

Abstract
We present a general framework for the generation, alignment, comparison, and hybridization of motion pathways between two known protein conformations. The framework, which is rooted in probabilistic motion-planning techniques in robotics, allows for the efficient generation of collision-free motion pathways, while considering a wide range of degrees of freedom involved in the motion. Within the framework, we provide the means to hybridize pathways, thus producing, the motion pathway of the lowest energy barrier out of the many pathways proposed by our algorithm. This method for comparing and hybridizing pathways is modular, and may be used within the context of molecular dynamics and Monte Carlo simulations. The framework was implemented within the Rosetta software suite, where the protein is represented in atomic detail. The K-channels switch between open and closed conformations, and we used the overall framework to investigate this transition. Our analysis suggests that channel-opening may follow a three-phase pathway. First, the channel unlocks itself from the closed state; second, it opens; and third, it locks itself in the open conformation. A movie that depicts the proposed pathway is available in the Supplementary Material (Movie S1) and at http://www.cs.tau.ac.il/∼angela/SuppKcsA.html.
INTRODUCTION
K-channels are found, in essence, in all kingdoms of life and all types of cells (1). They are best known for their function in excitable cells. For example, they are involved in the generation and propagation of nerve impulses in the synapse and neuron (1). Mutations in the proteins that form the channel may lead to diseases such as multiple sclerosis, cystic fibrosis, and cardiac arrhythmia (2). Because of their involvement in these and other channelopathies, i.e., channel-related diseases, they are major drug targets. The K-channels are diverse in their sequences and mechanisms of gating, i.e., the processes by which their pores open and close. However, they all share a similar pore structure and very similar ion permeability characteristics. Here, we focused on the pore domain. The structure of this domain was revealed by x-ray crystallography studies of the bacterial K-channel of Streptomyces lividans (KcsA) (3,4). Subsequently, several more K-channel structures of various sources have emerged (5–9), and we know the structure of some K-channels in their open conformation, and others in their closed conformation. For example, the voltage-dependent K-channels from Aeropyrum pernix (KvAP) (10) and Methanobacterium thermoautotrophicum (MthK) (11) are considered to be in open conformations, whereas KcsA is known in a closed conformation. We present a novel framework for exploring conformational changes in proteins, and use it to suggest a pathway between a closed state of the KcsA channel (4) and a model structure of the open state of the channel.
The pore-forming region in the K-channel is composed of four identical monomers that oligomerize around the channel pore (Figs. 1 and 2 a). Each monomer contributes a pair of transmembrane helices, TM1 (Fig. 1, blue) and TM2 (Fig. 1, green), that are connected by a reentrant loop. This loop, located at the extracellular end of the channel, contains the selectivity filter (Fig. 1, yellow), which is tuned, for example, to select potassium over sodium ions. The pore gate is found at the intracellular region of TM2 helices (Fig. 1).
FIGURE 1.

KcsA channel. Two opposing monomers of the tetrameric channel are shown. Each monomer contains two TM helices (TM1, blue; TM2, green). The TM helices are connected to each other by the turret loop (gray), pore helix (red), and selectivity filter (yellow). Two K+ ions within the selectivity filter are shown as purple spheres. Locations of intracellular and extracellular regions are indicated.
FIGURE 2.

Scheme of the overall algorithm: finding a motion pathway with a minimal energy barrier between open and closed conformations. (a) Two RRT conformation trees were grown simultaneously, starting from the initial (solid) and goal (open) conformations, until trees could be connected to create a full-motion pathway. The goal conformation (right) was based on the known x-ray structure of KvAP (10). (b) General outline of the methodology.
There is much evidence that the KcsA channel switches between open and closed conformations (12–17). The computational effort to provide molecular models of conformational changes includes coarse-grained normal-modes analysis (18). In one particularly interesting study, using the Gaussian-network method, Shrivastava and Bahar (19) suggested that channel-opening follows the corkscrew motion of intracellular regions of the channel. This suggestion recently gained strong experimental support from innovative single-molecule studies (20). The experimental results were interpreted to indicate a corkscrew motion of the intracellular ends of TM2 helices, and provided the first direct insights into the large-scale nature of the motion. However, experimental methods are still unable to give a detailed atomic account of the motion, which is of great interest in mutational analysis and the drug targeting of transition states.
The transition between the open and closed conformations involves major conformational changes and occurs within microseconds or more (21). The large amplitude of the motion, both in time and space, is beyond the reach of standard molecular-dynamics simulations, and external biasing was used to trace it. For example, Biggin and Sansom used steered molecular dynamics and simulated channel-opening by slowly inflating a sphere which was placed at the center of the (closed) gate (21). Tikhonov and Zhorov used a similar approach, by applying full-atom Monte Carlo simulations while iteratively inflating and deflating a cylinder at the pore axis (22).
In this study, we developed a very different framework to explore many putative conformational trajectories rapidly, align them using a novel dynamic-programming approach, and cluster them into representative motion pathways. Within this general framework, we found plausible low-energy trajectories of channel-opening, with and without a biasing constraint.
The basic motion-planning problem can be stated as follows. Given a robot moving in an environment cluttered with obstacles, and given start and goal configurations for the robot, find a collision-free path connecting these two configurations (“configuration” is the common robotics term for “conformation”). The motion-planning problem is fundamental, and was originally studied in robotics and computational geometry, but has implications in numerous other fields (23–25). A class of randomized-path planning methods, known as probabilistic roadmap (PRM) methods, was successfully applied to complicated high-dimensional problems (26–28).
A variant of probabilistic techniques focuses on single-query strategies, where the goal is typically to find a collision-free path between the two query configurations by exploring as little space as possible. Single-query strategies often build a new roadmap for each query by growing trees of sampled milestones rooted at the start and goal configurations (27). Rapidly exploring random trees (RRTs) were recognized as useful tools for designing efficient single-query paths in highly constrained spaces (29,30). We used RRTs here to predict plausible motion pathways of the KcsA channel.
