

Abstract
The product of yjeK in Escherichia coli is a homolog of lysine 2,3-aminomutase (LAM) from Clostridium subterminale SB4, and both enzymes catalyze the isomerization of (S)- but not (R)-α-lysine by radical mechanisms. The turnover number for LAM from E. coli is 5.0 min−1, 0.1% of the value for clostridial LAM. The reaction of E. coli LAM with (S)-α-[3,3,4,4,5,5,6,6- 2H8]lysine proceeds with a kinetic isotope effect of kH/kD = 1.4, suggesting that hydrogen transfer is not rate limiting. The product of the E. coli enzyme is (R)-β-lysine, the enantiomer of the clostridial product. β-Lysine-related radicals are observed in the reactions of both enzymes by electron paramagnetic resonance (EPR). The radical in the reaction of clostridial LAM has the (S)- configuration, whereas that in the reaction of E. coli LAM has the (R)-configuration. Moreover, the conformations of the β-lysine-related radicals at the active sites of E. coli and clostridial LAM are different. The nuclear hyperfine splitting between the C3-hydrogen and the unpaired electron at C2 shows the dihedral angle to be 6°, unlike the 77° reported for the analogous radical bound to the clostridial enzyme. Reaction of (S)-4-thialysine produces a substrate-related radical in the steady state of E. coli LAM, as in the action of the clostridial enzyme. While (S)-β-lysine is not a substrate for E. coli LAM, it undergoes hydrogen abstraction to form an (S)-β-lysine-related radical with the same stereochemistry of hydrogen transfer from C2 of (S)-β-lysine to the 5′-deoxyadenosyl radical as in the action of the clostridial enzyme. The resulting β-lysyl radical has a different conformation than that at the active site of clostridial LAM. All evidence indicates that the opposite stereochemistry displayed by the E. coli LAM is determined by the conformation of the lysine side chain in the active site. Stereochemical models for the actions of LAM from C. subterminale and E. coli are presented.
Lysine 2,3-aminomutase (LAM)1 from Clostridium subterminale SB4 is encoded by thegene kamA and catalyzes the interconversion of (S)-α-lysine and (S)-β-lysine (1–3). This enzyme plays a role in lysine metabolism in species such as C. subterminale and Porphyromonas gingivalis, whereas in such species as Streptomyces β-lysine produced by LAM is a constituent of a family of structurally diverse antibiotics (4–14). The action of LAM requires SAM, a [4Fe-4S] cluster, and PLP to catalyze the isomerization of (S)-α-lysine (15–17). LAM is a member of the “radical SAM” superfamily, a highly diverse family of enzymes, known members of which use SAM as a radical initiator in catalysis (18). This role of SAM is qualitatively unlike its traditional function as the principal biological methylating agent.
Scheme 1 illustrates the chemical mechanism by which clostridial LAM catalyzes the conversion of (S)-α-lysine into (S)-β-lysine (15–17,19,20). This mechanism is established by EPR spectroscopic analysis of the radical intermediates to characterize their structures and kinetic competencies (21–24). Reactions of lysine deuteriated at C3 proceed with substantial primary deuterium kinetic isotope effects, showing that hydrogen transfer is rate limiting in the mechanism (24). Inasmuch as radical 3 in Scheme 1 accumulates in the steady state, the rate limiting step is most likely the transfer of hydrogen from the methyl group of 5′-deoxyadenosine to C2 of radical 3, the product-related radical intermediate.
Scheme 1.

Currently, there are 53 prokaryotic proteins with amino acid sequences homologous to that of LAM from C. subterminale SB4. The aligned amino acid sequences of the E. coli protein encoded by the gene yjeK and of clostridial LAM shows 33 % identities, and the E. coli protein is one of the least similar to clostridial LAM. The active site residues, including those in the motif CxxxCxxC characteristic of radical SAM enzymes, are almost identical between the two enzymes. The residues in hydrogen bonded contact with (S)-α-lysine in clostridial LAM include Arg134, Asp293, and Asp330 (25). The identities between the enzymes from C. subterminale and E. coli include Arg134 and Asp293 of the clostridial LAM, but Asp330 is replaced with glutamate (Glu327) in the E. coli enzyme (1).
The significance of β-lysine in E. coli is not known. Unlike kamA in C. subterminale and Porphyromonas gingivalis, the gene yjeK does not appear in a gene cluster associated with lysine metabolism. In E. coli, yjeK is flanked by genes annotated as elongation factor protein, GroEL/ES chaperone proteins, membrane proteins and others that seem to be expressed under stress or in the stationary phase. Here we report that the product of yjeK is a LAM with low activity relative to clostrial LAM, and we show that it functions by an analogous but stereochemically variant mechanism.
Experimental Procedure
Materials
E. coli BL21 Rosetta cells were purchased from Novagen. PLP, ampicillin, chloramphenicol, cysteine, CM-cellulose, (S)-4-thialysine, ferric chloride dithionite, ferrous ammonium sulfate and pyridoxal hydrochloride were purchased from Sigma. SAM was obtained from Sigma as the p-toluenesulfonate salt and purified over CM-celluose (26). Terrific LB medium and (S)-α-lysine hydrochloride were purchased from Fisher Scientific. (S)-[3,3-2H2] 4-thialysine was prepared as described previously (22,23). (S)-[3,3,4,4,5,5,6,6-2H8]lysine, (S)-[2- 2H]lysine, (S)-[2-13C]lysine and (RS)-[α-15N,2H8]lysine were purchased from CDN Isotopes. IPTG and DTT were purchased from Inalco. Phenyl Sepharose was purchased from Amersham Bioscience.
Overexpression and purification of E.coli LAM
Expression of E. coli LAM was achieved in BL21 Rosetta cells. The cloning of the gene in pET23a has been described elsewhere (27). Cells were grown at 37 °C in Terrific LB medium containing 10 μM pyridoxal hydrochloride, 50 μM FeCl3, 100 μg/ml of ampicillin and chloramphenicol (30 μg/ml) to an OD600 of 0.9, induced by addition of 1 mM IPTG, and grown for an additional 3 h at 37 °C. The cells were harvested by centrifugation, frozen in liquid nitrogen, and stored at −70 °C.
