar]
- 9.Moss, R. L., G. G. Giulian, and M. L. Greaser. 1982. Physiological effects accompanying the removal of myosin LC2 from skinned skeletal muscle fibers. J. Biol. Chem. 257:8588–8591. [PubMed] [Google Scholar]
- 10.Brack, A. S., B. D. Brandmeier, R. E. Ferguson, S. Criddle, R. E. Dale, and M. Irving. 2004. Bifunctional rhodamine probes of myosin regulatory light chain orientation in relaxed skeletal muscle fibers. Biophys. J. 86:2329–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Houdusse, A., and C. Cohen. 1996. Structure of the regulatory domain of scallop myosin at 2Å resolution: implications for regulation. Structure. 4:21–32. [DOI] [PubMed] [Google Scholar]
- 12.Ferguson, R. E., Y.-B. Sun, P. Mercier, A. S. Brack, B. D. Sykes, J. E. T. Corrie, D. R. Trentham, and M. Irving. 2003. In situ orientations of protein domains: troponin C in muscle fibers. Mol. Cell. 11:865–874. [DOI] [PubMed] [Google Scholar]
- 13.Asenjo, A. B., N. Krohn, and H. Sosa. 2003. Configuration of the kinesin motor domains during ATP hydrolysis. Nat. Struct. Biol. 10:836–842. [DOI] [PubMed] [Google Scholar]
- 14.Julien, O., Y.-B. Sun, A. C. Knowles, B. D. Brandmeier, R. E. Dale, D. R. Trentham, J. E. T. Corrie, B. D. Sykes, and M. Irving. 2007. Towards protein structure in situ: comparison of two bifunctional rhodamine adducts of troponin C. Biophys. J. 93:1008–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sweeney, H. L. 1995. Function of the N-terminus of the myosin essential light chain of vertebrate striated muscle. Biophys. J. 68:112s–119s. [PMC free article] [PubMed] [Google Scholar]
- 16.Matthew, J. D., A. S. Khromov, K. M. Trybus, A. P. Somlyo, and A. V. Somlyo. 1998. Myosin essential light chain isoforms modulate the velocity of shortening propelled by nonphosphorylated cross-bridges. J. Biol. Chem. 273:31289–31296. [DOI] [PubMed] [Google Scholar]
- 17.Corrie, J. E. T., and J. S. Craik. 1994. Synthesis and characterization of pure isomers of iodoacetamidotetramethylrhodamine. J. Chem. Soc., Perkin Trans. 1. 2967–2974.
- 18.Sabido-David, C., B. Brandmeier, J. S. Craik, J. E. T. Corrie, D. R. Trentham, and M. Irving. 1998. Steady-state fluorescence polarization studies of the orientation of myosin regulatory light chains in single skeletal muscle fibers using pure isomers of iodoacetamidotetramethylrhodamine. Biophys. J. 74:3083–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ling, N., C. Shrimpton, J. Sleep, J. Kendrick-Jones, and M. Irving. 1996. Fluorescent probes of the orientation of myosin regulatory light chains in relaxed, rigor, and contracting muscle. Biophys. J. 70:1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dale, R. E., S. C. Hopkins, U. A. van der Heide, T. Marszalek, M. Irving, and Y. E. Goldman. 1999. Model-independent analysis of the orientation of fluorescent probes with restricted mobility in muscle fibers. Biophys. J. 76:1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Heide, U. A., S. C. Hopkins, and Y. E. Goldman. 2000. A maximum entropy analysis of protein orientations using fluorescence polarization data from multiple probes. Biophys. J. 78:2138–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Risal, D., S. Gourinath, D. M. Himmel, A. G. Szent-Györgyi, and C. Cohen. 2004. Myosin subfragment 1 structures reveal a partially bound nucleotide and a complex salt bridge that helps couple nucleotide and actin binding. Proc. Natl. Acad. Sci. USA. 101:8930–8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang, Y., S. Gourinath, M. Kovacs, L. Nyitray, R. Reutzel, D. H. Himmel, E. O'Neall-Hennessey, L. Reshetnikova, A. Szent-Györgyi, J. H. Brown, and C. Cohen. 2007. Rigor-like structures from muscle myosins reveal key mechanical elements in the transduction pathways of this allosteric motor. Structure. 15:553–564. [DOI] [PubMed] [Google Scholar]
- 24.Corrie, J. E. T., J. S. Craik, and V. R. N. Munasinghe. 1998. A homobifunctional rhodamine for labeling proteins with defined orientations of a fluorophore. Bioconjug. Chem. 9:160–167. [DOI] [PubMed] [Google Scholar]
- 25.Mercier, P., R. E. Ferguson, M. Irving, J. E. T. Corrie, D. R. Trentham, and B. D. Sykes. 2003. The NMR structure of a bifunctional rhodamine labeled N-domain of troponin C complexed with the regulatory ‘switch’ peptide from troponin I; implications for in situ fluorescence studies in muscle fibers. Biochemistry. 42:4333–4348. [DOI] [PubMed] [Google Scholar]
- 26.Moss, R. L., G. G. Giulian, and M. L. Greaser. 1985. The effects of partial extraction of TnC on the tension-pCa relationship in rabbit skinned skeletal muscle fibers. J. Gen. Physiol. 86:585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cook, W. J., L. J. Walter, and M. R. Walter. 1994. Drug binding by calmodulin: crystal structure of a calmodulin-trifluoperazine complex. Biochemistry. 33:15259–15265. [DOI] [PubMed] [Google Scholar]
- 28.Borejdo, J., D. S. Ushakov, R. Moreland, I. Akopova, Y. Reshetnyak, L. D. Saraswat, K. M. Kamm, and S. Lowey. 2001. The power stroke causes changes in orientation and mobility of the termini of essential light chain 1 of myosin. Biochemistry. 40:3796–3803. [DOI] [PubMed] [Google Scholar]
- 29.Borejdo, J., D. S. Ushakov, and I. Akopova. 2002. Regulatory and essential light chains of myosin rotate equally during contraction of skeletal muscle. Biophys. J. 82:3150–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holmes, K. C., I. Angert, J. Kull, W. Jahn, and R. R. Schroeder. 2003. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature. 425:423–427. [DOI] [PubMed] [Google Scholar]
- 31.Houdusse, A., A. G. Szent-Györgyi, and C. Cohen. 2000. Three conformational states of scallop myosin S1. Proc. Natl. Acad. Sci. USA. 97:11238–11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wendt, T., D. Taylor, K. M. Trybus, and K. Taylor. 2001. Three dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc. Natl. Acad. Sci. USA. 98:4361–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woodhead, J. L., F.-Q. Zhao, R. Craig, E. H. Egelman, L. Alamo, and R. Padron. 2005. Atomic model of a myosin filament in the relaxed state. Nature. 436:1195–1199. [DOI] [PubMed] [Google Scholar]
- 34.Zoghbi, M. E., J. L. Woodhead, R. L. Moss, and R. Craig. 2008. Three-dimensional structure of vertebrate cardiac muscle filaments. Proc. Natl. Acad. Sci. USA. 105:2386–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Linari, M., M. Caremani, C. Piperio, P. Brandt, and V. Lombardi. 2007. Stiffness and fraction of myosin motors responsible for active force in permeabilized muscle fibers from rabbit psoas. Biophys. J. 92:2476–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.