Probabilistic motion-planning techniques were applied in the context of molecular biology (31–34). For example, Cortes et al. (35) developed an algorithm to compute large-amplitude motions in flexible molecular models, using RRTs to compute protein loop conformational changes and ligand trajectories. Later, the same group integrated normal-modes analysis with path-planning techniques for the study of large conformational changes in proteins (36). Previously, we applied RRTs to predict stable conformations and motions in pairs of transmembrane helices (37).
Here we employ RRTs in a more demanding application, involving a full-fledged protein. We deal with a significantly larger number of degrees of freedom (dofs) and search in a much higher dimensional space. To handle the vast dimensionality, we build the open conformation of the KcsA channel by homology modeling, using the open conformation of KvAP as a template (Fig. 2 a, right). We then used the RRT technique to generate many collision-free pathways that connect the known structure of KcsA in its closed state (Fig. 2 a, left) to this (putative) conformation. Specifically, two conformation trees, rooted at each of the two KcsA states, were grown simultaneously, with the aim of merging these trees. The trees were grown using energetic considerations. Thus, each path in the merged tree, between the nodes that are the roots of the original trees, corresponds to a feasible motion pathway of KcsA.
The RRT method resembles Monte Carlo simulations, in that both generate conformations at random. However, RRT “looks back” at all previous conformations when generating new ones. Thus, it is biased toward unexplored regions of conformational space. In addition, RRT may yield more than one trajectory in each run. Overall, sampling using RRT usually provides a more complete picture of a conformational space. It is particularly suitable to searching in constrained environments, such as the torsion-angle space of a large protein, where motion is highly restricted because of ample steric clashes.
Our major innovation here involves the introduction of an efficient technique to cluster and hybridize multiple pathways between the initial and goal conformations. We are not aware of any similar feature within the context of path-planning. To this end, we developed a means to compare and align pairs of pathways, using dynamic programming. In this respect, the alignment of pathways is analogous to the alignment of sequences in BLAST (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). The fitting of paths is used here to cluster similar pathways meaningfully. The path-aligning technique is also used to hybridize paths as a means of enhancing the performance of the RRT search, to help find pathways that are short and energetically favorable.
The path-fitting algorithm that we propose is reminiscent of curve-fitting techniques previously used to match curves in two and three dimensions (38). The latter are close to the dynamic-programming solution to the longest common subsequence problem (39). Unlike the techniques used in these other problems, our path-alignment algorithm works in a very high-dimensional space, and requires various technical adaptations.
THEORY
Pathways clustering
Because of the random nature of the RRT algorithm, different runs produce different pathways. The pathways may partially overlap with each other, and it is challenging to analyze them in search of the shortest, energetically favorable, pathway between the initial and goal conformations among them. A first step toward this goal involves the clustering of pathways. We present a general-purpose approach to this end. Pathways are clustered based on their similarity, which is defined according to the distance between conformations along pathways and their sequence of appearance within a pathway. A dynamic programming algorithm is developed to that end. Pathways within each cluster are automatically hybridized to produce a unique, short, and representative pathway. The pathways are ranked using an energetic criterion, and the energetically most favorable ones are selected.
A dynamic-programming algorithm for measuring similarity between pathways
We use a variant of dynamic programming to find the best alignment between two pathways (Fig. 3 a). A pathway is defined as an ordered sequence of conformations, and its length is the number of conformations that it contains. For two pathways, P and Q, of lengths m and n, respectively, we define M as an m × n root mean-square deviation (rmsd) matrix, where the rmsd between the conformation at position i along pathway P and the conformation at position j along pathway Q is stored in entry Mi,j. The Mi,j values are normalized to be within the interval (0,1), and the normalized matrix is denoted by M′i,j, where M′i,j = 0 corresponds to the rmsd of zero between the corresponding conformations, and M′i,j = 1 corresponds to conformations that have the lowest resemblance, i.e., their rmsd is above a similarity threshold (here, 1.5 Å).
FIGURE 3.

Alignment between two pathways. Conformations along optimal pathway (i.e., pathway with the lowest energy barrier) are in cyan, and other conformations are in purple. (a) To align two pathways, we regard each pathway as a word with the constituting conformations as letters, which makes the problem analogous to that of the alignment of similar protein sequences. Thus, we can use dynamic programming for the aligning of two words, to align two pathways where two conformations from different pathways are matched, based on their rmsd. Zero or more gaps between conformations are allowed, to maximize the global alignment between two pathways. (b) In this example, the alignment can be used to shorten the optimal pathway (upper pathway) by removing the conformation that is aligned to a gap in the lower pathway. (c) The alignment can be used to hybridize pathways, to obtain an energetically favorable pathway by replacing subpathways. The first and last conformations along the two pathways are aligned with each other, and the other conformations are aligned with gaps. In other words, the two subpathways that are encircled by the ellipsoids cannot be matched. If the energy barrier of the purple subpathway is lower than that of the cyan subpathway, we may exchange between the two subpathways and obtain a better energy barrier for the optimal pathway.
Let p1, p2, …, pm and q1, q2, …, qn denote the conformations along pathways P and Q, respectively. We regard each pathway as a word, with the constituting conformations as letters, and we allow zero or more spacing between letters in each word (Fig. 3 a). A valid fitting between pathways P and Q is obtained by aligning the two respective words one on top of the other, so that in each column, at least one letter is not a space (depicted below as a dash). Here are examples of valid fittings of
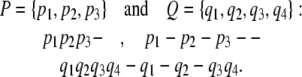 |
We next describe all possible valid fittings between two pathways P and Q, as above (Fig. 4). Let G = (V,E) be a directed graph, whose vertex set V consists of vertices vi,j (for i = 0 … m, j = 0 … n) arranged in a grid, and that has three outward directed edges from each vertex vi,j (i = 0 … m − 1, j = 0 … n − 1): 1), toward vi,j+1, 2), toward vi+1,j, and 3), toward vi+1, j+1. The vertices vm,j (j = 0 … n − 1) have one outward directed edge toward vm,j+1, and the vertices vi,n (i = 0 … m − 1) have one outward directed edge toward vi+1,n. Each directed path from v0,0 to vm,n describes a valid fitting of P and Q in the following manner: a vertical edge (vi,j, vi+1,j) corresponds to aligning the conformation pi with a gap, a horizontal edge (vi,j, vi,j+1) correspond to aligning the conformation qj with a gap, and the diagonal edge (vi,j, vi+1,j+1) corresponds to aligning the conformation pi with qj. It is possible to see that every valid fitting of the pathways P and Q corresponds to a directed path from v0,0 to vm,n in G, and vice versa (Fig. 4). We will use fittings and directed paths in G interchangeably.