All purification steps were carried out in a Coy anaerobic chamber. The recombinant protein was purified by the published procedure (27) except that the Phenyl Sepharose column was eluted with a step-wise rather than a linear gradient. LAM from C. subterminale and Bacillus subtilis were expressed in E. coli and purified as described (1,27).
Chemical characterization of E. coli LAM
The extinction coefficient at 280 nm was measured by quantitative analysis of PITC-amino acids (28) after complete acid hydrolysis of solutions of the enzyme obtained after reconstitution of the iron-sulfur clusters and measurement of A280. The procedure has been described for clostridial LAM (29). Iron, inorganic sulfide, and PLP in E. coli LAM were measured by published procedures (30–32).
Reconstitution of [4Fe–4S] clusters
Reconstitution of iron-sulfur clusters was based on a published method (33). To LAM at 10 mg/ml under anaerobic conditions, DTT, sodium sulfide and ferrous ammonium sulfate were added to final concentrations of 5 mM, 1.2 mM, and 1.2 mM, respectively, and incubated at ambient temperature for 3 h. The reconstituted protein was separated from iron and sulfide complexes by gel permeation chromatography over a column of Sephacryl S-200 equilibrated in 0.1 M EPPS at pH 8.
Reductive activation and assay
LAM was mixed with the reductive incubation buffer (200mM EPPS at pH 8.0, 36 mM ferrous ammonium sulfate, 24 mM PLP, and 10 mM (S)-cysteine) under anaerobic conditions in a ratio of 3:1 v/v and incubated for 3 h at 37 °C. Assay of activated LAM was by the procedure of Miller et al, in which the PITC derivative of (S)-β-lysine produced was separated from that of (S)-α-lysine by HPLC and measured spectrophotometrically (23).
Synthesis of (S)-β-[2-13C]lysine and (2S,3R)-β-[2-2H1]lysine
(S)-α-[2-13C]lysine (250 mM) was incubated with 30 μM LAM from C. subterminale, 150 μM SAM, 1 mM sodium dithionite, and 200 mM EPPS at pH 8.0 for 24 h at room temperature in the anaerobic chamber. Analysis by HPLC indicated 87% conversion of (S)-α-[2-13C]lysine into (S)-β-[2-13C]lysine at equilibrium. (S)-β-[2-13C]lysine was separated from (S)-α-[2-13C]lysine by chiral chromatography using a Chirobiotic T column (Astec) equilibrated in 50 mM sodium phosphate buffer at pH 3.1. The retention times for (S)-α- and (S)-β- [2-13C]lysine were 5.7 and 6.8 min, respectively, under the conditions employed. (S)-β-[2S-2H]lysine from (RS)-α-[2-2H]lysine was prepared by the same procedure. The retention time for (R)-α-[2-2H]lysine in this experiment was found to be 7.3 min.
Synthesis of [5′-13C]SAM and [5′-2H2]SAM
SAM with 13C in C5′ or deuterium bonded to C5′ was synthesized from the correspondingly labeled ATP and methionine by use of SAM synthetase as described elsewhere (34).
Synthesis and ultraviolet CD of PITC-(R)-β-lysine
LAM from E. coli (0.5 mM) was used to produce β-lysine by the procedure described above in 30 mM EPPS buffer at pH 8.0, 20 mM (S)-α-lysine, 5 mM sodium dithionite, and 3 mM SAM. The purified β-lysine was derivatized with PITC and purified by HPLC over a C18 column. A sample of PITC-(S)-β-lysine was produced in parallel using the LAM from C. subterminale. CD spectra of the purified PITC-β-lysines were recorded against water as a reference at 1 nm resolution between 215 and 350 nm in a quartz cell with 1 mm pathlength at 25 °C with an Aviv 62 DS CD spectrometer in the Biophysical Instrumentation Facility of the Department of Biochemistry.
EPR spectroscopy
Samples of LAM from E. coli at concentrations of 200–250 μM and with iron-sulfur clusters reconstituted were prepared in EPR tubes in the glove box. The samples were prepared in 200 mM EPPS (pH 8), 5 mM sodium dithionite, and 1.13 mM SAM. The reactions were initiated by addition of isotopically labeled or unlabeled (S)-α-lysine, (S)-β-lysine, or (S)- 4-thialysine. The EPR sample tubes were frozen within 20–40 seconds in isopentane cooled in a liquid nitrogen bath. After freezing, the EPR samples were removed from the anaerobic chamber and stored in liquid N2. For EPR analysis at 77 K at X-band on a Varian E3 spectrophotometer, the instrument settings were: microwave frequency, 9.1 GHz; modulation amplitude, 1.6 G; microwave power, 5.00 mW, unless noted other wise. A standard liquid nitrogen immersion dewar was used. The spectrometer was interfaced with a computer for data acquisition and analysis. Resolution enhancement and simulation of spectra were performed as described (21).
Results
Characterization of LAM from E. coli
Based on quantitative amino acid and spectrophotometric analyses, the extinction coefficient of E. coli LAM at 280 nm is 4.6 × 105 M−1 cm−1. The enzyme with reconstituted iron-sulfur clusters contains 3.7 mol of iron and 3.8 mol of sulfide per mol of subunits. The values of steady state kinetic parameters are kcat = 4.8 min−1, Km = 5 mM, and kcat/Km = 1 mM−1 min−1 at pH 8.0 and 25 °C. In terms of kcat-values, LAM from E. coli is 0.1% as active as clostridial LAM, but it displays a similar value of Km. Like the clostridial enzyme, LAM from E. coli will not accept (S)-ornithine or (R)-α-lysine as substrates. Unlike the clostridial enzyme, which displays a substantial deuterium kinetic isotope effect in the reaction of (S)-[3,3,4,4,5,5,6,6-2H]lysine, LAM from E. coli displays a small effect of kH/kD = 1.4 at either 40 mM or 90 mM substrate. Also unlike the clostridial enzyme, LAM from E. coli undergoes suicide inactivation in a substrate-dependent process over a period of 15–20 min. The chemical nature of the inactivation is not known; however, it is not accompanied by the destruction of SAM.