FIGURE 4.

Alignment graph for two pathways P = {p1, p2, p3} and Q = {q1, q2, q3, q4}. Two examples of alignments, as described in the text, are indicated by solid and dashed arrows. Each path in the graph suggests a pairwise alignment between two pathways.
We now assign a cost to a fitting by assigning weights to edges along the directed path in the graph. Horizontal and vertical edges are assigned equal weights, which we denote by GapPenalty. (In our experiments, GapPenalty = 1, for reasons to be clarified shortly.) A diagonal edge (vi,j, vi+1,j+1) is assigned the cost of the normalized rmsd M′i,j, as defined above. The normalized rmsd M′i,j measures the similarity between the conformations pi and qj that have been aligned, and is a real value between 0 (identity) and 1 (greatest dissimilarity). In our experiments, we chose to penalize gaps with the same score as that of greatest dissimilarity, although other choices can be incorporated.
Rephrased in terms of G, our best pathway-fitting problem is to find the path of minimum weight between v0,0 and vm,n in the directed graph. Let Pi denote the pathway consisting of the first i conformations of pathway P. We similarly define Qi. Our optimal pathway-fitting problem has the following optimal substructure property. The optimal fitting is either 1), the optimal fitting of Pm−1 and Qn−1 concatenated with the fitting of pm and qn, 2), the optimal fitting of Pm and Qn−1 concatenated with the fitting of a gap and qn, or 3), the optimal fitting of Pm−1 and Qn concatenated with the fitting of pm and a gap. The same arguments apply to any pair of subpathways Pi and Qi for i > 0, j > 0. This leads to the following dynamic programming formulation: DP is an (m + 1) × (n + 1) matrix, whose element DP[i,j] stores the score of the optimal fitting of the subpathways between Pi and Qj. Therefore, our goal is to compute DP[m,n], and deduce the optimal fitting.
TABLE 1.
| for i = 0…m DP [i, 0] = i × GapPenalty |
| for j = 0…n DP [0, j] = j × GapPenalty |
| for i = 1…m |
| for j = 1…n |
| if (Mi,j, ≤ SimilarityThreshold) |
| DP [i, j] = min (DP [i – 1, j – 1] + M′i,j, |
| DP [i – 1, j] +GapPenalty, |
| DP [i,j – 1] +GapPenalty) |
| else |
| DP [i, j] = min (DP [i – 1, j] +GapPenalty, |
| DP [i, j – 1] +GapPenalty) |
| return DP [m, n] |
The optimal fitting can be deduced easily from the table DP without extra storage, by tracing back the optimal solution from the last alignment backward. The matrix DP can be computed in O(mn) time, using O(mn) space (or less space, using standard techniques (39)).
Clustering
After defining a similarity score between pathways, one can cluster the pathways using hierarchical clustering (40). Given n objects, hierarchical clustering assigns each object to a cluster. It then applies a series of fusion steps to the n clusters until all the objects are clustered into a single cluster of size n. In each fusion step, the two most similar clusters are merged. There are several methods to define distance (i.e., dissimilarity) between clusters. Here we use the complete linkage clustering method. Distances between clusters of pathways are computed by the farthest-neighbor method. In other words, the distance between two clusters Ck and Cs is defined as d(Ck,Cs) = max{PD[i,j] | pathway i is in Ck, pathway j is in Cs}. Given n pathways, PD (distance pathway) is an n × n table, where PD[i,j] is the distance between pathways i and j, as defined by the dynamic-programming algorithm; clearly, PD[i,i] = 0 and PD[i,j] = PD[j,i].
In each fusion step of the hierarchical clustering procedure, the clusters Ck and Cs of minimal d(Ck,Cs) are merged. Clustering proceeds until the clusters are dissimilar from each other, based on a predefined criterion. Suppose that the clustering method attempts to merge clusters Ck and Cs. If two pathways exist, i.e., P of length m in Cs, and Q of length n in Cs, such that the number of matching conformations along them is below N × min(m,n), the hierarchical-clustering algorithm stops. A value of N = 0.7 is used here. In other words, each pair of pathways in the same cluster contains overlapping conformations (within an rmsd of 1.5 Å) of at least 70% of the shortest pathway. A preliminary examination showed that similar pathways are obtained using slightly different cutoff values.
Hybridization of pathways within a cluster, and selection of the best pathway
After the clustering step, the pathways are ranked, and a representative prominent pathway for each cluster is constructed. Intuitively, the representative pathway can be selected based on a min-max criterion, i.e., the pathway whose conformation of maximal energy is assigned the lowest value among all pathways. However, it may be possible to construct a better pathway by merging energetically preferable subpathways. To this end, we exploited the alignment that was produced by the dynamic programming step above.
A pathway-merger was built, in the search for a short pathway that comprises low-energy conformations (Fig. 3). There are two cases in the alignment between two pathways that can help in the production of a better representative pathway:
- An alignment of gaps in one pathway to a sequence of conformations on the other pathway (Fig. 3 b). For example, the following alignment was computed for pathways P and Q:
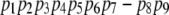
It is possible to shorten these pathways and obtain the following pathways: pq = {p1, p2, p3, p7, p8, p9}, and qp = {q1, q2, q3, q4, q6, q7}. These pathways are feasible, provided that the local planner successfully connects p3 to p7, and q4 to q6. If so, these pathways are preferable because they are shorter and are associated with energy that is equal to, or lower than, the energy of the original pathways. The merger computes the maximum energy potential of the conformations in these two short alternative-pathways, and selects the one with the lowest value. Based on the alignment, it is also possible to choose the best conformation at each step. For example, if energy (p1) < energy (q1), and energy (q2) < energy (p2), we assign p1 to the first conformation in the representative pathway and q2 to the second, provided that the local planner finds a transition between these conformations.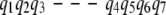
- Two consecutive gaps, one in each pathway (Fig. 3 c). For example, let us examine the following alignment:
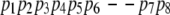
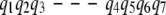
The following hybridized pathways may be produced: pq = {p1, p2, p3, q4, q5, p7, p8}, and qp = {q1, q2, q3, p4, p5, p6, q6, q7}. In other words, the two consecutive gaps, one on each pathway, suggest the possibility of choosing a subpathway within the representative pathway. Again, the merger selects the subpathway (among {p4, p5, p6} and {q4, q5}) that minimizes the maximal energy of its conformations.
In summary, it is possible to use the gaps in the best-fitting procedure according to the dynamic-programming approach, to detect short and energetically favorable pathways. In practice, a representative pathway was selected, iteratively aligned, and hybridized with all the other pathways, as described above.
METHODS
A scheme describing the overall approach is presented in Fig. 2. The different steps in the scheme are described below.
A model structure of the KcsA channel in its open state
The KcsA channel was solved in its closed conformation (Protein Data Bank identification 1k4c (4)). We modeled the open form of KcsA using the structure of KvAP (Protein Data Bank identification 1orq (10)), which was determined in an open conformation, as a template. Of the available templates, KvAP has the highest sequence similarity in the pore domain (∼30% sequence identity). The channel KvAP was previously suggested as a plausible template for modeling KcsA in its open form (41). More information about modeling and validation procedures, as well as the model structure (Fig. 2 a), is provided in the Supplementary Material (Data S1).
Types of motions that were considered
The KcsA tetramer includes 412 amino acids, each of which contributes two torsional backbone degrees of freedom to the overall dimensionality of the conformational space. In addition, side-chain flexibility throughout the gating phase is considered. Thus, the number of dofs is enormously high, and we used the following measures to reduce the computational load while maintaining an appropriate representation that accounts for relevant degrees of freedom. First, following a widespread modeling assumption, bond lengths and angles were held fixed throughout the search in conformational space (42).
Second, all known structures of potassium channels, including those of KcsA (Fig. 2 a) and KvAP, manifest a fourfold rotation symmetry around the pore axis. We assumed that this symmetry, which is anticipated for homotetramers, is retained throughout the pathway, thus reducing the number of dofs fourfold.
Third, the structures of the open and closed states of the channel are very different from each other, and the rmsd between their C-alpha atoms is 5.95 Å. However, the selectivity-filter regions of these structures are virtually the same (rmsd of 0.33 Å between the C-alpha atoms of amino acids T75VGYG79; see the Supplementary Material S2, Data S1). Thus, the backbone of the selectivity filter was held fixed. Following the same logic, we also fixed the backbone of the turret loop between ARG52 and THR61. This loop is located in the extracellular region farthest from the gate region, and connects the TM1 helix to the pore helix, which is in turn connected to the selectivity filter and TM2 helix (Fig. 1, gray). The two ends of this loop remain fixed between the open and closed conformations, and do not seem to relate to the orientation of interhelices (see the Supplementary Material S2, Data S1). Because the proper treatment of the motion would require sampling of many conformations of this loop, we also fixed this region. Significant differences between the structures are apparent only within TM1 and TM2. Hence, we allowed full movement of all backbone torsion angles of these helices, and of all the side chains, including regions with a fixed backbone.
In total, we allowed for 104 backbone degrees of freedom per monomer in our simulation (because of symmetry, this is also the total number of backbone dofs for the entire channel). In addition, all side-chain χ angles were allowed to rearrange, by sampling conformations from a backbone-dependent rotamer library (43). Each residue was represented by a number of dofs that corresponds to the number of χ angles needed to define the side-chain conformation. This number ranges from 0 for small amino acids, such as GLY and ALA, to 4 for large and flexible amino acids, such as LYS, defined by 4 χ angles. Because the side chains were not treated symmetrically, this adds to each of the four monomers 83 residues with sampled side-chain dofs, totaling 4 × 83 = 332 residues with sampled side-chain dofs.
Algorithm for motion-path generation
Pathways were generated based on a method described in our earlier study, within a different context (37). One major difference between these studies is that instead of constructing a single tree, rooted at the initial conformation (i.e., the native, closed conformation of KcsA), we followed a prevalent practice in robotics and constructed two trees, rooted in the initial and goal (Fig. 2 a) conformations. Two conformation trees were rapidly grown, starting from both initial and target conformations, and each path in the tree stands for a putative motion pathway. A single-tree version of the algorithm was also implemented, in which only the initial conformation was used as input.
The algorithm was implemented within the Rosetta modeling project (42). The energy score of generated conformations was calculated with a version of Rosetta score 12 that included attractive and repulsive van der Waals and hydrogen-bonding terms, and a statistical bias for prevalent Ramachandran torsion angles and side-chain rotamers. Side chains of initial and goal conformations were optimized using the Rosetta full-atom repack procedure, and side-chain intermediate conformations were optimized using the Rosetta greedy rotamer-trial procedure (44). A detailed description of the algorithm is provided in the Supplementary Material S3, Data S1.
RESULTS
Pathways between open and closed conformations of KcsA
We applied the RRT algorithm, described in detail in the Supplementary Material S3 (Data S1), to search for pathways between the (model of the) open and closed conformations of the KcsA channel (Fig. 2 a). The KcsA channel was represented in atomic detail, and the conformational space was explored and subjected to the Rosetta energy function, as described in Methods. Low-energy conformations were added to the tree to efficiently explore the free conformational space. Side-chain clashes were resolved, and their interactions were optimized, using the Rosetta greedy rotamer-trial procedure (44). Two motion pathways, one from the open and the other from the closed conformation, were generated and connected through a pair of similar conformations (Fig. 2 a, middle, red dashed line). The threshold of a C-alpha rmsd of 1.6 Å was used to connect nodes from the different trees. In practice, many pathways were connected between much closer conformations of up to 1.15 Å apart.