(R)-β-Lysine as the product
Certain properties of E. coli LAM detailed below, in particular the EPR spectra observed in its reactions with (S)-lysine and (S)-β-lysine, the product of clostridial LAM, suggested a structural difference in the products of the two enzymes. Inasmuch as the retention times for the PITC derivatives of the products upon reversed phase HPLC were identical, it was possible that the two might be enantiomeric, that is, that the E. coli product could be (R)-β-lysine. Because of the low activity of the E. coli enzyme and its suicide inactivation, it proved to be difficult to generate large amounts of the product for polarimetric analysis. In a more sensitive test, the Cotton effects associated with the chromophore in PITC-β-lysines allowed the configurations of the E. coli and clostridial products be compared by CD spectrophotometry. The resulting curves in Fig. 1 show that the two PITC derivatives display opposite Cotton effects, positive for PITC-(S)-β-lysine from C. subterminale LAM and negative for the PITC-β-lysine from E. coli. Therefore, inasmuch as the clostridial enzyme produces (S)-β-lysine (3), the E. coli enzyme must produce (R)-β-lysine.
Fig. 1.

UV circular dichroism spectra for PITC derivatives of (S)-β-lysine (—) and (R)-β-lysine produced in the reaction of E. coli LAM (–X–X–).
Stereochemical analysis of the product of LAM from B. subtilis by the above procedure led to the same Cotton effect as for β-lysine from clostridial LAM, showing that the B. subtilis product is (S)-β-lysine.
Characterization of a lysyl-free radical intermediate
A substrate-based radical bound to E. coli LAM is observed by EPR at 77 K after freeze-quenching in the steady-sate (Fig. 2A). The EPR spectra of samples prepared with (S)-α-[3,3,4,4,5,5,6,6-2H8]lysine show weak nuclear hyperfine splitting of at least one hydrogen from C3 to C6 of the lysyl side chain with the unpaired electron. The overall splitting pattern is similar to that of the unlabeled compound, while individual features are slightly narrowed (compare Figs. 2A & 2B). The principal location of the unpaired electron is C2 of the lysyl carbon skeleton, as shown by the spectra in Figs. 2C and 2D. The complex nuclear hyperfine splitting pattern is narrowed and simplified with the substitution of 2H for 1H at C2. Comparison of Fig. 2C with 2A or 2B shows that the unpaired electron is engaged in strong nuclear hyperfine coupling with C2(H), and its replacement with deuterium leads to a narrowed signal. The nuclear hyperfine splitting for deuterium is much smaller (~1/6th) than that for hydrogen (35). Further, samples prepared with (S)-[2-13C]lysine exhibit strong broadening of the EPR signals (Fig. 2D), owing to the large nuclear hyperfine splitting tensor of 13C at the center of unpaired spin (35). The results showing spin localized at C2 of lysine are consistent with the product-related free radical 3 in Scheme 1 being the principal radical intermediate in the steady state.
Fig. 2. EPR spectra of the radical from (S)-α-lysine at the active site of E. coli LAM.

The EPR samples were prepared under anaerobic conditions with 200 μM E. coli LAM, 1.1 mM SAM, 2.5 mM dithionite, and 200 mM EPPS buffer at pH 8.0. The EPR spectra were recorded at 77K. A, 40 mM α-(S)-lysine; B, 40 mM (S)-α-[3,3,4,4,5,5,6,6-2H8]lysine; C, 40 mM (S)-α-[2-2H]lysine; D, 40 mM [2-13C]lysine. The spectra show that the dominant radical in the steady state is the β-lysyl radical, 3 in Scheme 1.
The spectral envelope of the product-related radical (Fig. 2) is very different from that reported for the analogous radical at the active sites of clostridial and Bacillus LAM (21,27). Differences in the spectra arise differences in the conformations about the C2-C3 bond. The variant dihedral angles of the β-C3-substituents bring about different nuclear hyperfine splitting from the β-hydrogen and β-nitrogen. Simulations of the spectra for samples prepared with (RS)-[2-15N,2,3,3,4,4,5,5,6,6-2H9]lysine, (S)-[2-2H]lysine, and unlabeled (S)-lysine allowed the splitting constants to be evaluated by the procedures employed to characterize the corresponding radical in clostridial LAM (21). The spectra and simulations are shown in Fig. 3, and the splitting constants from the simulations are assembled in Table 1. The isotropic hyperfine splittings arising from β-substituents in π-radicals are strongly dependent on the dihedral angles, χ defined in Scheme 2. For β-proton splittings, aβH, the following relationship has been established: aβH = A1 + A2 cos2 χ (36). Values of A1 and A2 are empirically determined constants that have values of 0.92 G and 42.6 G, respectively, for unit spin density on Cα. The dihedral angles relating the spin-bearing p-orbital to the positions of H and N in the E. coli LAM (shown on the right of Scheme 2) are 6° and 50° respectively. In contrast, the corresponding angles in the enantiomeric radical bound to clostridial LAM (shown on the left in Scheme 2) are 77° (Hβ) and 17° (Nβ).
Figure 3. Simulated EPR spectra of β-lysyl radicals at the active site of E. coli LAM.

Experimental and simulated spectra of the β-lysyl radical are shown for the species generated with (S)-α-[2-2H]lysine, (RS)-α-[2-15N,2,3,3,4,4,5,5,6,6-2H9]lysine and unlabeled (S)-α-lysine in the active site of E. coli LAM. A. The spectrum (—) and simulation (- - -) of the radical generated upon addition of 40 mM (S)-α-lysine to 180 μM enzyme. B. The spectrum (—) and simulation (- - -) of the radical generated upon addition of 40 mM (S)-α-[2-2H]lysine. The samples were frozen within 30–40 seconds after addition of substrate. C. The spectrum (—) and simulation (- - -) of the radical generated upon addition of 80 mM (RS)-α-[2-15N,- 2,3,3,4,4,5,5,6,6-2H9]lysine. Parameters used for the calculation of simulated spectra are given in Table 1.
Table I.
Hyperfine Splitting Parameters Obtained from Spectral Simulations
| Principal values of hyperfine tensorb (G)
|
||||
|---|---|---|---|---|
| Nucleus | ao (G) | Axx | Ayy | Azz |
| 1Hα | 19.5 | 38.5 | 0 | 20 |
| 2Hα | 3.0 | 5.9 | 0 | 3.1 |
| 1Hβ | 13.5 | Isotropic | ||
| 2Hβ | 2.1 | Isotropic | ||
| 14Nβ | 7.8 | Isotropic | ||
| 15Nβ | 11 | Isotropic | ||
Scheme 2.

The spectrum shown in Fig. 3C contains flanking shoulders that are not included in the simulations. These shoulders are attributed to the presence of radicals elicited by contaminating unlabeled lysine: the commercial compound is only 90% deuteriated.
(S)-4-Thialysine and radical 1
(S)-4-Thialysine is an alternative substrate for C. subterminale LAM (22,23). The ultimate products from (S)- 4-thialysine are β-mercaptoethylamine and formyl acetate, owing to the chemical instability of β-4-thialysine. Figure 4 shows the EPR signal generated when (S)-4- thialysine or (RS)-4-thia[3,3-2H2]lysine is added to reductively activated E. coli LAM together with dithionite and SAM and then frozen in liquid N2 within 3 min. The hyperfine splitting in the labeled sample is narrower than in the unlabeled one, suggesting that the unpaired electron resides on C3. Thus, as in the case of clostridial LAM, the 4-thia analogue of radical 1 in Scheme 1 accumulates in the steady state of the reaction of (S)-4-thialysine.
Figure 4. EPR spectra of the radical induced at the active site of E. coli LAM by (S)-4- thialysine.

The spectrum (—) is generated upon addition of 80 mM (RS)-4-thialysine and the spectrum (- - -) is the radical formed upon addition of 80 mM (RS)-4-thia[3,3-2H2]lysine. The samples contain 2.5 mM dithionite, 1.5 mM SAM, and 200 mM EPPS at pH 8. Samples were frozen within one minute after addition of the substrate. The spectra indicate that the dominant radical in the steady state is the 4-thia analogue of radical 1 in Scheme 1.
Radical-formation with (S)-β-lysine
(S)-β-Lysine is the product of clostridial and Bacillus LAM, whereas (R)-β-lysine is the product of E. coli LAM. Nonetheless, addition of (S)-β-lysine to activated E. coli LAM leads to radical formation, as shown by the EPR spectra in Fig. 5. Figure 5A shows the resultant EPR spectrum upon addition of (S)-β-lysine to E. coli LAM. The splitting pattern of this radical is very different from that observed upon addition of (S)-α-lysine (Figs. 2 & 3). Within thirty minutes the radical signal diminishes. The radical is an isomer of the one generated from (S)-α-lysine as shown by the qualitative order of the hyperfine splittings in the spectrum generated with (S)-β-[2-13C]lysine (Figure 5B). The large 13C broadening and the additional features constitutes direct evidence that the unpaired electron resides on C2 of the carbon skeleton. The results strongly imply that the 5′-deoxyadenosyl radical derived from SAM abstracts a hydrogen atom from C2 of (S)-β-lysine. The resulting lysyl-radical and that generated from (S)-α-lysine in Figs. 2 and 3 must be isomers of radical 3 in Scheme 1. Radical 3 has a single optical center, that at C3, which has the (R)-configuration at the active site of E. coli LAM when it is generated from (S)-α-lysine (Fig. 1). The enantiomeric (S)-isomer of radical 3 must be produced in the reaction of (S)-β-lysine. There is currently no evidence that the reaction of (S)-β-lysine proceeds further in the reverse direction.
Figure 5. EPR spectra of the (S)-β-lysyl radical at the active site of E. coli LAM.

A. The radical generated upon addition of 70 mM (S)-β-lysine. B. Radical generated upon addition of 27 mM (S)-β-[2-13C]lysine. EPR samples were prepared with 200 μM enzyme, 2.5 mM dithionite, and 1.3 mM SAM in 200 mM EPPS at pH 8.0 and were frozen with 20–25 seconds after addition of substrate. C. The radical formed upon addition of 1 mM (S)-β-[2-2H]lysine. The samples contain 40 μM enzyme, 2.5 mM dithionite, 0.3 mM SAM, and 200 mM EPPS at pH 8.0 and were frozen within 20 seconds after addition of the substrate. The modulation amplitude was set at 8 G.
When the (S)-enantiomer of radical 3 at the active site of clostridial LAM abstracts a hydrogen atom from 5′-deoxyadenosine, the hydrogen atom enters the 2-pro-R position of C2 on the pathway to (S)-β-lysine. The stereochemistry of hydrogen transfer to the (R)-isomer of radical 3 at the active site of E. coli LAM can be addressed by preparing (S)-β-[2R-2H]lysine and determining whether hydrogen or deuterium is transferred to the 5′-deoxyadenosyl radical when added to E. coli LAM to generate the (S)-isomer of radical 3. If deuterium is transferred, the EPR signal will be the doublet in Fig. 5A; if hydrogen is transferred, the smaller nuclear hyperfine coupling of the remaining deuterium will lead to a broadened singlet spectrum. As shown in Fig. 5C, the signal is a broadened singlet, proving that deuterium is retained at C2 in the radical. Therefore, the stereochemistry of hydrogen transfer at C2 of (S)-β-lysine is the same for the E. coli and clostridial LAM.
Effects of [5′-13C]SAM and [5′-2H2]SAM on EPR spectra of (R)- and (S)-radical 3
The turnover number of E. coli LAM is 0.1% that of LAM from C. subterminale. Because hydrogen transfer from 5′-deoxyadenosine to radical 3 in Scheme 1 limits the rate in the action of the clostridial enzyme, and radical 3 accumulates to observable levels in the reactions of both enzymes, it is conceivable that slower turnover by the E. coli enzyme could be due to the absence of close contact between C2 in radical 3 and the methyl group of 5′-deoxyadenosine. These atoms are in van der Waals contact in the case of clostridial LAM (34).