Starting from the initial and goal conformations, we generated 100 pathways from 12 RRT trees. The overall run took <1 h to complete (on three dual-core dual CPU, AMD 2.2Ghz/1MB computers, on a Sun cluster). The RRT algorithm examined, on average, 50,082 conformations per tree, and the final tree comprised 28,300 feasible conformations, on average. Subsequently we aligned, merged, and clustered the pathways, using the hierarchical approach described in Theory. A final pathway of lower energy (Fig. 5 b) was generated by merging low-energy lags of motion from aligned input pathways. The overall procedure is outlined in Fig. 2 d.
FIGURE 5.

Putative three-phase motion pathway between closed and open conformations of KcsA. (a) Intracellular perspective of the KcsA channel in closed conformation (gray), with location of the Cα atom of residue V155 along the pathway marked in space-fill representation using different colors. The optimal pathway begins with Brownian motion (in gray). Phase I of the pathway, where the closed conformation is unlocked, is shown in cyan. Phase II, where the gate opens, is shown in purple, and phase III, where the channel is locked in its open conformation, is shown in red. (b) Energy profile of pathway. Arbitrary units of the Rosetta energy function were used (see Methods). The closed conformation (step 1) was assigned the lowest energy, and is the most stable. The first five steps are around the initial conformation, and are not an integral part of the opening pathway. The pathway from the closed conformation to the (model structure of the) open conformation (step 23) involves crossing an energy barrier (step 12). This is exactly the transition between phases I and II in a. (c and d) HOLE (45) analysis of changes in profile of the channel pore along the pathway. (c) Magnitude of the radius along the pore axis (Z). The green and red curves were obtained using the open and closed conformations, and the blue curves were obtained for intermediate conformations along the pathway between these two end-conformations. The gate is around Z = 20 Å, and the selectivity filter, which was held fixed, is around Z = 50–60 Å. (d) The green and red curves mark changes in the pore radius at Z = 35 Å and Z = 23 Å along the pathway, congruently. The black curve marks changes in pore radius at minimal constriction. The depth of the minimal constriction along the Z axis changes along the motion pathway.
A possible three-phase mechanism of channel-opening
In the simulation, the TM helices are readily observed following a curved rotational motion that resembles the opening of an iris, combined with a corkscrew motion of the intracellular ends of the helices to open the pore (Fig. 5 a and the Supplementary Material S4, Data S1). This is in agreement with previous computational predictions (19,22) and a recent single-molecule study that supported the suggested rotational mechanism for channel-opening (20). However, our analysis offers new insights. We suggest here that channel-opening follows a three-phase mechanism, involving safe-lock (Fig. 5). The suggested phases, described from an intracellular perspective, are as follows.
Following a phase of Brownian motion (conformations 1–5), the channel is unlocked from its closed state by a slight clockwise movement (conformations 6–12) that involves bridging over a significant energy barrier (Fig. 5 b).
Second, the channel opens through a bending motion, in which the intracellular part of the TM2 helix slides over the neighboring TM2 helix, moving counterclockwise and laterally away from the pore axis (conformations 13–18).
Finally, the channel is locked back in the open conformation by a counter motion (conformations 19–23).
Although the suggested mechanism obviously calls for further validation, it is interesting to examine its function as a control mechanism for channel-gating. By this mechanism, channels in their closed form are trapped in an energy minimum. Channel-opening can be triggered by an unlocking mechanism that involves a preemptive small movement, which allows a subsequent large-scale motion for opening of the channel. Such a “security mechanism” may reduce the frequency of unwanted events of channel-opening, which may have negative effects on a living cell.
To characterize the predicted pathway further, a detailed analysis of channel-opening in the gate region was performed. We used the program HOLE (45) to calculate the opening radii of the main axis of the channel. Analysis of two bottlenecks (Fig. 5, c and d) along the channel gate demonstrates the way that such a three-phase mechanism might work. The channel-opening throughout the motion pathway is plotted at two bottlenecks that determine the minimum constriction of the pore, at depths of 23 Å (red) and 35 Å (green). The importance of these regions in the predicted motion pathway is evident when comparing these plots to the minimal channel-opening over the whole gating region (below a depth of 40 Å), shown as a black curve, and representing the overall opening of the channel. After a phase of Brownian motion (conformations 1–5), the channel starts to unlock at depth of 35 Å (conformations 6–12). In agreement with our suggested mechanism, the widening of the cavity at a depth of 35 Å does not manifest itself in an overall opening of the channel (black curve). However, this cavity might provide the greater freedom of motion needed to allow the next phase, i.e., a sharp opening of a cavity at depth of 23 Å (conformations 13–18), opening a bottleneck, and allowing free flow of the ions.
From closed to open conformation, using a single RRT tree
We examined whether the independent growth of an RRT tree from its initial closed conformation can independently reach the open conformation. We applied the RRT method in its original context, using a single tree (37). After exploring 30,000 conformations in a 3-h run, a conformation that is <1.6 Å from the open conformation was detected by the algorithm, with a direct motion pathway from the initial conformation. Considering the vast number of degrees of freedom used here, we believe this demonstrates the efficiency of the algorithm and the energy function for sampling the conformational space. Hence, RRTs can be used to complement memory-less search procedures such as Monte Carlo simulations, and even procedures such as taboo-search (46) that use short-term memory.
DISCUSSION
We introduce a unique framework for the very rapid generation, alignment, and comparison of pathways between two known protein conformations. We also present an algorithmic implementation of these ideas, and we used it here to study conformational changes in the KscA channel. Pathways were generated and subjected to energetic considerations. The actual run took about 1 h. But it should be noted that the calculations begin with preparations, including the building of the open-state model structure of KcsA.
A putative safe-lock mechanism for the opening of the KcsA gate
Our studies suggest a plausible pathway between the open and closed states of the channel, which might suggest a three-phase safe-lock mechanism for channel-opening. If such mechanism is valid, it might lead to interesting implications, because ion channels take part in vital cellular processes (1). That said, as a computational model, it should be treated with appropriate caution, and subjected to further validations. Although the conformations along the pathway are stereochemically sound (Supplementary Material S1. Data S1), the pathway may best be regarded as a speculation that should be examined in wet-laboratory experiments. We make the pathway available, as a movie and a series of Protein Data Bank files, to facilitate the design of such experiments (Supplementary Material S4, Data S1). However, it is important to note that different runs using different parameters and energy functions might lead to different pathways. In this sense, we believe that the main power of the framework presented here is its capacity to explore a large number of putative pathways rapidly under varying constraints. These may be used to generate hypotheses that may be tested in experiments.