Comparisons of the EPR spectra in Figs. 6A and 6B for samples of the (R)-isomer of radical 3 bound to E. coli LAM prepared with (S)-α-lysine and SAM, [5′-2H2]SAM, or [5′-13C]SAM suggests that the methyl group of 5′-deoxyadenosine is in van der Waals contact with lysyl-C2 bearing the unpaired electron in radical 3. In the upper spectra of Fig. 6A the solid line spectrum is with SAM and the dashed line is with [5′-2H2]SAM. Distinct narrowing of the features with [5′-2H2]SAM indicates that C2 of radical 3 and deuterium of 5′-deoxy[5- 2H2]adenosine are in van der Waals contact. The smaller coupling constant for deuterium relative to hydrogen (35) leads to narrowed and sharpened features owing to through-space nuclear hyperfine coupling of deuterium with the unpaired electron at C2 in the (R)-isomer of radical 3. An analogous effect is observed when the (R)-isomer of radical 3 is generated with [5′-13C]SAM, as shown in the upper spectra of Fig. 6B. In this case, the spectral features are broadened by van der Waals contact of C2 with 13C in 5′-deoxy[5′-13C]adenosine, owing to the large value of the nuclear hyperfine coupling constant for 13C (35).2 The results do not support a hypothesis that the difference in activity between the E. coli and clostridial LAM can be attributed to a difference in contact-distance between 5′-deoxyadenosine and radical 3 in the mechanism.
Figure 6. Effects of [5′-13C]SAM and [5′-2H2]SAM on the EPR spectra of β-lysyl radicals at the active site of E. coli LAM.
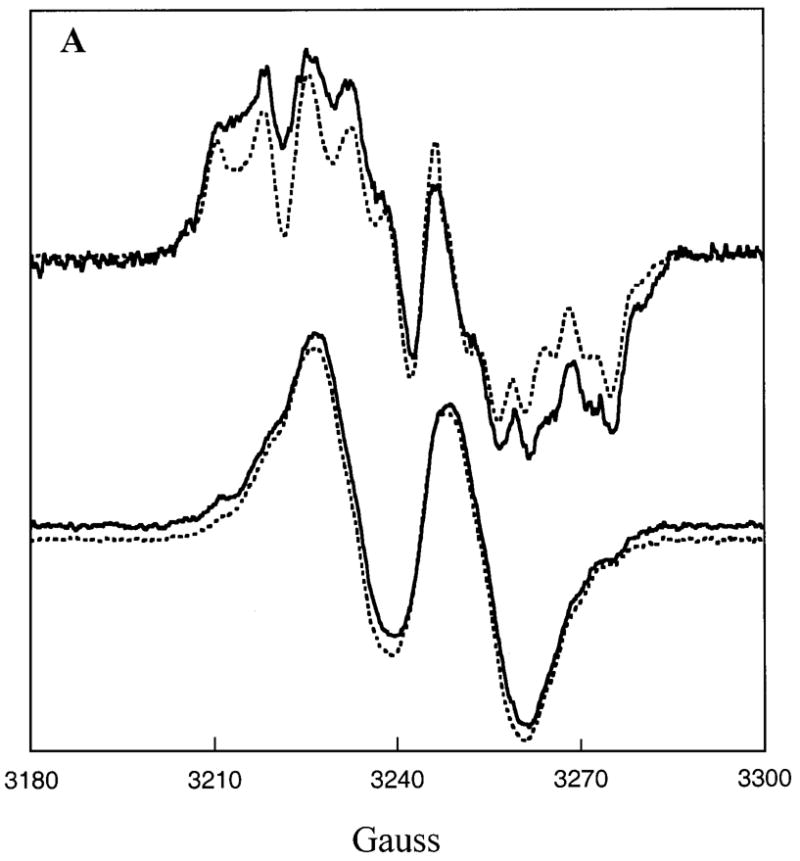
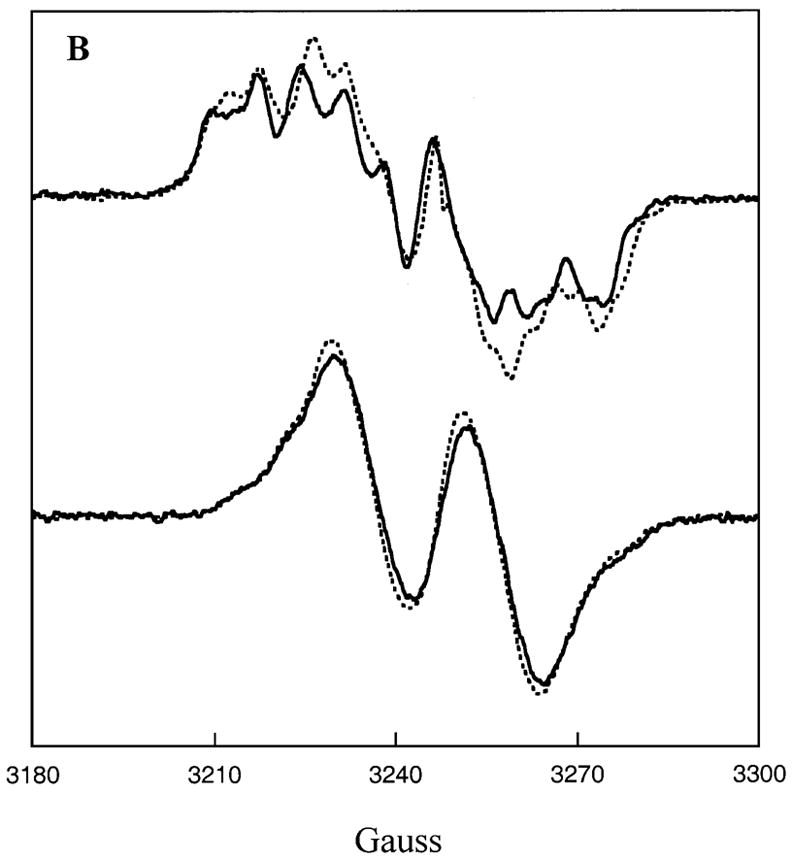
A. The upper spectra are of the (R)-β-lysyl radical formed with (S)-α-lysine in the presence of SAM (—) or of [5′-2H2]SAM (- - -). The lower spectra are of the (S)-β-lysyl radical formed with (S)-β-lysine in the presence of SAM (—) or [5′-2H2]SAM (- - -). B. The upper spectra are of the (R)-β-lysyl radical formed with (S)-α-lysine in the presence of SAM (—) or of [5′-13C]SAM (- - -). The lower spectra are of the (S)-β-lysyl radical formed with (Σ)-β-lysine in the presence of SAM (—) or [5′-13C]SAM (- - -).
The lower spectra in Figs. 6A and 6B shows the analogous EPR spectra when the (S)- isomer of radical 3 is generated with (S)-β-lysine and SAM, [5′-2H2]SAM, or [5′-13C]SAM at the active site of E. coli LAM. The spectra with 2H or 13C are hardly different and do not substantiate van der Waals contact between the radical and 5′-deoxyadenosine. The absence of van der Waals contact may be the reason that the reaction of (S)-β-lysine does not proceed further.
Discussion
Stereochemistry and mechanism
The central facts in comparing the actions of E. coli LAM with the corresponding enzymes from Cl. subtermiale and B. subtilis are that all three function by similar reaction mechanisms. β-Lysine-related free radicals are intermediates in the steady states of all three reactions (21,26,this work). and the E. coli and clostridial enzymes both produce the substrate-related radical 1 in Scheme 1 with (S)-4-thialysine as the substrate (22,23, this work).
Identical and variant stereospecificities characterize the actions of these enzymes. All three accept only (S)-stereoisomer of lysine, and the clostridial and Bacillus enzymes produce the (S)-stereoisomer of β-lysine; however, the E. coli enzyme produces (R)-β-lysine. The variant conformations of the steady state radicals in the reactions of the E. coli and clostridial LAMs in Scheme 2 are likely determined by the fact that they are enantiomers constrained to fit within similar active sites. In the structure of clostridial LAM, lysine is bound by three main interactions: the α-carboxylate makes an ionic contact with Arg134, the α- or β-amino group is bound as an imine to PLP, and the ε-aminium group makes ionic contacts with Asp293 and Asp330. The contact amino acids are conserved in the E. coli enzyme, except that Asp330 in clostridial LAM is a glutamate residue in E. coli LAM. If the amino acid contacts with the enantiomeric radicals 3 (Scheme 1) in the clostridial enzyme are conserved in the E. coli enzyme, the radicals must have different conformations, as they do. The identical stereochemistry of hydrogen abstraction from (S)-β-lysine by the clostridial and E. coli enzymes also requires an explanation.
The left side of Fig. 7 illustrates the stereochemical course of the action of clostridial LAM to produce (S)-β-lysine. The configuration at C3 of β-lysine is determined by the conformation of the lysyl side chain in the substrate-related radical 1 (Scheme 1). The analogous transformation to produce (R)-β-lysine illustrated in the right side of Fig. 7 shows that a variant conformation of the lysyl-side chain can lead to the (R)-configuration in the azacyclopropylcarbinyl radical 2 and in the product. The variant conformation is more sterically restricted, so that the side chain is less extended. The process naturally leads to different conformations in the product-related radicals, as observed and illustrated in Scheme 2. One can speculate that restricted binding of the lysyl side chain in the E. coli enzyme might be brought about by the steric bulk of a glutamate residue in place of Asp330 in the clostridial active site. The hypothetical conformations in the sequence on the right in Fig. 7 suggest that the action of E. coli LAM leads to abstraction of the 3-pro-S hydrogen from (S)-α-lysine, unlike the abstraction of the 3-pro-R hydrogen in the reaction of the clostridial enzyme (3).
Fig. 7. Stereochemical models for the mechanisms of reactions of LAM from C. subterminale and E. coli.

The sequence on the left depicts the mechanism and stereochemistry for the reaction of C. subterminale LAM, and that on the right depicts the hypothetical mechanism and stereochemistry of the action of E. coli LAM. Available evidence indicates that the basic chemistry is the same for the two enzymes, but the stereochemistry is different for the initial hydrogen abstraction by the 5′-deoxyadenosyl radical and cyclization to the azacyclopropylcarbinyl radical intermediate. Stereochemistry of hydrogen transfer to C2 of β-lysine is postulated, but not proven, to be the same for the two enzymes.
Both reaction sequences in Fig. 7 lead to the same stereochemistry of hydrogen transfer to C2 of (S)-β-lysine by clostridial LAM and C2 of (R)-β-lysine by E. coli LAM. This hypothetical stereochemistry is inspired by the likelihood that the binding interactions of the β-amino group to PLP and α-carboxylate to Arg134 control the stereochemistry. The fact that both E. coli and clostridial LAM catalyze abstraction of the 2-pro-R hydrogen from (S)-β-lysine supports but does not prove this model.
Activity of E. coli LAM
The low activity of E. coli LAM relative to the clostridial enzyme raises the question of whether (S)-α-lysine is the true substrate for LAM in E. coli. Because the functions of variant forms of the same enzyme in different organisms are often different, they frequently display different activities. Classic case is the alcohol dehydrogenases from horse liver and yeast, which differ by 100-fold in catalytic activity, yet they are structurally and apparently mechanistically similar. A similar difference in activity has been noted for LAM from C. subterminale and Bacillus subtilis (27), and LAM from E. coli LAM is even less active than the Bacillus enzyme.