A recent single-molecule study of the KcsA channel was interpreted to indicate an overall corkscrew rotational motion of the intracellular ends of TM2 helices (20). Our results, as well as the results of previous computational analysis using the Gaussian-network model (19), are in agreement with these new data. Moreover, we provide a (hypothetical) molecular interpretation of the single-molecule study.
Tikhonov and Zhorov (22) explicitly assumed a lateral outward movement of the channel by inflating and deflating the cylinder at the pore region, and then applying Monte Carlo simulations. Their predictions resemble ours, but only to a certain extent (compare our Fig. 5 a with Fig. 5 in Tikhonov and Zhorov (22)). A similar approach was used within the context of steered molecular dynamics in a detailed study by Biggin and Sansom (21). Unlike these two studies, we applied a symmetry operator over the four subunits of KcsA, and obtained a symmetrical motion. We assume that the motion was symmetrical because of the symmetry found in all potassium channels, but whether nonsymmetrical motion is a methodical artifact or a real property of the KcsA channel is not fully clear. Interestingly, as in the work of Biggin and Sansom (21), the helices did not disintegrate throughout our simulations, although the change in their internal coordinates was responsible for the movement.
All in all, this computational investigation complements experimental studies of KcsA using NMR (47), electron paramagnetic resonance (48), and single-molecule techniques (20). Ultimately, through the integration of experimental and computational results, we will further our understanding of channel-gating significantly.
Role of the selectivity filter
Here, our motion pathways were restricted to the gate region. The selectivity-filter region was examined exhaustively by molecular-dynamics simulations, as reviewed elsewhere (49). Cordero-Morales et al. (50) suggested that the filter undergoes conformation excursions. However, gating events, which are on the order of microseconds, cannot be simulated by pure molecular-dynamics simulations. Biggin and Sansom (21) used steered molecular dynamics to suggest possible open states for KcsA. In their simulations, the gate region underwent substantial conformational changes, whereas the selectivity-filter region was not altered significantly by the gate-opening, suggesting that gate-region dynamics are decoupled from those of the selectivity filter. Although the selectivity filter might change its conformation during the opening of the gate (50), these modifications are relatively minor (albeit of functional importance) compared with the conformational changes that occur in the gate region. In fact, the rmsd between the C-alpha atoms of the source (Protein Data Bank identification 1k4c) and target (a model of KcsA according to the KvAP structure) selectivity filters (T75VGYG79) is 0.33 Å, compared with 5.95 Å considering all the C-alpha atoms. Thus, generating motion pathways between these two conformations by RRTs will naturally lead to immense variations in the gate region rather than in the selectivity filter.
The energy function
Because this study involves a TM channel, we excluded solvation-related terms from the Rosetta scoring function. We used Rosetta's van der Waals potential, hydrogen-bonding score, and the statistical Ramachandran and Dunbrack rotamer scores. A designated energy function for TM proteins was recently introduced into Rosetta (51), and we look forward to using it. Unfortunately it was not publicly available as of this writing. It would be interesting to examine the sensitivity of the suggested motion pathway to different force fields. We previously (37) applied the RRT algorithm, using very simplistic energy functions that consisted of the Lennard-Jones term of either GROMOS (52) or CHARMM (53). We repeated these tests with KcsA. The pathways obtained using these simplistic energy functions differ from each other in certain aspects, and are similar in others. For instance, one of the pathways, observed when using the CHARMM Lennard-Jones potential alone, leads to a fuzzy lateral opening of the TM2 helices (data not shown). This is indicative of the importance of the choice of energy function. Because the quality of the Rosetta energy function is still questionable, our results should be taken with a grain of salt.
Interestingly, although we did not explicitly impose helix-favoring restraints, the helices retained their regular structures and, in essence, did not deform. This is attributable, in part, to the Rosetta energy function's inclusion of a bias toward prevalent Ramachandran torsion angles. When starting, as we did, from helical conformations and moving in small increments, it is difficult to deform the helices significantly. Further investigation showed that the low energy cutoff (of zero) that was used here essentially allows only tightly packed conformations of the type found in real proteins, and prevents significant helix deformations. Control runs that we conducted using higher-energy cutoff values (of 600 and 5000; energy is measured in Rosetta arbitrary units) showed the emergence of much less packed conformations that included deformed helices (data not shown).
Taking into account the solvation component of the free energy in an accurate way for a membrane channel, such as KcsA, is difficult, and perhaps even impossible. The standard treatment within the Rosetta energy function excludes the membrane environment, and involves a large and unnecessary desolvation free-energy penalty of all lipid-exposed residues. It also excludes contributions of the polar headgroup region of the membrane. Thus, it is very unsatisfying. Moreover, even if the presence of the membrane is taken into account implicitly, using some mean-field approximation, the treatment of the channel pore itself remains a major challenge. The small size of the pore may impose very specific arrangement of water molecules. Thus, it is not clear that a mean-field treatment, based on bulk water, would adequately describe desolvation in this region. To avoid all this, and because we believe that the motion is dominated by geometrical and packing restraints, we excluded the solvation term from the outset.
Following the same line of thinking, we also excluded the effects of lipids. Again, an explicit and accurate description of protein-lipid interaction is not feasible, and we suspect that any mean-field approximation may be erroneous. Thus, we eliminated these contributions altogether.
The motion between the open and closed conformations of the channel is driven by external effects, such as ligand-binding or changes in membrane potential. Here, such factors were not explicitly taken into account. Instead, we used two initial conformations and searched for a root that connects them. Namely, starting from the initial conformation, the effects of external perturbations were replaced by conducting a search in conformational space with a bias toward the goal conformation. This strategy is beneficial in that it bypassed the need to describe external effects in molecular detail. However, at the same time, we cannot address questions on the effects of external factors.
In general, the results, and the suggested pathway for channel-opening in particular, are sensitive to the choice of energy function.