The low activities of the Bacillus and E. coli enzymes may be related to their biological functions and the needs of these organisms. Clostridia use LAM in the metabolism of (S)-α-lysine as a source of carbon and nitrogen and so require a high activity. In other organisms, β-lysine is used in cellular defense mechanisms, such as the production of antibiotics (4–14). In E. coli and Bacilli, high activity may not be beneficial in the production of the few molecules of β-lysine required to produce a specialized molecule. Low activities are not uncommon in enzymes that produce specialized molecules such as antibiotics, examples being the tyrosine 2.3-aminomutase required for the biosynthesis of the antitumor antibiotic C-1027, and the lysine cyclodeaminase required in the biosynthesis of rapamycin (37,38). The latter enzymes display turnover numbers about 1/8th that of LAM from E. coli. It seems likely that if (S)-α-lysine is the unique substrate for E. coli LAM, the function of this enzyme in E. coli should be the production of a specialized molecule.
Footnotes
Supported by Grant DK28607 from the National Institute of Diabetes and Digestive and Kidney Diseases (P.A.F.), GM35752 from the National Institute of General Medical Sciences (G.H.R.), and by N.I.H. Predoctoral Training Grant T32 GM08293 (S.O.M).
Abbreviations: SAM, S-adenosyl-(S)-methionine; [5′-13C]SAM, S-[5′-13C]adenosyl-(S)-methionine; [5′-2H2]SAM; S-[5′-2H2]adenosyl-L-methionine; PLP, pyridoxal-5′-phosphate; DTT, dithiothreitol; PITC, phenylisothiocyanate; LAM, lysine 2,3-aminomutase; IPTG, β-D-isopropylthiogalactoside; EPPS, 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid ; EPR, electron paramagnetic resonance; HPLC, high performance liquid chromatography; EPR, electron paramagnetic resonance; CD, circular dichroism.
This effect also appears in samples prepared with the C. subterminale LAM (data not shown).
References
- 1.Ruzicka FJ, Lieder KW, Frey PA. Lysine 2,3-aminomutase from Clostridium subterminale SB4: mass spectral characterization of cyanogen bromide-treated peptides and cloning, sequencing, and expression of the gene kamA in Escherichia coli. J Bacteriol. 2000;182:469–76. doi: 10.1128/jb.182.2.469-476.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chirpich TP, Zappia V, Costilow RN, Barker HA. Lysine 2,3-aminomutase. Purification and properties of a pyridoxal phosphate and S-adenosylmethionine-activated enzyme. J Biol Chem. 1970;245:1778–1789. [PubMed] [Google Scholar]
- 3.Aberhart DJ, Gould SJ, Lin HJ, Thiruvengadam TK, Weiller BH. Stereochemistry of Lysine 2,3-Aminomutase Isolated from Clostridium subterminale Strain SB4. J Am Chem Soc. 1983;105:5461–5470. [Google Scholar]
- 4.Grammel N, Pankevych K, Demydchuk J, Lambrecht K, Saluz HP, Krugel H. A beta-lysine adenylating enzyme and a beta-lysine binding protein involved in poly beta-lysine chain assembly in nourseothricin synthesis in Streptomyces noursei. Eur J Biochem. 2002;269:347–357. doi: 10.1046/j.0014-2956.2001.02657.x. [DOI] [PubMed] [Google Scholar]
- 5.Singh K, Sehgal SN, Rakhit S, Vezina C. Isolation of a Streptomyces rochei idiotroph requiring beta-lysine for production of streptothricin. J Antibiot (Tokyo) 1983;36:1770–1773. doi: 10.7164/antibiotics.36.1770. [DOI] [PubMed] [Google Scholar]
- 6.Miyaki T, Tenmyo O, Numata K, Matsumoto K, Yamamoto H, Nishiyama Y, Ohbayashi M, Imanishi H, Konishi M, Kawaguchi H. Tallysomycin, a new antitumor antibiotic complex related to bleomycin. IV New biosynthetic derivatives of tallysomycin. J Antibiot (Tokyo) 1981;34:658–664. doi: 10.7164/antibiotics.34.658. [DOI] [PubMed] [Google Scholar]
- 7.Nomoto S, Teshima T, Wakamiya T, Shiba T. The revised structure of capreomycin. J Antibiot (Tokyo) 1977;30:955–959. doi: 10.7164/antibiotics.30.955. [DOI] [PubMed] [Google Scholar]
- 8.Sawada Y, Taniyama H. Studies on the beta-lysine peptide. IV Preparation of semi-synthetic racemomycins and their antimicrobial activities. Chem Pharm Bull (Tokyo) 1977;25:1302–1305. doi: 10.1248/cpb.25.1302. [DOI] [PubMed] [Google Scholar]
- 9.Carter JH, 2nd, Du Bus RH, Dyer JR, Floyd JC, Rice KC, Shaw PD. Biosynthesis of viomycin. II Origin of beta-lysine and viomycidine. Biochemistry. 1974;13:1227–1233. doi: 10.1021/bi00703a027. [DOI] [PubMed] [Google Scholar]
- 10.Seltmann G. Biochemical aspects of the resistance to nourseothricin (streptothricin) of Escherichia coli strains. J Basic Microbiol. 1989;29:547–559. doi: 10.1002/jobm.3620290820. [DOI] [PubMed] [Google Scholar]
- 11.Inamori Y, Amino H, Tsuboi M, Yamaguchi S, Tsujibo H. Biological activities of racemomycin-B, beta-lysine rich streptothricin antibiotic, the main component of Streptomyces lavendulae OP-2. Chem Pharm Bull (Tokyo) 1990;38:2296–2298. doi: 10.1248/cpb.38.2296. [DOI] [PubMed] [Google Scholar]
- 12.French JC, Bartz QR, Dion HW. Myomycin, a new antibiotic. J Antibiot (Tokyo) 1973;26:272–83. doi: 10.7164/antibiotics.26.272. [DOI] [PubMed] [Google Scholar]
- 13.Gould SJ, Martinkus KJ, Tann CH. Biocynthesis of Streptothricin F. 1 Observing the interaction of primary and secondary metabolism with [1,2-13C]acetate. J Am Chem Soc. 1981;103:2871–2872. [Google Scholar]
- 14.Gould SJ, Thiruvengadam TK. Studies of nitrogen metabolism using 13C NMR Spectroscopy. 3 Synthesis of DL-[3-13C,2-15N]lysine and its incorporation into Streptothricin F. J Am Chem Soc. 1981;103:6752–6754. [Google Scholar]
- 15.Frey PA, Magnusson OTh. S-Adenosylmethionine: a wolf in sheep’s clothing, or a rich man’s adenosylcobalamin? Chem Rev. 2003 Jun;103(6):2129–48. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 16.Frey PA, Booker SJ. Radical mechanisms of S-adenosylmethionine- dependent enzymes. Adv Protein Chem. 2001;58:1–45. doi: 10.1016/s0065-3233(01)58001-8. [DOI] [PubMed] [Google Scholar]
- 17.Frey PA. Radical mechanisms of enzymatic catalysis. Annu Rev Biochem. 2001;70:121–48. doi: 10.1146/annurev.biochem.70.1.121. [DOI] [PubMed] [Google Scholar]
- 18.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moss M, Frey PA. The role of S-adenosylmethionine in the lysine 2,3- aminomutase reaction. J Biol Chem. 1987;262:14859–14862. [PubMed] [Google Scholar]
- 20.Baraniak J, Moss ML, Frey PA. Lysine 2,3-aminomutase. Support for a mechanism of hydrogen transfer involving S-adenosylmethionine. J Biol Chem. 1989;264:1357–1360. [PubMed] [Google Scholar]
- 21.Ballinger MD, Frey PA, Reed GH. Structure of a substrate radical intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry. 1992;31:10782–10789. doi: 10.1021/bi00159a020. [DOI] [PubMed] [Google Scholar]
- 22.Wu W, Lieder KW, Reed GH, Frey PA. Observation of a second substrate radical intermediate in the reaction of lysine 2,3-aminomutase: a radical centered on the β-carbon of the alternative substrate, 4-thia-L-lysine. Biochemistry. 1995;34:10532–10537. doi: 10.1021/bi00033a027. [DOI] [PubMed] [Google Scholar]
- 23.Miller J, Bandarian V, Reed GH, Frey PA. Inhibition of lysine 2,3- aminomutase by the alternative substrate 4-thialysine and characterization of the 4-thialysyl radical intermediate. Arch Biochem Biophys. 2001;387:281–8. doi: 10.1006/abbi.2001.2261. [DOI] [PubMed] [Google Scholar]
- 24.Th Magnusson O, Reed GH, Frey PA. Characterization of an allylic analogue of the 5′-deoxyadenosyl radical: an intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry. 2001;40:7773–7782. doi: 10.1021/bi0104569. [DOI] [PubMed] [Google Scholar]
- 25.Lepore BW, Ruzicka FJ, Frey PA, Ringe D. The x-ray crystal structure of lysine-2,3-aminomutase from Clostridium subterminale. Proc Natl Acad Sci U S A. 2005;10(2):13819–24. doi: 10.1073/pnas.0505726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lieder KW, Booker S, Ruzicka FJ, Beinert H, Reed GH, Frey PA. S-Adenosylmethionine-dependent reduction of lysine 2,3-aminomutase and observation of the catalytically functional iron-sulfur centers by electron paramagnetic resonance. Biochemistry. 1998;37:2578–2585. doi: 10.1021/bi972417w. [DOI] [PubMed] [Google Scholar]
- 27.Chen D, Ruzicka FJ, Frey PA. A novel lysine 2,3-aminomutase encoded by the yodO gene of Bacillus subtilis: characterization and the observation of organic radical intermediates. Biochem J. 2000;348:539–49. [PMC free article] [PubMed] [Google Scholar]
- 28.Heinrikson RL, Meredith SC. Amino acid analysis by reverse-phase high-performance liquid chromatography: precolumn derivatization with phenylisothiocyanate. Anal Biochem. 1984;136:65–74. doi: 10.1016/0003-2697(84)90307-5. [DOI] [PubMed] [Google Scholar]
- 29.Chen D, Frey PA, Ruzicka FJ. Biochemistry. 2006 (submitted for publication) [Google Scholar]
- 30.Kennedy MC, Kent TA, Emptage M, Merkle H, Beinert H, Münck E. Evidence for the formation of a linear [3Fe-4S] cluster in partially unfolded aconitase. J Biol Chem. 1984;259:14463–14471. [PubMed] [Google Scholar]
- 31.Beinert H. Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal Biochem. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 32.Wada H, Snell EE. The enzymatic oxidation of pyridoxine and pyridoxamine phosphates. J Biol Chem. 1961;236:2089–2095. [PubMed] [Google Scholar]
- 33.Hewitson KS, Ollagnier-de Choudens S, Sanakis Y, Shaw NM, Baldwin JE, Münck E, Roach PL, Fontecave M. The iron-sulfur center of biotin synthase: site-directed mutants. J Biol Inorg Chem. 2002;7:83–93. doi: 10.1007/s007750100268. [DOI] [PubMed] [Google Scholar]
- 34.Lees NJ, Chen W, Walsby C, Frey PA, Hoffmann BM. J Am Chem Soc. 2006 In press. [Google Scholar]
- 35.Wertz JE, Bolton JR. Electron Spin Resonance. Chapman and Hall; NewYork: 1986. pp. 164–174. [Google Scholar]
- 36.Fischer H. In: Free Radicals. Kochi JK, editor. II. Wiley; NewYork: 1973. pp. 435–491. [Google Scholar]
- 37.Christenson SD, Wu W, Spies MA, Shen B, Toney MD. Kinetic analysis of the 4-methylideneimidazole-5-one-containing tyrosine aminomutase in enediyne antitumor antibiotic C-1027 biosynthesis. Biochemistry. 2003;42:12708–12718. doi: 10.1021/bi035223r. [DOI] [PubMed] [Google Scholar]
- 38.Gatto GJ, Boyne MT, Kelleher NL, Walsh CT. Biosynthesis of pipecolic acid by RapL, a lysine cyclodeaminase encoded in the rapamycin gene cluster. J Am Chem Soc. 2006;128:3838–3847. doi: 10.1021/ja0587603. [DOI] [PubMed] [Google Scholar]