Incorporation into the Rosetta package
We integrated our software into the Rosetta open-source project for modeling proteins (42), and examined pathways using Rosetta scoring, as described in Methods. After a certain period of testing for stability, our tool will be made available to the public.
General framework for generating, aligning, and comparing putative pathways
We emphasize that the framework presented here is independent of the particular choice of energy function. In particular, our approach to compare, align, and cluster the generated pathways, and rank them based on various criteria, may be useful in a much broader context. The pathways generated by the RRT algorithm comprise conformations in which each two consecutive conformations are close to each other in three dimensions, in both rmsd and internal-coordinates values. Thus, the dynamic-programming algorithm for aligning pathways is well-defined in this case. When using the approach within the context of other applications, the algorithm can be adapted by the generation of intermediate conformations that connect dissimilar consecutive conformations. We demonstrated that this approach can merge low-energy motion pathways into one or more representative pathways of higher quality, which can then be subjected to further analysis and comparison.
Future research
This study examined in detail the feasibility of our motion-prediction approach, and our studies of KcsA were sufficient for that purpose. A clear advantage of our approach is speed, such that a whole gamut of pathways can be generated, aligned, and compared in a matter of minutes or hours. We plan to examine more cases, and ultimately establish a database of feasible pathways between all proteins with two or more known conformations. In this respect, we will follow the pioneering work of Echols et al. (54). As experimental methodologies for observing proteins in motion are on the verge of becoming a reality (e.g., Shimizu et al. (20)), computational methods for motion prediction are as relevant as ever, and can both benefit from, and contribute to, our understanding of molecular motion.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
This study was supported by grants 222/04 and 611/07 from the Israel Science Foundation to N.B.-T. The research of D.H. is supported in part by the IST Program of the EU as a Shared-Cost RTD (FET Open) Project under contract IST-006413 (Algorithms for Complex Shapes), by the Israel Science Foundation (grant 236/06), and by the Hermann Minkowski-Minerva Center for Geometry at Tel Aviv University.
Editor: Klaus Schulten.
References
- 1.Hille, B. 2001. Ion Channels of Excitable Membranes, 3rd ed. Sinauer Associates, Sunderland, MA.
- 2.Ashcroft, F. M. 2000. Ion Channels and Disease: Channelopathies. Academic Press, London.
- 3.Doyle, D. A., J. Morais Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 4.Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]
- 5.Kuo, A., J. M. Gulbis, J. F. Antcliff, T. Rahman, E. D. Lowe, J. Zimmer, J. Cuthbertson, F. M. Ashcroft, T. Ezaki, and D. A. Doyle. 2003. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 300:1922–1926. [DOI] [PubMed] [Google Scholar]
- 6.Long, S. B., E. B. Campbell, and R. Mackinnon. 2005. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 309:897–903. [DOI] [PubMed] [Google Scholar]
- 7.Long, S. B., X. Tao, E. B. Campbell, and R. MacKinnon. 2007. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 450:376–382. [DOI] [PubMed] [Google Scholar]
- 8.Long, S. B., E. B. Campbell, and R. Mackinnon. 2005. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 309:903–908. [DOI] [PubMed] [Google Scholar]
- 9.Nishida, M., M. Cadene, B. T. Chait, and R. MacKinnon. 2007. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 26:4005–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B. T. Chait, and R. MacKinnon. 2003. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- 11.Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- 12.Perozo, E., D. M. Cortes, and L. G. Cuello. 1999. Structural rearrangements underlying K+-channel activation gating. Science. 285:73–78. [DOI] [PubMed] [Google Scholar]
- 13.Kelly, B. L., and A. Gross. 2003. Potassium channel gating observed with site-directed mass tagging. Nat. Struct. Biol. 10:280–284. [DOI] [PubMed] [Google Scholar]
- 14.Zimmer, J., D. A. Doyle, and J. G. Grossmann. 2006. Structural characterization and pH-induced conformational transition of full-length KcsA. Biophys. J. 90:1752–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwamoto, M., H. Shimizu, F. Inoue, T. Konno, Y. C. Sasaki, and S. Oiki. 2006. Surface structure and its dynamic rearrangements of the KcsA potassium channel upon gating and tetrabutylammonium blocking. J. Biol. Chem. 281:28379–28386. [DOI] [PubMed] [Google Scholar]
- 16.Webster, S. M., D. Del Camino, J. P. Dekker, and G. Yellen. 2004. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 428:864–868. [DOI] [PubMed] [Google Scholar]
- 17.del Camino, D., M. Holmgren, Y. Liu, and G. Yellen. 2000. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 403:321–325. [DOI] [PubMed] [Google Scholar]
- 18.Miloshevsky, G. V., and P. C. Jordan. 2007. Open-state conformation of the KcsA K+ channel: Monte Carlo normal mode following simulations. Structure. 15:1654–1662. [DOI] [PubMed] [Google Scholar]
- 19.Shrivastava, I. H., and I. Bahar. 2006. Common mechanism of pore opening shared by five different potassium channels. Biophys. J. 90:3929–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimizu, H., M. Iwamoto, T. Konno, A. Nihei, Y. C. Sasaki, and S. Oiki. 2008. Global twisting motion of single molecular KcsA potassium channel upon gating. Cell. 132:67–78. [DOI] [PubMed] [Google Scholar]
- 21.Biggin, P. C., and M. S. Sansom. 2002. Open-state models of a potassium channel. Biophys. J. 83:1867–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tikhonov, D. B., and B. S. Zhorov. 2004. In silico activation of KcsA K+ channel by lateral forces applied to the C-termini of inner helices. Biophys. J. 87:1526–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Latombe, J.-C. 1991. Robot Motion Planning. Kluwer Academic Publishers, Norwell, MA.
- 24.Latombe, J.-C. 1999. Motion planning: a journey of robots, molecules, digital actors, and other artifacts. Int. J. Robot. Res. 10:1119–1128. [Google Scholar]
- 25.Sharir, M. 2004. Algorithmic motion planning. In Handbook of Discrete and Computational Geometry, 2nd ed. J. E. Goodman and J. O'Rourke, editors. CRC Press, Boca Raton. 1037–1064.
- 26.Kavraki, L. E., P. Svestka, J. C. Latombe, and M. Overmars. 1996. Probabilistic roadmaps for path planning in high-dimensional configuration spaces. Proc. IEEE Trans. Robot. Automat. 12:566–580. [Google Scholar]
- 27.Hsu, D., J.-C. Latombe, and R. Motwani. 1999. Path planning in expansive configuration spaces. Int. J. Comput. Geom. Appl. 9:495–512. [Google Scholar]
- 28.Choset, H., K. M. Lynch, S. Hutchinson, G. Kantor, W. Burgard, L. E. Kavraki, and S. Thrun. 2005. Principles of Robot Motion: Theory, Algorithms, and Implementations. MIT Press, Cambridge, MA.
- 29.Lavalle, S. M., and J. J. Kuffner. 2001. Rapidly-Exploring Random Trees: Progress and Prospects. A. K. Peters, Wellesley, MA.
- 30.Lavalle, S. M. 2006. Planning Algorithms. Cambridge University Press, Cambridge, UK.
- 31.Amato, N. M., K. A. Dill, and G. Song. 2003. Using motion planning to map protein folding landscapes and analyze folding kinetics of known native structures. J. Comput. Biol. 10:239–255. [DOI] [PubMed] [Google Scholar]
- 32.Finn, P. W., D. Halperin, L. E. Kavraki, J.-C. Latombe, R. Motwani, C. Shelton, and S. Venkatasubramanian. 1996. Geometric Manipulation of Flexible Ligands. In Selected Papers from the Workshop on Applied Computational Geormetry, Towards Geometric Engineering. M. Lin and D. Manocha, editors. Springer-Verlag, Berlin. 1148:67–78.
- 33.Singh, A. P., J. C. Latombe, and D. L. Brutlag. 1999. A motion planning approach to flexible ligand binding. Proc. 7th International Conference on Intelligent Systems for Molecular Biology. 252–261. [PubMed]
- 34.Apaydin, M. S., A. P. Singh, D. L. Brutlag, and J. C. Latombe. 2001. Capturing molecular energy landscapes with probabilistic conformational roadmaps. Proc. IEEE Int. Conf. Robot. Automat. 1:932–939. [Google Scholar]
- 35.Cortes, J., T. Simeon, V. Ruiz de Angulo, D. Guieysse, M. Remaud-Simeon, and V. Tran. 2005. A path planning approach for computing large-amplitude motions of flexible molecules. Bioinformatics. 21:i116–i125. [DOI] [PubMed] [Google Scholar]
- 36.Kirillova, S., J. Cortes, A. Stefaniu, and T. Simeon. 2008. An NMA-guided path planning approach for computing large-amplitude conformational changes in proteins. Proteins. 70:131–143. [DOI] [PubMed] [Google Scholar]
- 37.Enosh, A., S. J. Fleishman, N. Ben-Tal, and D. Halperin. 2007. Prediction and simulation of motion in pairs of transmembrane alpha-helices. Bioinformatics. 23:e212–e218. [DOI] [PubMed] [Google Scholar]
- 38.Wolfson, H. J. 1990. On curve matching. IEEE Trans. Pattern Anal. Mach. Intell. 12:483–489. [Google Scholar]
- 39.Cormen, T. H., C. E. Leiserson, R. L. Rivest, and C. Stein. 2001. Introduction to Algorithms, 2nd ed. MIT Press, Cambridge, MA.
- 40.Johnson, S. C. 1967. Hierarchical clustering schemes. Psychometrika. 2:241–254. [DOI] [PubMed] [Google Scholar]
- 41.Jogini, V., and B. Roux. 2005. Electrostatics of the intracellular vestibule of K+ channels. J. Mol. Biol. 354:272–288. [DOI] [PubMed] [Google Scholar]
- 42.Rohl, C. A., C. E. Strauss, K. M. Misura, and D. Baker. 2004. Protein structure prediction using Rosetta. Methods Enzymol. 383:66–93. [DOI] [PubMed] [Google Scholar]
- 43.Canutescu, A. A., A. A. Shelenkov, and R. L. Dunbrack, Jr. 2003. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 12:2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang, C., O. Schueler-Furman, and D. Baker. 2005. Improved side-chain modeling for protein-protein docking. Protein Sci. 14:1328–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smart, O. S., J. M. Goodfellow, and B. A. Wallace. 1993. The pore dimensions of gramicidin A. Biophys. J. 65:2455–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cvijovicacute, D., and J. Klinowski. 1995. Taboo search: an approach to the multiple minima problem. Science. 267:664–666. [DOI] [PubMed] [Google Scholar]
- 47.Lange, A., K. Giller, S. Hornig, M. F. Martin-Eauclaire, O. Pongs, S. Becker, and M. Baldus. 2006. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature. 440:959–962. [DOI] [PubMed] [Google Scholar]
- 48.Perozo, E., L. G. Cuello, D. M. Cortes, Y. S. Liu, and P. Sompornpisut. 2002. EPR approaches to ion channel structure and function. Novartis Found Symp 245:146–165. [DOI] [PubMed]
- 49.Roux, B., and K. Schulten. 2004. Computational studies of membrane channels. Structure. 12:1343–1351. [DOI] [PubMed] [Google Scholar]
- 50.Cordero-Morales, J. F., L. G. Cuello, Y. Zhao, V. Jogini, D. M. Cortes, B. Roux, and E. Perozo. 2006. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 13:311–318. [DOI] [PubMed] [Google Scholar]
- 51.Barth, P., J. Schonbrun, and D. Baker. 2007. Toward high-resolution prediction and design of transmembrane helical protein structures. Proc. Natl. Acad. Sci. USA. 104:15682–15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oostenbrink, C., A. Villa, A. E. Mark, and W. F. van Gunsteren. 2004. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25:1656–1676. [DOI] [PubMed] [Google Scholar]
- 53.Neria, E., S. Fischer, and M. Karplus. 1996. Simulation of activation free energies in molecular systems. J. Chem. Phys. 105:1902–1921. [Google Scholar]
- 54.Echols, N., D. Milburn, and M. Gerstein. 2003. MolMovDB: analysis and visualization of conformational change and structural flexibility. Nucleic Acids Res. 31:478–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}